

ABSTRACT
Deficient (d) DNA mismatch repair (MMR) is a biomarker predictive of better response to PD-1 blockade immunotherapy in solid tumors. dMMR can be caused by mutations in MMR genes or by protein inactivation, which can be detected by sequencing and immunohistochemistry, respectively. To investigate the role of dMMR in diffuse large B-cell lymphoma (DLBCL), MMR gene mutations and expression of MSH6, MSH2, MLH1, and PMS2 proteins were evaluated by targeted next-generation sequencing and immunohistochemistry in a large cohort of DLBCL patients treated with standard chemoimmunotherapy, and correlated with the tumor immune microenvironment characteristics quantified by fluorescent multiplex immunohistochemistry and gene-expression profiling. The results showed that genetic dMMR was infrequent in DLBCL and was significantly associated with increased cancer gene mutations and favorable immune microenvironment, but not prognostic impact. Phenotypic dMMR was also infrequent, and MMR proteins were commonly expressed in DLBCL. However, intratumor heterogeneity existed, and increased DLBCL cells with phenotypic dMMR correlated with significantly increased T cells and PD-1+ T cells, higher average nearest neighbor distance between T cells and PAX5+ cells, upregulated immune gene signatures, LE4 and LE7 ecotypes and their underlying Ecotyper-defined cell states, suggesting the possibility that increased T cells targeted only tumor cell subsets with dMMR. Only in patients with MYC¯ DLBCL, high MSH6/PMS2 expression showed significant adverse prognostic effects. This study shows the immunologic and prognostic effects of genetic/phenotypic dMMR in DLBCL, and raises a question on whether DLBCL-infiltrating PD-1+ T cells target only tumor subclones, relevant for the efficacy of PD-1 blockade immunotherapy in DLBCL.
KEYWORDS: DNA mismatch repair, dMMR, PD-1, DLBCL, immune, MSH6, tumor microenvironment, multiplex IHC, mutation, gene expression profiling, MYC, p53, heterogeneity, genomic instability, HDAC
Introduction
DNA mismatch repair (MMR) is one of the five major DNA repair mechanisms and reverses base-base mismatches and insertion/deletion mutations that arise from DNA replication and damage, thereby protecting genomic integrity against cancer development and regulating the apoptotic threshold.1–7 Four proteins, MLH1, MSH2, MSH6, and PMS2, are essential for MMR in eukaryotic cells. MSH2 and MSH6 form the MutSα heterodimer, which recognizes mismatched bases and recruits the MutLα heterodimer comprising MLH1 and PMS2 to mediate subsequent MMR processes. However, MSH2 can also pair with MSH3 to form the MutSβ heterodimer involved in both small and large loop MMR; MLH1 can pair with PMS1 and MLH3 to form the MutLβ and MutLγ heterodimers with less well-understood role. In contrast, MSH6 and PMS2 cannot pair with other MMR proteins.4,8–10
Defective or deficient MMR (dMMR) can be caused by MMR gene mutations and protein inactivation, hypermethylation of the MLH1 gene, or other unclear abnormalities, leading to genomic instability and microsatellite instability (MSI) due to slippage of DNA polymerase, although tumors with MSH6 mutations can be microsatellite stable.4 Lynch syndrome (also known as hereditary non-polyposis colorectal cancer) commonly harbor MLH1 and MSH2 mutations, MSI-high, and less frequently, MSH6 and PMS2 mutations.11–13 dMMR can be detected by immunohistochemistry (IHC) of MMR proteins and/or MSI testing using polymerase chain reaction or next-generation sequencing (NGS).12,14 dMMR has variable prognostic effects for chemotherapeutic agents and is a favorable biomarker for PD-1 immune checkpoint blockade therapies in solid tumors.12,15 Mechanistically, genetic inactivation of MLH1 in mouse models increases the tumor mutation burden with dynamic mutational profiles and host immune surveillance, which can be enhanced by immune checkpoint blockade.16
Although lymphoma is not within the tumor spectrum of Lynch syndrome, homozygous or heterozygous MMR gene mutations in patient families have been associated with early onset of hematological malignancies,17,18 and in mouse models, MSH2 and/or MSH6 deficiency was associated with the development of lymphoid tumors.19–21 In 308 patients with diffuse large B-cell lymphoma (DLBCL), a germline single nucleotide polymorphism in MLH1 was associated with poor clinical outcome.22 A study sequenced 73 DNA repair genes including MSH6 and MSH2 in 22 patients with DLBCL and found that MMR genes were the most frequently mutated compared with the human genome reference (36%, more germline than somatic mutations) and associated with MSI and increased somatic mutations.23 Homozygous deletion of MSH6-MSH2 and heterozygous deletion of PMS2 were found in 2 of 70 DLBCL cases, associated with complete loss of MSH6/MSH2 and decreased PMS2 expression but not MSI.24 Interestingly, Duval et al. found that MSI-high was exclusively observed in immunodeficiency-related lymphomas.25 In two later studies,26,27 MSI-high was infrequent and lacking significant prognostic impact in DLBCL (3.2%, unknown immunodeficiency status) and hematological malignancies (none of 92 patients), respectively. How MMR abnormalities in DLBCL affected host immune responses were not analyzed in these previous studies.
In this study, the prognostic and immunologic effects associated with dMMR and the expression levels of MLH1, MSH2, MSH6, and PMS2 proteins were evaluated in a large cohort of DLBCL patients treated with standard chemoimmunotherapy.
Materials and methods
Mutation and copy number variation (CNV) analysis
Targeted NGS of 275–315 cancer-relevant genes, including MSH6, MSH2, MLH1, and PMS2, was successful in DNA samples of tumor tissues from 424 patients with de novo DLBCL, as part of the DLBCL Consortium Study Program described previously.28 Six patients had high-grade B-cell lymphoma (HGBCL) with MYC/BCL2 double-hit (DH),29,30 and the other cases were designated as DLBCL, not otherwise specified (NOS). Primary central nervous system DLBCL, primary cutaneous DLBCL, primary mediastinal large B-cell lymphoma, and HIV+ DLBCL were excluded. All patients were treated with standard rituximab, cyclophosphamide, doxorubicin, vincristine, prednisone (R-CHOP), or R-CHOP-like immunochemotherapy. This study was conducted in accordance with the principles of the Declaration of Helsinki and approved as minimal to no risk or exempt by the institutional review board of each participating institution.
NGS was performed on an Illumina NextSeq 550 System platform, using previously described methods.31 Mutations and CNVs in tumor samples without matching the normal samples were analyzed against the GRCh37 reference genome using the Illumina DRAGEN Somatic Pipeline tumor-only analysis.
Immunohistochemistry
IHC for MMR proteins was performed on tissue microarrays of 393 DLBCL-NOS cases and 10 HGBCL-MYC/BCL2-DH cases using the methods described in Ref.32 and corresponding antibodies: MSH6 (mouse IgG1 antibody, clone 44, Cell Marque Cat# 287 M–14, RRID:AB_1160603), MSH2 (mouse IgG1 antibody FE11, Millipore Cat# NA27, RRID:AB_2266524), MLH1 (mouse IgG2 antibody, clone G168–728, Cell Marque, Cat# 285 M–16, RRID:AB_1160581), and PMS2 (EPR3947, rabbit IgG antibody, Cell Marque).
Fluorescent multiplex immunohistochemistry (fmIHC)
FmIHC with antibodies against 13 immune markers was performed using the MultiOmyx platform (NeoGenomics Laboratories, Aliso Viejo, CA).33 The details of the fmIHC staining and quantification methods have been described previously.33 In this study, we examined the correlations of MMR gene mutations or protein expression with the abundance of immune cells (T cells, macrophages, and natural killer cells detected by antibodies: CD3, RRID:AB_2631163, CD68, RRID:AB_627158, and CD56, RRID:AB_2864402, respectively), the expression of immune checkpoint molecules in tumor cells and immune cells, and the average spatial distance between T cells and the nearest B (PAX5+) cells.
Statistical analysis
Gene expression profiling (GEP) data deposited in GSE3131234 were analyzed and visualized using previously described methods.28 Fisher’s exact test or Chi-square test was used to compare clinical and molecular features between the two groups. Unpaired (2-tailed) t-test or Mann-Whitney test was used to compare continuous variables between the two groups. Overall survival (OS) and progression-free survival (PFS) were compared using the Kaplan-Meier method using GraphPad Prism (RRID:SCR_000306) 8 software. Multivariate analysis was performed with a Cox proportional hazards regression model using the SPSS software (RRID:SCR_016479). p-values ≤0.05 were considered to be statistically significant.
Cell culture and materials
DLBCL cell lines, culture conditions, venetoclax (ABT-199), and tucidinostat have been described in detail previously.35 After the SUDHL-4 and DB cell lines were treated with drugs for 72 h, total RNAs were extracted from triplicate samples and sequenced using the DNBseq platform. Sequencing reads were filtered and aligned with the reference genome using the Bowtie 2 tool. The mRNA expression was normalized using the FPKM method, and gene expression was compared using the DEGseq2 algorithm.
Whole-cell extracts were subjected to sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) on a 4% to 15% gel (Bio-Rad, Hercules, CA) and then transferred onto PVDF membranes. After blocking nonspecific binding for 1 h using 1× TBS with 1% casein blocker (Bio-Rad, Hercules, CA, USA), the membranes were probed with specific primary antibodies overnight at 4°C and then incubated with a secondary HRP-conjugated monoclonal antibody (1:5000, Cell Signaling Technology, Danvers, MA) for 1 h at room temperature. Proteins were visualized using an ECL system (Amersham, Little Chalfont, UK). Antibodies against MSH6 (mouse, 1:1000), MSH2 (rabbit, 1:1000), MLH1 (mouse, 1:1000), Acetyl-Histone H4 (Lys8) (rabbit, 1:1000), Histone H4 (Lys5) (rabbit, 1:1000), Histone H4 (Lys12) (rabbit, 1:1000), and Histone H4 (rabbit, 1:1000) used for western blotting were purchased from Cell Signaling Technology (Danvers, MA, USA).
