

Abstract
Protein synthesis is crucial for regulating cell homeostasis and, when unrestricted, it can lead to tumorigenesis. Immunotoxins derived from Pseudomonas exotoxin are antibody–toxin fusion proteins that inhibit protein synthesis of mammalian cells via ADP-ribosylation of the eukaryotic elongation factor-2. Here we investigate the role of the Bcl-2 family proteins in the response of cancer cells to immunotoxin challenge. Besides the well-known reduction of the prosurvival Bcl-2 family member, Mcl-1, following inhibition of protein synthesis, we show for the first time that immunotoxins also reduce the levels of selected proapoptotic BH-3-only proteins. Among these, only Bim protein levels correlated with the ability of immunotoxins to induce an apoptotic response. To support our findings, we verified that a Bim knockout completely abolished immunotoxin-mediated apoptosis. Further, mice bearing either wild-type or Bid knockout tumors responded to immunotoxin treatment with a decrease in growth kinetics, whereas mice engrafted with Bim knockout tumors showed no reduction in tumor size or prolongation of survival following immunotoxin treatment. From these results, we conclude that Bim expression is a major susceptibility factor for tumor cell death and, as such, constitutes a potential biomarker that could be evaluated before immunotoxin treatment. In support of this hypothesis, clinically, we analyzed patient cells before immunotoxin treatment and report that samples of hairy cell leukemia with high levels of Bim protein responded with a greater decrease in leukemic cell count compared with those samples expressing a low level of Bim.
INTRODUCTION
The intrinsic pathway of apoptosis is a structured cascade of events, executed in the mitochondria, which eliminates damaged or unnecessary cells and is now appreciated as the major mechanism of cancer therapy-induced cell death.1 The Bcl-2 family proteins that regulate apoptosis include the anti-apoptotic members (Bcl-2, Bcl-xL, Bcl-W, Mcl-1 and Bfl1/A1), responsible for preserving mitochondrial integrity and the proapoptotic BH-3-only proteins (Bim, Puma, Bad, Bid, Bik, Bmf, Hrk and Noxa). BH-3-only proteins sense apoptotic stimuli and either neutralize Bcl-2, Bcl-xl or Mcl-1, or activate an additional group of proapoptotic proteins, Bax or Bak. As the effectors of the apoptosis, Bax and Bak, when activated, disrupt mitochondrial integrity, resulting in cytochrome c release into the cytosol leading to caspase activation.2 Tumor cells develop different strategies to reduce or suppress apoptosis such as overexpression of the anti-apoptotic regulators (Bcl-2, Bcl-xL and Mcl-1) or downregulation of proapoptotic proteins (Bax, Bim, Noxa, Bid or Puma) often related to the loss of the tumor suppressor, p53.3 Given the central role of the Bcl-2 proteins in determining the commitment to cell death, knowing their expression levels could be essential for designing successful anticancer therapies.
Immunotoxins are antibody–toxin chimeric proteins generated as therapeutics for targeting cancer-associated surface antigens and receptors. For immunotoxins derived from Pseudomonas exotoxin (PE) domains II and III of the toxin are joined to antibody fragments specific for antigens expressed on cancer cells. In susceptible cells, PE kills via the ADP-ribosylation of elongation factor 2, shutting down protein synthesis and leading to cell death.4 Immunotoxins are attractive as cancer treatment agents because of their potency, lack of natural inhibitors, their few debilitating side effects and the fact that they are not mutagenic.5 Clear clinical benefit has been achieved in patients when targeting CD22 expressed on two specific B-cell malignancies, hairy cell leukemia (HCL) and, to a lesser extent, B-cell acute lymphoblastic leukemia.6 However, success has not been universal7 and, when treating solid tumors, immunotoxins have fared less well in producing complete responses.8 Given the variety of responses, it would be of great value to identify biomarkers that predict either success or failure of immunotoxin treatment. Similarly, in designing combination treatments that overcome resistance barriers it would be important to choose compounds that addressed specific resistance phenotypes. We know from earlier work that some cancer cells are resistant to toxin-mediated apoptotic death despite protein synthesis inhibition.9 Immunotoxin treatment reduces the level of the anti-apoptotic Bcl-2 family member Mcl-1 (ref.10) but in resistant cells this was not enough to induce caspase activation and apoptosis.11,12
Here we undertook a survey of both anti- and proapoptotic Bcl-2 family proteins to investigate if the immunotoxin treatment had an effect on their cellular levels. From our analysis, we identified two BH-3-only proteins, Bim and Noxa, whose levels were reduced by immunotoxin treatment. In addition, we showed that Bim is crucial for the apoptotic response to immunotoxins. Based on gene knockout studies and responses of patients’ leukemia cells, we conclude that high Bim levels can serve as a biomarker for susceptibility to immunotoxin-mediated killing.
RESULTS
Protein synthesis inhibition is not sufficient to induce apoptosis in cells treated with PE-based immunotoxins
Different tumor cell lines exhibit a variety of sensitivities to the cytotoxic effects of PE-based immunotoxins.11,12 Therefore, we inquired about the contribution made by inhibiting protein synthesis in the induction of apoptosis leading to the loss of cell viability. Initially, we chose to characterize three epithelial tumor cell lines: KB3-1, DLD-1 and HCT116 and compared the decrease in protein synthesis at different time points after treating each cell line in a dose-dependent manner with an immunotoxin (HB21-PE40) targeting the transferrin receptor. We monitored responses during the first 8 h to record the early effects of toxin entry into the cells (Figure 1a). Inhibition of protein synthesis began as early as 2 h in DLD-1 and HCT116 cells, whereas in KB3-1 cells inhibition of protein synthesis occurred after 4 h. DLD-1 cells were the most sensitive cells with an IC50 around 1ng/ml after only 2 h of incubation and inhibition was almost complete by 8 h even at a low concentration of immunotoxin (Figure 1a). KB3-1 cells appeared to be the most resistant: after 2 h, no significant decrease was detected (Figure 1a). However, at 4 and 8 h, the rate of inhibition was comparable to the decrease observed in the HCT116 cells with IC50 values almost 10-fold higher than those of DLD-1. At concentrations of immunotoxin ≥ 10 ng/ml, protein synthesis inhibition was complete in all cell lines at 8 h (Figure 1a).
Figure 1.
