

Abstract
Sialidases or neuraminidases catalyze the hydrolysis of terminal sialic acid residues from sialyl oligosaccharides and glycoconjugates. Despite successes in developing potent inhibitors specifically against influenza virus neuraminidases, the progress in designing and synthesizing selective inhibitors against bacterial and human sialidases has been slow. Guided by sialidase substrate specificity studies and sialidase crystal structural analysis, a number of 2-deoxy-2,3-dehydro-N-acetylneuraminic acid (DANA or Neu5Ac2en) analogues with modifications at C9 or at both C5 and C9 were synthesized. Inhibition studies of various bacterial sialidases and human cytosolic sialidase NEU2 revealed that Neu5Gc9N32en and Neu5AcN39N32en are selective inhibitors against V. cholerae sialidase and human NEU2, respectively.
Graphical Abstract
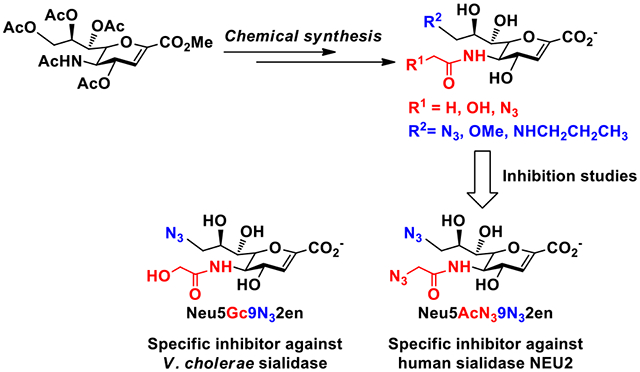
Two inhibitors with selectivity against V. cholerae sialidase and human cytosolic sialidase NEU2, respectively, were identified.
Introduction
Sialidases or neuraminidases (EC 3.2.1.18) are exo-glycosidases (EC 3.2.1.18) that catalyze the cleavage of terminal sialic acid residues from sialyl oligosaccharides, glycolipids, and glycoproteins. They are expressed by various organisms varying from bacteria and viruses to fungi, protozoa, animals, and humans.1, 2 Endo-sialidases (EC 3.2.1.129) that catalyze the hydrolysis of internal sialyl glycosidic bonds have also been reported.3 Based on their protein sequence similarities, all exo-sialidases known to date are mainly grouped into three glycoside hydrolase families (GH33, GH34, and GH83) in the Carbohydrate Active enZyme (CAZy) database (http://www.cazy.org).4 Bacterial, mammalian, and protozoan sialidases as well as trans-sialidases (EC 2.4.1.-) are grouped into GH33 family. Neuraminidases from influenza A and B viruses belonging to GH34 and other viral neuraminidases are grouped into GH83. In addition, a mouse klotho in GH1 has been found to have sialidase activity.5, 6 Several sialyltransferases in CAZy database, such as glycosyltransferase families GT427 and GT808–10 have also been shown to possess sialidase and/or trans-sialidase activities.11, 12 Despite their primary sequence differences, bacterial, viral, and human sialidases share a catalytic domain with a common canonical six-bladed β–propeller fold.13
Inhibitors specifically against neuraminidases of influenza viruses have been developed and applied efficiently for the treatment of flu.14 However, the emerge of drug-resistant viral strains and the increasing threat of viral pandemics15 urge the discovery of new neuraminidase inhibitors. Due to their important nutritional and pathogenic roles, bacterial sialidases, including those from Vibrio cholerae (causes cholera),16 Streptococcus pneumoniae (cause Otitis media in children),17 gram positive anaerobic bacteria Clostridium perfringens (cause gas gangrene disease), and Salmonella typhimurium LT2 (cause gastroenteritis)18, 19 are being considered as potential drug targets. Human sialidases, including lysosomal sialidase NEU1, cytosolic sialidase NEU2, and membrane-associated sialidase NEU3 and NEU420 have been shown to be related to a number of diseases such as cancer and type I and II sialidosis.21,22 They are currently being studied as important targets for inhibitor design.
The strategy of protein crystal structure-based rational design of sialidase inhibitors has been validated by the success of designing influenza virus neuraminidase inhibitors23–25 as clinically effective anti-influenza drugs [e.g., Zanamivir (Relenza)14 and Oseltamivir (Tamiflu)26]. The development of effective and selective inhibitors against bacterial and human sialidases has been less successful despite that an increasing number of bacterial sialidase crystal structures are becoming available. A number of derivatives of the common sialidase transition state analogue 2-deoxy-2,3-dehydro-N-acetylneuraminic acid (Neu5Ac2en or DANA, 1) have been synthesized as potential inhibitors against human NEU1,27 NEU2,28 and NEU3,29, 20, 30 as well as bacterial (e.g. V. cholerae31, 32 and C. perfringens32) sialidases. However, the inhibitory efficiency of the obtained inhibitors was usually similar to or even lower than Neu5Ac2en (1) (Figure 1).
Figure 1.
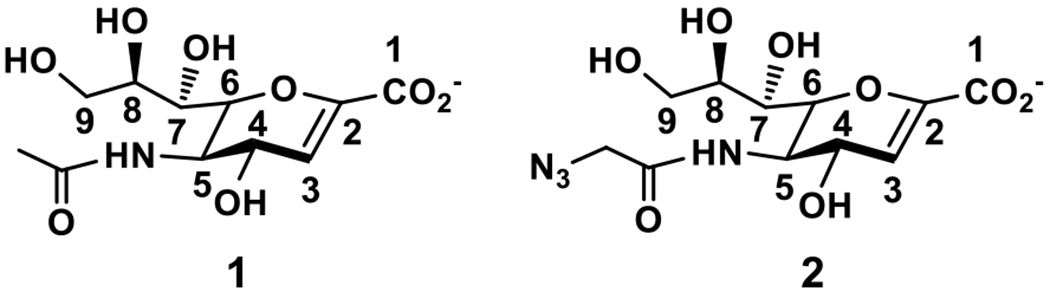
Structures of Neu5Ac2en 1 and Neu5AcN32en 2.
Recently, conformational flexible loops close to the catalytic site of sialidases were used as potential targets for inhibitor design.13, 33, 34 Based on information gained from mechanism-based labelling, a potent inhibitor selectively against V. cholerae sialidase with a Ki of 73 nM was identified by screening of a focused library.35
We have shown previously that results from sialidase substrate specificity studies can be used to guide the design of selective sialidase inhibitors.11 In this strategy, α2–3- and α2–6-linked sialyl galactosides containing different sialic acid forms or analogues are synthesized and used as sialidase substrates. The structural features of sialic acid forms in substrates selectively cleaved by certain sialidases can be introduced to Neu5Ac2en to obtain inhibitors that are selective against target sialidases. Indeed, sialidase substrate specificity studies using α2–3- and α2–6-linked sialyl galactosides with C5-modified sialic acid residues11, 36 helped to identify Neu5AcN32en (2) (a C5-derivative of Neu5Ac2en) (Figure 1) as a selective inhibitor against human NEU2.11 Herein, we explore the application of the strategy in designing additional selective sialidase inhibitors.
Results and discussion
Designing of sialidase inhibitors
To identify the logical positions for systematic modifications, the structures of sialidase-bound inhibitor Neu5Ac2en (a common transition-state analogue) in the reported crystal structures of bacterial and human sialidases were compared. Figure 2 shows that in addition to the N-acetyl group at C5, two significant differences of sialidase-bound Neu5Ac2en are the positions of C9 and the orientations of the hydroxyl groups attached to C9 (Figure 2).13, 37–42 Therefore, modifications at C9 or at both C9 and C5 will most likely provide selectivity for Neu5Ac2en-based sialidase inhibitors.
Figure 2.