Results
In this study, a molecularly well-characterized DLBCL cohort was further evaluated for dMMR status at the DNA and the protein levels, and analyzed for immunologic and prognostic impact. Numbers and overlaps of successfully evaluated DLBCL-NOS cases are illustrated in Figure 1(a).
Figure 1.
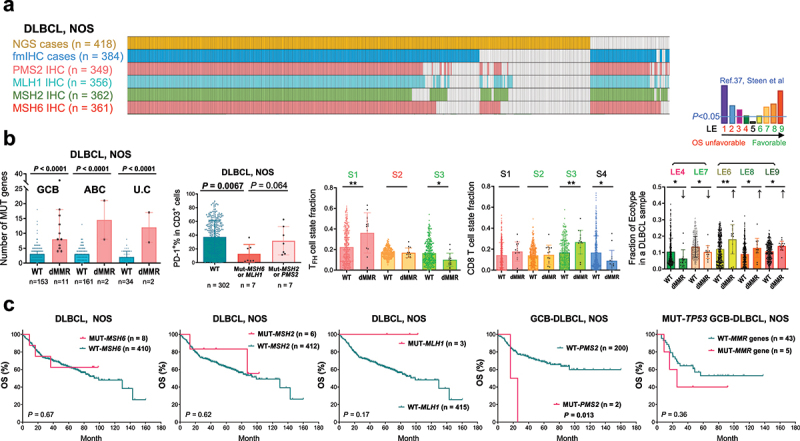
The DLBCL-NOS study cohort and analyses for mutations in four essential DNA mismatch repair (MMR) genes.
(a) Illustration of DLBCL-NOS case numbers successfully analyzed by next-generation sequencing (NGS), immunohistochemistry (IHC), and fluorescent multiplex IHC (fmIHC). Each column represents one patient. (b) Scatter plots showing the value for each patient (each dot) and mean values of patient groups (bars). MMR gene mutation (defective MMR, dMMR) was associated with increased non-silently mutated (MUT) gene numbers in GCB, ABC, and unclassifiable (U.C) subtypes of DLBCL classified by GEP. PD-1+ expression in T cells, which were quantified by fmIHC, was significantly lower in DLBCL cases with MSH6 or MLH1 mutation. Ecotyper analysis results showed that dMMR was associated with higher fractions of prognosis-favorable LE6, LE8, and LE9 ecotypes, TFH cell state S1 and CD8 T cell state S3, and lower fractions of prognosis-unfavorable LE4 ecotype, favorable LE7 ecotype, favorable TFH cell state S3, and CD8 T cell state S4. Greenish LE6-9, S1, S2 and S3 indicate favorable prognostic associations, and reddish LE4 and S2 indicate unfavorable prognostic associations according to the Ecotyper study in DLBCL by Steen et al. P values are by two-tailed unpaired t test. Significance: *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001. (c) MSH6, MSH2, and MLH1 mutations had no significant prognostic impact in patients with DLBCL-NOS. PMS2 mutations were associated with significant poorer OS in the GCB subtype (and the overall DLBCL-NOS cohort; figure not shown). In the subset of patients with GCB-DLBCL and TP53 gene mutation, combined dMMR patients showed a non-significant trend of adverse prognostic effect.
MMR gene mutations are associated with increased driver gene mutations and favorable immune microenvironment
Targeted NGS identified nonsynonymous MSH6, MSH2, MLH1, and PMS2 mutations in only 8, 6, 3, and 2 patients (one patient had concurrent MSH6 and MLH1 mutations), respectively, mostly with the germinal center B-cell-like (GCB) subtype of DLBCL-NOS (only one MSH6 mutated case and one MSH2 mutated case were ABC). Supplemental Figure S1 shows the position of mutations36 and the corresponding IHC results. Copy number analysis found only one GCB case with chromosome 2 MSH2/MSH6 loss and one GCB case with chromosome 2 EPAS1/MSH2/XPO1 gain. None of the six sequenced HGBCL-MYC/BCL2-DH cases had MMR gene mutations or CNVs.
Consistent with the role of MMR in genomic stability, mutations in each MMR gene were associated with significantly increased genomic cancer-relevant mutation numbers assessed by the targeted NGS panel (referred to as cancer driver mutations hereafter; Supplemental Figure S1). Combining cases with MSH6, MSH2, MLH1, or PMS2 mutations, dMMR cases had significantly higher mean and median numbers of cancer driver gene mutations and a higher proportion (two thirds) of cases with ≥ 6 mutated genes (a cutoff used in a previous study)31 than wild-type cases (Supplemental Figure S2A), independent of GCB and activated B-cell-like (ABC) subtypes (Figure 1(b)).
The tumor microenvironment characteristics in dMMR and non-dMMR cases were examined using bulk GEP data of 15 dMMR cases and 348 non-dMMR cases deposited in GSE31312 and EcoTyper software,37 which estimated the fractions of Ecotyper-defined cell states in each cell type and lymphoma ecotype (LE) in each DLBCL sample (LE1 to LE9 are ordered from associated adverse to favorable prognosis in four publicly available cohorts). dMMR cases showed an increase in B cell state S1 (germinal center B cells, in LE8) fractions and a prognosis-favorable immune microenvironment, as suggested by significant increases in LE6, LE8, and LE9 ecotypes and underlying cell states, including plasma cell state S3, CD4 follicular helper T (TFH) cell state S1, regulatory T cell (Treg) state S2, CD8 T cell state S3, mast cell state S2, neutrophil state S2, and monocyte/macrophage state S2, and a decrease in CD8 T cell state S4 (in LE4). On the other hand, the TFH cell state S3 and dendritic cell state S3 (in LE7), which are also prognostically favorable,37 decreased (Figure 1(b), Supplemental Figure S2B).
Immune cell infiltration and PD-1/PD-L1 protein expression in DLBCL tissues, which are relevant for the efficacy of PD-1 blockade immunotherapy, were analyzed by fmIHC and quantified digitally, as previously described.33 Cases with MSH6 or MLH1 mutations showed significant associations with lower PD-1% expression in T cells (Figure 1(b)) and in CD4 T cells and a non-significant trend of higher T cell densities (Supplemental Figure S2A).
Despite these associations, MMR gene mutations did not show a significantly favorable prognostic impact. Only MLH1 mutations showed a non-significant trend toward a favorable prognostic effect in DLBCL-NOS cases, whereas PMS2 mutations were associated with significantly worse OS and PFS in DLBCL, DLBCL-NOS, and the GCB subset (however, the mutation case number was too small). Combined dMMR cases (larger case number) had an OS rate similar to that of DLBCLs with all wild-type MMR genes (p = 0.64). Further subset analysis found only a non-significant trend of poorer survival of five dMMR cases in TP53-mutated GCB-DLBCLs (Figure 1(c)).
MMR proteins exhibit remarkably high percentage expression levels in DLBCL
IHC staining for MMR proteins revealed their high expression in both DLBCL and germinal centers of normal reactive tonsil samples, with higher expression intensities of MMR proteins and a higher median percentage expression level of MSH6 in DLBCL tumors than in normal controls (Figure 2). Moreover, median expression levels of MSH6 and MLH1, evaluated by percentage of positive tumor cells regardless of the staining intensity and nuclear/cytoplasmic expression, were higher in DLBCL samples than in solid tumor samples that were used as controls for staining (Supplemental Table S1). Most DLBCL cases exhibited positive MMR protein expression, and only 49 DLBCL patients had loss of at least one of the four MMR proteins, that is, IHC-defined dMMR. MSH6 had more negative cases than other MMR proteins (negative in 42 patients, including two double-hit cases, compared with 3, 4, and 8 patients without MSH2, MLH1, and PMS2 expression, respectively). There were no significant correlations between MMR gene mutations and the loss of MMR proteins. Nonetheless, MSH2 mutations were associated with a lower mean MSH2 expression level in GCB-DLBCL, NOS (Supplemental Figure S2C); the case with chromosome 2 MSH2/MSH6 loss also lost MSH2/MSH6 protein expression, and the case with chromosome 2 EPAS1/MSH2/XPO1 gain had high expression of four MMR proteins (all 100%). In contrast to the genetic dMMR, IHC-defined dMMR was not associated with the GCB subtype (incidence in ABC vs. GCB: 11.5% vs. 15%, p = 0.49).
Figure 2.
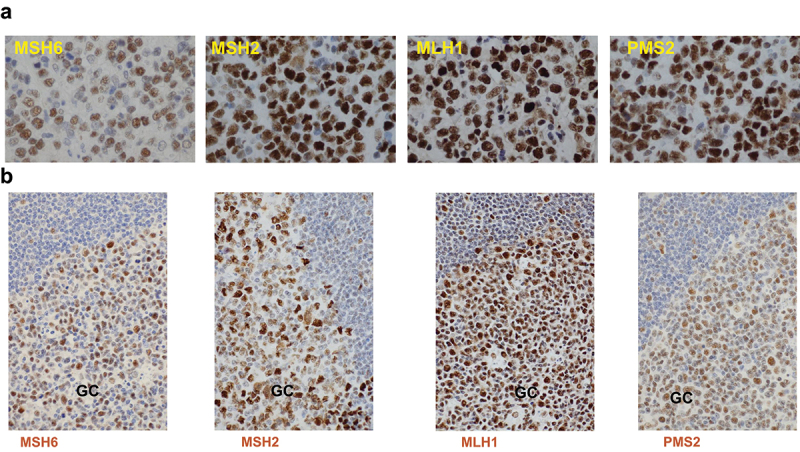
Representative immunohistochemistry staining images.
