

Abstract
Background
Infection of burn wounds is a serious problem because it can delay healing, increase scarring and invasive infection may result in the death of the patient. Antibiotic prophylaxis is one of several interventions that may prevent burn wound infection and protect the burned patient from invasive infections.
Objectives
To assess the effects of antibiotic prophylaxis on rates of burn wound infection.
Search methods
In January 2013 we searched the Wounds Group Specialised Register; The Cochrane Central Register of Controlled Trials (CENTRAL); Ovid MEDLINE; Ovid MEDLINE ‐ In‐Process & Other Non‐Indexed Citations (2013); Ovid EMBASE; EBSCO CINAHL and reference lists of relevant articles. There were no restrictions with respect to language, date of publication or study setting.
Selection criteria
All randomised controlled trials (RCTs) that evaluated the efficacy and safety of antibiotic prophylaxis for the prevention of BWI. Quasi‐randomised studies were excluded.
Data collection and analysis
Two review authors independently selected studies, assessed the risk of bias, and extracted relevant data. Risk ratio (RR) and mean difference (MD) were estimated for dichotomous data and continuous data, respectively. When sufficient numbers of comparable RCTs were available, trials were pooled in a meta‐analysis to estimate the combined effect.
Main results
This review includes 36 RCTs (2117 participants); twenty six (72%) evaluated topical antibiotics, seven evaluated systemic antibiotics (four of these administered the antibiotic perioperatively and three administered upon hospital admission or during routine treatment), two evaluated prophylaxis with non absorbable antibiotics, and one evaluated local antibiotics administered via the airway.
The 11 trials (645 participants) that evaluated topical prophylaxis with silver sulfadiazine were pooled in a meta analysis. There was a statistically significant increase in burn wound infection associated with silver sulfadiazine compared with dressings/skin substitute (OR = 1.87; 95% CI: 1.09 to 3.19, I2 = 0%). These trials were at high, or unclear, risk of bias. Silver sulfadiazine was also associated with significantly longer length of hospital stay compared with dressings/skin substitute (MD = 2.11 days; 95% CI: 1.93 to 2.28).
Systemic antibiotic prophylaxis in non‐surgical patients was evaluated in three trials (119 participants) and there was no evidence of an effect on rates of burn wound infection. Systemic antibiotics (trimethoprim‐sulfamethoxazole) were associated with a significant reduction in pneumonia (only one trial, 40 participants) (RR = 0.18; 95% CI: 0.05 to 0.72) but not sepsis (two trials 59 participants) (RR = 0.43; 95% CI: 0.12 to 1.61).
Perioperative systemic antibiotic prophylaxis had no effect on any of the outcomes of this review.
Selective decontamination of the digestive tract with non‐absorbable antibiotics had no significant effect on rates of all types of infection (2 trials, 140 participants). Moreover, there was a statistically significant increase in rates of MRSA associated with use of non‐absorbable antibiotics plus cefotaxime compared with placebo (RR = 2.22; 95% CI: 1.21 to 4.07).
There was no evidence of a difference in mortality or rates of sepsis with local airway antibiotic prophylaxis compared with placebo (only one trial, 30 participants).
Authors' conclusions
The conclusions we are able to draw regarding the effects of prophylactic antibiotics in people with burns are limited by the volume and quality of the existing research (largely small numbers of small studies at unclear or high risk of bias for each comparison). The largest volume of evidence suggests that topical silver sulfadiazine is associated with a significant increase in rates of burn wound infection and increased length of hospital stay compared with dressings or skin substitutes; this evidence is at unclear or high risk of bias. Currently the effects of other forms of antibiotic prophylaxis on burn wound infection are unclear. One small study reported a reduction in incidence of pneumonia associated with a specific systematic antibiotic regimen.
Keywords: Humans; Anti‐Bacterial Agents; Anti‐Bacterial Agents/adverse effects; Anti‐Bacterial Agents/therapeutic use; Antibiotic Prophylaxis; Antibiotic Prophylaxis/adverse effects; Antibiotic Prophylaxis/methods; Bandages; Burns; Burns/complications; Randomized Controlled Trials as Topic; Silver Sulfadiazine; Silver Sulfadiazine/adverse effects; Silver Sulfadiazine/therapeutic use; Skin, Artificial; Trimethoprim, Sulfamethoxazole Drug Combination; Trimethoprim, Sulfamethoxazole Drug Combination/adverse effects; Trimethoprim, Sulfamethoxazole Drug Combination/therapeutic use; Wound Infection; Wound Infection/chemically induced; Wound Infection/prevention & control
Plain language summary
Antibiotics to prevent burn wounds becoming infected
Burn injuries are a serious problem. They are associated with a significant incidence of death and disability, multiple surgical procedures, prolonged hospitalisation, and high costs of health care.
Various antibiotics are used with the aim of reducing the risk of infection in burn patients before it occurs. Some antibiotics are used locally on the skin (topical treatments), others are taken orally, or by injection, and affect the whole body (systemic treatments). It is not clear if prophylactic antibiotics are beneficial.
Thirty six studies involving 2117 participants are included in this review. The studies compared people with burns who were given antibiotics with people also with burns who received either an inactive treatment (placebo), no treatment, wound dressings, or another topical preparation or antibiotic. Twenty‐six trials (72%) evaluated topical antibiotics and smaller numbers evaluated antibiotics given orally, intravenously or via the airway. Most studies were small and of poor quality.
There was some evidence that a particular antibiotic (silver sulfadiazine) applied directly to the burn actually increases the rates of infection by between 8% and 80%. Otherwise there was not enough research evidence about the effects of antibiotics to enable reliable conclusions to be drawn.
Background
Description of the condition
The International Society for Burn Injuries defines a burn as an injury to the skin or other organic tissue caused by thermal trauma (Latarjet 1995). A skin burn is the destruction of some, or all, of the different layers of cells in the skin by a hot liquid (scald), a hot solid (contact burn), or a flame (flame burn). Skin injuries due to ultraviolet radiation, radioactivity, electricity or chemicals, as well as respiratory damage resulting from smoke inhalation, are also considered to be burns (Latarjet 1995; Peden 2002; Peden 2008; WHO 2006).
Burn injuries are a major source of morbidity and mortality; they represent a public health problem and a significant burden to the health care system (Church 2006; WHO 2006). Every year, more than 300,000 people worldwide die from fire‐related burns, most of them (i.e. 90%) occurring in low and middle‐income countries (Mock 2008; Peden 2002). However, burns also represent one of the main causes of injury‐related death in some high‐income countries, such as the USA and certain European countries (Church 2006; Hyder 2009; Mathers 2003; Miniño 2006; WHO 2006). Millions of burn victims suffer permanent disability and disfigurement, which is often stigmatising; it is estimated that 10 million disability‐adjusted life years are lost each year at a worldwide level through burn injury (Hyder 2009; Mock 2008; Peden 2008). Burns create a heavy economic burden for health services. Treatment costs depend upon the type and severity of the burn, as well as associated costs such as hospitalisation, the need for long‐term rehabilitation, the loss of schooling/absence from work, future unemployment, and social rejection. In spite of this, there are actually very few studies that provide evidence of the overall impact and cost of burns (Mock 2008; Peden 2008).
Infections are considered to be one of the most important and potentially serious complications in people with burns (Church 2006; Murray 2008). A report by the National Burn Repository of the United States mentions that in a 10‐year period there were 19,655 cases of complications in people with burns; 31% of these were pulmonary complications, 17% were related to the wound infection and cellulitis, and 15% were due to septicaemia and other infectious complications (Latenser 2007; Murray 2008). These data are supported by similar reports carried out in other countries (Alp 2012;SEMPSPH 2008; Soares 2006).
Infections generally arise in the acute period after the burn injury (Church 2006; Sheridan 2005). Burn wounds are highly susceptible to infection due to the loss of skin integrity and the reduction of immunity mediated by the cells. Once the physical barrier of the skin has been compromised, there is potential for the invasion of microbes into the body (Murray 2008; Sharma 2007). An area of dead tissue, with few or no blood vessels (avascular necrotic tissue (eschar)) replaces the skin and, eventually, will be colonised with micro‐organisms (De Macedo 2005; Erol 2004; Sharma 2007). The proliferation of micro‐organisms in the burn wound may be followed by tissue invasion, giving rise to burn wound infection (BWI) and invasive (systemic) infections. Common invasive infections in people with burns include pulmonary infections, urinary tract infection, bacteraemia and sepsis (Ansermino 2004; Church 2006; Pruitt 1998). Burn injury also has a severe impact on the host’s immune system, resulting in a general impairment of the host defences (Munster 1984; Sharma 2007).
Deciding whether a burn wound is infected can be difficult. Firstly, the inflammation resulting from the injury can mimic that seen with infection. Secondly, the interpretation of surface cultures is often difficult due to the extensive and rapid microbial colonisation of the wound (Ansermino 2004), with micro‐organisms coming from the persons skin or from external sources (Church 2006; Erol 2004; Wurtz 1995).
The nature and extent of the burn wound, together with the type and amount of colonising micro‐organisms can influence the risk of invasive infection. The spectrum of infective agents that can be present in the burn wounds varies. Nowadays, Gram‐positive bacteria such as Staphylococcus aureus, and Gram‐negative bacteria such as Pseudomona aeruginosa are the predominant pathogens. Nonetheless, other micro‐organisms, such as fungi, rickettsias and viruses, can also be implicated (Church 2006; Mayhall 2003; Polavarapu 2008; Sharma 2007). It should also be noted that multidrug‐resistant micro‐organisms, such as methicillin‐resistant Staphylococcus aureus (MRSA), are pathogens frequently identified in burns (Church 2006; DeSanti 2005; Mayhall 2003; Sharma 2007).
Burn wound infection (BWI) is a serious problem: it can delay wound healing, can increase the scarring and can favour the proliferation of micro‐organisms that may result in invasive infections (Church 2006; Edwards 2004; Singer 2002). Nowadays, after the initial resuscitation of burn victims, up to 75% of all deaths are a consequence of infection, rather than sudden cellular fluid imbalance (osmotic shock) and decreased volume of blood plasma (hypovolaemia) (Ansermino 2004; Bang 2002; Church 2006; Sharma 2007; Sheridan 2005).
Description of the intervention
Prevention of infection of burn wounds requires a team approach, and should be an early focus of the care of burned patients, with particular consideration given to infection‐control practices and long‐term rehabilitative care (Murray 2008).
A variety of interventions exists for preventing infections in burn wounds: namely, early removal of full‐thickness burned tissue (debridement); early definitive wound closure; strict enforcement of infection‐control procedures (hand washing, use of personal protective equipment, i.e. gown, gloves, and masks); and the use of antimicrobial prophylaxis (Church 2006; DeSanti 2005; Murray 2008; Weber 2002; Weber 2004). There is a wide variety of topical antimicrobial agents available for use as prophylaxis for BWI, such as silver nitrate and silver sulphadiazine (Ansermino 2004; Church 2006). Moreover, topical antimicrobials have been used together with systemic (whole body) antibiotics to prevent and treat infection. A range of antibiotics, and routes of administration have been evaluated for the prevention of systemic infection in people with burn wounds. For example, oral trimethoprim‐sulphamethoxazole prophylaxis and intravenous cephalothin prophylaxis (Alexander 1982; Kimura 1998).
To address complications of smoke inhalation, local antibiotic prophylaxis administered via the airway has been tested by using aerosolized antibiotics (Levine 1978). The most recent clinical practice guidelines, however, do not recommend the routine administration of prophylactic antibiotics in burned persons. Antibiotics are recommended only for patients with known infections (Alsbjörn 2007; Brychta 2011;Hospenthal 2011; NSW Severe Burn Injury Service 2008). Before the wide adoption of early excision and closure of deep wounds, infection was a frequent occurrence in the burn wound (Sheridan 2005). Nowadays, however, the early excision of eschar and avascularised tissues improves the perfusion of the burned tissues, and allows systemic antibiotics to reach adequate therapeutic levels in the burn wound (Church 2006; Kumar 2006; Mayhall 2003). Despite the fact that systemic infection, such as sepsis, is now less frequent, infection in people with burns continues to be a serious threat (Church 2006; Kumar 2006; Sheridan 2005).
This review will focus on the effects of antibiotic prophylaxis (oral (PO), parenteral (entry to body not via gastrointestinal tract) or topical antimicrobials) for preventing burn wound infections.
How the intervention might work
Improvements in recovery for seriously burned people have been attributed to medical advances in wound care and infection control practices (Church 2006; DeSanti 2005).
The efficacy of commonly‐used antimicrobial agents in burns units is dynamic due to the ability of micro‐organisms to develop resistance quickly (Church 2006; Mayhall 2003). The antibiotic regimen of choice is determined by the pathogen known, or suspected, to cause the infection (Church 2006). The use of an effective antimicrobial agent, however, could reduce substantially the microbial load in the open surface of the burn wound, and, therefore, reduce the risk of infection. Bearing the above in mind, antibiotic prophylaxis might be a useful way of protecting burn victims against wound, and invasive, infections.
Why it is important to do this review
The use of antibiotics has been considered useful in treatment of infections in burn victims (Polavarapu 2008). In some centres, patients with evidence of a positive microbiological culture from a burn site were given systemic antibiotic prophylaxis in an attempt to prevent wound infection and sepsis (Atoyebi 1992; Haq 1990; Lee 2009; Onuba 1987), though this is now controversial (Ansermino 2004). There is thought to be a paucity of high quality research evidence to determine the effectiveness and cost‐efficiency of antibiotic prophylaxis for preventing BWI (Avni 2010; Lee 2009; Ugburo 2004). Moreover, the use of prophylactic antibiotics may not be safe: it may increase the risk of diarrhoea due to overgrowth of toxigenic strains of Clostridium difficile and other secondary infections, allergic reactions to the drug or bone marrow suppression (Alexander 2009; Church 2006; Ergün 2004; Still 2002). Finally, it may also promote the emergence of resistant strains of micro‐organisms, making the treatment of infections even more difficult (Altoparlak 2004; Church 2006; Murphy 2003).
There is considerable debate concerning the use of antibiotic prophylaxis for the prevention of the BWI and therefore a Cochrane systematic review of the available evidence is warranted.
Objectives
To assess the effects of antibiotic prophylaxis on rates of burn wound infection.
Methods
Criteria for considering studies for this review
Types of studies
Randomised controlled trials (RCTs), published or unpublished, with allocation to interventions at the individual level (patient‐RCT) or at the group level (cluster‐RCT), testing the efficacy and safety of antibiotic prophylaxis for the prevention of burn wound infections. Quasi‐randomised studies were excluded.
Types of participants
People of any age or gender, with any type of burn injury to the epidermis, dermis, subcutaneous tissues, vessels, nerve, tendons, or bone; but not residual burn wounds (these type of wounds may have had previous infections or treatments) admitted to any unit in the hospital setting, or treated in an outpatient setting.
We included studies regardless of the severity of the burn (determined by either clinical evaluation or objective assessment, or both) or the type of burn injury (e.g. chemical, scald, or flame). We did not exclude studies depending on the presence of inhalation injury or co‐morbidity.
We excluded studies that contained mixed population, i.e. people with already infected wounds in addition to those without an infection (unless the data were presented separately).
Types of interventions
Prophylaxis was defined as the administration of antibiotics to patients without a documented infection, regardless of the signs of systemic inflammation, with the aim of preventing burn wound infection and invasive infection. Studies of the treatment of residual burn wounds was not included since the objective of this review is to assess the effect of first intention prophylaxis.