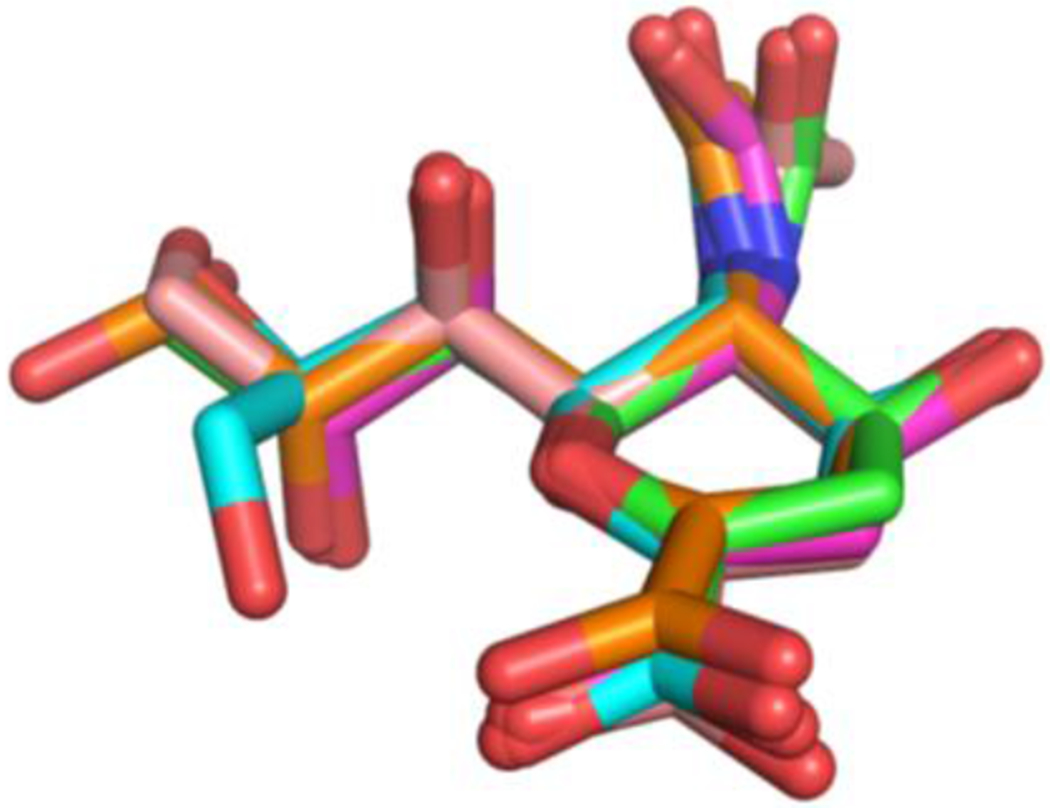
Overlay of sialidase-bound Neu5Ac2en structures in the reported crystal structures of sialidases11, 36–41 from S. pneumonia (NanB, carbons are shown in cyan), V. cholerae (carbons are shown in light pink), C. perfringens (NanI, carbons are shown in orange), S. typhimurium (carbons are shown in magenta), and human NEU2 (carbons are shown in green). Nitrogen atoms are shown in blue and oxygen atoms are shown in red. Neu5Ac2en structures are manually overlaid in PyMOL to allow the maximum overlap of the ring and C1 carboxyl of Neu5Ac2en.
Our recent substrate specificity studies of bacterial sialidases from C. perfringens, V. cholerae, S. typhimurium, and S. pneumoniae as well as human NEU2 using thirty α2–3/6-linked sialosides containing C9-modified sialic acid43 indicate that α2–3-linked sialoside containing Neu5Ac9N3 (Neu5Ac9N3α2–3GalβpNP) is a similarly good substrate as the sialoside containing non-modified Neu5Ac (Neu5Acα2–3GalβpNP) for bacterial sialidases. In contrast, α2–3-sialoside containing Neu5Gc9N3 (Neu5Gc9N3α2–3GalβpNP) is a similarly good or better substrate than sialoside containing non-modified Neu5Ac (Neu5Acα2–3GalβpNP) for human NEU2 and V. cholerae sialidase, but not for other bacterial sialidases tested including C. perfringens, S. typhimurium, and S. pneumoniae sialidases. Therefore, we hypothesized that Neu5Ac9N32en (3) would be a similarly good inhibitor as Neu5Ac2en against C. perfringens, V. cholerae, S. typhimurium, and S. pneumoniae bacterial sialidases but will be a weaker inhibitor, compared to Neu5Ac2en, for human NEU2. In addition, Neu5Gc9N32en (4) would be a similarly good inhibitor as Neu5Ac2en against human NEU2 and V. cholerae sialidase, but a worse inhibitor against C. perfringens, S. typhimurium, and S. pneumoniae sialidases. Furthermore, as we demonstrated before, 11 azido modification at C5-NHAc group on Neu5Ac2en provided an improved and selective inhibitor against human NEU2. Therefore, we predicted that combining the features of azido modifications at both C5-NHAc group and C9 of Neu5Ac2en will result in a selective inhibitor (Neu5AcN39N32en, 5) against human NEU2. Also, Neu5Ac9Me2en (6) with a methoxy at C9 of Neu5Ac2en may be a better or a similarly good inhibitor compared to Neu5Ac2en against S. typhimurium or S. pneumoniae sialidase based on our substrate specificity tests. To test these hypotheses, we synthesized a library of Neu5Ac2en analogues with modifications at C9 and both C5 and C9, including Neu5Ac9N32en (3), Neu5Gc9N32en (4), Neu5AcN39N32en (5), and Neu5Ac9Me2en (6) (Figure 3). In addition, a Neu5Ac2en derivative with a C9-propylamine modification (Neu5Ac9NPro2en, 7) was synthesized to test the effect of a larger C9-group in sialidase inhibitory activities.
Figure 3.
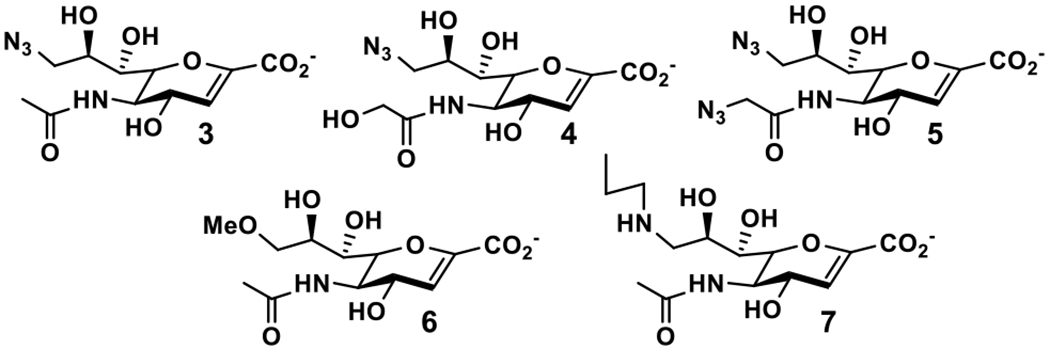
Structures of Neu5Ac9N32en 3, Neu5Gc9N32en 4, Neu5AcN39N32en 5, Neu5Ac9Me2en 6, and Neu5Ac9NPro2en 7.
Synthesis of sialidase inhibitors
Compounds 9-azido-9-deoxy-Neu5Ac2en (Neu5Ac9N32en, 3), 9-azido-9-deoxy-Neu5Gc2en (Neu5Gc9N32en, 4), 9-azido-5-(2-azidoacetamido)-5,9-dideoxy-Neu5Ac2en (Neu5AcN39N32en, 5), 9-O-methyl-Neu5Ac2en (Neu5Ac9Me2en, 6), and 9-aminopropyl-5,9-dideoxy-Neu5Ac2en (Neu5Ac9NPro2en, 7) were synthesized as shown in Scheme 1. Compound Neu5Ac9N32en (3) was generated using a reported method with optimization.27, 29 Neu5Gc9N32en (4) was synthesized from compound 8 by treating it with Boc2O44 to produce N-acetyl-N-Boc protected product 11, which was deacetyled under Zemplén condition to form 12. Selective tosylation of the primary hydroxyl group of 12 followed by azidation provided intermediate 13. After removal of the Boc group of 13 with TFA in water, the obtained amine was treated with acetoxyacetyl chloride in the presence of base DIPEA and coupling reagents HOBT and EDCI in CH2Cl2 followed by deprotection produced the desired compound Neu5Gc9N32en (4) in a high yield. To synthesize Neu5AcN39N32en (5), compound 13 was acetylated and the Boc protecting group was removed by the treatment with TFA in CH2Cl2. Coupling of the obtained amine with N3CH2COOH in the presence of EDCI/HOBT afforded compound 16 in a good yield. Deprotection of 16 followed by hydrolysis in aqueous NaOH afforded the desired product Neu5AcN39N32en (5). Compound Neu5Ac9Me2en (6) was obtained by base (NaOH in H2O)-catalyzed hydrolysis of compound 17 which was formed from intermediate 9 by gradual addition of a solution of NaOMe in MeOH to intermediate 9 followed by neutralization with Dowex (H+) resin to provide compound 17. Subsequent hydrolysis of 18 under basic condition provided Neu5Ac9Me2en (6). To prepare Neu5AcN39NPro2en (7), compound 10 was hydrogenated under atmospheric pressure to give amine 18. Reductive amination of 18 with propanal and subsequent demethylation provided the desired product 7 in a moderate yield.
Scheme 1.