(a) Expression of the four MMR proteins in DLBCL tissues. (b) Expression of the four MMR proteins in reactive tonsil tissues.
The distribution and median percentage of positive tumor cells in evaluated DLBCL-NOS cases are shown in Figure 3(a) and Supplemental Figure S3A. MSH6 had significantly lower mean and median expression levels (57% and 70% of tumor cells, respectively) than the other three MMR proteins (mean, 84%, 81%, and 77%; and median, 100%, 90%, and 90%, for MSH2, MLH1, and PMS2, respectively). Notably, the mean IHC scores of MSH2, MLH1, and PMS2 proteins in DLBCL-NOS cases were significantly higher than those of all other biomarkers that we previously stained in this cohort. In evaluated HGBCL-MYC/BCL2-DH cases, the median IHC scores were 15% for MSH6, 100% for MSH2, and 90% for MLH1 and PMS2.
Figure 3.
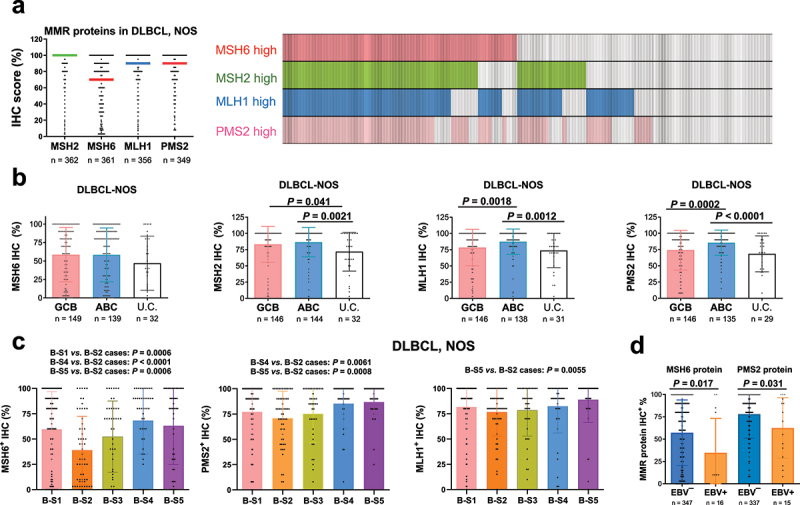
Immunohistochemistry (IHC) analysis for the four MMR proteins in the DLBCL-NOS cohort.
(a) Left: a scatter plot showing the distribution (each dot represents one patient) and median (indicated by horizontal lines) IHC scores (percentage of tumor cells) of the four MMR proteins in DLBCL-NOS cases. MSH6 had a significantly lower median expression score than other three MMR proteins. Right: a case distribution plot (each column represents one patient) showing the overlaps between MSH2high and MSH6high cases and between MLH1high and PMS2high cases in DLBCL-NOS cases scored for all the four MMR proteins. Cutoff for high expression of each MMR protein: the median IHC score. (b) Scatter plots for MMR protein IHC scores in the DLBCL cell-of-origin subtypes (subtyped by GEP). Each dot represents one patient with DLBCL-NOS and bars indicate mean values. The ABC subtype had significantly higher mean levels of MLH1 and PMS2 protein expression than the GCB subtype and unclassifiable (U.C) DLBCL-NOS. Unclassified cases had significantly lower mean MSH2 expression than both ABC and GCB subtypes. P values are by unpaired t test (two tailed). (c) MSH6, PMS2, and MLH1 expression in DLBCL-NOS cases showed differences between ecotyper-assigned subtypes based on the most abundant B cell state in each DLBCL sample. (c) EBV+ DLBCL had lower mean MSH6 and PMS2 IHC scores than EBV-negative DLBCL by unpaired t test.
The percentage expression of MMR proteins was significantly correlated with each other, especially between MSH2 and MSH6 (MutSα components) and between MLH1 and PMS2 (MutLα components) (r = 0.615 and 0.706, respectively; p < 0.0001, Supplemental Figure S3B). Figure 3(a) illustrates the overlaps of cases with high expression of MMR proteins using their median values as the cutoffs. MMR protein expression also showed significant associations with tumor p53 and MYC expression and higher Ki-67 scores evaluated by our previous studies29,38 (Supplemental Figure S4, Supplemental Table S2).
MMR protein expression levels were compared between the DLBCL subtypes. ABC, compared with the GCB subtype of DLBCL-NOS, had significantly higher mean levels of MLH1 and PMS2 expression. The cell-of-origin-unclassifiable cases were associated with significantly lower MSH2, MLH1, and PMS2 expression than ABC cases and lower MSH2 expression than GCB cases (Figure 3(b)). While MSH6 showed similar mean expression levels in the GCB and ABC subtypes, it was the most differentially expressed MMR protein among the EcoTyper subtypes37 assigned for DLBCL cases based on either the estimated most abundant B-cell state or CD4/CD8 T-cell state (Figure 3(c), Supplemental Figure S5A). Across B-cell state subtypes, the S2 (pre-memory) cell state subtype showed the lowest MSH6 expression. Among genetic subtypes assigned by LymphGen software,39 the A53 subtype showed high expression of all four MMR proteins. MSH6 differed from other MMR proteins in its low expression level across genetic subtypes, except for the A53 subtype (Supplemental Figure S5B). In addition, EBV+ DLBCLs were associated with lower MSH6 expression (t, U, and Fisher’s exact test), a lower mean PMS2 expression (t test, Figure 3(d)), a lower frequency of 100% MSH2 expression (3 out of 14; Fisher’s exact test), and a lower frequency of ≥ 90% MLH1 expression in ABC-DLBCL (3 out of 8. Data not shown).
Low MMR protein expression in DLBCL is associated with increased T cells and PD-1 expression
Similar to but less significant than genetically defined dMMR, IHC-defined dMMR was significantly associated with a higher mean number of cancer driver gene mutations in DLBCL (p = 0.019) and in the GCB (but not ABC) subset (p = 0.0027. Figure 4(a)). In ABC-DLBCL, IHC-dMMR showed association with CNVs (present in five of nine NGS cases; p = 0.026). Only in GCB-DLBCL was IHC-defined dMMR associated with increased PD-1+ T cells (higher mean and median; opposite to the correlation with genetic dMMR), a higher median T cell density (Figure 4(a)), and a higher mean LE5 ecotype fraction.
Figure 4.
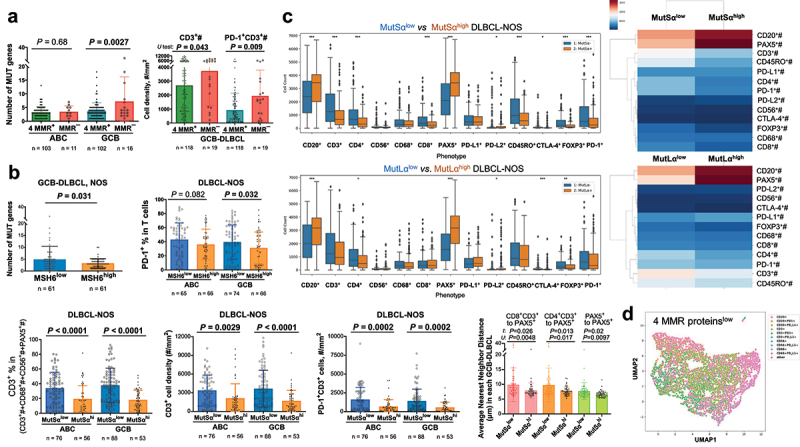
Phenotypic MMR protein deficiency.
(a) Correlative analyses for IHC-defined MMR deficiency (dMMR), 0% positive staining of a MMR protein. Each dot in the scatter plots represents one patient. In the GCB subset of the DLBCL cohort, patients with IHC-defined dMMR compared with patients with positive staining of all four MMR proteins had a higher mean number of mutated driver genes on the targeted NGS panel (bars in the left plot), a higher median T-cell density (bars in the right plot), and a higher median PD-1+ T cell density (also a higher mean, P = 0.002). (b) Top: In patients with GCB-DLBCL, NOS, low MSH6 expression (cutoff: ≤ 70%, the median in the cohort) was significantly associated with higher numbers of mutated driver genes and higher percentage of PD-1+ in T cells. P values in scatter plots are by unpaired t test (two tailed). Bottom: High MutSα (dual MSH6/MSH2) expression was significantly associated with lower T cell densities/proportion and PD-1+ T cell densities in both GCB and ABC DLBCL, as well as higher average nearest neighbor distance of T cells or PAX5+ cells to PAX5+ cells (each dot in the scatter plots represents one patient. Only spatial results in GCB-DLBCL are shown; in ABC-DLBCL, only P values by U test were significant). (c) Left: Scattered boxplots showing the distribution of absolute cell counts of 13 immune markers by fmIHC in patients with or without high expression of MutSα or dual MLH1/PMS2 (MutLα). Each dot represents one patient. Significant differences between two groups are marked by asterisks. *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001 by two-sided Mann-Whitney U test. Right: Z-score cluster maps to visualize the differences in mean cell counts between DLBCL groups with and without high expression of MSH6/MSH2 or MLH1/PMS2. The mean immunophenotypic count was computed within each group, and then the Z-score was computed across all groups. (d) Uniform Manifold Approximation and Projection (UMAP) plot generated from single-cell intensities of CD20, CD3, CD68, CD56, PD-1, and PD-L1 markers in a patient with low expression levels of four MMR proteins. Each data point represents a cell, color labeled according to phenotype.