We included any of the following antibiotic prophylaxis:
Systemic antibiotics given orally or parenterally (intravenously or via intramuscular injection).
Selective intestinal decontamination with antibiotics (non‐absorbable antibiotic therapy).
Topical antibiotics, such as topical antimicrobial dressings or ointments (Merriam‐Webster 2012).
Local airway prophylaxis, such as aerosolised antibiotics.
Eligible comparisons were placebo, no treatment, usual care or an alternative intervention. Alternative interventions could include non pharmacological measures such as isolation of the burn patient, surgical excision; or pharmacological measures, such as another antibiotic regimen. Trials comparing different antibiotics or different antibiotic dosages, routes of administration, timings or duration of administration were eligible for inclusion. Antibiotic prophylaxis could be given at any moment after admission ('general prophylaxis') or could be specifically given before surgical procedures ('perioperative prophylaxis'). We did not have a minimum duration of the intervention or of follow‐up as inclusion criteria.
We excluded studies evaluating antibiotic‐impregnated catheters; ointments or dressings that contained antimicrobials (iodine, chlorhexidine); and antifungals, since they are not considered to be antibiotic therapies. Dressings for superficial partial‐thickness burns are evaluated in another Cochrane review (Wasiak 2008), the principal objective of which was not the evaluation of antibiotic prophylaxis.
Types of outcome measures
Primary outcomes
Outcome 1:Burn wound infection: studies reporting an objective measure of burn wound infection. Diagnosis should rely on clinical examination (burn wound appearance) and culture data, if possible, however, burn wound infections diagnosed only by clinical examination were also eligible.
Outcome 2: Invasive infections, such as pneumonia, urinary tract infections (UTI), bacteraemia or blood infections (sepsis), or central venous catheter‐associated bloodstream infections. We admitted any measure for quantifying infections, such as incidence rate or incidence density rate.
Outcome 3: Infection‐related mortality: i.e. mortality due to infection of burn wounds, sepsis, or another infective complication.
Outcome 4: Adverse events: those considered by the study investigators to be related to antibiotic prophylaxis, such as toxicity, allergies, antibiotic‐associated diarrhoea due to the overgrowth of toxigenic strains of Clostridium difficile, etc.
Secondary outcomes
Outcome 5: Objective measures of wound healing rate: such as time to complete healing; proportion of wounds completely healed within a trial period; proportion of participants with completely healed wounds; or proportion of wounds partly healed in a specified time period.
Outcome 6: Antibiotic resistance: defined as the clinical infection or colonisation caused by bacteria resistant to one or more antibiotics (see Differences between protocol and review).
Outcome 7: All‐cause mortality: we tried to analyse this outcome according to the longest common time point of assessment among the included studies.
Outcome 8: Length of hospital stay (LOS).
Studies were eligible for inclusion even if they only reported secondary outcomes, as these outcomes are relevant to patients.
Search methods for identification of studies
Electronic searches
In January 2013 we searched the following electronic databases to find reports of relevant RCTs:
The Cochrane Wounds Group Specialised Register (searched 25 January 2013);
The Cochrane Central Register of Controlled Trials (CENTRAL) ‐ (The Cochrane Library 2012, Issue 12);
Ovid MEDLINE ‐ 1950 to January Week 3 2013;
Ovid MEDLINE ‐ In‐Process & Other Non‐Indexed Citations, January 23, 2013;
Ovid EMBASE ‐ 1980 to 2013 Week 03;
EBSCO CINAHL ‐ 1982 to 25 January 2013.
We searched the Cochrane Central Register of Controlled Trials (CENTRAL) using the following exploded MeSH headings and keywords:
#1 MeSH descriptor Burns explode all trees #2 (burn or burns or burned or scald*):ti,ab,kw #3 (thermal NEXT injur*):ti,ab,kw #4 (#1 OR #2 OR #3) #5 MeSH descriptor Anti‐Bacterial Agents explode all trees #6 MeSH descriptor Anti‐Infective Agents, Local explode all trees #7 (antibiotic* or amoxicillin or ampicillin* or bacitracin or cephalothin or cefazolin or cefotaxime or cefoperazone or ceftazidime or ceftriaxone or cefuroxime or chloramphenicol or ciprofloxacin or clarithromycin or clindamycin or cloxacillin or colistin or colymycin or erythromycin or flucloxacillin or furazolidone or "fusidic acid" or gentamicin or gramicidin or imipenem or "mafenide acetate" or mupirocin or natamycin or neomycin or nitrofurazone or oxacillin or penicillin or piperacillin or polymyxin or rifam* or "silver nitrate" or "silver sulfadiazine" or "sulfacetamide sodium" or tobramycin or amphotericin or tazocin or teicoplanin or tetracylcin or (trimethopri* NEXT sulfamethoxazole) or vancomycin):ti,ab,kw #8 (#5 OR #6 OR #7) #9 (#4 AND #8)
The search strategies for Ovid MEDLINE, Ovid EMBASE and EBSCO CINAHL can be found in Appendix 1. The Ovid MEDLINE search was combined with the Cochrane Highly Sensitive Search Strategy for identifying randomised trials in MEDLINE: sensitivity‐ and precision‐maximizing version (2008 revision) (Lefebvre 2011). The EMBASE and CINAHL searches were combined with the trial filters developed by the Scottish Intercollegiate Guidelines Network (SIGN 2011). There were no restrictions with respect to language, date of publication or study setting.
We searched in the following trials registers using the keywords: prophylaxis, antibiotic, and burn:
International Standard Randomized Controlled Trial Number Register (http://www.controlled‐trials.com/isrctn/) (last searched May 2012);
US National Institutes of Health trial registry (http://www.clinicaltrials.gov) (last searched May 2012).
Searching other resources
We searched the reference lists of all identified studies to find any further relevant trials.
Data collection and analysis
Selection of studies
Two review authors (LB and CJ) independently assessed all titles and abstracts of studies identified by the search strategy against the eligibility criteria in terms of their relevance and design. The full text versions of all potentially eligible studies were retrieved, and the two review authors independently assessed the eligibility of each study against the inclusion criteria.
The table of excluded studies provides details of all studies that appeared initially to meet our inclusion criteria, but which on closer examination did not, with the reasons for their exclusions. Any disagreements were resolved through discussion by the two review authors. Disagreements that could not easily be resolved were referred to a third review author (JL).
Data extraction and management
Data from the studies were extracted independently by two review authors (LB and CJ) using standardised forms. Details of included trials were extracted and summarised using a data extraction sheet. Data from trials published in duplicate were included only once, but maximal data extracted. All discrepancies were resolved by consensus among the review authors. When information within trial reports was not clear, we attempted to contact authors of the trial reports to request further details.
We extracted the following data:
Characteristics of the trial: study design, setting/location, country, period of study, method of randomisation, allocation concealment, blinding, unit of randomisation, unit of analysis, sample size calculation, use of Intention‐to‐treat analysis.
Participants: number, randomised, excluded (post‐randomisation), reasons for exclusion, participants assessed, withdrawals, reasons for withdrawals, age, gender, inclusion criteria, exclusion criteria, burned surface (% of total body surface area), full‐thickness burns, inhalation injury, time post‐burn, burn type, the state of the wounds at baseline, co‐morbidities.
Type of intervention: intervention group: antibiotic, dose, route, frequency, duration of treatment, co‐interventions. Control group: description of the intervention applied (if any).
Outcome data.
Source of funding, conflicts of interest.
Data were entered into Review Manager by one review author (LB) (RevMan 2011), and double checked by a second review author (JL).
Assessment of risk of bias in included studies
Two review authors (LB and CJ) independently assessed the risk of bias of each included study using the criteria outlined in the tool designed by the Cochrane Collaboration (Higgins 2011a) (see Differences between protocol and review). We considered the following domains:
Random sequence generation (selection bias).
Allocation concealment (selection bias).
Blinding for participants and personnel (performance bias).
Blinding of outcome assessment (detection bias).
Incomplete outcome data (attrition bias).
Selective outcome reporting (reporting bias).
-
Other sources of bias (considered in combination, that is, if at least one of these other sources of bias is considered to be 'high', this domain will be judged as 'high'):
for cluster‐randomised trials, we assessed these additional sources of bias: recruitment bias; baseline imbalance either across clusters or patients; loss of clusters and incorrect analysis (Higgins 2011b, Section 16.3.2);
for the trials where the unit of randomisation was the patient, we also assessed whether there were similar baseline characteristics between the study groups;
for all the included studies we also assessed whether there were baseline imbalances in factors that are strongly related to outcome measures, whether the analysis of time‐to‐event data was adequate, whether the study was stopped early due to some data‐dependent process, and whether there was any declared financial support.
We made assessments for each main outcome (or class of outcomes). We labelled each criterion as being at 'low ', 'high' or 'unclear' risk of bias. See Appendix 2 for details of criteria on which the judgements were based. We tried to obtain this information from the trial reports, but, when there was not enough information to make a judgement, we wrote to the trial authors for clarification. Disagreements were resolved by discussion and consensus. We included two figures in the review: a 'Risk of bias graph figure' (Figure 1) and a ‘Risk of bias summary figure’ (Figure 2). We assessed the overall risk of bias for each outcome (or class of similar outcomes) within each study. Each outcome (or class of outcomes) was defined as having a ‘low risk of bias’ only if it was at low risk of bias for all the domains; at ‘high risk of bias’ if it demonstrated high risk of bias for one or more of the domains; or at ‘unclear risk of bias’ if it demonstrated unclear risk of bias for at least one domain without any of the other domains being described as ‘high risk of bias’.
1.
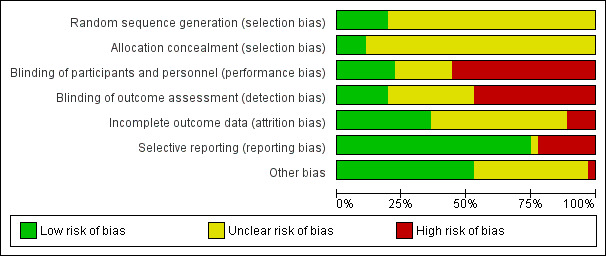
Risk of bias graph: review authors' judgements about each risk of bias item presented as percentages across all included studies.
2.
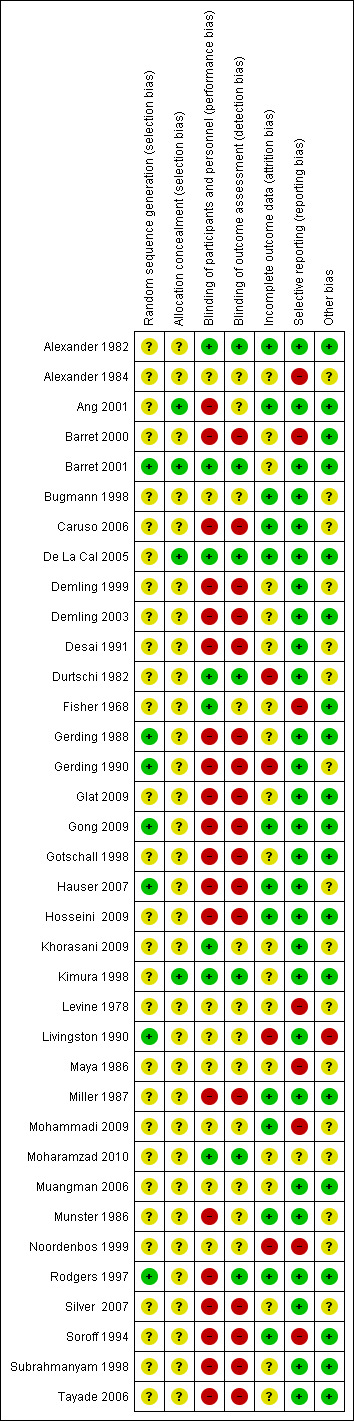
Risk of bias summary: review authors' judgements about each risk of bias item for each included study.
Finally, we incorporated the results of the risk of bias assessment into the review through systematic narrative description and commentary and we explored the effect of the risk of bias in the meta‐analysis by carrying out sensitivity analysis (see Effects of interventions).
Measures of treatment effect
We reported the risk ratio (RR) for dichotomous data (e.g. incidence of participants with infection), mean difference (MD) for continuous data (e.g. length of hospital stay) and hazard ratios (HR) for time‐to‐event data (e.g. time to healing) . All outcome effects are shown with their associated 95% confidence intervals (CI).
Unit of analysis issues
Although we did not expect that unit of analysis issues would arise in many eligible studies, a variety of them were found, including:
some trials reported on a per patient basis and other trials on a per burn basis; and
self‐controlled studies varied in the level where randomisation was done (some trials randomised adjacent wounds from the same arm or body part; others randomised non adjacent wounds from different parts of the body).
Effect measures adjusted by design were computed for these trials, namely Becker‐Balagtas odds ratios and corresponding confidence intervals (Curtin 2002). These trials provided data for the outcomes 'burn wound infection' and 'adverse effects', that were defined as generalized‐inverse variance in order to accommodate effect measures for both parallel and self‐controlled trials. The effect measures considered were either Mantel‐Haenzsel odds ratios, for parallel design trials, or Becker‐Balagtas odds ratios, for self‐controlled trials.
Dealing with missing data
We assessed and reported on missing outcome data for the included studies and contacted the authors of the primary studies where necessary (if we did not obtain this data, we documented this on the data extraction form and in the text of the review).
We carried out analyses on an intention‐to‐treat basis for all outcomes (i.e. to include all participants randomised to each group in the analyses, irrespective of what happened subsequently). There were some studies, however, that included participants whose outcomes were unknown. In these cases, we performed an ‘available case analysis’, where data are analysed for participants for whom outcome data were obtained.
We explored the impact on the overall treatment effect of missing data (>20% of for overall trial population or any trial arm) by using a worst‐case scenario sensitivity analysis (missing participants experienced a negative dichotomous outcome) (Sensitivity analysis)(see Differences between protocol and review).
Assessment of heterogeneity
Where possible we displayed the results of clinically and methodologically comparable studies graphically and assessed heterogeneity visually. We assessed heterogeneity between study results using the I² statistic (Higgins 2003) This examines the percentage of total variation across studies due to heterogeneity rather than chance. We judged the importance of the observed value of I² depending on the magnitude and direction of effects and the strength of evidence for heterogeneity (moderate to high heterogeneity will be defined as I² greater than, or equal to, 50%) (Deeks 2011).
Assessment of reporting biases
We planned to assess publication bias by means of a funnel plot for each outcome (a simple scatter plot of the intervention effect estimates from individual studies against some measure of each study’s size or precision (Sterne 2011)). Funnel plot asymmetry would be assessed statistically. If there was evidence of asymmetry, publication bias would be considered as only one of a number of possible explanations.
Data synthesis
Where sufficient numbers of comparable studies were available these were combined in a meta‐analysis to produce pooled RR for dichotomous data (e.g. incidence of peoples with infection), MD for continuous data (e.g. length of hospital stay (LOS)) and HR for time‐to‐event data (e.g. time to healing), with 95% CIs . We report outcome measurements for six different types of antibiotic prophylaxis addressed in the included trials.