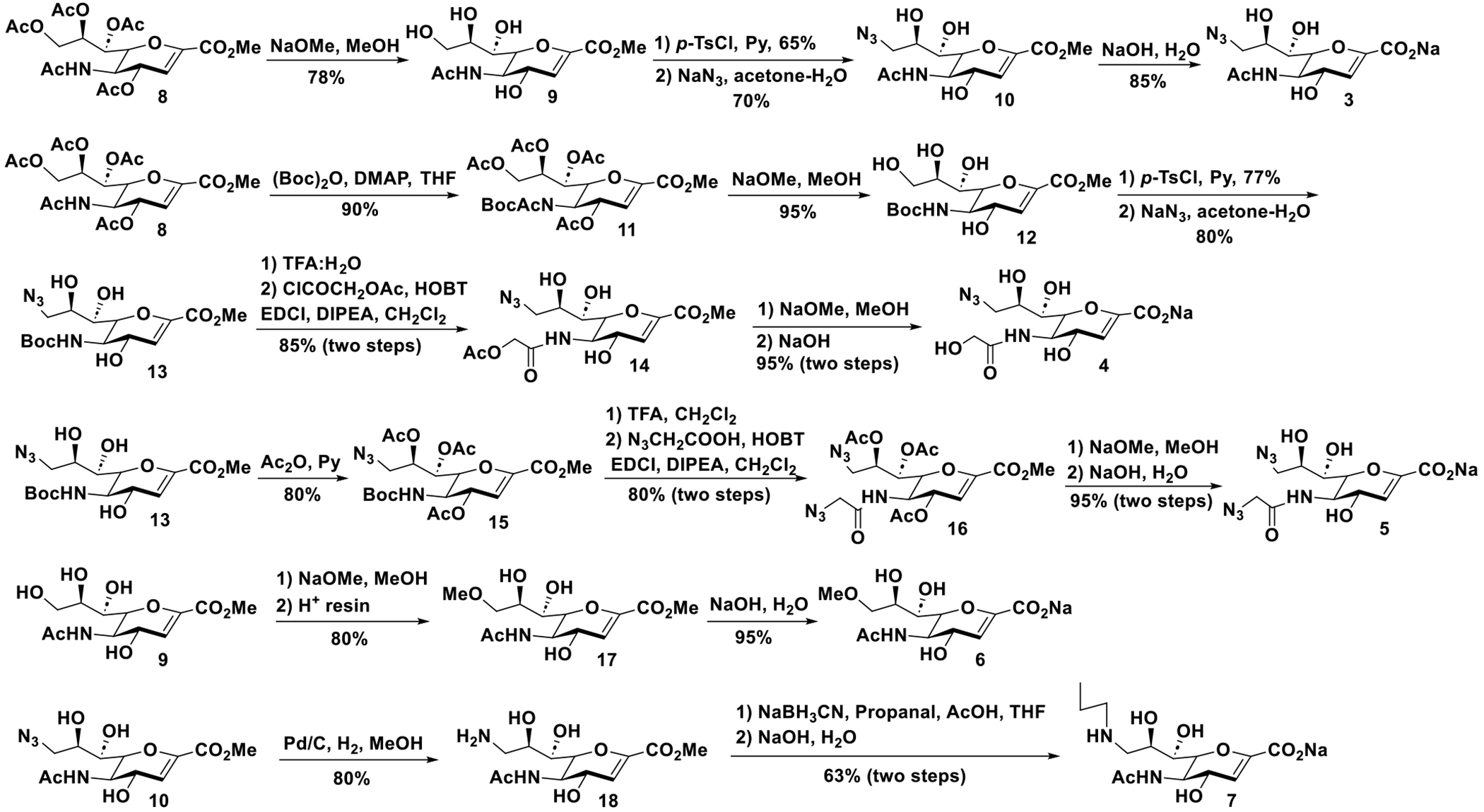
Synthesis of Neu5Ac2en derivatives.
Inhibition studies of bacterial sialidases and human NEU2
The IC50 values of the C9- as well as both C9- and C5-modified Neu5Ac2en derivatives 3–7 synthesized above were determined using human NEU2 and four commercially available bacterial sialidases from Salmonella typhimurium (Prozyme), Clostridium perfringens (Sigma), Vibrio cholerae (Sigma), and Streptococcus pneumoniae (NanB, Prozyme). Inhibition studies were conducted using Neu5Acα2–3GalBpNP or Neu5Acα2–6GalBpNP as sialidase substrate. As shown in Table 1, Neu5Ac9N32en (3) was a similarly effective inhibitor as Neu5Ac2en (1) against β2–3-specific S. typhimurium and S. pneumoniae sialidases as predicted. However, unlike the prediction, Neu5Ac9N32en (3) was 3-fold less efficient than Neu5Ac2en (1) against C. perfringens sialidase when either Neu5Acα2–3GalBpNP or Neu5Acα2–6GalBpNP was used as the substrate. For V. cholerae sialidase and human NEU2, Neu5Ac9N32en (3) was 2.3-fold or 2.4-fold less efficient than Neu5Ac2en (1) when Neu5Acα2–3GalBpNP was used as the substrate, while the IC50 values of these two inhibitors were similar when Neu5Acα2–6GalBpNP was used as the substrate. Neu5Ac9N32en (3) along with a series of C9-triazole derivatives of Neu5Ac2en have been tested previously as inhibitors against human NEU3.29 Neu5Ac9N32en (3) showed less inhibition activity against NEU3 than Neu5Ac2en (1). Nevertheless, slightly improved activity was observed using phenyl-, hexyl-, and phenoxymethyl-C9-triazole Neu5Ac2en derivatives (IC50 values were 20, 23, and 45 μM, respectively).29
| Sialidases | IC50 (μM) with Neu5Acα2–3GalβpNP as sialidase substrate | |||||
|---|---|---|---|---|---|---|
| Neu5Ac2en (1) | Neu5Ac9N32en (3) | Neu5Gc9N32en (4) | Neu5AcN39N32en (5) | Neu5Ac9Me2en (6) | Neu5Ac9NPro2en (7) | |
| C. perfringens | 20±1 | 59±5 | (1.9±0.4)×103 | (1.6±0.2)×103 | (4.6±0.6)×102 | >1×103 |
| V. cholerae | 8.6±1.0 [a] | 20±1 | 18±3 | (1.4±0.1)×102 | (1.3±0.3)×102 | >1×103 |
| S. typhimurium | (2.0±0.1)×102 [a] | (2.1±0.1)×102 | >1×104 | (1.6±0.4)×104 | (1.3±0.2)×102 | (2.3±0.3)×103 |
| S. pneumoniae | (3.7±0.6)×103 | (3.3±0.4)×103 | >1×104 | >1×103 | (3.0 ±0.6)×103 | (5.4±0.6)×103 |
| NEU2 | 18±1 [a] | 43 ±2 | (1.5±0.3)×102 | 13±4 | (1.1±0.1)×103 | >1×104 |
| IC50 (μM) with Neu5Acα2–6GalβpNP as sialidase substrate | ||||||
| C. perfringens | 13±1 | 40±4 | (1.4±0.4)×103 | (1.2±0.2) ×103 | (3.1±0.6)×102 | >1×103 |
| V. cholerae | 10±1 [a] | 8.7±1.0 | 13 ± 2 | (1.1±0.2)×102 | (1.2±0.3)×102 | >1×103 |
| NEU2 | 32±6 [a] | 31±6 | (1.9±0.7)×102 | 13± 3 | (4.1±0.6)×102 | (2.4±0.8)×103 |
Data are from Ref.11
As predicted from substrate specificity studies,43 Neu5Gc9N32en (4) was a weaker inhibitor than Neu5Ac2en (1) against C. perfringens, S. typhimurium, and S. pneumoniae sialidases. Disagreed with the prediction, Neu5Gc9N32en (4) was also a weaker inhibitor than Neu5Ac2en (1) against NEU2 and V. cholerae sialidase. Although with a mildly decreased IC50 value compared to Neu5Ac2en (1) in inhibiting V. cholerae sialidase, Neu5Gc9N32en (4) was shown to be a selective inhibitor against this enzyme.
Also as expected, Neu5AcN39N32en (5) was a selective inhibitor against human NEU2 with a slightly improved IC50 compared to Neu5Ac2en (1) when either Neu5Acα2–3GalβpNP or Neu5Acα2–6GalβpNP was used as the substrate. The inhibitory efficiency was comparable to other reported human sialidases inhibitors.27–29 These results indicate that Neu5AcN39N3-containing sialosides are most likely suitable substrates for human NEU2. A previous report using a library of octyl sialyllactosides with modifications at the Neu5Ac residue as sialidase substrates also showed that Neu5Ac with an azide at C5 or C9 could be cleaved by human membrane-associated sialidases NEU3.29
Neu5Ac9Me2en (6) and Neu5AcN39NPro2en (7) with a methoxy and a propylamine substitution, respectively, at C9 of Neu5Ac2en (1) were shown to be weak inhibitors against all five sialidases tested. The inhibitory activities of similar compounds as 7 with amide-linked C9-modified Neu5Ac2en analogues were studied against NEU1–NEU4.27 All the compounds were found to be poor inhibitors of the NEU2–NEU4. However, selective inhibitors with slightly improved inhibition compared to Neu5Ac2en were observed for those contain a methyl, propyl, butyl, or phenyl group.