For cases expressing MMR proteins, although generally high percentages of tumor cells expressed MMR proteins, other tumor cells did not exhibit positive staining. Such intratumoral heterogeneity could result in genomic instability in MMR-negative tumor cell subsets, which subsequently enhances tumor-reactive T-cell infiltration or proliferation in DLBCL. To examine this possibility, we first analyzed the effects of high/low percentage expression of each MMR protein, with their medians as cutoffs (>70% for MSH6high, 100% for MSH2high, ≥90% for MLH1high, and ≥ 90% for PMS2high), on numbers of NGS-detected cancer driver mutations. MSH6low percentages in tumors were found to be associated with a significantly higher mean number of mutated driver genes in the GCB subtype of DLBCL or DLBCL-NOS (Figure 4(b)). After excluding cases with 0% expression, other MSH6low cases continued to show a significant increase in mutated gene numbers in the GCB subtype of DLBCL-NOS (p = 0.043). In contrast, in ABC-DLBCL, cases with low percentage but > 0% expression of MSH6 had a significantly lower mean number of mutated driver genes (Supplemental Figure S2C). MSH2 expression and dual MSH6/MSH2 expression status showed similar results with less significant p values in GCB and more significant p values in ABC than MSH6 expression (Supplemental Figure S2C).
The effects on immune responses were then examined by correlating high/low MMR protein expression to immune biomarkers quantified by fmIHC. The results showed that lower percentage expression of MMR proteins was associated with significantly higher absolute counts of CD3+, CD4+, CD8+, CD45RO+, CTLA-4+, FOXP3+, and PD-1+ cells (Figure 4(c), Supplemental Figure S6), higher cell densities of T cells and PD-1+ T cells (in overall DLBCL and GCB/ABC subtypes; more significantly, associated with higher T cell percentage in the total count of PAX5+, CD3+, CD68+, and CD56+ in each DLBCL), higher PD-1+ percentage in T cells and CD4 T cells only in GCB-DLBCL, higher Treg (FOXP3+) percentage in CD4 T cells only in ABC-DLBCL, and higher average distance of each T cell or PAX5+ cell to its nearest PAX5+ cell in DLBCL tissues (more significant in GCB-DLBCL) (Figure 4(b), Supplemental Figure S7). Figure 4(d) shows a Uniform Manifold Approximation and Projection (UMAP) plot based on marker intensity for a DLBCL-NOS case with low MMR protein expression.
No significant differences were observed between MMR protein expression and the frequencies of normal tumor-infiltrating B-cell clonotypes, as determined in a recent study.40
GEP of DLBCLs with low MMR protein expression features immune gene signatures and higher fractions of cell states constituting LE4 and LE7 ecotypes
Genome-wide expression profiles were compared between patients with DLBCL-NOS with and without high MMR protein expression (data in GSE31312: 317 MSH6-evaluated cases, 320 MSH2-evaluated cases, 313 MLH1-evaluated cases, and 308 PMS2-evaluated cases) to identify significantly differentially expressed genes. Consistent with the negative effects of MMR proteins on T-cell infiltration revealed by immunophenotyping, prominent upregulation of immune-related genes, including those involved in T-cell receptor signaling, was demonstrated by GEP in DLBCLs with low percentage of MMR protein expression. Pathway analysis validated the enrichment of immune response genes and defense response genes upregulated in DLBCLs with low MMR protein expression, as well as the enrichment of mitotic cell cycle genes, DNA replication and chromosome cycle, and DNA metabolism genes upregulated in MMR expression-high cases, consistent with the associations with MYC/p53/Ki-67 high expression (Supplemental Table S3). Figure 5(a) and Table 1 show identified GEP signatures for dual MSH6/MSH2 or dual MLH1/PMS2 high expression status, and Supplemental Table S4 lists the significantly enriched pathways.
Figure 5.
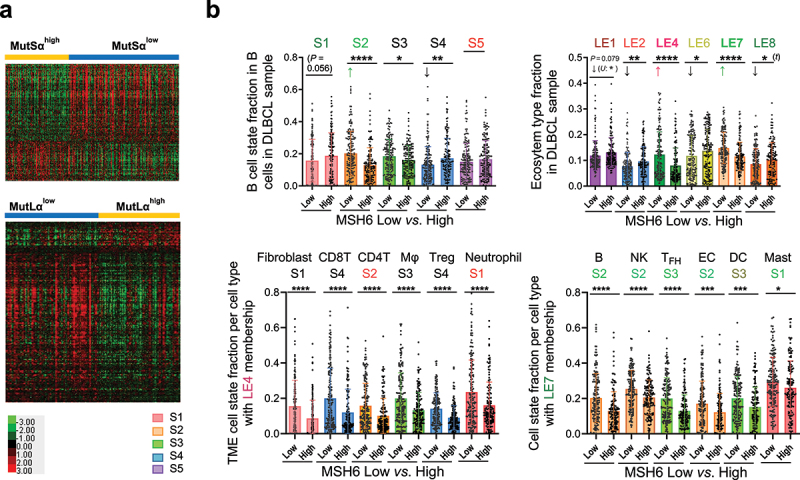
Gene-expression profiling analysis for high vs. low expression of MMR proteins in DLBCL-NOS.
(a) Top: Heatmap visualization for significantly downregulated genes (false discovery rate (FDR) < 0.01 and fold change ≥ 1.5) and upregulated genes in MutSαhigh DLBCLs (FDR <0.05 and fold change ≥ 1.4). Bottom: Heatmap to visualize significantly downregulated genes (FDR <0.01) and upregulated genes (FDR ≤0.02) in MutLαhigh DLBCLs. (b) Cell state and ecosystem subtype analysis by the Ecotyper software and comparisons for DLBCLs with low vs. high expression of MSH6 (cutoff for MSH6high: >70%). Top: Scatter plots for fractions of B-cell states and ecotypes in MSH6high and MSH6low DLBCLs. Bottom: Scatter plots for abundance of cell states comprising the LE4 and LE7 ecotypes. Colors of labels for cell states and ecotypes indicate their associated prognostic effects by Reference 37. Abbreviation: S, state; LE, lymphoma ecosystem type, MΦ, macrophage; Treg, regulatory T cell; NK, natural killer cell; TFH, follicular helper T cell; EC, endothelial cell; DC, dendritic cell. Reddish colors indicate unfavorable prognostic effects, greenish colors indicate favorable prognostic effects, and black color indicates no significant prognostic effect. Significance: *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001.
Table 1.
Significantly differentially expressed genes between DLBCL patient groups.
| MutSαhigh vs. MutSαlow | MutLαhigh vs. MutLαlow | |
|---|---|---|
| Upregulated genes | FDR <0.01, fold change ≥ 1.4: UHRF1, RFC4, KIF20A, ASF1B, PRKDC, SPC25, E2F8, NCAPG2, HELLS, RBBP8, WDR76, BCL11A, DKC1, TOP2A, RAB30, EFCAB3, SNRPD1, BRIP1, AURKA, SLC16A1, TMEM97, RAD51AP1, DANCR, PET117, C9orf40, GMNN, NET1, ANLN, BUB1B | FDR ≤0.02: RALGPS2, E2F5, PRO2964, LINC00665, SLC35B4, PMS2P5, RFC4, IRAK1BP1, RPIA, MTFP1, ACBD6, CD79A, PAX5, BRI3BP, EPM2A, GABRB2, NCAPG2, XKR6, LOC101928000, PPHLN1, RASGRP3, MRPS26, LINC00338, NUP205, OVOS2, CDK5RAP2, BZW2, ORC6, LOC100130458, MRS2, KIF11, PLCG2, C1orf220, ATG10, FCRLA, KIAA0040, FLJ37798, TMEM100 |
| Downregulated genes | FDR <0.01, fold change ≥ 1.5: CD3E, TRBC1, C1QB, GZMK, CD3G, GBP5, GZMB, GIMAP7, ITM2A, GZMA, SELL, CD3D, FYB, GBP2, CD2, UBD, CCR5, C1QC, STAT1, GBP3, GIMAP4, GBP1, FAM26F, GIMAP6, APOL3, FYN, FGL2, CECR1, GIMAP2, C1QA, TYROBP, GPR174, LAT, CD8A, SOD3, SLAMF8, GVINP1, TRAT1, CD96, CCL5, C1orf162, TXNIP, GGTA1P, TNFSF13B, SAMD9L, GBP4, PRF1, FCER1G, IFITM1, KLRK1, TNFSF10, LILRB2, WARS, TBC1D4, HVCN1, CST7, TRAC, IPCEF1, EPSTI1, SLC31A2, EOMES, SECTM1, CLEC2B, IL2RG, KCNJ10, APOL6, IL32, MAFB, LCP2, RAB27A, SIRPG, NLRC5, SLFN5, CSF1R, B2M, HLA-B, STAT4, HLA-A, CELF2, BCL11B, SAMHD1, NPC2, IL6ST, IGSF6, DAPK1, HLA-F, IRF1, AIF1, SLC7A7, FLJ32255, ITGAL, NTNG2, CPVL, RARRES3, GIMAP5, MAF, C5orf56, XAF1, GIMAP8, INPP1, ARL6IP5, TC2N, HLA-C, PATL2, ATHL1, MS4A6A, CTLA4, RNF213, SH2D1A, ARL4C, ITGA4, BTN3A3, SERPINA1, LYST, C19orf60, LEPROTL1, ATP2B4, SERPING1, ASCL2, RASAL3, PRKCH, SPOCK2, SKAP1, GPRIN3, CARD16, HLA-E, UBASH3A, DYSF, ZAP70, CD300LF, BTN3A1, CASP1, GSDMD, STOM, UTRN, CORO1B, DOK2, CARD9, LOC100507419, C5orf20, IL12RB1, IGFLR1 | FDR <0.01: CD3E, ITM2A, TRBC1, GZMK, GBP2, GIMAP7, GBP1, GBP5, CD3G, GBP3, FYB, APOL3, STAT1, CD2, CD3D, IFITM1, CEBPB, FGL2, GIMAP4, CCR5, GIMAP6, TNFSF13B, C1orf162, STAT4, ANKRD22, CCL5, CD8A, EPSTI1, SLAMF8, FAM26F, FCER1G, CST7, LILRB2, PRF1, FYN, HSPA1A, C1QA, GVINP1, TBC1D4, TNFSF10, CD96, TXNIP, IL6ST, EOMES, GBP4, ITGAL, WARS, RAB27A, TNFAIP3, DDX60L, APOL6, IPCEF1, XAF1, TYROBP, SAMSN1, TC2N, PLSCR1, CELF2, ITM2B, BCL11B, CXCL12, CLEC2B, DAPK1, SLFN5, LCP2, MAFB, GGTA1P, TMEM66, THEMIS2, B2M, SAMHD1, GNS, IRF1, LEPROTL1, MEIS3P1, GIMAP8, GPRIN3, SRGN, LAT, SERPING1, MAF, IGF2R, TNFRSF1A, ATHL1, DENND2D, SERPINA1, SIRPG, ARL6IP5, LYST, HLA-F, EPB41L3, RNF213, GIMAP5, PRKCH, SLC2A3, PRKCQ-AS1, NEAT1, STOM, KLF3, HLA-A, THEMIS, SATB1, AIF1, ASCL2, CD300LF, CTSA, CLIP4, MS4A6A, NTNG2, IL6R, ARL4C, ATXN1, TTC39B |
Abbreviation: FDR, false discovery rate.