When there were results from different follow‐up points within the same study, we considered shorter follow‐up periods for the meta‐analysis. We made this decision on the grounds that it was more likely that we would obtain measurements over a short period. Results for subsequent follow‐up periods were presented in narrative form only. For the outcome 'all‐cause mortality' we considered results concerning the end of the follow‐up period. We used a random‐effects model to pool data, although we assessed by means of a sensitivity analysis the influence of a fixed‐effect model. In the event that relevant statistical heterogeneity was detected (I² greater than, or equal to, 50%), or if the meta‐analysis was inappropriate for any other reason, we presented a narrative analysis of eligible studies, providing a descriptive presentation of the results, grouped by intervention and study design, with supporting tables. All outcome effects were shown with their associated 95% confidence intervals (CI).
We performed the analyses using Review Manager 5.1 (RevMan 2011), the statistical package provided by the Cochrane Collaboration.
Subgroup analysis and investigation of heterogeneity
We planned to carry out subgroup analyses:
age of participants: children (aged between 0 and 18 years) compared with adults (over18 years); and,
severity of burn: burns involving less than 20% total body surface area (TBSA) versus burns involving more than 20% TBSA.
It was not possible to perform these subgroup analyses, however, because of the paucity of studies providing the necessary data.
Sensitivity analysis
We conducted sensitivity analyses to assess for:
The effect of including studies with high or unclear risk of bias (as defined above), by excluding these trials from a comparative analysis.
The effect of missing data, by performing a comparative analysis excluding studies with high levels of missing data (more than 20% of missing data for the overall trial population, or for any of the trial arms) ).
The impact of withdrawals, by performing a comparative analysis (per protocol analysis 'available case analysis' and intention‐to‐treat analysis). We also performed a worst case scenario sensitivity analysis (considering missing data as negative events).
The effect of the allocation/analysis unit (burn wounds or patients) by performing a comparative analysis restricted to self‐controlled studies (post hoc sensitivity analysis).
All other analyses planned in the protocol were not performed for a variety of reasons, see 'Differences between protocol and review' for details.
Results
Description of studies
See: Characteristics of included studies; Characteristics of excluded studies; Characteristics of studies awaiting classification.
Results of the search
Searches for this review yielded 835 references. After eliminating duplicates, two review authors (LB and CJ) independently assessed the identified references against the inclusion criteria. During the scrutiny of titles and abstracts we identified 72 potentially‐relevant references, and the full text of each was retrieved. Of these, two articles have been designated as awaiting classification whilst contact with the trial author is made in order to obtain further information (Maghsoudi 2011; Panahi 2012) Thirty six studies (37 publications) met the inclusion criteria for this review (information on methods, participants, interventions, and outcomes of each one of these trials can be found in the Characteristics of included studies table). The search strategy identified one duplicate publication (Ang 2001). See Figure 3 flow diagram.
3.
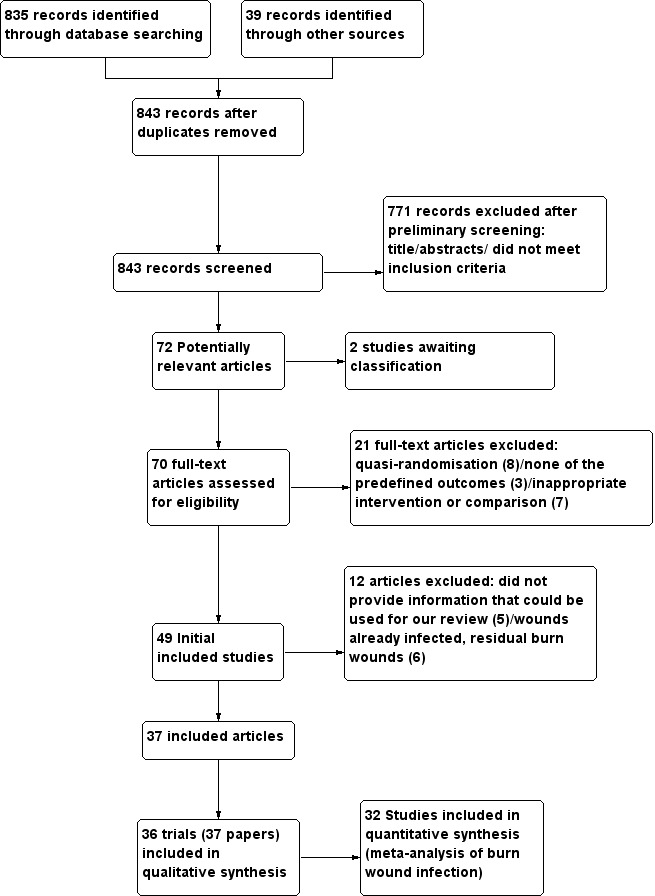
Flow diagram.
Excluded studies
The Characteristics of excluded studies table provides reasons for the exclusion of each study. Nine studies were quasi‐randomised (Cason 1966; Deutsch 1990; Hunter 1976; Lowbury 1968; Malik 2010; Manuskiatti 1999; Munster 1989; Proctor 1971; Waffle 1988). Three studies did not evaluate any of the review outcomes (Ahuja 2009; Mashhood 2006; Varas 2005). In nine studies, the interventions did not allow evaluation of the effectiveness of the antibiotic because the antibiotic was given in both arms of the study (i.e. topical silver sulfadiazine (SSD) alone versus SSD combined with cerium nitrate (SSD–CN); 1% silver sulfadiazine plus 0.2% chlorhexidine digluconate cream versus 1% silver sulfadiazine) (Abdel‐Razek 2000; Branski 2008; De Gracia 2001; Donati 1994; Fang 1987; Inman 1984; Miller 1990; Oen 2012; Ostile 2012). Seven trials were excluded either because the wounds were already infected, or because they involved residual burn wounds (Baghel 2009; Carneiro 2002; Huang 2006; Huang 2007; Li XL 2006; Ramos 2008; Subrahmanyam 1991); five studies did not provide information that could be used for our review (Afilalo 1992; Grippaudo 2010; Piel 1985; Steer 1997; Ugburo 2004).
Included studies
This review included a total of 36 trials, published between September 1968 and July 2010. Thirty‐four articles were published in English, one in German (Hauser 2007), one in Spanish (Maya 1986) and one in Chinese (Gong 2009). Fifteen studies included adult and paediatric participants (Alexander 1984; Ang 2001; Caruso 2006; Gerding 1988; Gerding 1990; Hauser 2007; Hosseini 2009; Kimura 1998; Levine 1978; Mohammadi 2009; Moharamzad 2010; Muangman 2006; Noordenbos 1999; Subrahmanyam 1998; Tayade 2006); 11 included only adults (De La Cal 2005; Demling 1999; Demling 2003; Durtschi 1982; Gong 2009; Khorasani 2009; Livingston 1990; Miller 1987; Munster 1986; Silver 2007; Soroff 1994), and 10 included only children (Alexander 1982; Barret 2000; Barret 2001; Bugmann 1998; Fisher 1968; Desai 1991; Glat 2009; Gotschall 1998; Maya 1986; Rodgers 1997).
Study country
Twenty trials were conducted in the USA (Alexander 1982; Alexander 1984; Barret 2000; Barret 2001; Caruso 2006; Demling 1999; Desai 1991; Durtschi 1982; Gerding 1988; Gerding 1990; Glat 2009; Gotschall 1998; Levine 1978; Livingston 1990; Miller 1987; Munster 1986; Noordenbos 1999; Rodgers 1997; Silver 2007; Soroff 1994), four in Iran (Hosseini 2009; Khorasani 2009; Mohammadi 2009; Moharamzad 2010), two in China (Ang 2001; Gong 2009), and two in India (Subrahmanyam 1998; Tayade 2006). There was one trial that did not specify the country or region where the study took place (Demling 2003). The remaining seven trials were conducted in Switzerland, Spain, South Africa, Germany, Japan, Mexico and Thailand (Bugmann 1998; De La Cal 2005; Fisher 1968; Hauser 2007; Kimura 1998; Maya 1986; Muangman 2006).
Setting
Trials were conducted in emergency departments (Gerding 1990; Kimura 1998), operating rooms (Bugmann 1998; Rodgers 1997; Subrahmanyam 1998), intensive care units (De La Cal 2005), ambulatory care units (Tayade 2006) or burns care facilities (Alexander 1982; Alexander 1984; Ang 2001; Barret 2000; Barret 2001; Caruso 2006; Demling 1999; Desai 1991; Durtschi 1982; Gerding 1988; Glat 2009; Khorasani 2009; Livingston 1990; Miller 1987; Mohammadi 2009; Muangman 2006; Munster 1986). For twelve trials there was no specific information about the place where the trial took place, but the trial authors did mention that the trials were conducted in a hospital setting (Demling 2003; Fisher 1968; Gong 2009; Gotschall 1998; Hauser 2007; Hosseini 2009; Levine 1978; Maya 1986; Moharamzad 2010; Noordenbos 1999; Silver 2007; Soroff 1994).
Sample size
Four trials reported a sample size calculation (Barret 2000; Barret 2001; Caruso 2006; De La Cal 2005). The size of the studies varied between 14 and 249 participants, with a total of 2117 participants included in the review. Only eleven trials (30%) included more than 70 participants (Alexander 1982; Ang 2001; Bugmann 1998; Caruso 2006; De La Cal 2005; Durtschi 1982; Fisher 1968; Gong 2009; Hosseini 2009; Mohammadi 2009; Moharamzad 2010).
Unit allocation
In 30 trials (83%) the unit of allocation was the individual participant (Alexander 1982; Alexander 1984; Ang 2001; Barret 2000; Barret 2001; Bugmann 1998; Caruso 2006; De La Cal 2005; Demling 1999; Demling 2003; Desai 1991; Durtschi 1982; Fisher 1968; Glat 2009; Gong 2009; Gotschall 1998; Hosseini 2009; Kimura 1998; Levine 1978; Livingston 1990; Maya 1986; Miller 1987; Mohammadi 2009; Moharamzad 2010, Muangman 2006; Munster 1986; Rodgers 1997; Silver 2007; Subrahmanyam 1998; Tayade 2006). In the six remaining trials, the unit of allocation was the wound, with each participant serving as his, or her, own control (Gerding 1988; Gerding 1990; Hauser 2007; Khorasani 2009; Noordenbos 1999; Soroff 1994). Matched wounds in the same participant (areas with similar burns) were randomised to both modalities of treatment (see Unit of analysis issues and Allocation (selection bias)).
The analysis of six studies included in the review, however, did not take into account the level at which randomisation occurred (thus incurring 'Unit of analysis issues`) (Gerding 1988; Gerding 1990; Hauser 2007; Khorasani 2009; Noordenbos 1999; Soroff 1994):
Two trials mostly randomised a single burn for each participant (Gerding 1988; Gerding 1990), but included some data for different burns in some participants. These trials randomised 43 and 52 participants, and analysed 50 and 56 burns, respectively. The degree of correlation introduced was considered to be low.
Four trials randomised different parts of the body to receive different interventions (Hauser 2007; Khorasani 2009; Noordenbos 1999; Soroff 1994), that is, for each participant two different burn wounds were randomised to either the treatment or to the control group. These trials provided data for outcomes such as 'burn wound infection' and 'adverse effects', that were analyzed as described in Unit of analysis issues.
Economic support
Eight trials reported that they had received economic support from pharmaceutical companies, foundations, or public institutions (Caruso 2006; De La Cal 2005; Demling 1999; Hosseini 2009; Khorasani 2009; Kimura 1998; Miller 1987; Munster 1986). One trial reported that authors had not received any economic support (Noordenbos 1999), while the remaining 29 articles did not report any information on this matter.
Conflicts of interest
Only three trials reported potential conflicts of interest (Ang 2001; Bugmann 1998; Hauser 2007). In the Hauser 2007 study, one of the authors worked with financial support from BG‐Kliniken Bergmannsheil Bochum and Mundipharma, who also provided some of the medications used in the study. The authors highlighted that, despite the potential conflict of interest, they had conducted the trial in an independent manner. Foundations and public institutions supported four trials (De La Cal 2005; Demling 1999; Hosseini 2009; Khorasani 2009). There was no information about sources of funding for the remaining studies. None of the included studies was judged to be at high risk of bias due to funding.
Characteristics of the burn wounds
Source of burn
Fourteen studies included participants with burns caused by a variety of sources: fire, hot liquids (scalds), hot solids (contact burns), electrical, chemical and other agents (Alexander 1982; Ang 2001; Bugmann 1998; Caruso 2006; Demling 2003; Durtschi 1982; Gerding 1988; Levine 1978; Livingston 1990; Maya 1986; Muangman 2006; Munster 1986; Rodgers 1997; Soroff 1994). Seven studies included participants with burns caused by fire and hot liquids (Barret 2000; Fisher 1968; Gong 2009; Hosseini 2009; Kimura 1998; Subrahmanyam 1998; Tayade 2006). Three studies included participants with burns caused exclusively by fire (Demling 1999; Desai 1991; Mohammadi 2009), and two studies included participants with burns caused exclusively by hot liquids (Gerding 1990; Gotschall 1998). Ten studies did not specify the source of the burn (Alexander 1984; Barret 2001; De La Cal 2005; Glat 2009; Hauser 2007; Khorasani 2009; Miller 1987; Moharamzad 2010; Noordenbos 1999; Silver 2007).
Thickness
Partial‐thickness and superficial burns were the most prevalent types of burn and featured in 27 studies (Ang 2001; Barret 2000; Bugmann 1998; Caruso 2006; De La Cal 2005; Demling 1999; Demling 2003; Desai 1991; Fisher 1968; Gerding 1988; Gerding 1990; Glat 2009; Gong 2009; Gotschall 1998; Hauser 2007; Hosseini 2009; Khorasani 2009; Livingston 1990; Maya 1986; Moharamzad 2010, Muangman 2006; Noordenbos 1999; Rodgers 1997; Silver 2007; Soroff 1994; Subrahmanyam 1998; Tayade 2006), followed by full‐thickness burns in five studies (Barret 2001; Kimura 1998; Levine 1978; Miller 1987; Mohammadi 2009). Four studies did not describe the thickness of burns (Alexander 1982; Alexander 1984; Durtschi 1982; Munster 1986).
Burned surface
There was considerable variation among studies regarding the size of the reported burn area in terms of total body surface area (TBSA) which varied from one percent to 91% (average values per group).
Time post‐burn
Twelve trials included people with burns acquired less than 24 hours earlier at the time of enrolment in the study (Barret 2000; Bugmann 1998; Caruso 2006; Fisher 1968; Gerding 1988; Hauser 2007; Hosseini 2009; Khorasani 2009; Maya 1986; Moharamzad 2010; Subrahmanyam 1998; Tayade 2006). In the remaining trials, limits were: less than 36 hours (Glat 2009), 48 hours (Durtschi 1982), 72 hours (Alexander 1982; De La Cal 2005; Desai 1991; Levine 1978; Miller 1987), four days (Barret 2001), and six days (Kimura 1998). Fifteen trials did not provide this information (Alexander 1984; Ang 2001; Demling 1999; Demling 2003; Gerding 1990; Gong 2009; Gotschall 1998; Livingston 1990; Mohammadi 2009; Muangman 2006; Munster 1986; Noordenbos 1999; Rodgers 1997; Silver 2007; Soroff 1994).
Type of antibiotic prophylaxis evaluated
The studies evaluated the following types of antibiotic prophylaxis:
1. Topical antibiotic prophylaxis (26 trials)
Comparison 1: neomycin, bacitracin, and polymyxin B versus inactive control (no intervention or placebo).
Comparison 2: silver sulfadiazine (SSD) versus polymyxin B/bacitracin.