Inhibition studies of compounds 1, 3, 4, 5, 6, and 7 against β-galactosidase were also carried out. Using 1 mM of GalβpNP as the substrate for β-galactosidase, no inhibitory effect was observed for any of the compounds with a final concentration of 100 µM or 1 mM (data not shown). This confirms that the selective inhibitory activities of Neu5Gc9N32en (4) and Neu5AcN39N32en (5) against V. cholerae sialidase and human NEU2, respectively, are not due to inhibition of the compounds against β-galactosidase in the assays.
Docking studies of compounds 3, 4, and 5 in the crystal structures of human NEU2 and V. cholerae sialidase
To help the understanding of the selectivity of Neu5Gc9N32en (4) and Neu5AcN39N32en (5) in inhibiting V. cholerae sialidase and human NEU2, respectively, structure of Neu5Ac9N32en (3), Neu5Gc9N32en (4), or Neu5AcN39N32en (5) was docked into the active site of human NEU2 and V. cholerae sialidase using FRED. Most docking programs are usually effective but highly inconsistent, while the pose of the ligand structure is predictable and reproducible and the virtual screening is accurate using docking program of FRED.45 Three structure-based scoring functions PLP (Piecewise Linear Potential),46 chemgauss3,47 and Oechemscore,48 implemented in the program FRED were used to optimize the multiple docked poses. The best scoring pose was selected as the final position of the ligand in the crystal of protein.
As shown in Figures 4A3 and 4A5, the C9-azido group in Neu5Ac9N32en (3) and Neu5AcN39N32en (5) fits well in the deep pocket of human NEU2 and points out towards the neighbouring cavity close to C9 of the inhibitors. However, replacing the C5-N-acetyl group in Neu5Ac9N32en (3) by the N-glycolyl group in Neu5Gc9N32en (4) causes a 180 degree flipping of the inhibitor (Figures 4A4 and S1). This may be due to the less favorable interaction between the inner neutral cavity (mainly composed of Met85, Ile103, Ile105, and Ala158) and the N-glycolyl. The bigger outside positive cavity (mainly contributed by Arg83) may allow more favorable interaction with the N-glycolyl group11 (Figure S1). Switching the positions of C5 and C9 groups may lead to lower inhibition efficiency of Neu5Gc9N32en (4) against human NEU2. Compared to Neu5Ac9N32en (3) in Figure 4A3 (IC50 = 43±2 μM when Neu5Acα2–3GalβpNP was used as sialidase substrate and IC50 = 31±6 μM when Neu5Acα2–6GalβpNP was used as sialidase substrate), Neu5AcN39N32en (5) with an N3-substitution at C5-N-acetyl group in Figure 4A5 fits the binding pocket of NEU2 better, which explains an improved IC50 value for human NEU2 by Neu5AcN39N32en (5) (IC50 = 13±4 μM when Neu5Acα2–3GalβpNP was used as sialidase substrate and IC50 = 13±3 μM when Neu5Acα2–6GalβpNP was used as sialidase substrate).
Figure 4.
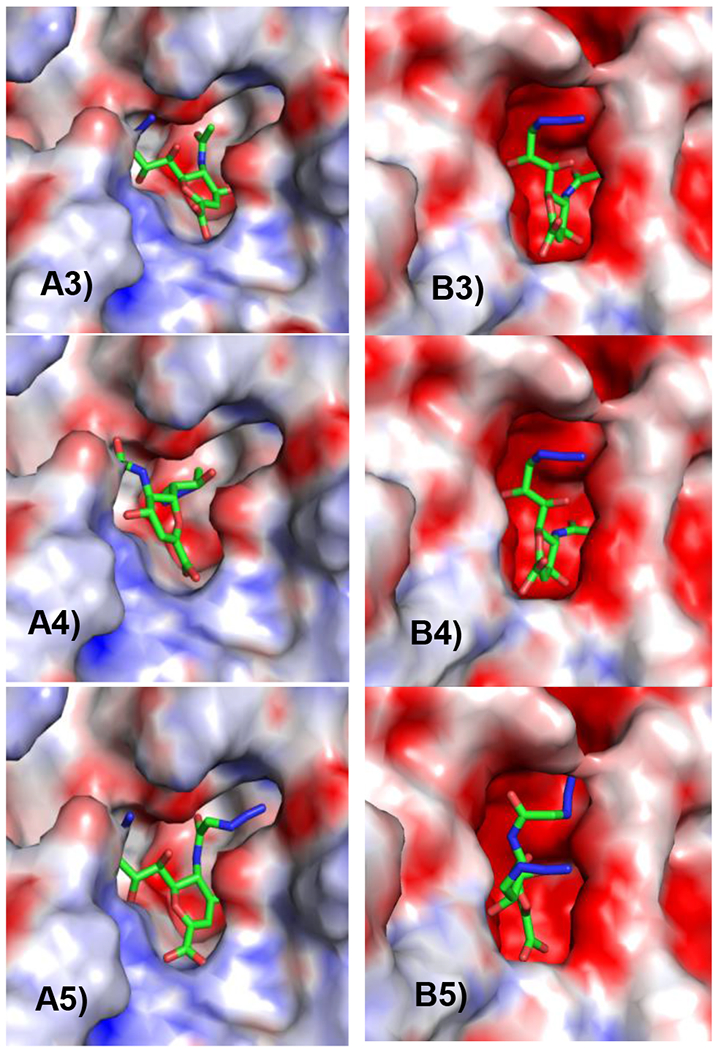
Docking studies of Neu5Ac9N32en (3), Neu5Gc9N32en (4), and Neu5AcN39N32en (5) in the crystal structures of human NEU2 (A) and V. cholerae sialidase (B). The surface is coloured according to the electrostatic potential. Positive potentials are shown in blue, negative potentials are in red, and neutral are in white.
For V. cholerae sialidase, both Neu5Ac9N32en (3) and Neu5Gc9N32en (4) fit the substrate binding pocket of the enzyme similarly well (Figures 4B3 and 4B4), which explains similar IC50 values of these two inhibitors. In comparison, the extra N3-substitution at C5-N-acetyl group in Neu5AcN39N32en (5) causes flipping and ill fitting of the inhibitor (Figures 4B5 and S1), which increases the IC50 values for about 10-fold when either Neu5Acα2–3GalβpNP or Neu5Acα2–6GalβpNP was used as sialidase substrate.
Conclusions
In conclusion, guided by sialidase substrate specificity studies and sialidase crystal structural analysis, several C9- or both C9- and C5-modified Neu5Ac2en analogues were synthesized and their inhibitory activities were tested. Neu5Gc9N32en (4) and Neu5AcN39N32en (5) were identified as selective inhibitors against V. cholerae sialidase and human NEU2, respectively, using Neu5Acα2–3GalβpNP or Neu5Acα2–6GalβpNP as the substrate for human cytosolic sialidase NEU2 and four commercially available bacterial sialidases. Nevertheless, their IC50 values were not improved compared to Neu5Ac2en. Docking of Neu5Ac9N32en (3), Neu5Gc9N32en (4), or Neu5AcN39N32en (5) to the crystal structures of human NEU2 and V. cholerae sialidase helped to illustrate the selectivity of sialidase inhibitors 4 and 5. Therefore, substrate specificity study of various sialidases is a valuable tool for identifying unique structural features of selective inhibitors. However, drastic improvement of inhibitory activity of bacterial and human sialidases will require more significant structural changes which could be facilitated by screening of focused compound libraries.
Experimental
Materials
All chemicals were obtained from commercial suppliers and used without further purification unless otherwise noted. Anhydrous solvents were used to carry out organic reactions under inert argon or nitrogen environment. Neu5Acα2–3GalβpNP and Neu5Acα2–6GalβpNP were synthesized as reported previously.49 1H NMR (400 and 600 MHz) and 13C NMR (151 MHz) spectra were recorded on a Varian Inova 400 and 600 MHz spectrometer. High resolution electrospray ionization (ESI) mass spectra were obtained at the Mass Spectrometry Facility in the University of California, Davis. Optical rotation values were recorded on an Autopol IV Automatic polarimeter at 589 nm wavelength. Infrared spectra were recorded on a PerkinElmer Spectrum 100 ATR-FTIR. Silica gel 60 Å (200–425 mesh, Fisher Chemical) was used for flash column chromatography. Thin-layer chromatography (TLC) was performed on silica gel plates 60 GF254 (Sorbent technologies) using anisaldehyde sugar stain for detection. Clostridium perfringens sialidase (type VI), Vibrio cholerae sialidase (type III), and β-galactosidase from Aspergillus oryzae were purchased from Sigma. Salmonella typhimurium sialidase and Streptococcus pneumoniae sialidase NanB were bought from Prozyme. All of these enzymes were used without further purification. NEU2 was expressed in E. coli and purified as described previously.17 384-Well plates for sialidase assays were from Fisher Biotech.