Furthermore, the fractions of Ecotyper-defined cell states in each cell type and the fractions of LE1–9 ecotypes in each DLBCL sample were correlated with the expression of each MMR protein. MSH6high expression had a significant impact on the distribution of 12 cell types and 5 lymphoma ecotypes (Figure 5(b), Supplemental Figures S8 and S9). CD8 T cell state S4 showed the most substantial difference between the MSH6low and MSH6high groups (a difference of 9.4% between median fractions and a 8% between mean fractions), followed by fibroblast cell state S1, TFH cell state S3 (8.5% between median fractions and 7% between mean fractions), and B-cell state S2 (pre-memory; 6.8% between median fractions and 7.3% between mean fractions). CD8 T cell state S4 is characterized by the expression of both co-inhibitory (e.g., LAG3, TIM3, PD1, TIGIT, CTLA4, CD300A) and co-stimulatory (e.g., BTNL8, CD84, CD2, CD244, SLAMF7, CD70, CD27) molecules; TFH cell state S3 expresses more co-stimulatory molecules (e.g., CD28, ICOS, CD48, OX40, CD70, CD84) than co-inhibitory molecules.37 Other MMR proteins showed similar associations with slight differences (Supplemental Figure S10).
LE4 and LE7 ecotype fractions were significantly increased in MSH6low cases, oppositely to the decrease associated with genetic dMMR. MSH6low DLBCLs had significant increases in all the six cell states constituting the LE4 ecotype (p < 0.0001), including three immunoreactive T cell states (CD8 cell state S4, CD4 T cell state S2, Treg state S4), M2-like macrophages (S3), fibroblast state S1, and neutrophil state S1 (Figure 5(b)), and all the six cell states in LE7 (B cell state S2, NK cell state S2, TFH cell state S3, p < 0.0001, endothelial cell state S2, dendritic cell state S3, p < 0.001, and mast cell state S1, p < 0.05). According to a previous Ecotyper study in publicly available DLBCL cohorts by Steen et al.,37 the LE4 ecotype was characterized by a higher abundance of immune reactive T cells and was associated with the ABC subtype and unfavorable OS with borderline significance; CD4 T-cell state S2 and neutrophil state S1 had significant associations with unfavorable prognosis. In contrast, the LE7 ecotype was elevated in tumors with high stromal content, independent of GCB/ABC, and associated with favorable OS in publicly available cohorts (as well as the six cell states).37
High MSH6 or PMS2 expression is associated with significantly poorer survival of patients with low MYC expression
Finally, MMR protein expression status was correlated with patient survival. Similar to genetic dMMR, IHC-defined dMMR (0% of any MMR protein) did not show a significant prognostic effect in DLBCL. High expression of MMR proteins (cutoffs, medians) also did not show significant effects on OS in the overall cohort (p = 0.069, 0.12, 0.75, and 0.20, for MSH6, MSH2, MLH1, and PMS2, respectively). However, high MSH6 expression showed a significant adverse effect on OS in HGBCL-MYC/BCL2-DH cases (Figure 6(a)) and in patients with MYC¯ DLBCL-NOS (MYC IHC < 40%; 37 patients with MSH6high and 79 with MSH6low; Figure 6(b)). Although MSH6low was associated with increased T-cell densities significantly only in MYC¯ patients (Figure 6(c)) and low T-cell densities were associated with a non-significant trend of poorer OS only in MYC+ patients (Supplemental Figure S11A), the favorable prognostic effects of MSH6low in MYC¯ patients could not be attributable to its associated higher T-cell numbers, since high T-cell densities were actually associated with poorer survival in MSH6high and MSH6low subsets of MYC¯ patients, likely caused by increased PD-1+CD8+ T cells (Figure 6(b), Supplemental Figure S11A).
Figure 6.
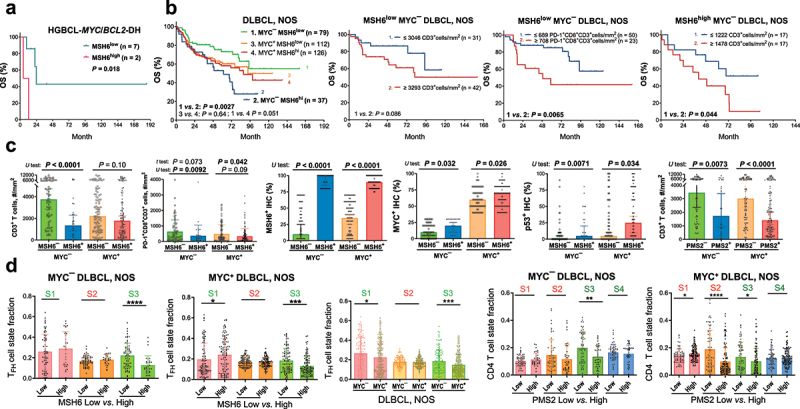
Prognostic effects of high MMR protein expression and correlative analyses.
(a) High MSH6 expression (cutoff, >70%) was associated with significantly poorer overall survival (OS) in evaluated MYC/BCL2 double-hit cases. (b) High MSH6 expression was associated with significantly poorer OS only in patients with MYC¯ (<40%) DLBCL-NOS. In MYC¯ patients with low MSH6 expression, high T cell densities were associated with a non-significant trend of poorer OS, and high cell densities of PD-1+CD8+ T cells were associated with significantly poorer OS. In MYC¯ patients with high MSH6 expression, high T cell densities were associated with significantly poorer OS. (c) Scatter plots for comparisons between MSH6low and MSH6high or between PMS2low and PMS2high patients with MYC¯ or MYC+ DLBCLs. Low and high expression were denoted with negative and positive signs, respectively. U test and median values (bars) were shown because in MYC+ patients, comparing between MSH6low and MSH6high DLBCLs, the mean T-cell densities were not significantly different (P = 0.32 by t test). For comparisons in the third to sixth plots, the differences were significant by both t and U tests. (d) Only in MYC+ patients, MSH6high expression was associated with significantly higher fractions of TFH cell state S1, PMS2high expression (cutoff: ≥90%) was associated with significantly higher fractions of CD4 T cell state S1 and lower fractions of CD4 T-cell state S2 (featuring co-inhibitory and stimulatory molecules). In cell state labeling, red color indicates unfavorable prognostic effect, and green color indicates favorable prognostic effect according to Reference 37. MYC+ compared with MYC¯ patients had significantly lower fractions of TFH cell states S1 and S3.
GEP-based Ecotyper cell states and ecotypes were correlated with MSH6high status separately in patients with MYC¯ or MYC+ DLBCL-NOS (Supplemental Figures S12–13). The associations shown in overall DLBCL were largely similarly seen in both MYC¯ and MYC+ patients, although p values differed, most likely due to the case number difference. Only in MYC¯ patients, MSH6high expression was significantly associated with increased B-cell state S1 (GC-like) fraction. Only in MYC+ patients, MSH6high was significantly associated with increased prognosis-favorable LE9 ecotype, TFH state S1 (in LE637; Figure 6(d)), and prognosis-unfavorable dendritic cell state S4 (in LE1).
Similarly, PMS2high expression showed a significant unfavorable prognostic effect only in MYC¯ patients (Supplemental Figure S11B). In both MYC¯ and MYC+ patients, PMS2high expression was significantly associated with lower T-cell densities and higher MYC expression (Figure 6(c), Supplemental Figure S11B). Only in MYC+ patients, PMS2low expression was significantly associated with increased CD4 T cell state S2 (unfavorable,37 in LE4) (Figure 6(d)).
Combined HDAC/BCL2 inhibitor treatment decreases MSH6 expression in DLBCL cells
Currently, there are no MMR-targeting agents available. Interestingly, the expression of MMR genes (MSH6, MSH2, MLH1, and PMS2) was positively correlated with histone deacetylase (HDAC) HDAC1 and HDAC3 expression and negatively correlated with HDAC10 expression in our cohort (Supplemental Figure S14). Tucidinostat (chidamide) is an HDAC inhibitor that specifically inhibits HDAC1, 2, and 3 (class I HDACs, but also HDAC10, class II HDAC) activity. In literature, HDAC inhibitors synergize with PD-1/L1 blockade by enhancing T-cell infiltration in mouse models of solid cancers.41–44 As lower MMR protein expression correlated with enhanced T-cell infiltration in our study, we tested whether tucidinostat inhibited MMR protein expression in DLBCL cells.