Comparison 3: SSD versus dressings or skin substitutes.
Comparison 4: SSD versus any topical preparation of natural products (traditional medicine).
Comparison 5: other topical antibiotics versus dressings or skin substitutes.
Comparison 6: antibiotic prophylaxis versus other treatments.
2. Systemic antibiotic prophylaxis (general) (3 trials)
3. Systemic antibiotic prophylaxis (perioperative) (4 trials)
Comparison 1: antibiotic prophylaxis versus control/placebo.
Comparison 2: cephazolin versus another antibiotic.
4. Non‐absorbable antibiotic prophylaxis (selective decontamination of the digestive tract (SDD)) (2 trials)
Comparison 1: non‐absorbable antibiotic prophylaxis versus placebo.
Comparison 2: non‐absorbable antibiotic prophylaxis and cefotaxime versus placebo.
5. Local antibiotic prophylaxis (administered by airway) (1 trial)
Outcomes reported
Primary outcomes
With the exception of four trials (Alexander 1984; Kimura 1998; Levine 1978; Soroff 1994), all trials reported on the frequency of burn wound infection, however, the definitions of infection and the methods of diagnosis were heterogeneous. Five trials (14%) defined burn wound infection as the presence of over 105 organisms per gram of tissue (Barret 2001; Livingston 1990; Munster 1986; Rodgers 1997; Subrahmanyam 1998); 10 trials (28%) accepted a positive bacterial culture of wound samples as proof of infection (Ang 2001; De La Cal 2005; Demling 2003; Fisher 1968; Gerding 1988; Gong 2009; Gotschall 1998; Hauser 2007; Miller 1987; Muangman 2006); 11 trials (30%) determined burn wound infection through the clinic evaluation of signs and symptoms (Alexander 1982; Barret 2000; Demling 1999; Desai 1991; Durtschi 1982; Glat 2009; Hosseini 2009; Khorasani 2009; Maya 1986; Mohammadi 2009; Tayade 2006), while six trials (17%) did not define burn wound infection (Bugmann 1998; Caruso 2006; Gerding 1990; Moharamzad 2010; Noordenbos 1999; Silver 2007).
In 14 trials (39%) information was provided about other types of infection present in the burned person, specifically pneumonia, urinary tract Infection, bacteraemia, and sepsis (Alexander 1984; Ang 2001; Barret 2000; Barret 2001; Caruso 2006; De La Cal 2005; Durtschi 1982; Fisher 1968; Kimura 1998; Livingston 1990; Miller 1987; Mohammadi 2009; Munster 1986; Rodgers 1997); definitions for each of these can be found in Table 1.
1. Definition of the outcomes assessed.
| Study‐Year | Wound infection | Sepsis | Bacteraemia | Pneumonia | Urinary tract infection | Adverse events | Time to complete healing |
| Alexander 1982 | Discharge of pus in the graft site, associated with graft loss. | |
|
|
|
Any adverse event related to the administration of the antibiotic or the placebo. | |
| Alexander 1984 | |
|
Did not define bacteraemia, but assessed the total number of episodes of bacteraemia per days at risk | |
|
|
|
| Ang 2001 | Clinical evaluation (presence of fever and/or redness of the wound) and qualitative bacteriological examination of samples from the wound. | |
Bacterial infection was evaluated by bacteriological examination of blood. | Presence of respiratory infection was determined by qualitative bacteriological examination of sputum. | Determined the presence of urinary tract infection by qualitative bacteriological examination of urine. | |
Wound was declared healed when 75% of the total surface had healed. |
| Barret 2000 | Determined BWI through the assessment of clinical data. | |
Did not define bacteraemia, but reported data on this result. | |
|
|
Defined wound cicatrisation as closing of all affected areas in the initial wound. |
| Barret 2001 | Burn wound biopsy with more than 105 organism/g tissue and/or histologic evidence of viable tissue invasion. | Presence of a septic source: (1) burn wound biopsy with more than 105 organism/g
tissue and/or histologic evidence of viable tissue
invasion, (2) positive blood culture, (3) urinary tract infection with 105 organism/ml urine; and (4) pulmonary infection with positive bacteria and white
cells on a class III, or better sputum specimen. In addition to the identification of a septic source, five or more of the following criteria had to be met: tachypnoea (> 40 breaths/minute); prolonged paralytic ileus; hyper‐ or hypothermia (< 36.5°C or > 38.5°C); altered mental status; thrombocytopenia (< 50 000 platelets/mm3); leucocytosis or leukopenia (< 3.5 or > 15.0 cells/mm3); unexplained acidosis; or hyperglycaemia. |
|
Pulmonary infection with positive bacteria and white cells on a class III, or better sputum specimen. | |
Did not define adverse events, but registered diverse complication. | Did not define cicatrisation, but reported time to complete healing. |
| Bugmann 1998 | Did not define BWI, but reported data on this result. | |
|
|
|
Reported allergies and bleeding as adverse events. | A wound was considered cicatrised when it had healed completely. |
| Caruso 2006 | Did not define BWI, but reported data on this result. | |
|
|
Any new adverse event (including infection) or any adverse event that had worsened during the study. | A wound was considered cicatrised when there was 100% re‐epithelisation, including the small residual crusts, blisters and open areas of < 1 cm in an area that had been re‐epithelialized completely. | |
| De La Cal 2005 | BWI determined by microbiological testing of samples from the surface of the wound, performed upon admission and then twice a week. | |
Bloodstream infections were diagnosed according to CDC definitions for nosocomial infections. | Presence of new (or progressive) pulmonary infiltrates persisting for > 48 h on chest X‐ray, in addition to at least 2 of the following criteria: (1) fever ≥ 38.5°C or hypothermia < 35.0°C; (2) leucocytosis ≥ 10,000/mm3 or leukopenia < 3000/mm3; (3) isolation of potential pathogens in high concentration of ≥ 4 x 107 cfu/ml using semi quantitative culture, from unprotected purulent tracheal aspirates. | Urinary tract infections were diagnosed according to CDC definitions for nosocomial infections. | |
|
| Demling 1999 | BWI determined by clinical assessment of signs (increased exudate and surrounding cellulitis). | |
|
|
|
|
Determined that the wounds were healed when re‐epithelialization had reached ≥ 90%. |
| Demling 2003 | Accepted quantitative culture of samples from the wound as evidence of infection. | |
|
|
|
|
Determined that the wounds were healed when re‐epithelialization had reached 95%. |
| Desai 1991 | Determined BWI through the clinical evaluation of signs and symptoms. | |
|
|
|
|
|
| Durtschi 1982 | Wound infection was considered when had cellulitis. The cellulitis was clinically defined as an area of warm, spreading, cutaneous erythema, accompanied by local pain and fever. Cellulite was determined by positive culture of samples from the surface of the wound. The samples for the culture were taken upon admission, and the samples for follow‐up, at days 5 and 7. |
Syndrome resulting from the presence of > 100,000 organisms/g biopsied wound tissue, associated with variable temperature and leucocyte count, blood chemistry abnormalities, and occasionally ‐ but not invariably ‐ accompanied by positive blood cultures. | Did not define bacteraemia, but reported data on bacteraemia by beta‐haemolytic streptococcal. | |
Did not define urinary tract infection, but reported data on this outcome. | |
|
| Fisher 1968 | Determined that the presence of "local purulence" constituted infection. Additionally, accepted a positive bacterial culture of wound samples as proof of infection. | Did not define the term sepsis, but reported patients who had presented with purulence with septicaemia. | Presence of systemic disease. | |
|
|
Did not define cicatrisation, but reported time to healing. |
| Gerding 1988 | Infection determined by semi‐quantitative cultures of surface samples. | |
|
|
|
|
Completely re‐epithelialized wounds. Wounds considered to be treatment failures if had not healed within 21 days or had required skin grafts. |
| Gerding 1990 | Did not define BWI, but reported data on this outcome. | |
|
|
|
|
Healing time defined as the time required to achieve full epithelialisation of the burned surface. |
| Glat 2009 | Determined BWI through the assessment of clinical data. | |
|
|
|
Did not define adverse events, but reported data on possible adverse effects. | |
| Gong 2009 | Infection determined by semi‐quantitative cultures of wound surface samples. | |
|
|
Did not define adverse events, but reported data on adverse reactions. | Did not report the definition of cicatrisation, but assessed the percentage of wound healing at different times, up to 21 days and to the time healing. | |
| Gotschall 1998 | BWI defined by the presence of clinical data in conjunction with cultures from the wound surface. | |
|
|
|
|
Did not define cicatrisation, but reported time to healing. |
| Hauser 2007 | Infection determined by semi‐quantitative cultures of wound surface samples. | |
|
|
|
|
Determined that the wounds were healed when re‐epithelialization had reached 95%‐100%. |
| Hosseini 2009 | Determined BWI through the assessment of clinical data. | |
|
|
|
|
|
| Kimura 1998 | |
|
|
Patients satisfying all of the following criteria: (1) infiltration of lung fields on chest X‐ray films;
(2) fever (> 38°C) for at least 3 consecutive days; (3) peripheral white blood cell count > 104/mm3. (4) Pathogenic bacteria (> 103 cfu/ml) detected in airway secretions. |
|
Did not define adverse events, but assessed the associated secondary effects during the period of antibiotic administration. | |
| Khorasani 2009 | |
|
|
Did not define urinary tract infection but reported data on this outcome. |
Determined by clinical assessment of the wound (assessment of the nature of epithelialization ‐ percentage and healing time). |
||
| Levine 1978 | |
Did not define sepsis, but reported that blood cultures were performed three times a week and upon suspicion of sepsis. | |
|
|
|
|
| Livingston 1990 | More than 105 organisms/g of tissue in both the nonadherent graft and recipient site. | Did not define sepsis, but reported data on this outcome. | |
|
|
|
|
| Maya 1986 | BWI determined by daily assessment of signs and symptoms. | |
|
|
|
|
|
| Miller 1987 | Defined wound infection as cellulitis. Bacterial cultures were performed when there was suspicion of infection and at the end of the study. |
|
|
Did not define pneumonia, but reported data on this outcome. | Did not define urinary tract infection, but reported data on this outcome. Conducted routine urine analysis. | Did not define adverse events, but reported data on adverse reactions. | |
| Mohammadi 2009 | Defined wound infection through the daily evaluation of signs and symptoms. | When there were symptoms and signs of hypothermia, hypotension, abrupt hyperglycaemia, decreased urine output, thrombocytopenia and diet intolerance, a thorough check‐up including blood culture and urine culture was done. | |
|
|
|
|
| Moharamzad 2010 | Did not define BWI, but reported data on this outcome. | |
|
|
|
|
Did not define cicatrisation, but reported data on time to healing. |
| Muangman 2006 | Determined wound infection by clinical data in conjunction with cultures from the wound surface. |
|
|
|
|
|
|
| Munster 1986 | Presence of clinical data together with burn wound biopsy with > 105 organisms/g tissue. Biopsies were taken twice a week. | Determined sepsis through two parameters: (1) presence of a positive blood culture, and the presence or absence of standard signs of sepsis such as hypothermia, disorientation and paralytic ileus; or (2) presence of a quantitative biopsy on one or more occasions of ≥ 105 organisms coupled with any of the clinical parameters mentioned above. | |
|
|
Did not define adverse events, but reported data on possible adverse effects. | |
| Noordenbos 1999 | Did not define BWI, but reported data on this outcome. | |
|
|
|
|
Determined that wounds were healed when there had been epithelial closing of 90% of the site of the wound. |
| Rodgers 1997 | BWI determined by quantitative culture of tissue biopsies. Performed colony count; results expressed as cfu/g of tissue. A culture was considered positive when growth was more than 105 cfu/g of tissue. |
|
Did not define bacteraemia, but reported data on this outcome. Blood cultures were performed for the isolation and identification of pathogenic organisms. | |
|
|
|
| Silver 2007 | Did not define BWI, but reported data on this outcome. | |
|
|
|
Did not define adverse events, but reported data on possible adverse effects. | |
| Soroff 1994 | |
|
|
|
|
Did not define adverse events, but reported data on this outcome. | Determined that the wounds were healed when there was a new layer of epithelium. |
| Subrahmanyam 1998 | Presence of burn wound biopsy with > 105 organisms/g tissue. | |
|
|
|
|
|
| Tayade 2006 | Defined wound infection through the evaluation of clinical data. | |
|
|
|
Any adverse effects related to medication (allergic or hypersensitivity reactions) or a worsening of symptoms or complications (infection, wound infection). | Did not define cicatrisation, but reported data on time to healing. |
Abbreviations
< = less than > = more than ≥ = more than or equal to BWI = burn wound infection CDC = Centers for Disease Control cfu = colony forming units h = hour(s)
Infection‐related mortality was reported in four trials (Ang 2001; Durtschi 1982; Livingston 1990; Munster 1986).
Twelve trials presented information on adverse events related to antibiotic prophylaxis (Alexander 1982; Barret 2001; Bugmann 1998; Caruso 2006; Glat 2009; Gong 2009; Kimura 1998; Miller 1987; Munster 1986; Tayade 2006; Silver 2007; Soroff 1994).
Secondary outcomes
The most frequently reported secondary outcomes were time to complete wound healing (Ang 2001; Barret 2000; Barret 2001; Bugmann 1998; Caruso 2006; Demling 1999; Demling 2003; Fisher 1968; Gerding 1988; Gerding 1990; Gong 2009; Gotschall 1998; Hauser 2007; Khorasani 2009; Moharamzad 2010; Noordenbos 1999; Soroff 1994; Tayade 2006), and length of hospital stay (LOS) (Alexander 1982; Barret 2000; Barret 2001; De La Cal 2005; Desai 1991; Durtschi 1982; Hosseini 2009; Livingston 1990; Maya 1986; Mohammadi 2009; Muangman 2006; Tayade 2006). Definitions of outcomes reported can be found in Table 1.
Risk of bias in included studies
Risk of bias is summarised in Figure 1 and Figure 2. The Risk of bias graph (Figure 1) illustrates the proportion of studies with each of the judgements (‘low risk’, 'high risk’, ‘unclear risk’ of bias) for each domain in the tool, while the Risk of bias summary (Figure 2) presents all the judgements in a cross‐tabulation of study by domain. We also presented a descriptive analysis of each domain of the risk of bias tool. In total, eight trials were deemed to be at unclear risk of bias (Alexander 1982; Barret 2001; Bugmann 1998; De La Cal 2005; Khorasani 2009; Kimura 1998; Moharamzad 2010; Muangman 2006), with the remainder deemed to be at high risk (28 studies).
Allocation
All included studies reported that the allocation sequence was generated randomly, but only seven trials described the method used in sufficient detail. In these seven trials, adequate sequence generation methods were applied: i.e. computer‐generated codes (Gerding 1988; Gerding 1990); random‐number table (Barret 2001; Gong 2009; Hauser 2007; Rodgers 1997), and shuffled cards (Livingston 1990).
Allocation concealment was described in four studies. Centralised randomisation was performed at a central trial office (Ang 2001), and an hospital pharmacy department (Barret 2001; De La Cal 2005; Kimura 1998).