Chemical synthesis of sialidase inhibitors
5-Acetamido-2,6-anhydro-9-azido-3,5,9-trideoxy-D-glycero-D-galacto-non-2-enonic acid (Neu5Ac9N32en, 3)
To a suspension of 850 (1.12 g, 2.36 mmol) in dry MeOH (15 mL) was added a catalytic amount of NaOMe and the mixture was stirred at room temperature for 3 h. The reaction was neutralized with Dowex (H+) resin, filtrated, concentrated, and purified by silica gel flash column chromatography (EtOAc:MeOH:H2O = 8:1:0.5) to afford 9 (0.56 g, 78%). 1H NMR (600 MHz, D2O) δ 6.04 (s, 1H), 4.50 (d, J = 9.0 Hz, 1H), 4.27 (d, J = 11.4 Hz, 1H), 4.08 (t, J = 9.6 Hz, 1H), 3.91–3.85 (m, 2H), 3.81 (s, 3H), 3.66–3.63 (m, 2H), 2.06 (s, 3H); 13C NMR (151 MHz, D2O) δ 177.56, 167.08, 146.16, 115.30, 78.83, 72.67, 70.74, 69.79, 65.69, 55.64, 52.30, 24.82.
A stirred solution of 9 (0.56 g, 1.83 mmol) in anhydrous pyridine (10 mL) was treated with a solution of p-toluenesulfonyl chloride (0.52 g, 2.75 mmol) at 0 °C and the mixture was stirred for overnight at room temperature. The reaction was stopped by adding MeOH (2 mL) and concentrated. The compound was purified by column chromatography (EtOAc:MeOH = 9:1) to give 9-OTs Neu5Ac2en (0.54 g, 65%) which was then dissolved in water (10 mL) and acetone (10 mL) containing NaN3 (0.76 g, 11.75 mmol) and stirred at 70 °C for overnight. The mixture was concentrated and purified by silica gel flash column chromatography (EtOAc:MeOH = 9.5:0.5) to give product 10 (0.27 g, 70%). 1H NMR (600 MHz, D2O) δ 6.04 (s, 1H), 4.51 (d, J = 9.1 Hz, 1H), 4.27 (d, J = 8.4 Hz, 1H), 4.08–4.07 (m, 2H), 3.81 (s, 3H), 3.65–3.63 (m, 2H), 3.51 (s, 1H), 2.06 (s, 3H); 13C NMR (151 MHz, D2O) δ 177.56, 167.08, 146.16, 115.30, 78.83, 72.67, 70.74, 69.79, 65.69, 55.64, 52.30, 24.82.
Compound 10 (0.27 g, 0.82 mmol) was dissolved in an aqueous solution of NaOH (10 mL, 0.1 M) and stirred at room temperature for 1 h. The reaction was neutralized with Dowex (H+) resin, filtrated, concentrated, and purified using silica gel column chromatography (EtOAc:MeOH:H2O = 8:3:0.5) to give final product 3 (0.22 g, 85%). = +39.00 (c = 1.0 H2O). Wavenumbermax (film)/cm−1 3205 (m, broad, OH, NH, C–H alkene, COOH), 2101 (s, N3), 1646 (s, CO carboxylic acid), 1557 (m, CO amide, CC alkene), 1145 (s, C–N). 1H NMR (600 MHz, D2O) δ 5.67 (s, 1H, H-3), 4.41 (d, J = 8.4 Hz, 1H, H-6), 4.15 (d, J = 10.8 Hz, 1H, H-4), 4.03–3.97 (m, 2H, H-5 and H-7), 3.58–3.54 (m, 2H, H-8 and H-9), 3.44 (dd, J = 12.6, 5.4 Hz, 1H, H-9′), 2.00 (s, 3H, NHCOMe); 13C NMR (151 MHz, D2O) δ 174.83 (COONa), 169.16 (NHCOMe), 147.39 (C-2), 108.51 (C-3), 75.32 (C-6), 68.69 (2C, C-7 and C-8), 67.63 (C-4), 53.81 (C-5), 49.92 (C-9), 22.26 (NHCOMe). HRMS (ESI) calculated for C11H16N4NaO7 (M+H) 339.0917, found 339.0910.
2,6-Anhydro-9-azido-3,5,9-trideoxy-5-(2-hydroxyacetamido)-D-glycero-D-galacto-non-2-enonic acid (Neu5Gc9N32en, 4)
To a solution of compound 8 (2.47 g, 5.21 mmol) in dry THF (70 mL) was added (Boc)2O (4.55 g, 20.86 mmol) and 4-dimethylaminopyridine (DMAP) (0.19 g, 1.56 mmol) and the mixture was refluxed for 4 h. The reaction mixture was cooled to room temperature, diluted with CH2Cl2, washed with HCl (0.5 M), water, saturated NaHCO3, and water. The solution was dried, concentrated, and purified by flash column chromatography (EtOAc:Hexane = 3:7) to afford 11 (2.69 g, 90%). 1H NMR (600 MHz, CDCl3) δ 5.99 (s, 1H), 5.87 (d, J = 2.4 Hz, 1H), 5.25–4.60 (m, 5H), 4.12–4.04 (m, 2H), 3.75 (s, 3H), 2.32 (s, 3H), 2.01–1.95 (m, 12 H), 1.54 (s, 9H); 13C NMR (151 MHz, CDCl3) δ 170.75, 170.31, 169.93, 161.93, 146.46, 109.25, 85.38, 71.10, 67.63, 66.97, 62.03, 52.73, 50.55, 28.08, 20.95.
Compound 11 (2.69 g, 4.69 mmol) was dissolved in dry MeOH (30 mL) containing a catalytic amount of NaOMe. The mixture was stirred at room temperature for 2 h and neutralized using Dowex (H+) resin. The resulted suspension was filtrated and concentrated. Silica gel column chromatography (EtOAc:MeOH = 9.5:0.5) of the crude product afforded product 12 (1.62 g, 95%). 1H NMR (600 MHz, CD3OD) δ 5.91 (d, J = 1.8 Hz, 1H), 4.35 (dd, J = 8.4, 1.8 Hz, 1H), 4.11 (d, J = 10.8 Hz, 1H), 3.89–3.82 (m, 2H), 3.76 (s, 3H), 3.70–3.62 (m, 3H), 1.45 (s, 9H); 13C NMR (151 MHz, CD3OD) δ 164.41, 159.32, 145.13, 113.68, 81.01, 78.60, 71.11, 70.19, 67.94, 64.98, 52.75, 52.52, 28.69.