Two DLBCL cell lines, SUDHL-4 and DB, were treated with tucidinostat. RNA-seq analysis revealed that treatment with tucidinostat alone or in combination with venetoclax significantly downregulated MSH6 gene expression in both cell lines, as well as MLH1 gene expression in the SUDHL-4 cell line (Figure 7(a), Supplemental Figure S14). Immunoblotting results showed that tucidinostat treatment decreased MSH6, MSH2, and MLH1 protein levels in SUDHL-4 cells, and the combined tucidinostat/venetoclax treatment significantly decreased MSH6 protein expression in both SUDHL-4 and DB cells (Figure 7(b)).
Figure 7.
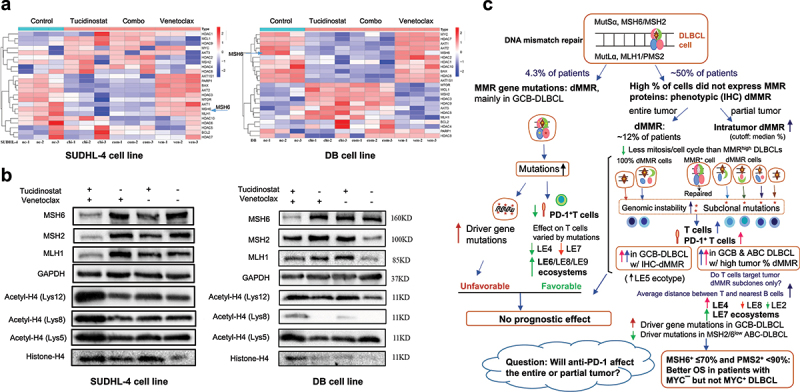
Experiments in two DLBCL cell lines, SUDHL-4 and DB, and graphic summary.
(a) Heatmap for RNA-seq results of genes of interest in triplet DLBCL cell samples treated with a HDAC inhibitor tucidinostat alone, a BCL2 inhibitor venetoclax alone, or in combination. (b) Western blotting results for histone acetylation, MSH6, MSH2, and MLH1 expression before and after treatment with tucidinostat/venetoclax as single agent or in combination. (c) Schematic summary and implications of the findings in DLBCL patients.
Discussion
In this study, we investigated the incidence and clinical significance of genetic dMMR (MSH6, MSH2, MLH1, or PMS2 mutations) and high expression of MMR proteins in a large cohort of DLBCL patients treated with R-CHOP therapy, and correlated them with genomic gene mutation numbers, tumor immune microenvironment characteristics, and gene expression profiles to gain immunological insights. Therapeutic inhibition of MMR expression was also briefly explored in DLBCL cell lines.
Genetic dMMR due to mutations in any MMR gene was infrequent in our DLBCL cohort (4.3% of DLBCL-NOS cases), compared to 9% (2 of 22 DLBCLs) in a previous smaller-scale study.23 Similarly, IHC-defined phenotypic dMMR, identified by IHC staining being 100% negative expression of at least one MMR protein (mainly MSH6¯), was detected in only 12% of DLBCL-NOS cases (not associated with MMR gene mutations).14 High percentage expression of MMR proteins was common in DLBCL, likely due to the fast growth of tumor cells or its germinal center origin, since MMR proteins play an important role in class switch.45 Genetic dMMR significantly correlated with increased mutated gene numbers by targeted NGS in overall DLBCL and GCB/ABC/unclassifiable subsets, whereas IHC-defined dMMR only showed effect on NGS mutations in the GCB subset. Differentially, genetic dMMR was correlated with significantly decreased PD-1+ T cells, whereas in GCB-DLBCLs with IHC-defined dMMR, PD-1+ T cells were significantly increased along with the overall T cells. Neither mutation-defined nor IHC-defined dMMR showed significant prognostic effects in our DLBCL cohort. For a plausible explanation, genomic instability possibly has two sides of effects on prognosis: favorable effect by T cell activation and unfavorable effect due to increased oncogenic driver mutations (which were associated with poorer survival in GCB-DLBCL with wild-type TP53 in our previous study).31
Although dMMR was infrequent, the majority of DLBCL cases had subsets of tumor cells without MMR protein expression. Tumors with high proportions of IHC-negative cells exhibited lower IHC scores of MYC, p53, and Ki-67, as well as downregulation of GEP signatures involved in the cell cycle, mitosis, DNA replication, and proliferation, consistent with the observation of a previous study on MSH2 induction in peripheral blood lymphocytes and thymocytes.46 However, in our in vitro experiments,35 downregulation of MYC/p53 after drug treatment was not consistently accompanied by MMR protein downregulation, which may suggest that p53/MYC are not crucial regulators of MMR protein expression. Potential genomic instability in MSH6low or MSH2low GCB-DLBCL cases is suggested by the increased cancer driver mutations (however, in ABC-DLBCL, MSH6low or MSH2low was associated with lower numbers of mutated NGS genes, unlike genetic dMMR). Intratumor subclonal dMMR, potentially leading to subclonal antigens, significantly correlated with increased T cell counts quantified by fmIHC and upregulation of immune gene signatures by GEP analysis, significantly in both GCB and ABC subtypes and independent of high/low expression of MYC, p53, and Ki-67 (data not shown). These remarkable associations suggest that both clonal and subclonal driver/passenger mutations can contribute to an increase in tumor-infiltrating T cells, as well as the possibility that the mounted T cell responses targeted only subsets of neoantigen-producing tumor cells. Moreover, increased PD-1+ CD8 T cell cells and high PD-1 expression in T cells had adverse prognostic effects in DLBCL,33 and Ecotyper-defined cell states with co-inhibitory molecules were increased in MSH6low cases, suggesting that T cell activation could be suboptimal or checked.47 Likely due to these reasons, intratumoral dMMR also did not show significantly favorable prognostic effects in the overall cohort despite the increased T cell abundance. A following question can be raised: can PD-1 blockade immunotherapy improve the prognosis of patients with DLBCL or MSH6low DLBCL? If DLBCL-infiltrating PD-1+ T cells in MSH6low patients only target a subset of MSH6¯ but not MSH6+ tumor cells, the antitumor efficacy of PD-1 blockade releasing the brake on T cell responses may still be limited. A previous phase II clinical trial of immune checkpoint blockade immunotherapy showed very low efficacy in relapsed/refractory systemic DLBCL.48 Hence, the cause of PD-1 expression in DLBCL and the antigen-specificity of PD-1+ T cells warrant further studies.
Among patients with MYC¯ (<40%) DLBCL, high percentages of MSH6¯ or PMS2¯ cells were associated with significantly better survival, which, however, appeared not to stem from enhanced T-cell infiltration (Figure 6(b)). Our recent study on tumor-infiltrating B cells also showed that T-cell infiltration alone was not sufficient to predict DLBCL prognosis.40 The unique prognostic effects in MYC¯ but not MYC+ DLBCL could be attributable to interactions of multiple factors. For example, the favorable effects of low MMR expression might be weakened or changed in MYC+ DLBCL with higher proliferation and unfavorable tumor microenvironment (in our cohort, MYC+ was associated with higher abundance of prognosis-unfavorable ecotypes and lower abundance of prognosis-favorable ecotypes, but not LE4 and LE7 ecotypes; data not shown). Tumor MYC may crosstalk with the MMR pathway; however, the role of MYC in DNA repair was contradictory in previous studies.49–51 The number of mutated driver genes associated with MSH6high/PMS2high showed opposite trends in MYC¯ and MYC+ patients (Supplemental Figure S2C).
In summary (Figure 7(c)), in a large cohort of DLBCL cases, genetic dMMR is uncommon in DLBCL (predominantly in GCB-DLBCL) and is associated with increased cancer driver mutations and low PD-1 expression in T cells but not prognostic impact, whereas intratumor partial dMMR phenotype is common in DLBCL and is associated with higher T-cell infiltration with increased PD-1 expression (significant in both GCB and ABC subtypes). Only in DLBCL with low MYC expression, higher tumor proportion of MSH6 or PMS2 protein deficiency was associated with significantly better survival of DLBCL patients. To our knowledge, this is the first study that systemically analyzed genetic and IHC-defined clonal and subclonal dMMR and their differential effects on clinical outcome, fmIHC-quantified T cell infiltration, PD-1 expression, and the tumor microenvironment characteristics estimated by bulk GEP. The comprehensive analysis showed the different effects of genetic and phenotypic MMR deficiency on genomic instability and immune responses in DLBCL, exposed the intratumoral heterogeneity, and suggested the subclonal specificity of PD-1 expressing T cells in DLBCL, which have implications for understanding the antigen-specificity of T-cell responses and immunotherapy efficacy in DLBCL.
Supplementary Material
Acknowledgments
We thank the patients, their family members, caregivers, technicians and the volunteer student Kelly Au who contributed to this study.
Funding Statement
This study was funded by grants R01CA233490, R01CA187415, and R01CA138688 from the National Cancer Institute/National Institutes of Health. KHY was also supported by The Duke University Institutional Research Grant Award and the Hagemeister Lymphoma Foundation.
Disclosure statement
D.O., Q.A., and H.N. are employees of NeoGenomics Laboratories, Inc. The NeoGenomics Laboratories, Inc. had no role in the design of the study, in the writing of the manuscript, or in the decision to publish the results.
Data availability statement
Microarray data are available at GEO under the accession number GSE31312. For other original data, please refer to ken.young@duke.edu.
Ethics approval statement
The study was conducted in accordance with the Declaration of Helsinki, and data collection protocols were approved as being of minimal to no risk or exempt by the Institutional Review Board of Duke University (protocol ID Pro00104078, date of approval Oct 24, 2019) and each participating institution.