Blinding
Seven trials were open (Caruso 2006; Gerding 1990; Glat 2009; Gong 2009; Hauser 2007; Miller 1987; Silver 2007), and one was partially blinded (Rodgers 1997). The Rodgers 1997 trial report did not provide enough information about the blinding of participants, but the authors reported that the professional who administered the intervention was not blinded. In eighteen trials, although not explicitly stated, it appeared that neither participants, personnel nor outcome assessors were blinded (Alexander 1984; Ang 2001; Barret 2000; Bugmann 1998; Demling 1999; Demling 2003; Desai 1991; Gerding 1988; Gotschall 1998; Hosseini 2009; Livingston 1990; Maya 1986; Muangman 2006; Munster 1986; Noordenbos 1999; Soroff 1994; Subrahmanyam 1998; Tayade 2006).
In ten trials it was possible that participants and personnel were blinded (Alexander 1982; Barret 2001; De La Cal 2005; Durtschi 1982; Fisher 1968; Khorasani 2009; Kimura 1998; Levine 1978; Mohammadi 2009; Moharamzad 2010).
Seven trials reported the methodology used to assess outcomes with sufficient detail to establish that this had been done in a blinded manner, and this methodology was considered adequate (Alexander 1982; Barret 2001; De La Cal 2005; Durtschi 1982; Kimura 1998; Moharamzad 2010; Rodgers 1997). Four trials that blinded participants and personnel, did not appear to blind outcome assessment (Fisher 1968; Khorasani 2009; Levine 1978; Mohammadi 2009).
Incomplete outcome data
Four trials reported post‐randomisation losses greater than 20% during the study (Durtschi 1982; Gerding 1990; Livingston 1990; Noordenbos 1999); eleven studies reported losses less than 20% during the study (Alexander 1982; Ang 2001; Bugmann 1998; Caruso 2006; De La Cal 2005; Hauser 2007; Hosseini 2009; Miller 1987; Mohammadi 2009; Rodgers 1997; Soroff 1994); and two trials reported no losses (Gong 2009; Munster 1986). For the remaining studies, the magnitude of such losses could not be determined. Fourteen trials reported the reasons for these losses (Alexander 1982; Ang 2001; Bugmann 1998; Caruso 2006; De La Cal 2005; Durtschi 1982; Gerding 1990; Gong 2009; Hosseini 2009; Miller 1987; Mohammadi 2009; Munster 1986; Rodgers 1997; Soroff 1994).
Intention‐to‐treat (ITT) analysis
Twenty‐two trials (61%) used intention‐to‐treat analysis (ITT). ITT was not implemented in ten studies (Bugmann 1998; Caruso 2006; Durtschi 1982; Gerding 1990; Hosseini 2009; Livingston 1990; Miller 1987; Noordenbos 1999; Rodgers 1997; Soroff 1994), and it was unclear, or there was not enough information to determine whether ITT had been implemented, in the remaining four (Gerding 1988; Gotschall 1998; Mohammadi 2009; Moharamzad 2010).
Incomplete outcome data
The overall assessment for incomplete outcome data was that the risk of bias was low for 13 studies (Alexander 1982; Ang 2001; Bugmann 1998; Caruso 2006; de La Cal 2005; Gong 2009; Hauser 2007; Hosseini 2009; Miller 1987; Mohammadi 2009; Munster 1986; Rodgers 1997; Soroff 1994), high in four studies (Durtschi 1982; Gerding 1990; Livingston 1990; Noordenbos 1999), and unclear for the remaining studies.
Selective reporting
We carried out a search for the protocols of the included studies (searched 1995 to March 2012), but none was identified. Nonetheless, 27 (75%) of the included studies presented all the results that had been specified in the methods section of the article, and, therefore, we assumed there was no selective reporting.
Other potential sources of bias
Randomisation unit
Six studies included randomisation of more than one burn on the same participant: (Gerding 1988, Gerding 1990, Hauser 2007, Khorasani 2009, Noordenbos 1999, Soroff 1994). Two trials generally randomised only a single burn for each participant (Gerding 1988, Gerding 1990), but also included some participants with more than one burn wound, where wounds were randomised to different treatments (control and experimental). These trials randomised 43 and 52 participants, and analysed 50 and 56 burns, respectively. Four trials included only participants with at least two burn wounds that were randomised to different treatments (control and experimental) (Hauser 2007; Khorasani 2009; Noordenbos 1999; Soroff 1994).
Baseline imbalance
No study reported relevant baseline differences regarding factors that could influence results (for example, sociodemographic variables, size of the burn, aetiology, and post‐burn time).
Early stopping
One study (Livingston 1990), originally designed to include 90 patients, was stopped early due to the occurrence of poor results, and data were evaluated after 45 patients had completed the study. For this reason, the 'other potential sources of bias' domain for this study was judged to be at high risk of bias.
Effects of interventions
The 36 included trials evaluated different antibiotic interventions for prevention of infection in people with burn wounds. For each comparison between antibiotic prophylaxis and the corresponding control group, we present results for the primary and secondary outcomes of the review, if they had been evaluated in the study, and where information was available.
The results are summarized in narrative form and, where possible and appropriate, through the corresponding meta‐analysis. Meta‐analysis, however, could not be performed for all comparisons, either because studies used different outcome measures, or because they did not provide all the information required. No studies were included that used a cluster randomized design (studies with group‐level allocation of interventions).
It is inappropriate to analyse data for the time to an event, such as time to healing, with the methods used for continuous outcomes (e.g. using the mean time to the event), since pertinent times are known only in the subset of participants in which the event occurred (e.g. healing). The incorrect analysis of outcome data may introduce bias in the interpretation of results. All studies analysed the time to healing as a continuous quantitative variable, except Ang 2001. Given that the estimate of effect carried out in this manner may not have been appropriate, we decided not to conduct a meta‐analysis for this outcome.
The results of the studies were grouped according to the type of antibiotic prophylaxis evaluated: topical, systemic (general and perioperative), non‐absorbably, local and antibiotic prophylaxis with unspecified regimens. Outcomes were reported variably across the trials, therefore, where an outcome is absent from a comparison, this was not reported in any of the trials of that comparison.
1. Topical antibiotic prophylaxis
Twenty‐six trials (1329 participants) evaluated topical antibiotics compared with either an active or inactive control intervention.
Comparison 1: Neomycin, bacitracin, and polymyxin B compared with inactive control (no intervention or placebo)
Two trials (99 participants) evaluated a topical antibiotic compared with an inactive control (Fisher 1968; Livingston 1990). Fisher 1968 had three arms: Polybactrin spray (combination of neomycin, bacitracin and polymyxin B) (33 participants), Dermoplast spray (benzocaine 4.5%, benzethonium chloride1.1%, menthol 0.5%, methyl paraben 2% and 8‐hydroxyquinoline 0.83 %) (33 participants), and control (no spray) (33 participants). Livingston 1990 also had three arms: neomycin plus bacitracin (bacitracin/polymyxin B) (18 participants), 0.5% silver nitrate (19 participants) and placebo (Ringer's lactate) (15 participants).
Outcome 1: Burn wound infection
Pooled data showed there was no significant difference in the number of participants with burn wound infection between Polybactrin or neomycin plus bacitracin and control group (OR = 0.75; 95% CI: 0.32 to 1.73), (I2 = 0%) (Analysis 1.1). The overall risk of bias for this outcome was high for both trials. Follow‐up was complete for Fisher 1968, but, overall, there was a high rate of post‐randomisation exclusions in Livingston 1990. The incidence of three invasive infections were measured.
1.1. Analysis.
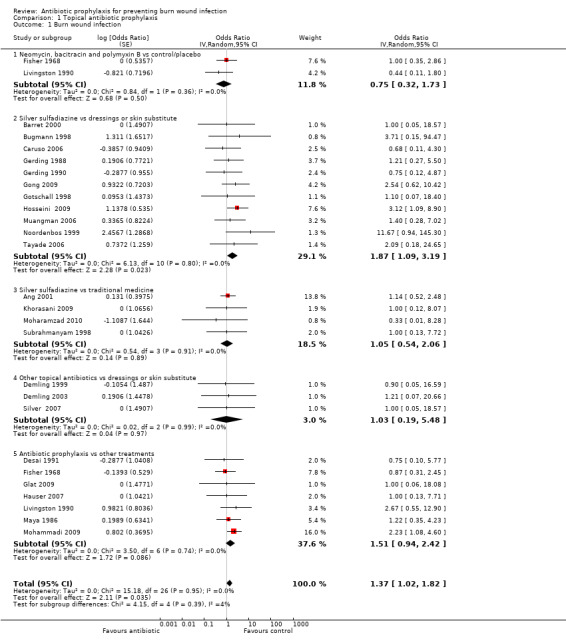
Comparison 1 Topical antibiotic prophylaxis, Outcome 1 Burn wound infection.
Outcome 2: Infections in the burned people
Livingston 1990 (33 participants) reported that four participants developed sepsis in the neomycin plus bacitracin group compared with none in the control group however this difference was not statistically significant (RR = 7.58; 95% CI: 0.44 to 130.38) (Analysis 1.2); the overall risk of bias was high. No participant developed pneumonia.
1.2. Analysis.
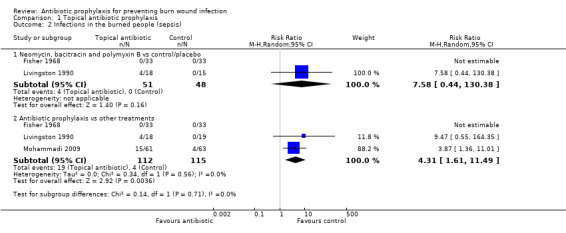
Comparison 1 Topical antibiotic prophylaxis, Outcome 2 Infections in the burned people (sepsis).
Fisher 1968 (66 participants) reported that none of the participants developed sepsis; two participants in the Polybactrin group developed bacteraemia, compared with five in the control group, but this difference was not statistically significant (RR = 0.40; 95% CI: 0.08 to 1.92) (Analysis 1.3); there was a high risk of bias for this latter outcome.
1.3. Analysis.
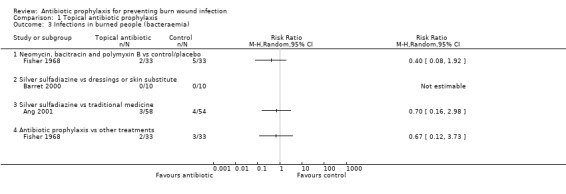
Comparison 1 Topical antibiotic prophylaxis, Outcome 3 Infections in burned people (bacteraemia).
Outcome 3: Infection‐related mortality
In Livingston 1990, four participants in the neomycin plus bacitracin group died as a consequence of sepsis and multiple organ failure, compared with none in the Ringer’s lactate group, but this difference was not statistically significant (RR = 7.58; 95% CI: 0.44 to 130.38) (Analysis 1.7). Results for this trial are presented as an available data analysis, since there was a high rate of post‐randomisation exclusions in this study and only 52 of 90 randomised participants were included in the analysis. The overall risk of bias for this outcome was high.
1.7. Analysis.
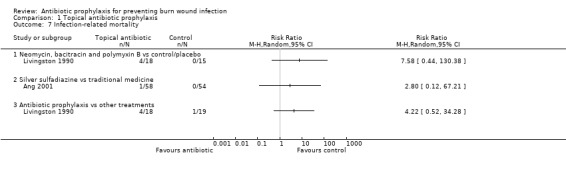
Comparison 1 Topical antibiotic prophylaxis, Outcome 7 Infection‐related mortality.
Outcome 5: Objective measures of wound healing
Fisher 1968: the mean time of healing of burn wounds was 22 days in the Polybactrin group, and 24 days in the control group (Table 2). According to the study authors, these differences were not significant (P value not reported). There was an overall high risk of bias for this outcome.
2. Time to complete wound healing.
| Study or Subgroup | Antibiotic | Control | P value |
Hazard Ratio (HR) |
||||
| Mean | SD | N | Mean | SD | N | |||
| Neomycin, bacitracin and polymyxin B vs control/placebo | ||||||||
| Fisher 1968 | 22.0 | — | 33 | 24.0 | — | 33 | — | |
| Silver sulfadiazine vs polymyxin B/bacitracin | ||||||||
| Soroff 1994 | 15.0 | 20.3 | 15 | 10.0 | 4.6 | 15 | P value 0.0007 | |
| Silver sulfadiazine vs dressings (skin substitute) | ||||||||
| Barret 2000 | 16.1 | 0.6 | 10 | 9.7 | 0.7 | 10 | P value < 0.001 | |
| Bugmann 1998 | 11.26 | 6.02 | 35 | 7.58 | 3.12 | 41 | P value < 0.01 | |
| Caruso 2006 | 17.0 | — | 42 | 16.0 | — | 42 | P value 0.517 | |
| Gerding 1988 | 21.3 | 2.3 | 23 | 13.7 | 1.3 | 27 | P value < 0.01 | |
| Gerding 1990 | 15.0 | 1.2 | 26 | 10.6 | 0.8 | 30 | P value < 0.01 | |
| Gong 2009 | 17.3 | 4.56 | 52 | 13.1 | 3.5 | 52 | P value < 0.05 | |
| Gotschall 1998 | 27.6 | — | 30 | 10.5 | — | 33 | P value 0.0002 | |
| Noordenbos 1999 | 18.1 | 6.05 | 14 | 11.1 | 4.37 | 14 | P value 0.002 | |
| Tayade 2006 | 18.44 | — | 25 | 12.64 | — | 25 | — | |
| Silver sulfadiazine vs any topical preparation of natural products (traditional medicine) | ||||||||
| Ang 2001 | 20.0 | — | 58 | 17.0 | — | 57 | P value 0.11 | 0.67 |
| Khorasani 2009 | 18.73 | 2.65 | 30 | 15.9 | 2.0 | 30 | P value < 0.0001 | |
| Moharamzad 2010 | 9.7 | 3.5 | 55 | 12.8 | 1.8 | 56 | P value < 0.05 | |
| Topical antibiotic prophylaxis vs other treatments | ||||||||
| Fisher 1968 | 22.0 | — | 33 | 23.0 | — | 33 | — | |
| Hauser 2007 | 11.3 | 4.9 | 47 | 9.9 | 4.5 | 47 | P value 0.015 | |
| Other topical antibiotics vs dressings (skin substitute) | ||||||||
| Demling 1999 | 13.0 | 3.5 | 11 | 8.0 | 1.5 | 10 | P value < 0.05 | |
| Demling 2003 | 10.5 | — | 20 | 8.5 | — | 24 | P value < 0.05 | |
| Non‐absorbable antibiotic prophylaxis vs placebo | ||||||||
| Barret 2001 | 40.0 | 8.0 | 11 | 33.0 | 4.0 | 12 | — | |
Abbreviations
< = less than vs = versus
Only Ang 2001 presented this outcome as a time‐to‐event outcome, not as continuous data, therefore, a pooled estimate was not produced.
Outcome 6: Antibiotic resistance
Livingston 1990 found no statistically significant difference between the frequency of participants with methicillin‐resistant Staphylococcus aureus (MRSA) in the neomycin plus bacitracin group (2/18) and the placebo group (3/15) (RR = 0.56; 95% CI: 0.11 to 2.90) (Analysis 1.8). The overall risk of bias for this outcome was high.
1.8. Analysis.
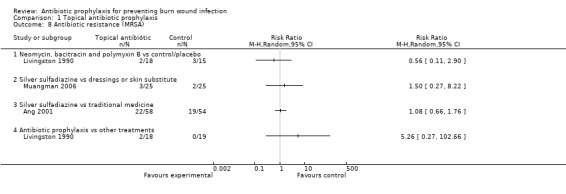
Comparison 1 Topical antibiotic prophylaxis, Outcome 8 Antibiotic resistance (MRSA).