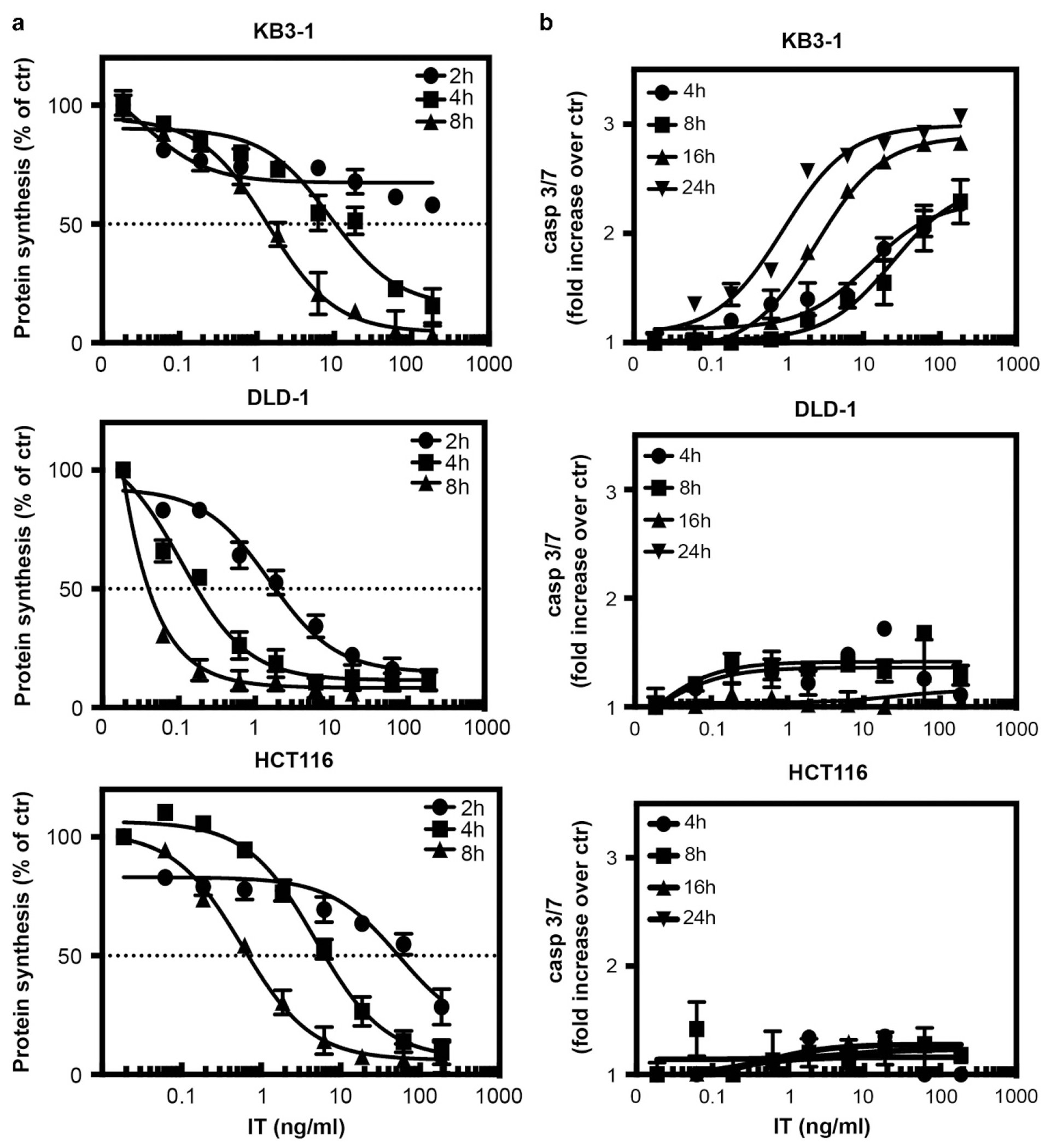
Immunotoxin-mediated inhibition of protein synthesis and activation of caspase 3/7. (a) Following the addition of various concentrations of immunotoxin to KB3-1, DLD-1 or HCT116 cells for 2, 4 or 8 h, inhibition of protein synthesis was determined by measuring the incorporation of 3H-leucine into cells. Protein synthesis was determined by measuring the incorporation of 3H-leucine into cells and the values are presented as percent compared with untreated cells. (b) KB3-1, DLD-1 and HCT116 cells were treated for 4, 8, 16 or 24 h with increasing concentrations of immunotoxin. Caspase 3/7 activity was determined using the Caspase-Glo3/7 kit. The luminescence of each well was measured and the values are presented as a fold increase relative to untreated cells. The mean values were determined from three different experiments where each point in each experiment was derived from wells in triplicate. The error bars represent the s.e.m.
Previously, it was reported that protein synthesis inhibition triggers apoptosis.13 Consequently, we anticipated detecting the induction of apoptosis shortly after complete inhibition of protein synthesis had been achieved. We treated each cell line with the same immunotoxin concentrations used in the protein synthesis assay and determined caspase 3/7 activation at different time points after treatment. Surprisingly, there was no evidence of apoptosis in DLD-1 or HCT116 cells over a period of 24 h, not even at high concentrations of immunotoxin (Figure 1b). Moreover, after immunotoxin addition, DLD-1 and HCT116 cells were viable and no significant reduction in cell proliferation was detected (Supplementary Figure 1), suggesting also that no other death mechanism had been induced. By contrast, KB3-1 cells showed a clear caspase 3/7 activation starting at 4 h, which increased in a dose- and time-dependent manner (Figure 1b). At 24 h after immunotoxin addition, most of the cells were apoptotic and viability was reduced substantially (IC50 = 2.669 ng/ml) (Supplementary Figure 1). Treating the cells with staurosporin confirmed that the cells could undergo apoptosis; but this program was not engaged in DLD-1 and HCT116 cells upon treatment with immunotoxin. Thus, immunotoxin-mediated shut-down of the translational machinery was not sufficient to kill either DLD-1 or HCT116 cells in relatively short periods of time. We conclude that the cytotoxic effect of PE-based immunotoxins does not depend exclusively on the ability of the toxin to inactivate eukaryotic elongation factor-2; suggesting that other pathways may be involved in determining the fate of tumor cells.
The level of the Bcl-2 family proteins regulates the response to the immunotoxin treatment
The failure to induce apoptosis within 24 h of immunotoxin treatment in either HCT116 or DLD-1 cells prompted us to investigate the fate of Bcl-2 family members following immunotoxin treatment. First, we established baseline levels of both the prosurvival and proapoptotic proteins in each of the three untreated cell lines (Figure 2). The prosurvival proteins Bcl-xl and Bcl-2 were present at comparable levels in all three lines, whereas Mcl-1 appeared to be lower in DLD-1 and HCT116 cells. The apoptotic effectors Bax and Bak appeared to be expressed less abundantly in HCT116 cells compared with KB3-1 and DLD-1 cells. There was no difference in the BH-3-only proteins Puma, Bid and Bad, whereas Noxa appeared poorly expressed in DLD-1. Noticeably, Bim was most abundant in KB3-1.
Figure 2.

Cellular level of the Bcl-2 prosurvival family members. The expression level of various Bcl-2 family proteins was determined from lysates of each cell line, as indicated, by western blot. Blots are representative of three experiments.
Then, we asked if the level of any of these proteins changed during the first 24 h of treatment with the immunotoxin (Figure 3). Among the proapoptotic members, the level of two BH-3-only proteins, Bim and Noxa, were reduced in all three cell lines after treatment in a dose- and time-dependent manner (Figure 3 and Supplementary Figure 2). We noted that in DLD-1 cells, Bim was lost by 4 h (Figure 3b and Supplementary Figure 2). In HCT116 cells, the diminution started at 8 h when cells were treated at the highest concentration of immunotoxin and was complete at 16 h at all concentrations (Figure 3c and Supplementary Figure 2). In contrast to HCT116 and DLD-1 cells, Bim levels in KB3-1 cells were largely unaltered for the first 16 h and a decrease in Bim was observed only after 24 h (Figure 3a and Supplementary Figure 2).
Figure 3.