N otes on contributors
Conceptualization, Z.Y.X-M., L.Y., and K.H.Y.; methodology, Z.Y.X-M., C.L., Y.L., A.S., D.O., Q.A., H.N., K.H.Y.; investigation, Z.Y.X-M, C.L., L.Y., Y.L., X.F., A.S., N.T.W., A.F.Y., W.S., D.O., Q.A., H.N., K.H.Y.; resources, G.B., A.T., C.V., K.D., A.C., W.T., Y.Z., E.D.H., A.M.P., H.G., M.B.M., B.M.P, S.M-M., M.P., A.J.M.F., A.R.S., J.S.A., B.X., K.H.Y.; writing – original draft preparation, Z.Y.X-M.; writing – review and editing, C.L., L.Y., Y.L., G.B., A.T., C.V., X.F., K.D., A.S., A.F.Y., N.T.W., A.C., W.T., Y.Z., E.D.H., A.M.P., W.S., D.O., Q.A., H.N., H.G., M.B.M., B.M.P, S.M-M., M.P., A.J.M.F., A.R.S., J.S.A., B.X., K.H.Y.; visualization, Z.Y.X-M., C.L., H.N.; supervision, L.Y., K.H.Y.; project administration, K.H.Y.; funding acquisition, Y.L., K.H.Y. All authors have read and agreed to the published version of the manuscript.
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/2162402X.2024.2384667
References
- 1.Jiricny J. The multifaceted mismatch-repair system. Nat Rev Mol Cell Biol. 2006;7(5):335–13. doi: 10.1038/nrm1907. [DOI] [PubMed] [Google Scholar]
- 2.Bellacosa A. Functional interactions and signaling properties of mammalian DNA mismatch repair proteins. Cell Death Differ. 2001;8(11):1076–1092. doi: 10.1038/sj.cdd.4400948. [DOI] [PubMed] [Google Scholar]
- 3.Li GM. Mechanisms and functions of DNA mismatch repair. Cell Res. 2008;18(1):85–98. doi: 10.1038/cr.2007.115. [DOI] [PubMed] [Google Scholar]
- 4.Martin SA, Lord CJ, Ashworth A. Therapeutic targeting of the DNA mismatch repair pathway. Clin Cancer Res. 2010;16(21):5107–5113. doi: 10.1158/1078-0432.CCR-10-0821. [DOI] [PubMed] [Google Scholar]
- 5.Bernstein C. DNA repair/pro-apoptotic dual-role proteins in five major DNA repair pathways: fail-safe protection against carcinogenesis. Mutat Res/Rev In Mutat Res. 2002;511(2):145–178. doi: 10.1016/S1383-5742(02)00009-1. [DOI] [PubMed] [Google Scholar]
- 6.Mac Partlin M, Homer E, Robinson H, McCormick CJ, Crouch DH, Durant ST, Matheson EC, Hall AG, Gillespie DA, Brown R. Interactions of the DNA mismatch repair proteins MLH1 and MSH2 with c-MYC and MAX. Oncogene. 2003;22(6):819–825. doi: 10.1038/sj.onc.1206252. [DOI] [PubMed] [Google Scholar]
- 7.Hickman MJ, Samson LD. Role of DNA mismatch repair and p53 in signaling induction of apoptosis by alkylating agents. Proc Natl Acad Sci USA. 1999;96(19):10764–10769. doi: 10.1073/pnas.96.19.10764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wu X, Platt JL, Cascalho M. Dimerization of MLH1 and PMS2 limits nuclear localization of MutLα. Molec Cell Biol. 2003;23(9):3320–3328. doi: 10.1128/MCB.23.9.3320-3328.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen PC, Dudley S, Hagen W, Dizon D, Paxton L, Reichow D, Yoon SR, Yang K, Arnheim N, Liskay RM, et al. Contributions by MutL homologues Mlh3 and Pms2 to DNA mismatch repair and tumor suppression in the mouse. Cancer Res. 2005;65(19):8662–8670. doi: 10.1158/0008-5472.CAN-05-0742. [DOI] [PubMed] [Google Scholar]
- 10.Kadyrova LY, Kadyrov FA. Endonuclease activities of MutLα and its homologs in DNA mismatch repair. DNA Repair (Amst). 2016;38:42–49. doi: 10.1016/j.dnarep.2015.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lynch HT, Snyder CL, Shaw TG, Heinen CD, Hitchins MP. Milestones of lynch syndrome: 1895-2015. Nat Rev Cancer. 2015;15(3):181–194. doi: 10.1038/nrc3878. [DOI] [PubMed] [Google Scholar]
- 12.Luchini C, Bibeau F, Ligtenberg MJL, Singh N, Nottegar A, Bosse T, Miller R, Riaz N, Douillard JY, Andre F, et al. ESMO recommendations on microsatellite instability testing for immunotherapy in cancer, and its relationship with PD-1/PD-L1 expression and tumour mutational burden: a systematic review-based approach. Ann Oncol. 2019;30(8):1232–1243. doi: 10.1093/annonc/mdz116. [DOI] [PubMed] [Google Scholar]
- 13.Latham A, Srinivasan P, Kemel Y, Shia J, Bandlamudi C, Mandelker D, Middha S, Hechtman J, Zehir A, Dubard-Gault M, et al. Microsatellite instability is associated with the presence of lynch syndrome pan-cancer. J Clin Oncol. 2019;37(4):286–295. doi: 10.1200/JCO.18.00283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sarode VR, Robinson L. Screening for lynch syndrome by immunohistochemistry of mismatch repair proteins: significance of indeterminate result and correlation with mutational studies. Arch Pathol Lab Med. 2019;143(10):1225–1233. doi: 10.5858/arpa.2018-0201-OA. [DOI] [PubMed] [Google Scholar]
- 15.Wensink E, Bond M, Kucukkose E, May A, Vink G, Koopman M, Kranenburg O, Roodhart J. A review of the sensitivity of metastatic colorectal cancer patients with deficient mismatch repair to standard-of-care chemotherapy and monoclonal antibodies, with recommendations for future research. Cancer Treat Rev. 2021;95:102174. doi: 10.1016/j.ctrv.2021.102174. [DOI] [PubMed] [Google Scholar]
- 16.Germano G, Lamba S, Rospo G, Barault L, Magrì A, Maione F, Russo M, Crisafulli G, Bartolini A, Lerda G, et al. Inactivation of DNA repair triggers neoantigen generation and impairs tumour growth. Nature. 2017;552(7683):116–120. doi: 10.1038/nature24673. [DOI] [PubMed] [Google Scholar]
- 17.Krüger S, Kinzel M, Walldorf C, Gottschling S, Bier A, Tinschert S, von Stackelberg A, Henn W, Görgens H, Boue S, et al. Homozygous PMS2 germline mutations in two families with early-onset haematological malignancy, brain tumours, HNPCC-associated tumours, and signs of neurofibromatosis type 1. Eur J Hum Genet. 2008;16(1):62–72. doi: 10.1038/sj.ejhg.5201923. [DOI] [PubMed] [Google Scholar]
- 18.Wang X, Song Y, Chen W, Ding N, Liu W, Xie Y, Wang Y, Zhu J, Zeng C. Germline variants of DNA repair genes in early onset mantle cell lymphoma. Oncogene. 2021;40(3):551–563. doi: 10.1038/s41388-020-01542-2. [DOI] [PubMed] [Google Scholar]
- 19.Reitmair AH, Schmits R, Ewel A, Bapat B, Redston M, Mitri A, Waterhouse P, Mittrücker HW, Wakeham A, Liu B, et al. MSH2 deficient mice are viable and susceptible to lymphoid tumours. Nat Genet. 1995;11(1):64–70. doi: 10.1038/ng0995-64. [DOI] [PubMed] [Google Scholar]
- 20.Gu X, Booth CJ, Liu Z, Strout MP. AID-associated DNA repair pathways regulate malignant transformation in a murine model of BCL6-driven diffuse large B-cell lymphoma. Blood. 2016;127(1):102–112. doi: 10.1182/blood-2015-02-628164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Peled JU, Sellers RS, Iglesias-Ussel MD, Shin DM, Montagna C, Zhao C, Li Z, Edelmann W, Morse HC, Scharff MD. Msh6 protects mature B cells from lymphoma by preserving genomic stability. Am J Pathol. 2010;177(5):2597–2608. doi: 10.2353/ajpath.2010.100234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rossi D, Rasi S, Di Rocco A, Fabbri A, Forconi F, Gloghini A, Bruscaggin A, Franceschetti S, Fangazio M, De Paoli L, et al. The host genetic background of DNA repair mechanisms is an independent predictor of survival in diffuse large B-cell lymphoma. Blood. 2011;117(8):2405–2413. doi: 10.1182/blood-2010-07-296244. [DOI] [PubMed] [Google Scholar]
- 23.de Miranda NF, Peng R, Georgiou K, Wu C, Falk Sörqvist E, Berglund M, Chen L, Gao Z, Lagerstedt K, Lisboa S, et al. DNA repair genes are selectively mutated in diffuse large B cell lymphomas. J Exp Med. 2013;210(9):1729–1742. doi: 10.1084/jem.20122842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Couronné L, Ruminy P, Waultier-Rascalou A, Rainville V, Cornic M, Picquenot JM, Figeac M, Bastard C, Tilly H, Jardin F. Mutation mismatch repair gene deletions in diffuse large B-cell lymphoma. Leuk Lymphoma. 2013;54(5):1079–1086. doi: 10.3109/10428194.2012.739687. [DOI] [PubMed] [Google Scholar]