Outcome 8: Length of hospital stay
Livingston 1990 reported that the mean length of hospital stay (LOS) was 36.33 days in the neomycin plus bacitracin group and 40 days in the placebo group. There were no statistically significant differences between the groups (MD = ‐3.67 days; 95% CI: ‐9.46 to 2.12) (Analysis 1.10). The overall risk of bias for this outcome was high.
1.10. Analysis.
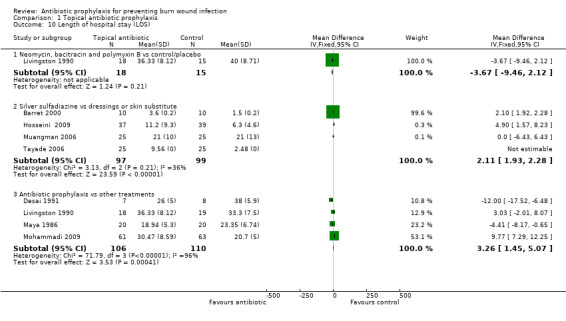
Comparison 1 Topical antibiotic prophylaxis, Outcome 10 Length of hospital stay (LOS).
Comparison 2: Silver sulfadiazine compared with polymyxin B/bacitracin
One trial (15 participants, 30 burn wounds) compared silver sulfadiazine (SSD) cream with topical polymyxin B sulphate/bacitracin spray and collagenase ointment, with participants acting as their own control (there were two non‐contiguous wounds per participant of similar size and acuteness) (Soroff 1994).
Outcome 4: Adverse events
Soroff 1994 found no adverse events in the group allocated to silver sulfadiazine (SSD), while there were three adverse events in the polymyxin B sulfate/bacitracin‐collagenase group (no statistically significant difference; OR = 0.20; 95% CI: 0.02 to 2.16) (Analysis 1.6). Denominator values suggested complete follow‐up, but the overall risk of bias for this outcome was high.
1.6. Analysis.
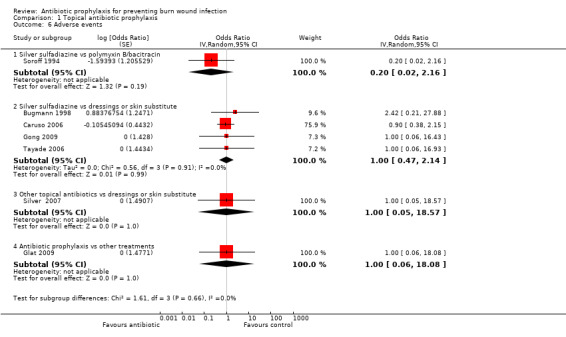
Comparison 1 Topical antibiotic prophylaxis, Outcome 6 Adverse events.
Outcome 5: Objective measures of wound healing
Soroff 1994 presented data on mean healing time (complete epithelialization) with this comparison. Results reported that time to healing was significantly shorter with polymyxin B sulphate/bacitracin plus collagenase (10 days) than with SSD (15 days) (P value 0.007) (Table 2). This time‐to‐event outcome was presented as continuous data and, therefore, we have presented the data in narrative form only. The overall risk of bias for this outcome was high.
Comparison 3: Silver sulfadiazine compared with dressings or skin substitute
Eleven trials (645 participants) compared 1% SSD cream with some kind of synthetic, or biosynthetic, dressing or skin substitute (Barret 2000; Bugmann 1998; Caruso 2006; Gerding 1988; Gerding 1990; Gong 2009; Gotschall 1998; Hosseini 2009; Muangman 2006; Noordenbos 1999; Tayade 2006). Two studies compared SSD cream with silicone‐coated nylon (Mepitel) (Bugmann 1998; Gotschall 1998). Six trials compared SSD cream with biosynthetic skin substitute dressing. The commercial brands studied were Biobrane (Smith & Nephew) (Barret 2000; Gerding 1988; Gerding 1990), Transcyte (Smith & Nephew) (Noordenbos 1999), Xenoderm (Medical Biomaterial Products, Germany) (Hosseini 2009), and Kollagen sheet (Tayade 2006). Three trials compared SSD compared with a silver‐impregnated dressing. Products studied were Acticoat (Smith & Nephew) (Muangman 2006), AQUACEL (ConvaTec, a Bristol‐Myers Squibb company) (Caruso 2006), and the ionic silver dressing combined with hydrogel (Gong 2009).
Outcome 1: Burn wound infection
Meta‐analysis of the 11 trials (645 participants) indicated a statistically significant increase in infection among patients receiving SSD compared with patients receiving dressing/skin substitute (OR = 1.87; 95% CI: 1.09 to 3.19, I2 = 0% ) (Analysis 1.1).
Interpretation of these results needs to take the overall risk of bias of the analysed trials into account. Only two trials had complete follow‐up, and an overall unclear risk of bias for this outcome (Bugmann 1998; Muangman 2006). The remaining nine trials had an overall high risk of bias for this outcome (Barret 2000; Caruso 2006; Gerding 1988; Gerding 1990; Gong 2009; Gotschall 1998; Hosseini 2009; Noordenbos 1999; Tayade 2006), although in some trials there was almost complete follow‐up (Gong 2009; Tayade 2006; Hosseini 2009; Caruso 2006).Barret 2000
Outcome 2: Infections in the burned people
Barret 2000 (20 participants) reported this outcome, and no participants developed bacteraemia. Denominator values suggested complete follow‐up, and the overall risk of bias for this outcome was high.
Outcome 4: Adverse events
The results of four trials, with 302 participants, were pooled (Bugmann 1998; Caruso 2006; Gong 2009; Tayade 2006); there was no statistically significant difference in adverse event rates between SSD and its comparators (dressings or skin substitute) (OR = 1.00; 95% CI: 0.47 to 2.14; I2 = 0%) (Analysis 1.6) .
Bugmann 1998 mentioned that three participants reported bleeding, and this outcome had an overall unclear risk of bias. Caruso 2006 reported that at least 45% of participants developed one or more adverse events in both trial arms, and had an overall high risk of bias for this outcome. Gong 2009 and Tayade 2006 reported no adverse events in either group.
Outcome 5: Objective measures of wound healing
Nine studies provided data on time to wound healing (Barret 2000; Bugmann 1998; Caruso 2006; Gerding 1988; Gerding 1990; Gong 2009; Gotschall 1998; Noordenbos 1999; Tayade 2006), however, while all authors reported the mean time to healing in each group, they did not provided complete data for time‐to‐event analysis. Therefore, we could not pool the trial data to estimate the hazard ratio, and the lack of a standard deviation around the mean in several trials meant we could not produce a pooled estimate of mean difference either. The results of each trial are presented in Table 2.
All trials showed an overall high risk of bias for this outcome except for Bugmann 1998, where the risk of bias for this outcome was unclear.
Outcome 6: Antibiotic resistance
Muangman 2006 was the only trial that reported on antibiotic resistance. The results from this trial showed a lack of precision around the point estimate, so there were no statistically significant differences in the risk of development of MRSA between the SSD (3/25 participants) and the dressing impregnated with silver (2/25 participants) groups (RR = 1.50; 95% CI: 0.27 to 8.22) (Analysis 1.8). The overall risk of bias for this outcome was unclear.
Outcome 7: All‐cause mortality
Two studies (132 participants) reported data on mortality, with only one death in one study (Caruso 2006), and none in the other (Muangman 2006). Meta‐analysis showed an important lack of precision in estimations and it was not possible to determine whether there were differences in mortality between SSD and the silver‐impregnated dressings (RR = 0.35; 95% CI: 0.01 to 8.34) (Analysis 1.9). In Caruso 2006 there was an overall high risk of bias for this outcome.
1.9. Analysis.
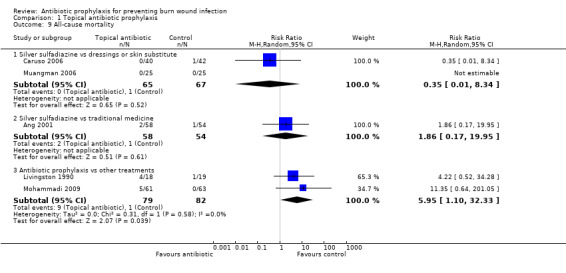
Comparison 1 Topical antibiotic prophylaxis, Outcome 9 All‐cause mortality.
Outcome 8: Length of hospital stay
Four studies (196 participants) provided data on length of hospital stay (LOS) (Barret 2000; Hosseini 2009; Muangman 2006; Tayade 2006). Tayade 2006 was not included in the meta‐analysis because no information was provided on the standard deviation around the mean. The meta‐analysis included the other three trials and showed a statistically significantly greater LOS among participants treated with SSD than for those given standard dressings (MD = 2.11 days; 95% CI: 1.93 to 2.28; I2 = 36%) (Analysis 1.10). Two trials had an overall high risk of bias for this outcome (Barret 2000; Hosseini 2009), and one had an overall unclear risk of bias (Muangman 2006).
Comparison 4: Silver sulfadiazine compared with any topical preparation of natural products (traditional medicine)
Four trials (333 participants) compared 1% SSD cream with any topical preparation of natural products (traditional medicine) (Ang 2001; Khorasani 2009; Moharamzad 2010; Subrahmanyam 1998). The natural products tested were an oil‐based ointment (MEBO) with sesame oil, beta‐sisterol, berberine and small concentrations of other herbal ingredients (Ang 2001), Aloe vera cream (Khorasani 2009), a herbal cream with Aloe vera, Geranium robertianum, and Lavandula stoechas (Moharamzad 2010), and unprocessed undiluted honey obtained from hives (Subrahmanyam 1998).
Outcome 1: Burn wound infection
All trials (333 participants) provided data for this outcome (Ang 2001; Khorasani 2009; Moharamzad 2010; Subrahmanyam 1998). These trials reported burn infection at different time points. None of these trials individually found a statistically significant difference in rates of burn wound infection between SSD and the natural product. Meta‐analysis of these results showed no overall statistically significant difference in the incidence of infection (OR = 1.05; 95% CI: 0.54 to 2.06; I2 = 0%) (Analysis 1.1). Overall risk of bias for this outcome was either high (Ang 2001; Subrahmanyam 1998), or unclear (Moharamzad 2010).
Outcome 2: Infections in burned people
Only Ang 2001 (112 participants) reported on invasive infections: There was no statistically difference between groups in: the incidence of bacteraemia in the first (RR = 0.70; 95% CI: 0.16 to 2.98) (Analysis 1.3) or second (one participant in each group developed bacteraemia) week of follow‐up; the incidence of respiratory tract infection during the first (RR = 2.80; 95% CI: 0.12 to 67.21) (Analysis 1.4) or second (2/58 participants in the SSD group and 1/54 patients in the MEBO group developed pneumonia) week of follow‐up; or the incidence of UTI during the first (RR = 0.47; 95% CI: 0.04 to 4.99) (Analysis 1.5) or second (one participant in each group developed UTI) week of follow‐up. There was an overall high risk of bias for all invasive infection outcomes.
1.4. Analysis.
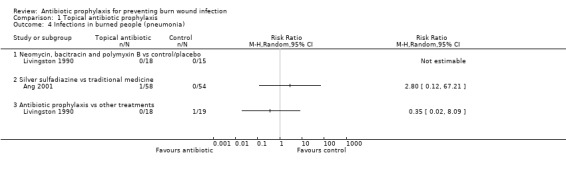
Comparison 1 Topical antibiotic prophylaxis, Outcome 4 Infections in burned people (pneumonia).
1.5. Analysis.
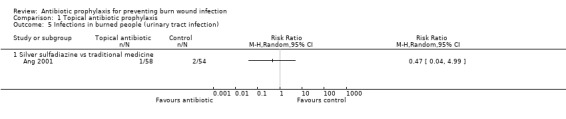
Comparison 1 Topical antibiotic prophylaxis, Outcome 5 Infections in burned people (urinary tract infection).
Outcome 3: Infection‐related mortality
Ang 2001 reported that one participant in the SSD group died due to infection. There was no statistically significant in mortality but given only one death this comparison lacks statistical power (RR = 2.80, 95% CI: 0.12 to 67.21). The overall risk of bias for this outcome was high.
Outcome 5: Objective measures of wound healing
Ang 2001, Khorasani 2009, and Moharamzad 2010 reported on this outcome. In Ang 2001, the mean time needed for 75% epithelisation of the wound was 20 days in the SSD group and 17 days in the MEBO group (no statistically significant difference, hazard ratio [HR]: 0.67; 95%CI: 0.41 to 1.11; P value 0.11) (Table 2). In Khorasani 2009, the mean time to wound healing was significantly longer in the SSD group than in the cream of Aloe vera group. In Moharamzad 2010, the mean time to healing was significantly longer in the cream made from herbs group than in the SSD group (Table 2).
The overall risk of bias was unclear except for Khorasani 2009, where it was high.
Outcome 6: Antibiotic resistance
Ang 2001 reported on the test results for detection of MRSA performed 14 days after treatment. The difference in MRSA incidence between the SSD and the silver‐coated dressing groups was not statistically significant (RR = 1.08; 95% CI: 0.66 to 1.76). (Analysis 1.8). The overall risk of bias was high.
Outcome 7: All‐cause mortality
Ang 2001 reported no significant differences between SSD and MEBO groups on the risk of mortality (RR = 1.86; 95% CI: 0.17 to 19.95) (Analysis 1.9). The overall risk of bias for this outcome was high.
Comparison 5: Other topical antibiotics compared with dressings or skin substitute
Three trials (85 participants) evaluated a non‐SSD topical antibiotic (bacitracin or mafenide acetate) compared with a synthetic or biosynthetic dressing or a skin substitute (Demling 1999; Demling 2003; Silver 2007). Two trials compared the bacitracin ointment with a biosynthetic skin substitute dressing, TransCyte (Demling 1999; Demling 2003). Silver 2007 evaluated mafenide acetate solution (Sulfamylon 5%) applied with a dressing (Exu‐Dry, Smith & Nephew) compared with a silver dressing (Acticoat, Smith & Nephew).
Outcome 1: Burn wound infection
None of the participants included in the three trials developed burn wound infection. We did not perform meta‐analysis with these studies because all of them presented no events in both arms.
Outcome 4: Adverse events
Silver 2007 (20 participants) reported that no participant presented with serious adverse events.
Outcome 5: Objective measures of wound healing
In Demling 1999 the mean time to wound healing (defined as 90% or more re‐epithelization) was significantly longer in the bacitracin group than in the biosynthetic dressing group. In Demling 2003, the mean time to healing (cicatrisation; defined as 95% or more re‐epithelisation) was significantly longer in the bacitracin group than in the biosynthetic dressing group (Table 2).
Comparison 6: Topical antibiotic prophylaxis compared with other treatments
Seven trials (353 participants) evaluated a topical antibiotic compared with other treatments of topical administration (Desai 1991; Fisher 1968; Glat 2009; Hauser 2007; Livingston 1990; Maya 1986; Mohammadi 2009). Desai 1991 evaluated gentamicin cream 1% applied by iontophoresis compared with routine care (cleaning and change of dressings). Fisher 1968 had three treatment arms: neomycin spray, Polybactrin, Dermoplast spray, and control (no spray). Glat 2009 compared SSD cream (Silvadene) with silver ions hydrogel (SilvaSorb Gel). Hauser 2007 compared SSD cream (Flammazine, Solvay Arzneimittel GmbH, Hannover, Deutschland) with a hydrosome gel (Repithel, Mundipharma GmbH, Limburg/Lahn, Deutschland). Livingston 1990 had three treatment arms: neomycin, bacitracin plus (bacitracin/polymyxin B) , silver nitrate 0.5% and placebo (Ringer's lactate). Maya 1986 compared rifamycin and amniotic membranes with amniotic membranes alone. Lastly, Mohammadi 2009 compared either SSD or mafenide acetate with amniotic membranes.