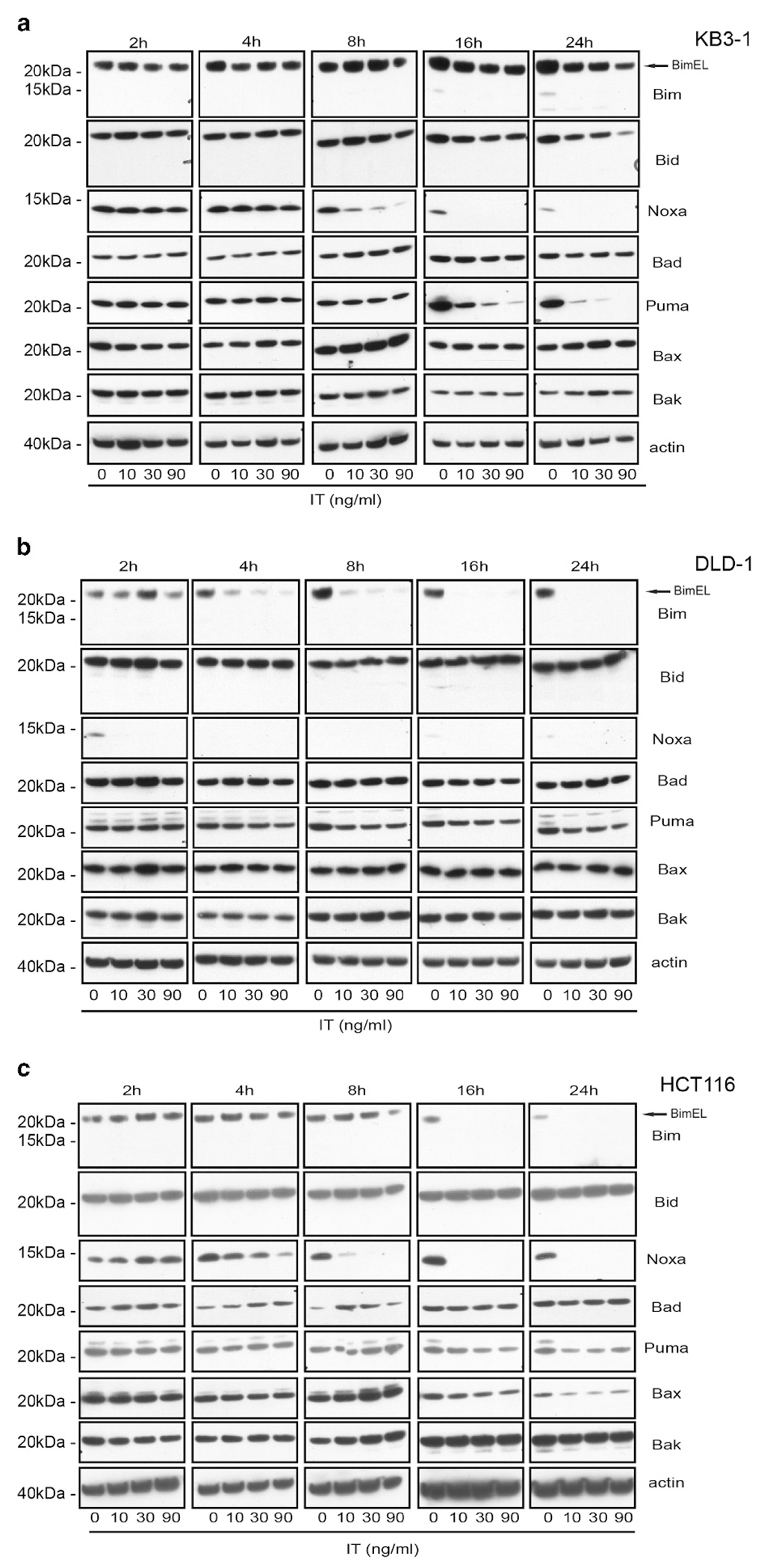
Cellular levels of the proapoptotic members of the Bcl-2 protein family after immunotoxin treatment. KB3-1 (a), DLD-1 (b) or HCT116 (c) cells were exposed for the indicated times to the immunotoxin in a concentration-dependent manner. The levels of the proapoptotic Bcl-2 proteins from lysates were visualized by western blot as indicated. Blots are representative of three experiments and are quantified in Supplementary Figure 2.
Noxa levels dropped comparably between 4 and 8 h in KB3-1 and HCT116 (Figures 3a and c and Supplementary Figure 2), which was expected as they expressed similar levels of the protein in untreated cells (Figure 2). At 8 h, Noxa was completely gone in HCT116 (Figure 3c and Supplementary Figure 2), whereas traces were still visible in KB3-1 cells (Figure 3a and Supplementary Figure 2), possibly because the rate of protein synthesis inhibition was slower in KB3-1 compared with HCT116 cells (Figure 1a). After only 2 h of treatment, Noxa disappeared in DLD-1 suggesting that the low initial level of protein (Figure 2b and Supplementary Figure 2) together with the high rate of protein synthesis inhibition (Figure 1a) led to a rapid loss. In addition, we noticed a general decline in Puma level, more pronounced in KB3-1 cells (Figure 3 and Supplementary Figure 2); leading us to speculate that this drop was caused not only by the inhibition of protein synthesis but also by the natural cycle of protein turnover since, at 16 and 24 h, Puma expression was low even in untreated cells (Figure 3a). As expected, among the prosurvival members of the Bcl-2 family, only Mcl-1 was affected by treatments with the immunotoxin, decreasing in a dose-dependent manner starting at 8 h (Supplementary Figure 3). To exclude that the alteration of the protein levels of Bim or Noxa was associated with regulatory changes at the transcription level, we determined if immunotoxin treatments caused changes in the mRNA level of these genes (Supplementary Figure 4). We monitored and compared the levels of each mRNA from 2 to 24 h of treatment with immunotoxin (90 ng/ml). Results clearly indicated that immunotoxin treatment did not reduce transcript levels for either gene. Rather, we observed a slight increase in Bim mRNA in KB3-1 and a more pronounced increase in DLD-1 cells. Increase in Noxa mRNA in DLD-1 and HCT116, was also noted, both probably induced by the stress caused by immunotoxin treatment.9 As the level of the Bim transcript was not diminished during the immunotoxin treatment, we wished to confirm that loss of Bim resulted from protein turnover following inhibition of protein synthesis. Therefore, we treated HCT116 with cycloheximide and noted a similar result to immunotoxin treatment (Supplementary Figure 5).
In summary, we report that following immunotoxin addition, protein synthesis was rapidly inhibited in all three cell lines rendering them incapable of making new proteins. Consequently, Bim, Noxa and Mcl-1 were degraded. However, although Noxa and Mcl-1 disappeared after immunotoxin treatment in all three cell lines, the fate of Bim of was different. Untreated HCT116 and DLD-1 cells had a low level of Bim protein compared with KB3-1, where Bim seemed rather abundant. In addition, exposure to immunotoxin caused a rapid loss of this protein in HCT116 and DLD-1 but not in KB3-1 cells where Bim levels were only partially reduced at 24 h post-treatment. Assessing the role of each protein, we concluded that Noxa did not have a pivotal role in the apoptotic response because its marked loss from KB3-1 cells did not negate the activation of caspase 3/7. Rather, we argue that low Bim levels coupled with its rapid loss accounted for the lack of apoptosis in DLD-1 and HCT116 cells, whereas its abundance and relative stability in KB3-1 cells allowed for caspase 3/7 activation.