A stirred solution of 12 (1.62 g, 4.46 mmol) in anhydrous pyridine (50 mL) was treated with a solution of p-toluenesulfonyl chloride (2.55 g, 13.37 mmol) at 0 °C and the mixture was stirred for overnight at room temperature. The reaction was stopped by adding MeOH (3 mL) and concentrated. Purification of the residue by flash column chromatography (EtOAc:Hexane = 7:3) afforded 9-tosylated product (1.77 g, 77%) which was then dissolved in water (15 mL) and acetone (15 mL) containing NaN3 (2.22 g, 34.20 mmol) and stirred at 70 °C for overnight. The mixture was concentrated and purified by silica gel flash column chromatography (EtOAc:Hexane = 7:3) to give compound 13 (1.06 g, 80%). 1H NMR (600 MHz, CD3OD) δ 5.83 (d, J = 3.0 Hz, 1H), 4.28 (dd, J = 9.0, 3.0 Hz, 1H), 4.01–3.95 (m, 2H), 3.68 (s, 3H), 3.59 (dd, J = 10.8, 8.4 Hz, 1H), 3.55 (d, J = 9.0 Hz, 1H), 3.46 (dd, J = 12.6, 2.4 Hz, 1H), 3.35 (dd, J = 13.2, 6.0 Hz, 1H), 1.38 (s, 9H); 13C NMR (151 MHz, CD3OD) δ 164.31, 159.35, 144.99, 113.82, 81.10, 78.33, 70.53, 70.08, 67.79, 55.67, 52.81, 52.49, 28.65. Compound 13 (1.06 g, 2.73 mmol) was dissolved in a mixture of CH2Cl2 (30 mL) and an aqueous solution of trifluoroacetic acid (90%, 10 mL) and stirred at room temperature for 30 min. The mixture was concentrated, coevaporated with toluene, and dried. To a solution of the resulted mixture in CH2Cl2 (20 mL) was added hydroxybenzotriazole (HOBT) (0.44 g, 1.2 mmol), 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDCI) (0.51 g, 1.2 mmol), and N,N-diisopropylethylamine (DIPEA) (0.57 mL, 1.2 mmol) at 0 °C. Acetoxyacetyl chloride (0.23 mL, 2.12 mmol) was added drop-wisely and the reaction mixture was stirred at room temperature for 2.5 h. The reaction solution was concentrated and diluted with CH2Cl2, washed with water, saturated NaHCO3, and water then dried and concentrated. Silica gel column chromatography (EtOAc:MeOH:H2O = 9:2:0.5) afforded compound 14 (0.91 g, 85%). To a suspension of 14 (0.91 g, 2.32 mmol) in dry MeOH (10 ml) was added NaOMe (40 mg) and the mixture was stirred for 1 h at room temperature. Water was then added to the solution and pH was adjusted to about 9 by adding NaOMe. The reaction mixture was stirred at room temperature for 1 h. The solution was neutralized with Dowex (H+) resin, filtrated, concentrated, and purified by silica gel flash column chromatography (EtOAc:MeOH:H2O = 7.5:3:0.5) to afford product 4 (0.77 g, 95%). = +16.50 (c = 1.0 H2O). Wavenumbermax (film)/cm−1 3184 and 3058 (s, broad, OH, NH, C–H alkene, COOH), 2101 (s, N3), 1665 (s, CO carboxylic acid), 1570 (s, CO amide, CC alkene), 1153 (s, C–N). 1H NMR (400 MHz, D2O) δ 5.69 (d, J = 1.6 Hz, 1H, H-3), 4.55 (dd, J = 6.4, 1.6 Hz, 1H, H-6), 4.31 (d, J = 7.2 Hz, 1H, H-4), 4.15–4.09 (m, 4H, NHCOCH2OH, H-5, and H-7), 3.66–3.59 (m, 2H, H-9 and H-8), 3.50 (dd, J = 8.8, 4.0 Hz, 1H, H-9′); 13C NMR (151 MHz, D2O) δ 175.75 (COONa), 165.58 (NHCOCH2OH), 143.72 (C-2), 112.86 (C-3), 75.78 (C-6), 68.87 (C-7), 68.47 (C-8), 67.19 (C-4), 61.18 NHCOCH2OH), 53.81 (C-5), 49.37 (C-9). HRMS (ESI) calculated for C11H16N4NaO8 (M+H) 355.0866, found 355.0858.
2,6-Anhydro-9-azido-5-(2-azidoacetamido)-3,5,9-trideoxy-D-glycero-D-galacto-non-2-enonic acid (Neu5AcN39N32en, 5)
To a solution of compound 13 (0.21 g, 0.56 mmol) in pyridine (10 mL) was added acetic anhydride (0.7 mL) at 0 °C and the mixture was stirred at room temperature for overnight. The solvent was removed under diminished pressure and the crude product was purified using silica gel column chromatography (EtOAc:Hexane = 3:7) to give compound 15 (0.22 g, 80%). 1H NMR (600 MHz, CDCl3) δ 5.93 (d, J = 4.2 Hz, 1H), 5.53–5.48 (m, 1H), 5.20–5.17 (m, 1H), 4.79 (d, J = 15.6 Hz, 1H), 4.29 (dd, J = 15.0, 4.2 Hz, 1H), 4.10–4.01 (m, 1H), 3.89 (dd, J = 20.4, 3.6 Hz, 1H), 3.78 (s, 3H), 3.45 (dd, J = 20.4, 12.0 Hz, 1H), 2.11 (s, 3H), 2.07 (s, 3H), 2.05 (s, 3H), 1.37 (s, 9H); 13C NMR (151 MHz, CDCl3) δ 170.97, 170.57, 169.96, 161.72, 155.13, 145.10, 108.64, 80.51, 77.36, 72.93, 69.21, 68.34, 52.71, 50.29, 47.93, 28.20, 20.99, 20.91, 20.79.
To a solution of compound 15 (0.22 g, 0.43 mmol) in CH2Cl2 (6 mL) was added 90% aqueous trifluoroacetic acid (2 mL) at 0 °C. The reaction mixture was stirred at room temperature for 30 min. It was then concentrated, co-evaporated with toluene, and dried under vacuum. To the resulted ammonium salt in CH2Cl2 (3 mL) was added N3CH2COOH (52 μL, 0.51 mmol), HOBT (0.08 g, 0.51 mmol), EDCI (0.10 g, 0.51 mmol), and DIPEA (0.22 mL, 1.29 mmol) at 0 °C and the reaction mixture was stirred at room temperature for 2 h. The mixture was diluted with CH2Cl2, washed with water, saturated NaHCO3, and water then dried and concentrated. Silica gel column chromatography (EtOAc:Hexane = 4:6) afforded compound 16 (0.17 g, 80%). 1H NMR (600 MHz, CDCl3) δ 6.79 (d, J = 9.6 Hz, 1H), 5.93 (d, J = 1.8 Hz, 1H), 5.63 (dd, J = 8.4, 3.0 Hz, 1H), 5.47 (t, J = 3.0 Hz, 1H), 5.17–5.15 (m, 1H), 4.46–4.39 (m, 2H), 3.93 (dd, J = 13.2, 1.8 Hz, 1H), 3.83 (d, J = 5.4 Hz, 2 H), 3.79 (s, 3H), 3.46 (dd, J = 13.8, 8.4 Hz, 1H), 2.13 (s, 3H), 2.10 (s, 3H), 2.07 (s, 3H); 13C NMR (151 MHz, CDCl3) δ 171.28, 170.97, 170.50, 167.54, 161.54, 145.32, 108.68, 77.17, 73.41, 68.75, 68.03, 52.78, 52.61, 50.20, 46.41, 20.88.
To a suspension of 16 (0.17 g, 0.35 mmol) in dry MeOH (10 ml) was added NaOMe (40 mg) and the mixture was stirred for 2 h at room temperature. After adding water to the reaction mixture, the pH was adjusted to 9 and the solution was stirred for 1 h at room temperature. The reaction was neutralized with Dowex (H+) resin, filtrated, concentrated, and purified by silica gel flash column chromatography (EtOAc:MeOH:H2O = 8:3:0.5) to afford 5 (0.12 g, 95%). = +35.50 (c = 1.04 H2O). Wavenumbermax (film)/cm−1 3293 (s, broad, OH, NH, C–H alkene, COOH), 2109 (s, N3), 1596 (s, CO carboxylic acid), 1542 (s, CO amide, CC alkene), 1072 (s, C–N). 1H NMR (600 MHz, D2O) δ 5.68 (d, J = 1.8 Hz, 1H, H-3), 4.50 (dd, J = 8.4, 3.0 Hz, 1H, H-6), 4.28 (d, J = 11.4 Hz, 1H, H-4), 4.14–4.07 (m, 4H, NHCOCH2N3, H-5, and H-7), 3.63 (dd, J = 13.2, 2.4 Hz, 1H, H-9), 3.59 (d, J = 9.6 Hz, 1H, H-8), 3.49 (dd, J = 13.2, 6.0 Hz, 1H, H-9′); 13C NMR (151 MHz, D2O) δ 171.08 (COONa), 169.65 NHCOCH2N3), 148.04 (C-2), 107.91 (C-3), 75.03 (C-6), 68.76 (2C, C-7 and C-8), 67.65 (C-4), 53.86 (NHCOCH2N3), 52.15 (C-5), 50.13 (C-9). HRMS (ESI) calculated for C11H15N7NaO7 (M+H) 380.0931, found 380.0924.
5-Acetamido-2,6-anhydro-3,5-dideoxy-9-O-methyl-D-glycero-D-galacto-non-2-enonic acid (Neu5Ac9Me2en, 6)
A solution of NaOMe (0.17 g, 3.14 mmol) in MeOH (5 mL) was prepared. A 2 mL portion of the prepared solution was added to compound 9 (0.22 g, 0.48 mmol) and stirred at room temperature for 20 min. The second portion of NaOMe in MeOH (2 mL) was added to the reaction mixture and stirred at room temperature for 30 min. Dowex (H+) resin was added to the solution to adjust the pH to about 2 and the mixture was stirred at room temperature for 2 h. The reaction was neutralized using Et3N, filtrated, and washed with MeOH and H2O. The concentrated crude product was purified by silica gel flash column chromatography (EtOAc:MeOH = 9:1) to afford compound 17 (0.12 g, 80%). 1H NMR (600 MHz, D2O) δ 6.01 (d, J = 2.4 Hz, 1H), 4.47 (dd, J = 8.4, 2.4 Hz, 1H), 4.25 (d, J = 10.8 Hz, 1H), 4.06 (dd, J = 10.8, 9.0 Hz, 1H), 3.99–3.97 (m, 1H), 3.79 (s, 3H), 3.69 (dd, J = 10.8, 2.4 Hz, 1H), 3.61 (d, J = 9.0 Hz, 1H), 3.55 (dd, J = 10.8, 6.0 Hz, 1H), 3.69 (s, 3H), 2.03 (s, 3H); 13C NMR (151 MHz, D2O) δ 174.79, 164.40, 143.51, 112.69, 76.12, 73.75, 68.39, 68.09, 67.26, 58.65, 53.04, 49.67, 22.23.