Outcome 1: Burn wound infection
Pooling the seven trials indicated no statistically significant difference in rates of burn wound infection between the antibiotic prophylaxis and control groups (OR = 1.51; 95% CI: 0.94 to 2.42, with no statistical heterogeneity (I² = 0%)) (Analysis 1.1). The overall risk of bias for this outcome was high for all the pooled trials.
Outcome 2: Infections in burned people
Three trials (227 participants) provided data on purulence with septicaemia (Fisher 1968), and sepsis (Livingston 1990; Mohammadi 2009). Meta‐analysis of these three trials showed a significantly greater incidence of sepsis among participants receiving antibiotic prophylaxis than for the group given other treatments (RR = 4.31; 95% CI: 1.61 to 11.49), with no statistical heterogeneity (I² = 0%) (Analysis 1.2). The overall risk of bias for this outcome was high for the three trials (Fisher 1968; Livingston 1990; Mohammadi 2009).
Fisher 1968 (66 participants) did not show a statistically significant difference in the incidence of bacteraemia between the Polybactrin group and the Dermoplast group (RR = 0.67; 95% CI: 0.12 to 3.73) (Analysis 1.3). Denominator values suggested complete follow‐up, but the overall risk of bias was high.
In Livingston 1990 (37 participants), one participant developed pulmonary sepsis in the silver nitrate group, but the difference was not statistically significant (RR = 0.35; 95% CI: 0.02 to 8.09) (Analysis 1.4). The overall risk of bias was high.
Outcome 3: Infection‐related mortality
In Livingston 1990, four out of 18 participants in the neomycin plus bacitracin group and one out of 19 participants in the silver nitrate group died due to sepsis and multiple organ failure. The estimations of the effect of interventions on these outcomes showed an important lack of precision that does not allow valid conclusions to be drawn (RR = 4.22; 95% CI: 0.52 to 34.28) (Analysis 1.7). Due to the high rate of post‐randomisation exclusions in this study (only 52 of 90 randomised participants were included in the analysis), the overall risk of bias was high.
Outcome 4: Adverse events
Glat 2009 (24 participants) was the only trial that reported on adverse events. The results from this trial stated that no participants developed adverse events during the study.
Outcome 5: Objective measures of wound healing
In Fisher 1968, there was no statistical difference in mean healing time of wounds between the Polybactrin group and the Dermoplast group. In Hauser 2007, burns were reviewed and evaluated by two independent investigators and time to complete healing was found to be longer in the SSD group than in the Repithel cream group; this difference was statistically significant (Table 2). The time‐to‐event outcome was presented as continuous data, so we have presented the data in narrative form only.
Outcome 6: Antibiotic resistance
In Livingston 1990 there were no statistically significant differences regarding the frequency of participants with MRSA between the two groups (neomycin, plus bacitracin group 2/18; and the silver nitrate group 0/19) (RR = 5.26; 95% CI: 0.27 to 102.66) (Analysis 1.8). The overall risk of bias for this outcome was high.
Outcome 7: All‐cause mortality
Two trials (181 participants) provided data on all‐cause mortality for neomycin plus bacitracin compared with silver nitrate (Livingston 1990), and SSD or mafenide acetate compared with amniotic membrane (Mohammadi 2009). The meta‐analysis of these two trials showed a statistically significantly higher incidence of mortality among participants treated with antibiotic prophylaxis than for the group that received other treatments (RR = 5.95; 95% CI: 1.10 to 32.33), with no statistical heterogeneity (I2 = 0%) (Analysis 1.9). The overall risk of bias for this outcome was high for both trials.
Additionally, the report of Livingston 1990 indicated that four more participants died during the study (two from myocardial infarction and two from pulmonary emboli), but the group(s) to which these participants belonged was not specified. Because of this, it was not possible to include this data in the main analysis. A sensitivity analysis was carried out with this data (see Dealing with missing data, Sensitivity analysis) which did not find differences with respect to the main analysis.
Outcome 8: Length of hospital stay
Four trials (216 participants) provided data on LOS for this comparison (Desai 1991; Livingston 1990; Maya 1986; Mohammadi 2009).
In Desai 1991, the mean time for LOS was shorter in the gentamicin cream group than in the routine care group. This difference was statistically significant (MD = ‐12.00 days; 95% CI: ‐17.52 to ‐6.48). Livingston 1990 did not find a statistically significant difference in length of stay between neomycin/bacitracin and silver nitrate (MD = 3.03; 95% CI: ‐2.01 to 8.07). In Maya 1986, the mean LOS was significantly shorter in the rifamycin and amniotic membranes group than in the group with amniotic membranes alone (MD = ‐4.41 days; 95% CI: ‐8.17 to ‐0.65). Mohammadi 2009 reported that the mean LOS was significantly longer in the SSD arm compared with amniotic membranes (MD = 9.77; 95% CI: 7.29 to 12.25) (Analysis 1.10).
The meta‐analysis of these four trials showed a high statistical heterogeneity (I² = 96%) (Analysis 1.10); therefore, its results are not presented. The risk of bias was high for the four trials.
2. Systemic antibiotic prophylaxis (general)
Three trials (119 participants) evaluated systemic antibiotics administered at admission or during routine treatment (Durtschi 1982; Kimura 1998; Munster 1986). All three studies compared an antibiotic administered orally, or intravenously, with no treatment or placebo. Durtschi 1982 evaluated penicillin (penicillin V (250 mg) orally every six hours or sodium penicillin 1.2 million units intravenously every 12 hours) compared with placebo. Most people received the medication or placebo orally for five days. Kimura 1998 compared trimethoprim‐sulfamethoxazole (TMP‐SMX) 1.0 g (400 mg SMX/TMP 80 mg) with placebo (lactose 1.0 g), both administered orally or by nasogastric tube three times a day. Other antibiotics such as ampicillin, cephazolin, cephamandole, cefmetazole, and flomoxef were administered in combination with TMP‐SMX or placebo when the attending physician deemed it necessary. Munster 1986 evaluated polymyxin B compared with an inactive control (received no antibiotic prophylaxis). The antibiotic regimen was as follows: 5000 units/kg intravenously on the first day of the study, subsequently, doses were reduced by 500 units/kg per day until 1500 units was reached on the last day.
Outcome 1: Burn wound infection
Neither of the two trials comparing the effects of systemic antibiotics on burn wound infection identified a statistically significant difference in rates of burn infection. In Durtschi 1982 11 out of 25 people in the penicillin V group developed a burn wound infection compared with 7 out of 26 in the placebo group (RR = 1.63; 95% CI: 0.75 to 3.54) (Analysis 2.1). In Munster 1986one person out of 15 in the polymixin B group developed a burn wound infection compared with five out of 13 in the no treatment group (RR = 0.17; 95% CI: 0.02 to 1.30) (Analysis 2.1).
2.1. Analysis.
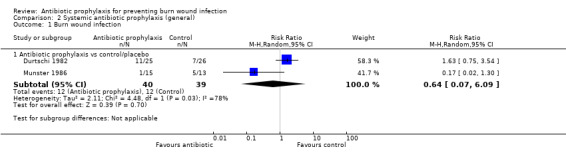
Comparison 2 Systemic antibiotic prophylaxis (general), Outcome 1 Burn wound infection.
Meta analysis of these studies was inappropriate due to the high statistical heterogeneity (I² = 78%) (Analysis 2.1). Overall risk of bias for this outcome was either high (Munster 1986), or unclear (Durtschi 1982).
Outcome 2: Infections in burned people
Two trials (59 participants) assessed incidence of sepsis with penicillin (Durtschi 1982) and polymyxin B (Munster 1986). Pooling the two trials (Durtschi 1982; Munster 1986) did not indicate a statistically significant difference in the number of participants with sepsis between the antibiotic prophylaxis group and its comparators (RR = 0.43; 95% CI: 0.12 to 1.61 (P value 0.21)), with no statistical heterogeneity (I2 = 0%) (Analysis 2.2) however statistical power is low with only 10 events. Overall risk of bias for this outcome was either high (Munster 1986), or unclear (Durtschi 1982).
2.2. Analysis.
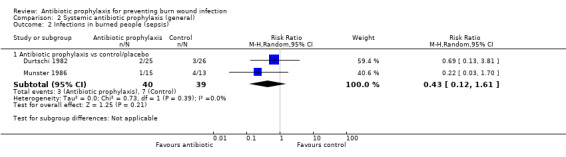
Comparison 2 Systemic antibiotic prophylaxis (general), Outcome 2 Infections in burned people (sepsis).
Durtschi 1982 (51 participants) reported data for beta‐haemolytic streptococcal bacteraemia: one participant developed bacteraemia in the penicillin group, but there were no statistically significant differences (RR = 3.12; 95% CI: 0.13 to 73.06) (Analysis 2.3).
2.3. Analysis.
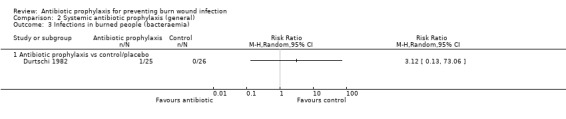
Comparison 2 Systemic antibiotic prophylaxis (general), Outcome 3 Infections in burned people (bacteraemia).
In Kimura 1998 (40 participants), a significantly lower number of participants developed pneumonia in the trimethoprim‐sulfamethoxazole (TMP‐SMX) group than in the placebo group (RR = 0.18; 95% CI: 0.05 to 0.72) (Analysis 2.4).
2.4. Analysis.
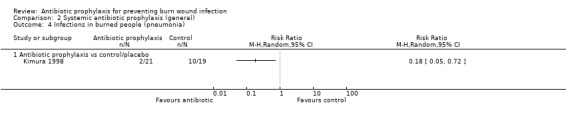
Comparison 2 Systemic antibiotic prophylaxis (general), Outcome 4 Infections in burned people (pneumonia).
In Durtschi 1982, one participant developed a UTI in the placebo group, but there were no statistically significant differences between the groups (RR = 0.35; 95% CI: 0.01 to 8.12) (Analysis 2.5). The overall risk of bias for all the invasive infection outcomes was unclear.
2.5. Analysis.
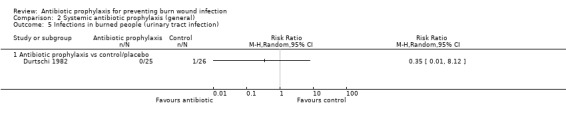
Comparison 2 Systemic antibiotic prophylaxis (general), Outcome 5 Infections in burned people (urinary tract infection).
Outcome 3: Infection‐related mortality
In Durtschi 1982, one participant in the penicillin group died due to infection compared with three participants in the placebo group. In Munster 1986, two participants in the control group died during the study as a consequence of sepsis. Pooling the two trials demonstrated a no significant difference regarding infection‐related mortality between comparison groups (RR = 0.27; 95% CI: 0.05 to 1.58), with no statistical heterogeneity (I2 = 0%) (Analysis 2.6). Overall risk of bias for this outcome was either high (Munster 1986), or unclear (Durtschi 1982).
2.6. Analysis.
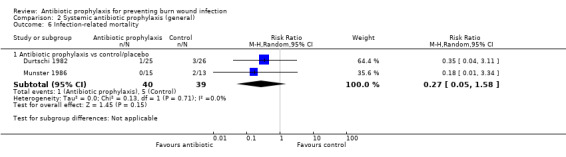
Comparison 2 Systemic antibiotic prophylaxis (general), Outcome 6 Infection‐related mortality.
Outcome 4: Adverse events
In Durtschi 1982, data on adverse events were not reported. Kimura 1998 and Munster 1986 reported that none of the participants developed adverse events and, consequently, there were no dropouts attributable to adverse effects. There was an overall high risk of bias.
Outcome 6: Antibiotic resistance
Kimura 1998 reported that there was a statistically significantly lower frequency of patients with MRSA in those treated with TMP‐SMX than in those treated with placebo (RR = 0.13; 95% CI: 0.02 to 0.96) (Analysis 2.7). There was an unclear overall risk of bias.
2.7. Analysis.
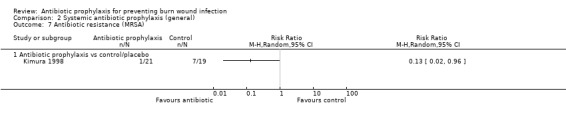
Comparison 2 Systemic antibiotic prophylaxis (general), Outcome 7 Antibiotic resistance (MRSA).
Outcome 7: All‐cause mortality
Three trials (109 participants) presented data on all‐cause mortality, comparing penicillin (Durtschi 1982), TMP‐SMX (Kimura 1998) or polymyxin B (Munster 1986) each with placebo or an inactive control. On pooling the results of these three trials (I= 0%) there was no statistically significant difference (RR = 0.41; 95% CI: 0.17 to 1.02) (Analysis 2.8). Overall risk of bias for this outcome was either high (Munster 1986), or unclear (Durtschi 1982; Kimura 1998).
2.8. Analysis.
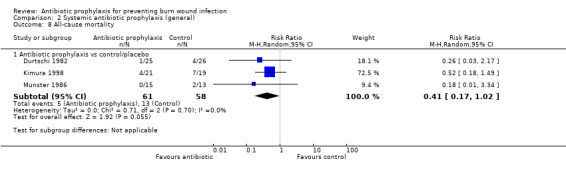
Comparison 2 Systemic antibiotic prophylaxis (general), Outcome 8 All‐cause mortality.
Outcome 8: Length of hospital stay
No significant difference was observed in LOS in Durtschi 1982 between the penicillin and placebo groups (MD = 0.80 days; 95% CI: ‐1.47 to 3.07) (Analysis 2.9). There was an overall high risk of bias.
2.9. Analysis.
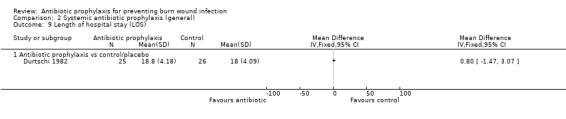
Comparison 2 Systemic antibiotic prophylaxis (general), Outcome 9 Length of hospital stay (LOS).
3. Systemic antibiotic prophylaxis (perioperative)
Four trials (390 participants) compared perioperative systemic antibiotics with an inactive control (no intervention or placebo) or another antibiotic (Alexander 1982; Alexander 1984; Miller 1987; Rodgers 1997).
Comparison 1: Antibiotic prophylaxis compared with no intervention or placebo (inactive control)
Three trials compared a systemic antibiotic with no treatment, or placebo, to prevent burn wound infection (Alexander 1982; Alexander 1984; Rodgers 1997). Alexander 1982 compared cephalothin 15 mg/kg intravenously with placebo (equal volume). Three doses were administered throughout the perioperative process. In Alexander 1984, a systemic antibiotic administered for debridement and skin graft (perioperative) was compared with a control group without antibiotic prophylaxis. Selection of the antibiotic was based on the antibiotic sensitivity of the dominant organism, and on the cultures of the most recent wound. Participants who received therapeutic antibiotics did not receive further antibiotic prophylaxis. All antibiotics were administered intravenously. Rodgers 1997 had four arms: where burns were less than 35% TBSA, cephazolin versus placebo and, where burns were more than 35% TBSA, cephazolin versus specific antibiotics. Cephazolin was administered at a dose of 25 mg/kg every six hours intravenously for 24 hours; the consultant for infectious diseases selected antibiotics specifically on the basis of the results of the most recent cultures.