Bim regulates the immunotoxin cytotoxicity in vitro and in vivo
To determine if Bim was critical to the apoptotic response, CRISP/Cas9 technology was used to create Bim KO cell lines in both the KB3-1 and HCT116 backgrounds (Supplementary Figure 6). To address the issue of specificity, we also knocked out the Bid gene because, like Bim, Bid binds all the prosurvival Bcl-2 proteins14 and can directly activate both Bax and Bak.15 We first measured caspase 3/7 activity and viability in KB3-1-wild-type (WT), KB3-1-empty vector and both KO cells at 16 and 24 h after treatment because at 16 h (in KB3-1-WT cells) we observed a peak of caspase 3/7 activity and at 24 h a consistent decrease in cell viability (Figure 1). Following immunotoxin treatment, KB3-1-Bim KO cells showed no significant increase in caspase 3/7 activity at 16 h and a negligible increase (~1.5-fold) at 24 h compared with the WT cells (Figure 4). KB3-1-Bim KO cells retained approximately 80% viability at 24 h. In contrast, KB3-1-Bid KO and the empty vector cells had a response similar to the WT cells with a peak in the casp3/7 activity at 16 h followed by an almost 60% reduction in viability at 24 h confirming that Bid was not a major contributor to immunotoxin-mediated apoptosis. For KB3-1-WT cells, we confirmed that they exhibited a threefold increase in casp3/7 activity at 16 h and the casp3/7 activity was still high at 24 h but had started to diminish because of loss of viable cells (almost 60% loss) (Figure 4). Conducting similar experiments with HCT116 cells revealed that immunotoxin did not induce apoptosis in WT, empty vector, Bim KO or Bid KO cells after 24 h of treatment (Supplementary Figure 7a).
Figure 4.
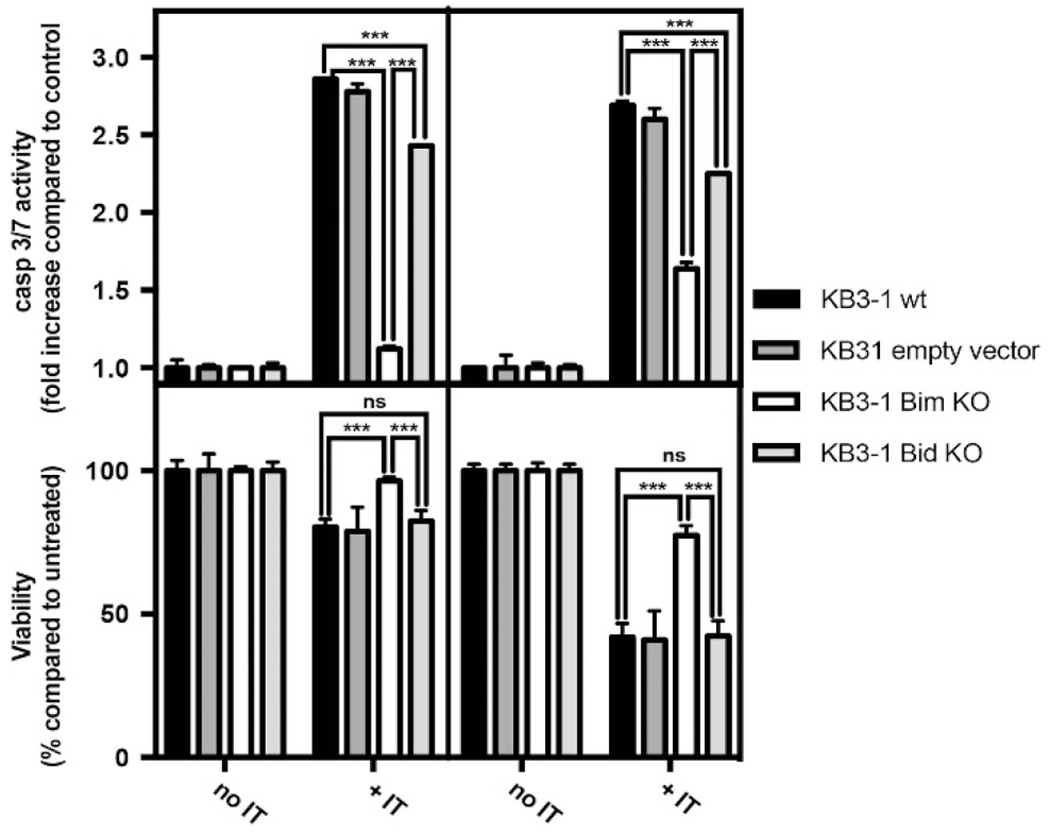
Bim deficiency protects cells from immunotoxin-mediated cell death. Caspase 3/7 activity and cell viability were determined in KB3-1 WT, empty vector, Bim KO and Bid KO cells after 16 or 24 h of treatment with immunotoxin at 10 ng/ml. Caspase 3/7 activity was determined using the Caspase-Glo3/7 kit. The luminescence of each well was measured and the values are presented as a fold increase relative to untreated cells. Viability was determined using the CellTiter-Glo Luminescent Assay kit. The luminescence of each well was measured and the values are presented as a percentage relative to untreated cells. Data are from at least three independent experiments in triplicate. Error bars display s.d. value. Two tailed unpaired t-tests were performed with ***P < 0.001.
To determine if Bim has a role in immunotoxin-mediated antitumor effects in vivo, we established tumor xenografts from both KB3-1 and HCT116 cells. Xenografts included WT and Bim or Bid KO tumors. For each cell line, tumors were readily established in athymic nude mice, confirming that loss of Bid or Bim did not interfere with tumorigenesis. Immunotoxin treatment was then initiated as tumors grew to approximately 100 mm3. Remarkably, tumors derived from KB3-1 Bim KO cells were completely unresponsive to immunotoxin treatment with no change in the median survival between treated and untreated mice (Figure 5b). In contrast, tumors grown from KB3-1 WT cells or Bid KO cells and treated with the immunotoxin showed reduced growth rates with an increase in median survival of >10 days (Figures 5a and c). Tumors grown from HCT116wt or Bid KO cells were not very sensitive to the immunotoxin (Supplementary Figure 7b). However, loss of Bim (via the Bim KO) eliminated any effect of the immunotoxin. We conclude that a Bim knockout severely impairs the apoptotic process and eliminates immunotoxin-mediated tumor regression, supporting the hypothesis that Bim expression is required for immunotoxin cytotoxicity both in vitro and in vivo.
Figure 5.