Compound 17 (0.12 g, 0.37 mmol) was dissolved in an aqueous solution of NaOH (10 mL, 0.05 M) and stirred at room temperature for 30 min. The solution was neutralized with Dowex (H+) resin, filtrated, concentrated, and purified by silica gel flash column chromatography (EtOAc:MeOH:H2O = 8:3:0.7) to afford product 6 (107 mg, 95%). = +25.30 (c = 1.0 H2O). Wavenumbermax (film)/cm−1 3278 (s, broad, OH, NH, C–H alkene, COOH), 1590 (s, broad, CO carboxylic acid, CO amide, CC alkene), 1092 (s, C–N). 1H NMR (600 MHz, D2O) δ 5.69 (d, J = 2.4 Hz, 1H, H-3), 4.45 (dd, J = 9.0, 2.4 Hz, 1H, H-6), 4.20 (d, J = 11.4 Hz, 1H, H-4), 4.07–4.03 (m, 2H, H-5 and H-7), 3.73 (dd, J = 12.0, 2.4 Hz, 1H, H-9), 3.61–3.57 (m, 2H, H-8 and H-9′), 3.40 (s, 3H, OMe), 2.06 (s, 3H, NHCOMe); 13C NMR (151 MHz, D2O) δ 174.85 (COONa), 169.72 (NHCOMe), 148.01 (C-2), 107.91 (C-3), 75.40 (C-6), 73.91 (C-9), 68.41 (C-7), 68.28 (C-8), 67.79 (C-4), 58.66 (OMe), 50.01 (C-5), 22.30 (NHCOMe). HRMS (ESI) calculated for C12H19NNaO8 (M+H) 328.1008, found 328.1005.
5-Acetamido-9-aminopropyl-2,6-anhydro-3,5,9-trideoxy-D-glycero-D-galacto-non-2-enonic acid (Neu5Ac9NPro2en, 7)
Compound 10 (0.33 g, 1.0 mmol) was dissolved in MeOH (20 mL) and Pd/C (100 mg) was added. The resulted mixture was hydrogenated for 1.5 h with stirring under H2 at atmospheric pressure and then the catalyst was removed by filtration over Celite and washed with MeOH and water. The concentrated product was purified by flash column chromatography (EtOAc:MeOH:H2O = 5:4:3) to afford 18 (0.26 g, 80%). To compound 18 (42 mg, 0.14 mmol) in dry THF (15 mL) was added NaBH3CN (52 mg, 0.83 mmol), acetic acid to adjust pH to about 5, and propanal (50 μL, 0.7 mmol). The mixture was stirred at room temperature for overnight. The mixture was concentrated and purified by silica gel flash column chromatography (EtOAc:MeOH:H2O = 5:3:1) to afford the target compound intermediate (33.5 mg, 70%) which was then dissolved in an aqueous solution of NaOH (10 mL, 0.05 M) and stirred at room temperature for 30 min. The solution was neutralized with Dowex (H+) resin, filtrated, concentrated, and purified by silica gel flash column chromatography (EtOAc:MeOH:H2O = 5:3:1) to afford product 7 (30.8 mg, 90%). = +33.20 (c = 1.0 H2O). Wavenumbermax (film)/cm−1 3255 (s, broad, OH, NH, C–H alkene, COOH), 1583 (s, broad, CO carboxylic acid, CO amide, CC alkene), 1148 (s, C–N). 1H NMR (600 MHz, D2O) δ 5.70 (d, J = 1.8 Hz, 1H, H-3), 4.48 (dd, J = 9.0, 2.4 Hz, 1H, H-6), 4.33 (t, J = 9.0 Hz, 1H, H-4), 4.21 (d, J = 10.8 Hz, 1H, H-5), 4.05–4.02 (m, 1H, H-7), 3.57–3.54 (m, 2H, H-8 and H-9), 3.21–3.17 (m, 3H, CH2-1 propyl, H-9′), 2.07 (s, 3H, NHCOMe), 1.78–1.72 (m, 2H, CH2-2 propyl), 0.97 (t, J = 7.8 Hz, 3H, CH3 propyl); 13C NMR (151 MHz, D2O) δ 175.07 (COOH), 169.77 (NHCOMe), 148.06 (C-2), 108.08 (C-3), 75.20 (C-6), 70.57 (C-7), 67.61 (C-8), 64.52 (C-4), 56.17 (C-5), 55.39 (C-9), 50.00 (CH2-1 propyl), 22.26 (NHCOMe), 16.92 (CH2-2 propyl), 10.31 (CH3 propyl). HRMS (ESI) calculated for C14H24N2O7 (M+H) 333.1662, found 333.1670.
Inhibition assays of bacteria sialidases and human NEU2
All sialidase assays were carried out at 37 °C in duplicates in 384-well plates in a final volume of 20 μL containing Neu5Acα2–3GalβpNP or Neu5Acα2–6GalβpNP (0.3 mM), and the β-galactosidase (12 μg, 126 mU) with or without inhibitors. The amount of the β-galactosidase required to completely hydrolyze the GalβpNP within the time frame of the assay was predetermined and confirmed by assays with GalβpNP (0.3 mM). The assay conditions for various sialidases were as follows: C. perfringens sialidase (1.6 mU), MES buffer (100 mM, pH 5.0); V. cholerae sialidase (0.5 mU), NaCl (150 mM), CaCl2 (10 mM), and sodium acetate buffer (100 mM, pH 5.5); S. typhimurium sialidase (0.6 mU), NaCl (100 mM) and sodium acetate buffer (100 mM, pH 5.5); S. pneumoniae sialidase NanB (0.2 mU), sodium acetate buffer (100 mM, pH 6.0); NEU2 (6 μg for Neu5Acα2–3GalβpNP and 9.6 μg for Neu5Acα2–6GalβpNP), MES buffer (100 mM, pH 5.0). The reactions were carried out for 30 min. The assay was stopped by adding CAPS buffer (N-cyclohexyl-3-aminopropane sulfonic acid, 40 μL, 0.5 M, pH 10.5). The amount of the para-nitrophenolate formed was determined by measuring the A405 nm of the reaction mixtures using a microplate reader. Reactions without inhibitors were used as controls.
IC50 values were obtained by varying the concentration of inhibitors from 0 to 50 mM (twelve concentrations of each inhibitor were used to determine an IC50) with a fixed concentration (0.3 mM) of Neu5Acα2–3GalβpNP or Neu5Acα2–6GalβpNP. Typical concentration-response plots were obtained from the average values of duplicate assay results. The values of IC50 were obtained by fitting the averages to the Michaelis-Menten equation using Grafit 5.0.
Docking studies
The monomeric structure of NEU2 was modelled based on the dimeric structure of NEU2 (pdb: 1VCU).13 Neu5Ac9N32en (3), Neu5Gc9N32en (4), or Neu5AcN39N32en (5) was docked into the active sites of human NEU2 and Vibrio cholerae sialidase (pdb: 1W0O), respectively, using molecular building and optimizing software Gaussview 5.0 and Gaussview 09W and docking software OpenEye. The designation of the protein active site was performed using Fred Receptor 2.2.5. Docking the molecule into the designated active site was achieved using Fred as well as 3D displaying and analyzing software Vida 4.0.0. The electrostatic potential maps were built by PyMOL.
Supplementary Material
Acknowledgements
This work was supported by NIH grant 1R43AI092874. X.C. is a Camille Dreyfus Teacher-Scholar and a UC-Davis Chancellor’s Fellow.