- 25.Duval A, Raphael M, Brennetot C, Poirel H, Buhard O, Aubry A, Martin A, Krimi A, Leblond V, Gabarre J, et al. The mutator pathway is a feature of immunodeficiency-related lymphomas. Proc Natl Acad Sci USA. 2004;101(14):5002–5007. doi: 10.1073/pnas.0400945101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tian T, Li J, Xue T, Yu B, Li X, Zhou X. Microsatellite instability and its associations with the clinicopathologic characteristics of diffuse large B-cell lymphoma. Cancer Med. 2020;9(7):2330–2342. doi: 10.1002/cam4.2870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bødker JS, Sønderkær M, Vesteghem C, Schmitz A, Brøndum RF, Sommer M, Rytter AS, Nielsen MM, Madsen J, Jensen P, et al. Development of a precision medicine workflow in hematological cancers, aalborg university hospital, Denmark. Cancers (Basel). 2020;12(2). doi: 10.3390/cancers12020312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xu-Monette ZY, Wei L, Fang X, Au Q, Nunns H, Nagy M, Tzankov A, Zhu F, Visco C, Bhagat G, et al. Genetic subtyping and phenotypic characterization of the immune microenvironment and MYC/BCL2 double expression reveal heterogeneity in diffuse large B-cell lymphoma. Clin Cancer Res. 2022;28(5):972–983. doi: 10.1158/1078-0432.CCR-21-2949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hu S, Xu-Monette ZY, Tzankov A, Green T, Wu L, Balasubramanyam A, Liu WM, Visco C, Li Y, Miranda RN, et al. MYC/BCL2 protein coexpression contributes to the inferior survival of activated B-cell subtype of diffuse large B-cell lymphoma and demonstrates high-risk gene expression signatures: a report from the international DLBCL rituximab-CHOP consortium program. Blood. 2013;121(20):4021–4031; quiz 4250. doi: 10.1182/blood-2012-10-460063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tzankov A, Xu-Monette ZY, Gerhard M, Visco C, Dirnhofer S, Gisin N, Dybkaer K, Orazi A, Bhagat G, Richards KL, et al. Rearrangements of MYC gene facilitate risk stratification in diffuse large B-cell lymphoma patients treated with rituximab-CHOP. Mod Pathol. 2014;27(7):958–971. doi: 10.1038/modpathol.2013.214. [DOI] [PubMed] [Google Scholar]
- 31.You H, Xu-Monette ZY, Wei L, Nunns H, Nagy ML, Bhagat G, Fang X, Zhu F, Visco C, Tzankov A, et al. Genomic complexity is associated with epigenetic regulator mutations and poor prognosis in diffuse large B-cell lymphoma. Oncoimmunol. 2021;10(1):1928365. doi: 10.1080/2162402X.2021.1928365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Khoury JD, Wang WL, Prieto VG, Medeiros LJ, Kalhor N, Hameed M, Broaddus R, Hamilton SR. Validation of immunohistochemical assays for integral biomarkers in the NCI-MATCH EAY131 clinical trial. Clin Cancer Res. 2018;24(3):521–531. doi: 10.1158/1078-0432.CCR-17-1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xu-Monette ZY, Xiao M, Au Q, Padmanabhan R, Xu B, Hoe N, Rodriguez-Perales S, Torres-Ruiz R, Manyam GC, Visco C, et al. Immune profiling and quantitative analysis decipher the clinical role of immune-checkpoint expression in the tumor immune microenvironment of DLBCL. Cancer Immunol Res. 2019;7(4):644–657. doi: 10.1158/2326-6066.CIR-18-0439. [DOI] [PubMed] [Google Scholar]
- 34.Visco C, Li Y, Xu-Monette ZY, Miranda RN, Green TM, Li Y, Tzankov A, Wen W, Liu WM, Kahl BS, et al. Comprehensive gene expression profiling and immunohistochemical studies support application of immunophenotypic algorithm for molecular subtype classification in diffuse large B-cell lymphoma: a report from the International DLBCL Rituximab-CHOP consortium program study. Leukemia. 2012;26(9):2103–2113. doi: 10.1038/leu.2012.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Luo C, Yu T, Young KH, Yu L. HDAC inhibitor chidamide synergizes with venetoclax to inhibit the growth of diffuse large B-cell lymphoma via down-regulation of MYC, BCL2, and TP53 expression. J Zhejiang Univ Sci B. 2022;23(8):666–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tang D, Chen M, Huang X, Zhang G, Zeng L, Zhang G, Wu S, Wang Y, Yin Y. SRplot: a free online platform for data visualization and graphing. PLoS One. 2023;18(11):e0294236. doi: 10.1371/journal.pone.0294236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Steen CB, Luca BA, Esfahani MS, Azizi A, Sworder BJ, Nabet BY, Kurtz DM, Liu CL, Khameneh F, Advani RH, et al. The landscape of tumor cell states and ecosystems in diffuse large B cell lymphoma. Cancer Cell. 2021;39(10):1422–1437. e1410. doi: 10.1016/j.ccell.2021.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xu-Monette ZY, Moller MB, Tzankov A, Montes-Moreno S, Hu W, Manyam GC, Kristensen L, Fan L, Visco C, Dybkaer K, et al. MDM2 phenotypic and genotypic profiling, respective to TP53 genetic status, in diffuse large B-cell lymphoma patients treated with rituximab-CHOP immunochemotherapy: a report from the international DLBCL rituximab-CHOP consortium program. Blood. 2013;122(15):2630–2640. doi: 10.1182/blood-2012-12-473702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wright GW, Huang DW, Phelan JD, Coulibaly ZA, Roulland S, Young RM, Wang JQ, Schmitz R, Morin RD, Tang J, et al. A probabilistic classification tool for genetic subtypes of diffuse large B cell lymphoma with therapeutic implications. Cancer Cell. 2020;37(4):551–568 e514. doi: 10.1016/j.ccell.2020.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Xu-Monette ZY, Li Y, Snyder T, Yu T, Lu T, Tzankov A, Visco C, Bhagat G, Qian W, Dybkaer K, et al. Tumor-infiltrating normal B cells revealed by immunoglobulin repertoire clonotype analysis are highly prognostic and crucial for antitumor immune responses in DLBCL. Clin Cancer Res. 2023;29(23):4808–4821. doi: 10.1158/1078-0432.CCR-23-1554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zheng H, Zhao W, Yan C, Watson CC, Massengill M, Xie M, Massengill C, Noyes DR, Martinez GV, Afzal R, et al. HDAC inhibitors enhance T-Cell chemokine expression and augment response to PD-1 immunotherapy in lung adenocarcinoma. Clin Cancer Res. 2016;22(16):4119–4132. doi: 10.1158/1078-0432.CCR-15-2584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Que Y, Zhang XL, Liu ZX, Zhao JJ, Pan QZ, Wen XZ, Xiao W, Xu BS, Hong DC, Guo TH, et al. Frequent amplification of HDAC genes and efficacy of HDAC inhibitor chidamide and PD-1 blockade combination in soft tissue sarcoma. J Immunother Cancer. 2021;9(2):e001696. doi: 10.1136/jitc-2020-001696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Woods DM, Sodre AL, Villagra A, Sarnaik A, Sotomayor EM, Weber J. HDAC inhibition upregulates PD-1 ligands in melanoma and augments immunotherapy with PD-1 blockade. Cancer Immunol Res. 2015;3(12):1375–1385. doi: 10.1158/2326-6066.CIR-15-0077-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bretz AC, Parnitzke U, Kronthaler K, Dreker T, Bartz R, Hermann F, Ammendola A, Wulff T, Hamm S. Domatinostat favors the immunotherapy response by modulating the tumor immune microenvironment (TIME). J Immunother Cancer. 2019;7(1):294. doi: 10.1186/s40425-019-0745-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schrader CE, Vardo J, Stavnezer J. Role for mismatch repair proteins Msh2, Mlh1, and Pms2 in immunoglobulin class switching shown by sequence analysis of recombination junctions. J Exp Med. 2002;195(3):367–373. doi: 10.1084/jem.20011877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Marra G, Chang CL, Laghi LA, Chauhan DP, Young D, Boland CR. Expression of human MutS homolog 2 (hMSH2) protein in resting and proliferating cells. Oncogene. 1996;13(10):2189–2196. [PubMed] [Google Scholar]
- 47.Xu-Monette ZY, Zhang M, Li J, Young KH. PD-1/PD-L1 blockade: have we found the key to unleash the antitumor immune response? Front Immunol. 2017;8:1597. doi: 10.3389/fimmu.2017.01597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ansell SM, Minnema MC, Johnson P, Timmerman JM, Armand P, Shipp MA, Rodig SJ, Ligon AH, Roemer MGM, Reddy N, et al. Nivolumab for relapsed/refractory diffuse large B-Cell lymphoma in patients ineligible for or having failed autologous transplantation: a single-arm, phase II study. J Clin Oncol. 2019;37(6):481–489. doi: 10.1200/JCO.18.00766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ning JF, Stanciu M, Humphrey MR, Gorham J, Wakimoto H, Nishihara R, Lees J, Zou L, Martuza RL, Wakimoto H, et al. Myc targeted CDK18 promotes ATR and homologous recombination to mediate PARP inhibitor resistance in glioblastoma. Nat Commun. 2019;10(1):2910. doi: 10.1038/s41467-019-10993-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li Z, Owonikoko TK, Sun SY, Ramalingam SS, Doetsch PW, Xiao ZQ, Khuri FR, Curran WJ, Deng X. c-myc suppression of DNA double-strand break repair. Neoplasia. 2012;14(12):1190–IN35. doi: 10.1593/neo.121258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bucci B, D’Agnano I, Amendola D, Citti A, Raza GH, Miceli R, De Paula U, Marchese R, Albini S, Felsani A, et al. Myc down-regulation sensitizes melanoma cells to radiotherapy by inhibiting MLH1 and MSH2 mismatch repair proteins. Clin Cancer Res. 2005;11(7):2756–2767. doi: 10.1158/1078-0432.CCR-04-1582. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Microarray data are available at GEO under the accession number GSE31312. For other original data, please refer to ken.young@duke.edu.