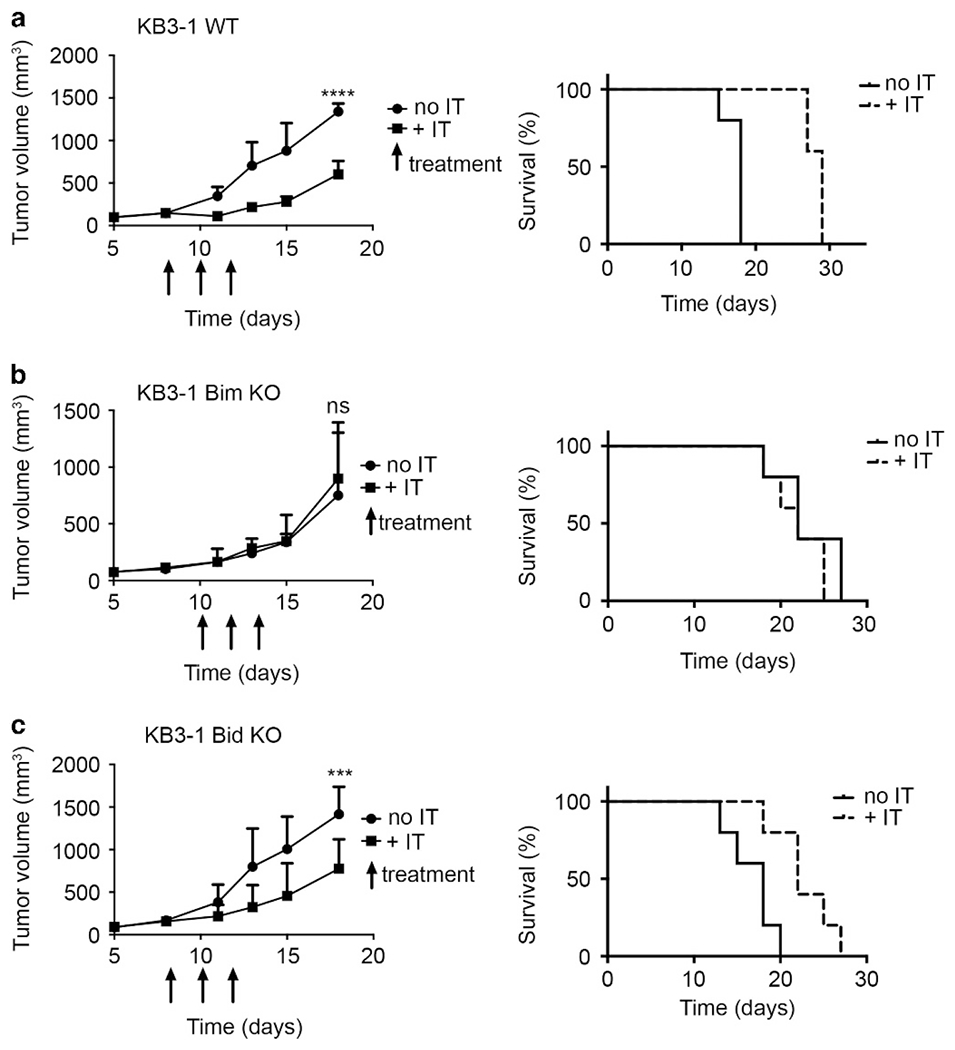
Bim determines the response to immunotoxin treatment in vivo. KB3-1 WT (a), Bim KO (b) and Bid KO (c) cells were implanted in the left flank of athymic nude mice. Mice were treated with HB21-PE40 (n = 5), or vehicle (n = 5) 3 × qod, indicated by arrows. Tumor volume was calculated as (0.5 × L × W2). Tumor volumes were compared by two-way analysis of variance test. ***P<0.01, ****P<0.0001. For the Kaplan–Meier plot, showing time to experimental endpoint for each KB3-1 tumor-bearing mouse, mice were killed once tumor volume reached 1200 mm3 or tumors became necrotic. NS, nonsignificant.
Bim protein level influences the response to immunotoxin in hematological tumors
Next, we sought to determine if Bim functioned as major determinant of immunotoxin activity in other cell types, in particular in hematological malignancies. This was especially important because the anti-CD22 immunotoxin, moxetumomab pasudotox (previously known as HA22), has achieved significant efficacy including complete remissions during phase I testing16 and is currently completing pivotal multi-center phase III testing.17 We evaluated one leukemic cell line (Nalm6) and two lymphoma cell lines (Raji and CA46). Each showed an equivalent IC50 for protein synthesis inhibition when treated in a dose-dependent manner for 20 h with moxetumomab pasudotox.18,19 We compared the caspase 3/7 activity induced by immunotoxin in the three cell lines and noted a substantial increase in Raji cells at 16 h (>10-fold increase compared with the 2-fold increase in Nalm and Raji). Raji cell proliferation started to decrease at 16 h and most cells were dead by 24 h. In contrast, Nalm6 and CA46 cells showed little activation of caspase and were mostly viable at 24 h (Figure 6a). Checking the level of the Bcl-2 family proteins revealed that Bim was most abundant in Raji cells (Figure 6b). As a consequence of immunotoxin treatment for 16 h, Bim disappeared in CA46 and Nalm6 cells, but was still present in Raji even if slightly reduced (Figure 6c). No other changes in Bcl-2 protein levels were noted (data not shown).
Figure 6.

Response of hematological cell lines to an immunotoxin directed to CD22. (a) Caspase 3/7 activity and cell viability were determined in Nalm6, Raji and CA46 cells after 16 or 24 h treated with immunotoxin in a dose-dependent manner. Caspase 3/7 activity was determined using the Caspase-Glo3/7 kit. The luminescence of each well was measured and the values are presented as a fold increase relative to untreated cells. Viability was determined using the CellTiter-Glo Luminescent Assay kit. The luminescence of each well was measured and the values are presented as a percentage relative to untreated cells. Data are from three independent experiments in triplicate. Error bars display s.d. value. (b) The expression level of various Bcl-2 family proteins was determined from lysates of each cell line, as indicated, by western blot. Blots are representative of two experiments. (c) CA46, Nalm6 and Raji cells were exposed for 16 h to immunotoxin in a concentration-dependent manner. The levels of Bim and Mcl-1 from lysates were visualized by western blot as indicated. Blots are representative of three experiments.
The above results suggested that Bim levels may predict the apoptotic response in a clinical setting that included leukemic patients undergoing immunotoxin treatment. HCL is an indolent B-cell malignancy that characteristically expresses high levels of surface CD22. Therefore, we sought to determine if pretreatment Bim levels would predict clinical responses to the immunotoxin. Accordingly, the pretreatment mononuclear fraction of each patient was assessed for protein levels of Bim, Bad and Bid. Following quantification for each protein, a median level was determined for a total of 16 HCL patient samples. Next, we analyzed the data available about the number of leukemic cells in the peripheral blood mononuclear cells of those patients 1 week after immunotoxin treatment and looked for correlations between response and the presence of each of the three BH-3-only proteins. We decided to consider the number of leukemic cells after a week of treatment because the clinical status of the patients before treatment was very complex and long-term outcomes did not depend exclusively on immunotoxin action. Our data indicated that there was a significant difference in the percentage of leukemic cells remaining after a week of treatment that depended specifically on relative Bim levels (Figure 7 and Supplementary Figure 8). In patients with high Bim, no leukemic cells were detectable after a week of treatment, whereas a variable percentage (on average, 20%) of leukemic cells remained in low Bim patients. No significant difference in responses was found for Bid levels. Regarding Bad levels, there appeared to be an inverse correlation: with samples containing high Bad levels showing incomplete killing, whereas low Bad levels were associated with the absence of tumor cells (Figure 7). We conclude that an assessment of Bim protein levels can be used to predict the initial response to immunotoxin treatment in patients, at least those with HCL.
Figure 7.

Response to immunotoxin treatment. Bim, Bid and Bad levels were quantified by densitometry, normalized to Glyceraldehyde 3-phosphate dehydrogenase and plotted against the percentage of remaining leukemic cells after a week of immunotoxin treatment. Protein level values are the mean of at least three western blots.
DISCUSSION
Immunotoxins are promising cytotoxic agents deployed as cancer treatment agents. Their value lies in the specificity of the antibody portion (of the molecule) combined with a highly toxic bacterial toxin that disrupts cell homeostasis through general protein synthesis inhibition. Unfortunately, there is a heterogeneous response among tumor cells treated with PE-based immunotoxins. In this study, we report that immunotoxin-mediated inhibition of protein synthesis is not always cytotoxic for tumors because it does not universally lead to caspase activation (Figure 1). Considering our earlier published work describing the cytotoxic synergy of ABT-737 (a BH-3-only mimetic) and immunotoxins in several cell lines,9,11,12 we speculated that the Bcl-2 family of proteins could have a major role in the immunotoxin mechanism of action. We determined that cellular level of Bim was critical for the outcome of the immunotoxin treatment. Bim is normally degraded by the proteasome.18,19 Further, for the first time, we report that immunotoxin, as a translation inhibitor, strongly reduces the level of two proapoptotic BH-3 only proteins: Bim and Noxa. In tumor cells, such as DLD-1 and HCT116, with low pretreatment levels (Figure 2), Bim was rapidly lost and could not be replenished because of immunotoxin-mediated inhibition of protein synthesis (Figure 1b). However, in cells with an abundant level of Bim, the decrease in protein synthesis was not rapid enough to eliminate Bim and prevent apoptosis. To confirm this hypothesis, we knocked out Bim in the most immunotoxin sensitive cell line, KB3-1, and showed that the absence of Bim negated caspase activation and consistently delayed cell killing. The effect was remarkable in vivo where lack of Bim completely eliminated any antitumor effect of the immunotoxin (Figure 5). Even in tumors derived from cells with low Bim expression, knocking out the expression of Bim abolished the minimal response seen with immunotoxin treatments (Supplementary Figure 6).