Footnotes
Electronic Supplementary Information (ESI) available: 1H and 13C NMR spectra for compound 4–8 . See DOI: 10.1039/b000000x/
Notes and References
- 1.Taylor G, Curr. Opin. Struct. Biol, 1996, 6, 830. [DOI] [PubMed] [Google Scholar]
- 2.Buschiazzo A and Alzari PM, Curr. Opin. Chem. Biol, 2008, 12, 565. [DOI] [PubMed] [Google Scholar]
- 3.Stummeyer K, Dickmanns A, Muhlenhoff M, Gerardy-Schahn R and Ficner R, Nat. Struct. Mol. Biol, 2005, 12, 90. [DOI] [PubMed] [Google Scholar]
- 4.Cantarel BL, Coutinho PM, Rancurel C, Bernard T, Lombard V and Henrissat B, Nucleic Acids Res, 2009, 37, D233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kuro-o M, Biochim. Biophys. Acta, 2009, 1790, 1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Camilli TC, Xu M, O’Connell MP, Chien B, Frank BP, Subaran S, Indig FE, Morin PJ, Hewitt SM and Weeraratna AT, Pigment Cell Melanoma Res, 2011, 24, 175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cheng J, Yu H, Lau K, Huang S, Chokhawala HA, Li Y, Tiwari VK and Chen X, Glycobiology, 2008, 18, 686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cheng J, Huang S, Yu H, Li Y, Lau K and Chen X, Glycobiology, 2010, 20, 260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yu H, Chokhawala H, Karpel R, Yu H, Wu B, Zhang J, Zhang Y, Jia Q and Chen X, J. Am. Chem. Soc, 2005, 127, 17618. [DOI] [PubMed] [Google Scholar]
- 10.Yamamoto T, Hamada Y, Ichikawa M, Kajiwara H, Mine T, Tsukamoto H and Takakura Y, Glycobiology, 2007, 17, 1167. [DOI] [PubMed] [Google Scholar]
- 11.Li Y, Cao H, Yu H, Chen Y, Lau K, Qu J, Thon V, Sugiarto G and Chen X, Mol. BioSyst, 2011, 7, 1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen X and Varki A, ACS Chem. Biol, 2010, 5, 163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chavas LM, Tringali C, Fusi P, Venerando B, Tettamanti G, Kato R, Monti E and Wakatsuki S, J. Biol. Chem, 2005, 280, 469. [DOI] [PubMed] [Google Scholar]
- 14.Vonitzstein M, Wu WY, Kok GB, Pegg MS, Dyason JC, Jin B, Phan TV, Smythe ML, White HF, Oliver SW, Colman PM, Varghese JN, Ryan DM, Woods JM, Bethell RC, Hotham VJ, Cameron JM and Penn CR, Nature, 1993, 363, 418. [DOI] [PubMed] [Google Scholar]
- 15.von Itzstein M, Nat. Rev. Drug Discov, 2007, 6, 967. [DOI] [PubMed] [Google Scholar]
- 16.Mann MC, Thomson RJ, Dyason JC, McAtamney S and von Itzstein M, Bioorg. Med. Chem, 2006, 14, 1518. [DOI] [PubMed] [Google Scholar]
- 17.Diven WF, Doyle WJ and Vietmeier B, Ann. Oto. Rhinol. Laryn, 1988, 97, 6. [DOI] [PubMed] [Google Scholar]
- 18.Kawano M, Bando H, Yuasa T, Kondo K, Tsurudome M, Komada H, Nishio M and Ito Y, Virology, 1990, 174, 308. [DOI] [PubMed] [Google Scholar]
- 19.Corfield T, Glycobiology, 1992, 2, 509–521. [DOI] [PubMed] [Google Scholar]
- 20.Albohy A, Li MD, Zheng RB, Zou C and Cairo CW, Glycobiology, 2010, 20, 1127. [DOI] [PubMed] [Google Scholar]
- 21.Monti E, Bassi MT, Bresciani R, Civini S, Croci GL, Papini N, Riboni M, Zanchetti G, Ballabio A, Preti A, Tettamanti G, Venerando B and Borsani G, Genomics, 2004, 83, 445. [DOI] [PubMed] [Google Scholar]
- 22.Caciotti A, Di Rocco M, Filocamo M, Grossi S, Traverso F, d’Azzo A, Cavicchi C, Messeri A, Guerrini R, Zammarchi E, Donati MA and Morrone A, J. Neurol, 2009, 256, 1911. [DOI] [PubMed] [Google Scholar]
- 23.Colman PM, Varghese JN and Laver WG, Nature, 1983, 303, 41. [DOI] [PubMed] [Google Scholar]
- 24.Mitrasinovic PM, Curr. Drug Targets, 2010, 11, 315. [DOI] [PubMed] [Google Scholar]
- 25.Varghese JN, McKimm-Breschkin JL, Caldwell JB, Kortt AA and Colman PM, Proteins, 1992, 14, 327. [DOI] [PubMed] [Google Scholar]
- 26.Kim CU, Lew W, Williams MA, Liu HT, Zhang LJ, Swaminathan S, Bischofberger N, Chen MS, Mendel DB, Tai CY, Laver WG and Stevens RC, J. Am. Chem. Soc, 1997, 119, 681. [DOI] [PubMed] [Google Scholar]
- 27.Magesh S, Moriya S, Suzuki T, Miyagi T, Ishida H and Kiso M, Bioorg. Med. Chem. Lett, 2008, 18, 532. [DOI] [PubMed] [Google Scholar]
- 28.Magesh S, Savita V, Moriya S, Suzuki T, Miyagi T, Ishida H and Kiso M, Bioorg. Med. Chem, 2009, 17, 4595. [DOI] [PubMed] [Google Scholar]
- 29.Zou Y, Albohy A, Sandbhor M and Cairo CW, Bioorg. Med. Chem. Lett, 2010, 20, 7529. [DOI] [PubMed] [Google Scholar]
- 30.Albohy A, Mohan S, Zheng RB, Pinto BM and Cairo CW, Bioorg. Med. Chem, 2011, 19, 2817. [DOI] [PubMed] [Google Scholar]
- 31.Mann MC, Thomson RJ, Dyason JC, McAtamney S and von Itzstein M, Bioorg. Med. Chem, 2006, 14, 1518. [DOI] [PubMed] [Google Scholar]
- 32.Nguyen PH, Nguyen TN, Kang KW, Ndinteh DT, Mbafor JT, Kim YR and Oh WK, Bioorg. Med. Chem, 2010, 18, 3335. [DOI] [PubMed] [Google Scholar]
- 33.Hinou H and Nishimura S, Curr. Top. Med. Chem, 2009, 9, 106. [DOI] [PubMed] [Google Scholar]
- 34.Russell RJ, Haire LF, Stevens DJ, Collins PJ, Lin YP, Blackburn GM, Hay AJ, Gamblin SJ and Skehel JJ, Nature, 2006, 443, 45. [DOI] [PubMed] [Google Scholar]
- 35.Hinou H, Miyoshi R, Takasu Y, Kai H, Kurogochi M, Arioka S, Gao XD, Miura N, Fujitani N, Omoto S, Yoshinaga T, Fujiwara T, Noshi T, Togame H, Takemoto H and Nishimura S, Chem. Asian J, 2011, 6, 1048. [DOI] [PubMed] [Google Scholar]
- 36.Cao H, Li Y, Lau K, Muthana S, Yu H, Cheng J, Chokhawala HA, Sugiarto G, Zhang L and Chen X, Org. Biomol. Chem, 2009, 7, 5137. [DOI] [PubMed] [Google Scholar]
- 37.Crennell S, Garman E, Laver G, Vimr E and Taylor G, Structure, 1994, 2, 535. [DOI] [PubMed] [Google Scholar]
- 38.Xu G, Potter JA, Russell RJ, Oggioni MR, Andrew PW and Taylor GL, J. Mol. Biol, 2008, 384, 436. [DOI] [PubMed] [Google Scholar]
- 39.Xu G, Li X, Andrew PW and Taylor GL, Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun, 2008, 64, 772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Newstead SL, Potter JA, Wilson JC, Xu G, Chien CH, Watts AG, Withers SG and Taylor GL, J. Biol. Chem, 2008, 283, 9080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Crennell SJ, Garman EF, Philippon C, Vasella A, Laver WG, Vimr ER and Taylor GL, J. Mol. Biol, 1996, 259, 264. [DOI] [PubMed] [Google Scholar]
- 42.Xu X, Zhu X, Dwek RA, Stevens J and Wilson IA, J. Virol, 2008, 82, 10493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Khedri Z, Muthana MM, Li Y, Muthana SM, Yu H, Cao H and Chen X, Chem. Commun, 2012, 48, 3357. [DOI] [PubMed] [Google Scholar]
- 44.Gregar TQ and Gervay-Hague J, J. Org. Chem, 2004, 69, 1001. [DOI] [PubMed] [Google Scholar]
- 45.McGann M, J. Chem. Inf. Model, 2011, 51, 578. [DOI] [PubMed] [Google Scholar]
- 46.Verkhivker GM, Bouzida D, Gehlhaar DK, Rejto PA, Arthurs S, Colson AB, Freer ST, Larson V, Luty BA, Marrone T and Rose PW, J. Comput. Aided Mo.l Des, 2000, 14, 731. [DOI] [PubMed] [Google Scholar]
- 47.McGann MR, Almond HR, Nicholls A, Grant JA and Brown FK, Biopolymers, 2003, 68, 76. [DOI] [PubMed] [Google Scholar]
- 48.Eldridge MD, Murray CW, Auton TR, Paolini GV and Mee RP, J. Comput. Aided Mol. Des, 1997, 11, 425. [DOI] [PubMed] [Google Scholar]
- 49.Chokhawala HA, Yu H and Chen X, Chembiochem, 2007, 8, 194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ercegovic T and Magnusson G, J Org Chem, 1995, 60, 3378–3384. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.