Together, our pre-clinical data suggested that the expression level of Bim was critical for determining the response of cells to immunotoxin treatment: lack of Bim expression precluded cells from undergoing apoptosis, which our results support as essential for successful immunotoxin outcomes. Further, our analysis points to the value of monitoring the level of Bim protein rather than Bim mRNA;because loss of Bim protein did not correlate with changes in the level of Bim mRNA.
Our initial data were generated with an immunotoxin targeting the transferrin receptor expressed on epithelial cell lines and tumors derived from these lines. We then expanded our study to include hematologic lines expressing CD22 as their surface target. Here we characterized one leukemic line and two lymphoma lines. Similar to results with epithelial lines, cells (Raji) expressing high levels of Bim protein exhibited caspase activation and loss of viability (within 24 h) when treated with immunotoxin. In contrast, lines (Nalm6 and CA46) with lower Bim levels exhibited only a slight reduction in viability and only minimal activation of caspase. With both epithelial and hematologic cell data supporting a role of Bim, we sought to evaluate its role in determining patient responses.
For patients harboring epithelial tumors, it is difficult to get pre- and post-treatment samples that contain uniform populations of tumor cells. However, in patients with hematologic tumors it is often possible to obtain samples from peripheral blood that are highly enriched in tumor cells. We noted that in a series of 16 HCL patients, high Bim expression was associated with a better response to immunotoxin treatment than low Bim. That association was not seen with either Bid or Bad.
In considering roles for other BH-3-only proteins following immunotoxin treatment, we note that Noxa reduction was not linked to a poor apoptotic response as loss of this protein was noted even in KB3-1 cells, which responded with caspase 3/7 activation and apoptosis. Moreover, we speculate that Noxa downregulation annuls any possibility of improving the apoptotic response because of the concomitant reduction of its prosurvival antagonist Mcl-1;13,20 supporting the contention that reductions of Mcl-1 do not have a pivotal role in apoptotic cell death caused by translation inhibitors.13 Similarly, another BH-3 protein, Bid, seems to be dispensable for the apoptotic response induced by the immunotoxin treatment (Figure 5 and Supplementary Figure 7), suggesting that the extrinsic pathway of apoptosis was not involved. Given the complexity of cancer, it would be extremely unlikely that low Bim expression is the only reason for poor responses or defective apoptosis in patients treated with the immunotoxin. However, we propose that Bim expression may contribute to the heterogeneity of responses observed in the clinic and therefore Bim should be monitored closely in pretreatment biopsies to determine its predictive value as a biomarker.
Finally, strategies will need to be developed to overcome a poor apoptotic response by cells that express low Bim. Most likely, combination therapy will be used19 and the use of large chemical libraries for screening will likely be useful, especially if apoptosis is used as the final read out.
MATERIALS AND METHODS
Reagents and cell lines
The immunotoxin, HB21scFv-PE40 (HB21-PE40) was produced recombinantly in Escherichia coli as described previously.21 Cycloheximide was from Sigma-Aldrich (St Louis, MO, USA).
The cervical adenocarcinoma cell line, KB3-1, was obtained from Michael Gottesman (National Cancer Institute, Bethesda, MD, USA). The cells were grown in Dulbecco’s modified Eagle’s medium (Thermofisher, Waltham, MA, USA) plus 10% heat-inactivated fetal bovine serum (Thermofisher). The colorectal carcinoma cell line DLD-1 (CCL-221) was obtained from American Type Culture Collection (ATCC, Manassas, VA, USA) and it was cultured in RPMI-1640 medium (Thermofisher) with glutamax (Thermofisher) and 10% heat-inactivated fetal bovine serum. The colorectal carcinoma cell line HCT116 was obtained from Richard Youle (National Institute of Neurological Disorders and Stroke, National Institutes of Health (NINDS, NIH, Bethesda, MD, USA)). The cells were grown in McCoys medium (Thermofisher) with glutamax and 10% heat-inactivated fetal bovine serum. The media for all the three cell lines were supplemented with non-essential amino acids (Thermofisher). The B-cell precursor leukemia cell line Nalm6 was obtained from Alan Wayne (National Cancer Institute). The Burkitt’s lymphoma cell lines, CA46 (CRL-1648) and Raji (CCL-86) were purchased from ATCC. They were grown in RPMI-1640 plus 10% fetal bovine serum.
Cytotoxicity assays
KB3-1, DLD-1 and HCT116 (10 000 cells per well) were plated in a 96-well format. After 24 h, the recombinant immunotoxin was added at different concentrations and incubated for various times (as indicated). Inhibition of protein synthesis was then measured by the addition of 3H-leucine (1 μCi/ml) in leucine-free RPMI media for 30 min. 3H-leucine was measured with a Wallac MicroBeta Trilux plate reader (Perkin Elmer, Whaltam, MA, USA). Counts per minute data were normalized and presented as a percentage of control. Data from at least three independent experiments are presented and were derived from triplicate samples plus or minus one s.d. Viability was determined using the CellTiter-Glo Luminescent Cell Viability Assay kit (Promega, Madison, WI, USA). This assay quantifies the amount of ATP present, which signals the presence of metabolically active cells. ATP was measured as luminescence produced by the monooxygenation of luciferin catalyzed by the Ultra-Glo-luciferase. Caspase 3/7 protease activity was measured using the Caspase-Glo3/7 Assay kit (Promega) that quantifies the cleavage of the substrate, Ac-DEVD-pNA.
Immunoblot
Treated or control cells were collected, washed with phosphate-buffered saline (PBS), and solubilized in RIPA buffer (Thermo Scientific, Waltham, MA, USA) with protease and phosphatase inhibitors (Thermo Scientific). Protein concentrations were determined using the Nanodrop2000c Spectrophotometer (Thermo Scientific). Equal amounts of protein were loaded onto NuPAGE 4-12% Bis-Tris gels (Thermofisher) and transferred to nitrocellulose membranes (Thermofisher). The following primary antibodies were used: Bim (cat# 2933), Bid (cat# 2002), Puma (cat# 4976), Bak (cat#12105), Bcl-xL (cat# 2764), and Bcl-2 (cat#2876) (Cell Signaling Technology, Danvers, MA, USA); Mcl-1 (Santa Cruz Biotechnology, Santa Cruz, CA, USA; cat# sc-819) and Bad (Santa Cruz Biotechnology, cat# sc-8044); Noxa (Novus Biological, Littleton, CO, USA, cat# NB600-1159); Bax (EMD Millipore, Billerica, MA, USA, cat# ABC11); actin (BD Biosciences, San Jose, CA, USA; ca# 612656); Glyceraldehyde 3-phosphate dehydrogenase (Abcam, Cambrigde, MA, USA, cat# 9485). Primary antibodies were routinely detected with donkey anti-mouse horseradish peroxidase or donkey anti-rabbit horseradish peroxidase (Jackson ImmunoResearch, West Grove, PA, USA, cat# 715-035-150 and cat# 711-035-152) using the SuperSignal West Pico Chemiluminescent Substrate kit (Thermo Scientific). Densitometric quantification of immunoblot images from the three different experiments was performed using an analysis program ImageJ (NIH, Bethesda, MD, USA) (developed by W Rasband, Research Services Branch of the National Institute of Mental Health). Each band intensity was normalized to the correspondent actin band and at each time point, each treatment band was compared with the untreated.
Quantitative PCR
KB3-1, DLD-1 and HCT116 cells were treated with HB21-PE40 90 ng/ml for different times. RNA was isolated from treated cells and purified using the Qiagen RNeasy Mini kit (Valencia, CA, USA) and was reverse-transcribed and amplified using superscript First-strand cDNA synthesis (Thermofisher). Bim and Noxa transcript levels were monitored in real time on a StepOnePlus Real Time PCR system using a double-quenched probe contain a 5’-fluorophore FAM, a 3′ IBFQ quencher and an internal ZEN quencher Integrated DNA Technologies (IDT, Coralville, IA, USA). The primers used were: BIM forward (5′-CTTACATCAGAAGGTTGCTTTGC-3′) and BIM reverse (5′-CCCTTTCTTGGCCCTTGTTC-3′), Noxa forward (5′-GAGCAGAAGAGTTTGGATATCAGA-3′) and Noxa reverse, (5′-GCAAGAACTCTCAACCGA-3′). The endogenous control for each cell line was determined using a TaqMan array 96-well FAST plate, with RNA isolated from cells from each cell line treated with HB21-PE40 (90 ng/ml) for 24 h. We found that the level of mRNA for the GUSB1 gene did not change compared with untreated cells in KB3-1 or DLD-1 cells. The primers used were GUSB1 forward (5′-GTTCAAACAGATCACATCCACA-3′) and reverse (5′-CACCTAGAATCTGCTGGCTAC-3′). For HCT116 we used HPRT1 forward (5′-GTATTCATTATAGTCAAGGGCATATCC-3′) and HPRT1 reverse (5′-AGATGGTCAAGGTCGCAAG-3′). Relative level of of mRNA was determined as the relative expression = 2−(ΔΔCt), where ΔΔCt = ΔCt gene of interest in IT treated cells – ΔCt gene of interest in untreated cells. ΔCt gene of interest was determined as Ct gene of interest – Ct endogenous control at each time point.
Generation of the Bim KO and Bid KO cells
Constitutive Cas9 and inducible guide RNA vectors have been described previously.22 The guide RNA sequences are described in Supplementary Figure 1. Lentiviruses were produced using third-generation lentivirus packaging plasmids (pMDLg/pRRE, pRSV RRE23,24 and pCMV VSV-G,24 Addgene, Cambridge, MA, USA) transiently transfected into HEK293T cells along with the sgRNA constructs (Effectene Transfection Reagent, Qiagen). Virus containing supernatants were filtered through a 0.45 μm filter and stored at 4 °C until used. Cell lines were infected with lentiviruses constructs encoding CAS9 mCherry and sgRNA GFP. Double-positive mCherry GFP cells were isolated by flow cytometry cell sorting (Aria, BD Biosciences). Doxycycline (1 μg/ml, Sigma, St Louis, MO, USA) was added to tissue culture medium to induce expression of sgRNA constructs and after 72 h the cells were cloned by serial dilution in 96-well plates. After 96 h, wells that contained single colonies were selected and expanded. Cell lysates were prepared from cloned cells and the expression of Bim or Bid was determined by western blot using rabbit anti-Bim polyclonal antibody (ADI-AAP-330, Enzo Biosciences, Farmingdale, NY, USA) or the rat anti-Bid monoclonal antibody (4C1-12, Walter and Eliza Hall Institute of Medical Reserch (WEHI, Parkville, VIC, Australia)) (Supplementary Figure 1a). We chose clone#1 for Bim KO and clone#1 for Bid KO. Mutation of each gene was confirmed by sequencing of PCR amplified DNA from each clone.22 Genomic DNA was isolated (DNAeasy kit, Qiagen) and amplified using sgRNA-targeting region PCR primers (described in Supplementary Figure 1b) and sequenced using Illumina MiSeq technology (San Diego, CA, USA).
Murine xenograft models
All animal experiments were performed in accordance with NIH guidelines and approved by the NCI Animal Care and Use Committee. KB3-1 or HCT116 tumors were grown in female nude athymic mice (Charles River Laboratories, Wilmington, MA, USA). In all, 5 × 106 cells per mouse in serum-free Dulbecco’s modified Eagle’s medium were mixed with Matrigel (Corning, Corning, NY, USA) (4 mg/ml) and injected into the left rear flank of mice weighing 20–25 g. After the average tumor volume had reached approximately 150 mm3, mice were randomized into groups and treated with vehicle alone (PBS+0.2% human serum albumin) or HB21-PE40 (2 μg per mouse per injection). Three injections were performed qod via tail vein for each mouse. Tumor volume and animal weight were measured at least three times weekly. Tumor volume was calculated as 0.5 × (L × W2). Animals were killed by CO2 inhalation once tumors reached 1200 mm3 or became necrotic. Time to death was displayed as a Kaplan–Meier plot and statistical significance calculated by log-rank test. **P < 0.01; ***P < 0.001.
Statistical analysis
All experimental error bars display s.d., with all P-values calculated for 95% confidence intervals. All statistics were performed using Graphpad Prism 6 software (La Jolla, CA, USA). Two-way analysis of variance were performed for each xenograft, comparing tumor volumes of immunotoxin-treated and vehicle-treated mice for each cell line. Statistically significant differences in tumor volumes between vehicle-treated and HB21-PE40-treated mice are indicated. HB21-PE40-treated KB3-1-Bim KO or HCT116-Bim KO tumors were not significantly different from vehicle-treated tumors at any time point for either xenograft model. *P < 0.05; **P < 0.01; ***P < 0.001.
Patient cells
Sixteen samples containing pretreatment cells from HCL patient were obtained from blood collected in sodium heparin-containing tubes, after giving informed consent approved by the Institutional Review Board of the National Cancer Institute. Blood was diluted 1:1 with PBS and centrifuged over Ficoll. The mononuclear layer was collected and washed by centrifuging in PBS and finally RPMI containing 10% fetal bovine serum. For the flow cytometry analysis, the specimens were stained within 24 h of collection with a panel of antibodies. HCL cells were bright-positive for CD20, CD22 and CD11c and positive for CD19 and CD103, and all patients except the patients with variant HCL expressed CD25. When present, erythrocytes were lysed by incubating with lysing solution (150 mm NH4Cl, 10 mm KHCO3 and 0.1 mm EDTA) for 10 min at room temperature at a ratio of 1:9 (volume of sample: volume of lysing solution). Specimens were then washed with PBS before determining cell number. Cells were stained for 30 min at room temperature with various antibodies (antibody concentrations used per manufacturer’s recommendations) according to Clinical Laboratory Standards Institute document H43-A recommendations. All cells were fixed in 1.0% paraformaldehyde post staining and stored at 4 °C for up to 12 h before acquisition. Specimens were acquired with six-parameter four-color FC on the FACSCalibur (BD Biosciences) using CellQuest Pro software (BD Biosciences) (sensitivity of fluorescent detectors monitored using standard beads according to the manufacturer’s recommendations). Data (collected in list mode) were analyzed with CellQuest Pro (BD Biosciences) or FCS Express version 3 (De Novo Software, Los Angeles, CA, USA). Bright expression was determined as higher than normal, whereas dim expression was determined as lower than that observed in normal lymphocytes of the same lineage, in concordance with the 1997 U.S.-Canadian consensus guidelines. The peripheral blood mononuclear cells used in this experiment contained at least 95% HCL cells.
The protein fraction was extracted from cells using RIPA buffer and the level of Bim, Bid and Bad proteins were assessed by western blot. The amount of each protein was quantified by densitometry analysis using ImageJ. The relative amount of protein was expressed as the logarithm in base two of the ratio between the intensity of the BH-3-only protein band and the intensity of the Glyceraldehyde 3-phosphate dehydrogenase band. For each BH-3-only protein, we defined low protein level as any value below the median and high protein level as any amount above the median. The error bar represents 95% confidence interval. *P < 0.05.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank R Anderson and W Welch for the gift of reagents and Jianan Gong and Stephen Wilcox for assistance with sequence validation of CRISPR/Cas9 knockout cell lines. We thank E Arons for advice on the RT–PCR. This work was funded by the intramural programs of the National Cancer Institute, NIH and is supported by fellowships, and grants from the Australian National Health and Medical Research Council (research fellowships to DCSH; project grant 1057742 (DCSH); program grant 1016701; and Independent Research Institutes Infrastructure Support Scheme (grant 9000220)), the Cancer Council Victoria (grant-in-aid to DCSH), the Leukemia and Lymphoma Society (Specialized Centers of Research grants 7001-13), the Australian Cancer Research Foundation, a Victorian State Government Operational Infrastructure Support grant.
Footnotes
CONFLICT OF INTEREST
The authors declare no conflict of interest.
Supplementary Information accompanies this paper on the Oncogene website (http://www.nature.com/onc)
REFERENCES
- 1.Adams JM, Cory S. Bcl-2-regulated apoptosis: mechanism and therapeutic potential. Curr Opin Immunol 2007; 19: 488–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chipuk JE, Moldoveanu T, Llambi F, Parsons MJ, Green DR. The BCL-2 family reunion. Mol Cell 2010; 37: 299–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hata AN, Engelman JA, Faber AC. The BCL2 family: key mediators of the apoptotic response to targeted anticancer therapeutics. Cancer Discov 2015; 5: 475–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Weldon JE, Pastan I. A guide to taming a toxin--recombinant immunotoxins constructed from Pseudomonas exotoxin A for the treatment of cancer. FEBS J 2011; 278: 4683–4700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Antignani A, Fitzgerald DImmunotoxins: the role of the toxin. Toxins (Basel). 2013; 5: 1486–1502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.FitzGerald DJ, Wayne AS, Kreitman RJ, Pastan I. Treatment of hematologic malignancies with immunotoxins and antibody-drug conjugates. Cancer Res 2011; 71: 6300–6309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wayne AS, Fitzgerald DJ, Kreitman RJ, Pastan I. Immunotoxins for leukemia. Blood 2014; 123: 2470–2477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Alewine C, Hassan R, Pastan I. Advances in anticancer immunotoxin therapy. Oncologist 2015; 20: 176–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Antignani A, Sarnovsky R, FitzGerald DJ. ABT-737 promotes the dislocation of ER luminal proteins to the cytosol, including Pseudomonas exotoxin. Mol Cancer Ther 2014; 13: 1655–1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Andersson Y, Juell S, Fodstad O. Downregulation of the antiapoptotic MCL-1 protein and apoptosis in MA-11 breast cancer cells induced by an anti-epidermal growth factor receptor-Pseudomonas exotoxin a immunotoxin. Int J Cancer 2004; 112: 475–483. [DOI] [PubMed] [Google Scholar]
- 11.Traini R, Ben-Josef G, Pastrana DV, Moskatel E, Sharma AK, Antignani A et al. ABT-737 overcomes resistance to immunotoxin-mediated apoptosis and enhances the delivery of Pseudomonas exotoxin-based proteins to the cell cytosol. Mol Cancer Ther 2010; 9: 2007–2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hollevoet K, Antignani A, Fitzgerald DJ, Pastan I. Combining the antimesothelin immunotoxin SS1P with the BH3-mimetic ABT-737 induces cell death in SS1P-resistant pancreatic cancer cells. J Immunother 2014; 37: 8–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lindqvist LM, Vikstrom I, Chambers JM, McArthur K, Ann Anderson M, Henley KJ et al. Translation inhibitors induce cell death by multiple mechanisms and Mcl-1 reduction is only a minor contributor. Cell Death Dis 2012; 3: e409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Willis SN, Fletcher JI, Kaufmann T, van Delft MF, Chen L, Czabotar PE et al. Apoptosis initiated when BH3 ligands engage multiple Bcl-2 homologs, not Bax or Bak. Science 2007; 315: 856–859. [DOI] [PubMed] [Google Scholar]
- 15.Ren D, Tu HC, Kim H, Wang GX, Bean GR, Takeuchi O et al. BID, BIM, and PUMA are essential for activation of the BAX- and BAK-dependent cell death program. Science 2010; 330: 1390–1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kreitman RJ, Tallman MS, Robak T, Coutre S, Wilson WH, Stetler-Stevenson M et al. Phase I trial of anti-CD22 recombinant immunotoxin moxetumomab pasudotox (CAT-8015 or HA22) in patients with hairy cell leukemia. J Clin Oncol 2012; 30: 1822–1828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kreitman RJ, Pastan I. Immunoconjugates in the management of hairy cell leukemia. Best Pract Res Clin Haematol 2015; 28: 236–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Salvatore G, Nagata S, Billaud M, Santoro M, Vecchio G, Pastan I. Generation and characterization of novel monoclonal antibodies to the Ret receptor tyrosine kinase. Biochem Biophys Res Commun 2002; 294: 813–817. [DOI] [PubMed] [Google Scholar]
- 19.Antignani A, Mathews Griner L, Guha R, Simon N, Pasetto M, Keller J et al. Chemical screens identify drugs that enhance or mitigate cellular responses to antibody-toxin fusion proteins. PLoS ONE 2016; 11: e0161415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen L, Willis SN, Wei A, Smith BJ, Fletcher JI, Hinds MG et al. Differential targeting of prosurvival Bcl-2 proteins by their BH3-only ligands allows complementary apoptotic function. Mol Cell 2005; 17: 393–403. [DOI] [PubMed] [Google Scholar]
- 21.Buchner J, Pastan I, Brinkmann U. A method for increasing the yield of properly folded recombinant fusion proteins: single-chain immunotoxins from renaturation of bacterial inclusion bodies. Anal Biochem 1992; 205: 263–270. [DOI] [PubMed] [Google Scholar]
- 22.Aubrey BJ, Kelly GL, Kueh AJ, Brennan MS, O’Connor L, Milla L et al. An inducible lentiviral guide RNA platform enables the identification of tumor-essential genes and tumor-promoting mutations in vivo. Cell Rep 2015; 10: 1422–1432. [DOI] [PubMed] [Google Scholar]
- 23.Dull T, Zufferey R, Kelly M, Mandel RJ, Nguyen M, Trono D et al. A third-generation lentivirus vector with a conditional packaging system. J Virol 1998; 72: 8463–8471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stewart SA, Dykxhoorn DM, Palliser D, Mizuno H, Yu EY, An DS et al. Lentivirus-delivered stable gene silencing by RNAi in primary cells. RNA 2003; 9: 493–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.