Outcome 1 Burn wound infection
In Alexander 1982 (249 participants), there was no statistically significant difference in the incidence of burn wound infection between the cephalothin and placebo groups (RR = 0.14; 95% CI: 0.02 to 1.10). There was an unclear overall risk of bias for this outcome. In Rodgers 1997 (20 participants), there was no statistically significant difference in the incidence of burn wound infection between the cephazolin group and the placebo group (RR = 2.00; 95% CI: 0.21 to 18.69).
Meta‐analysis for this outcome was inappropriate due to high heterogeneity (I² = 67%) (Analysis 3.1).There was an overall high risk of bias for this outcome.
3.1. Analysis.
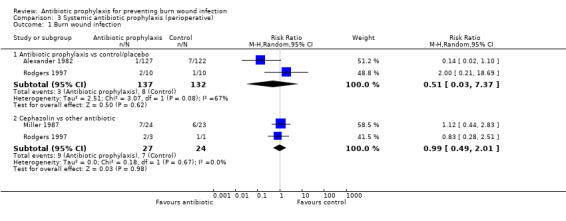
Comparison 3 Systemic antibiotic prophylaxis (perioperative), Outcome 1 Burn wound infection.
Outcome 2: Infections in burned people
Two trials (89 participants) reported the rates of bacteraemia (Alexander 1984; Rodgers 1997). There was no statistically significant difference in bacteraemia between treatment groups (RR = 1.32; 95% CI: 0.31 to 5.60), with no statistical heterogeneity (I2 = 0%) (Analysis 3.2). There was an overall high risk of bias for this outcome for both trials.
3.2. Analysis.
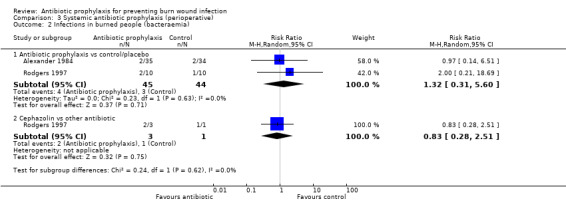
Comparison 3 Systemic antibiotic prophylaxis (perioperative), Outcome 2 Infections in burned people (bacteraemia).
Outcome 4: Adverse events
Alexander 1982 one participant in each group presented scattered areas of cutaneous erythema (RR = 0.96; 95% CI: 0.06 to 15.19) (Analysis 3.5). There was an unclear risk of bias.
3.5. Analysis.
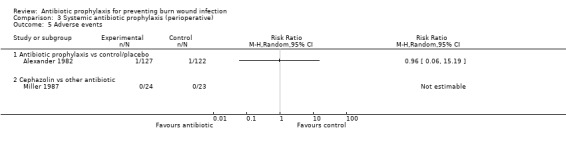
Comparison 3 Systemic antibiotic prophylaxis (perioperative), Outcome 5 Adverse events.
Outcome 7: All‐cause mortality
In Alexander 1984 the difference between groups for all‐cause mortality was not statistically significant (RR = 1.62; 95% CI: 0.42 to 6.25) (Analysis 3.6). There was an overall high risk of bias for this outcome.
3.6. Analysis.
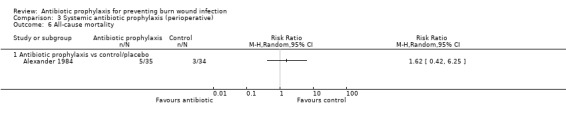
Comparison 3 Systemic antibiotic prophylaxis (perioperative), Outcome 6 All‐cause mortality.
Outcome 8: Length of hospital stay
In Alexander 1982, the mean LOS was 12.38 days in the cephalothin group and 13.66 days in the placebo group; this difference was statistically significant (MD = ‐1.28; 95% CI: ‐2.64 to 0.08; P value less than 0.02) (Analysis 3.7). There was an unclear risk of bias .
3.7. Analysis.
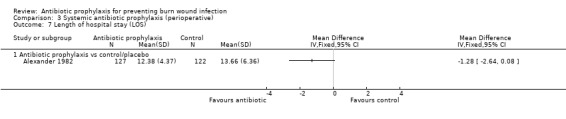
Comparison 3 Systemic antibiotic prophylaxis (perioperative), Outcome 7 Length of hospital stay (LOS).
Comparison 2: Cephazolin compared with another antibiotic
Two trials (51 participants) compared cephazolin with another antibiotic (Miller 1987; Rodgers 1997). Miller 1987 compared cephazolin 1g intravenously (three doses, the first one hour before surgery and two more doses six and 12 hours after the first dose) with ceforanide 1g intravenously applied one hour before surgery. Rodgers 1997 had four treatment arms; cephazolin versus placebo (where burns were less than 35% TBSA), and cephazolin versus specific antibiotics (burns more than 35% TBSA). Cephazolin was administered intravenously at a dose of 25 mg/kg every six hours for 24 hours.
Outcome 1: Burn wound infection
Two trials provided data for this outcome, either in comparison with ceforanide (Miller 1987), or with a specific, targeted antibiotic (Rodgers 1997). Pooling of data failed to demonstrate significant differences in burn wound infection between comparison groups (RR = 0.99; 95% CI: 0.49 to 2.01, with no statistical heterogeneity (I2 = 0%) (Analysis 3.1). The overall risk of bias for this outcome was high for both trials.
Outcome 2: Infections in burned people
Rodgers 1997 (four participants) showed no statistically significant differences in bacteraemia between cephazolin and the specific antibiotic group (RR = 0.83; 95% CI: 0.28 to 2.51) (Analysis 3.2).
Miller 1987 (47 participants) reported that one participant developed pneumonia during the study in the ceforanide group, but the difference was not statistically significant (RR = 0.32; 95% CI: 0.01 to 7.48) (Analysis 3.3).
3.3. Analysis.
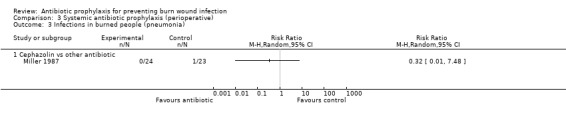
Comparison 3 Systemic antibiotic prophylaxis (perioperative), Outcome 3 Infections in burned people (pneumonia).
Miller 1987 reported that one participant developed urinary tract infection during the study in the cephazolin group, but this difference was not statistically significant (RR = 2.88; 95% CI: 0.12 to 67.29) (Analysis 3.4). There was an overall high risk of bias for all invasive infection outcomes.
3.4. Analysis.
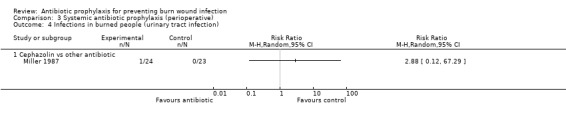
Comparison 3 Systemic antibiotic prophylaxis (perioperative), Outcome 4 Infections in burned people (urinary tract infection).
Outcome 4: Adverse events
Miller 1987 reported that no participant in the cephazolin or ceforanide groups presented with adverse events.
4. Non‐absorbable antibiotic prophylaxis (selective decontamination of the digestive tract (SDD))
Two trials (140 participants) evaluated non absorbable antibiotic prophylaxis using selective decontamination of the digestive tract (SDD) compared with placebo (Barret 2001; De La Cal 2005). Barret 2001 evaluated polymyxin E suspension (100 mg), tobramycin (100 mg), and amphotericin B (500 mg) by nasogastric tube four times a day compared with physiologic isotonic solution (Ringer's lactate). Additionally, systemic antibiotics (vancomycin, amikacin, and piperacillin) were administered preoperatively in both study arms in order to prevent sepsis and bacteraemia. De La Cal 2005 evaluated (1) polymyxin E (100 mg), tobramycin (100 mg) and amphotericin B (500 mg) administered orally four times a day; (2) cefotaxime 1 g iv 8‐hourly for 4 days; (3) non absorbable polymyxin E, tobramycin and amphotericin B 0.5 g of a 2% paste, topical application in the oropharynx 4 times/day, compared to (1) placebo solution administered orally; (2) placebo solution, isotonic 0.9% saline iv; (3) placebo paste, topical application in the oropharynx. The placebo solution was indistinguishable from the test drug with respect to colour, smell, and consistency. Other systemic antibiotics, such as vancomycin, ceftazidime and aminoglycosides, were administered empirically in both study arms when people developed clinical signs of infection; antibiotic treatment in these people was adjusted according to microbiological results.
Comparison 1: non‐absorbable antibiotic prophylaxis versus placebo
One trial with 23 participants compared non‐absorbable antibiotic prophylaxis, through selective decontamination of the digestive tract, against placebo (Barret 2001).
Outcome 2: Infections in burned people
There was no statistically significant difference in sepsis between the non‐absorbable antibiotic group and the placebo group for Barret 2001 (23 participants) (RR = 2.18; 95% CI: 0.49 to 9.65) (Analysis 4.2).
4.2. Analysis.
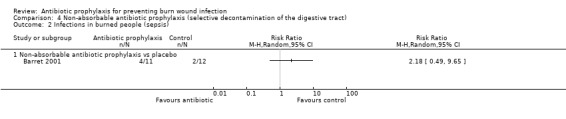
Comparison 4 Non‐absorbable antibiotic prophylaxis (selective decontamination of the digestive tract), Outcome 2 Infections in burned people (sepsis).
Barret 2001 (23 participants) only one participant developed pneumonia in the non absorbable antibiotic group , the difference was not statistically significant (RR= 3.25; 95% CI: 0.15 to 72.36) (Analysis 4.4) The overall risk of bias was unclear for both outcomes.
4.4. Analysis.
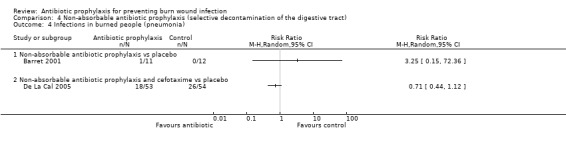
Comparison 4 Non‐absorbable antibiotic prophylaxis (selective decontamination of the digestive tract), Outcome 4 Infections in burned people (pneumonia).
Outcome 4: Adverse events
In Barret 2001 (23 participants), significantly more participants in the non‐absorbable antibiotics group developed adverse events (diarrhoea or gastrointestinal bleeding) than in the placebo group (RR = 3.64; 95% CI: 1.34 to 9.86) (Analysis 4.6). The overall risk of bias was unclear.
4.6. Analysis.
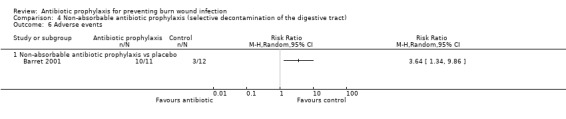
Comparison 4 Non‐absorbable antibiotic prophylaxis (selective decontamination of the digestive tract), Outcome 6 Adverse events.
Outcome 5: Objective measures of wound healing
In Barret 2001 there was no statistically significant difference in mean time to wound healing 40 ±8 days in the antibiotic group compared with 33±4 days in the placebo group (Table 2). There was an overall unclear risk of bias.
Outcome 7: All‐cause mortality
Barret 2001 reported that two out of 11 participants in the non‐absorbable antibiotic group died from respiratory distress and one out of 12 participants in the placebo group died of a systemic fungal infection. There was no statistically significant difference in mortality between the groups (RR = 2.18; 95% CI: 0.23 to 20.84) (Analysis 4.8). The overall risk of bias for this outcome was unclear.
4.8. Analysis.
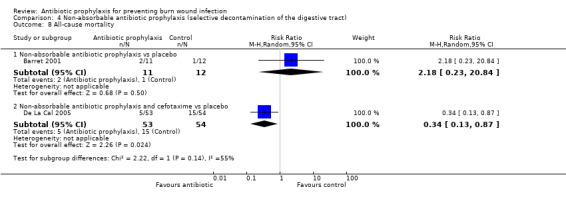
Comparison 4 Non‐absorbable antibiotic prophylaxis (selective decontamination of the digestive tract), Outcome 8 All‐cause mortality.
Outcome 8: Length of hospital stay
In Barret 2001, the mean LOS was significantly longer in the non‐absorbable antibiotic group (42 days) compared with the placebo group (35 days) (MD = 7.00 days; 95% CI: 3.28 to 10.72) (Analysis 4.9), and there was an overall low risk of bias for this outcome. The overall risk of bias for this outcome was unclear.
4.9. Analysis.
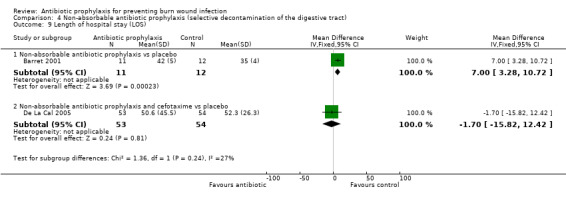
Comparison 4 Non‐absorbable antibiotic prophylaxis (selective decontamination of the digestive tract), Outcome 9 Length of hospital stay (LOS).
Comparison 2: non‐absorbable antibiotic prophylaxis and cefotaxime versus placebo.
One trial with 117 participants evaluated non‐absorbable antibiotic prophylaxis and cefotaxime, against placebo, however data are presented for 107 participants as there 10 post randomisation losses (De La Cal 2005).
Outcome 1: Burn wound infection
In De La Cal 2005 (107 participants), there was no statistically significant difference between the number of participants with burn wound infection in the non‐absorbable antibiotics and cefotaxime group (10/53) compared with the placebo group (11/54) (RR = 0.93; 95% CI: 0.43 to 2.00) (Analysis 4.1).
4.1. Analysis.
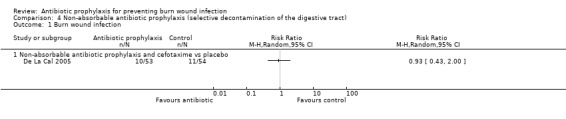
Comparison 4 Non‐absorbable antibiotic prophylaxis (selective decontamination of the digestive tract), Outcome 1 Burn wound infection.
Outcome 2: Infections in burned people
There was no statistically significant difference in occurrence of bacteraemia between the non‐absorbable antibiotic and cefotaxime group and the placebo group for De La Cal 2005 (107 participants) (RR = 1.14; 95% CI: 0.67 to 1.94) (Analysis 4.3).
4.3. Analysis.
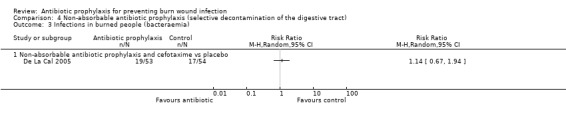
Comparison 4 Non‐absorbable antibiotic prophylaxis (selective decontamination of the digestive tract), Outcome 3 Infections in burned people (bacteraemia).
In De La Cal 2005 18/53 participants in the non absorbable antibiotic and cefotaxime group developed pneumonia during the study, compared with 26/54 participants in the placebo group, the difference was not statistically significant (RR= 0.71; 95% CI: 0.44 a 1.12) (Analysis 4.4).
There were no statistically significant differences in urinary tract infection between groups for De La Cal 2005 (RR = 0.44; 95% CI: 0.18 to 1.05) (Analysis 4.5). The overall risk of bias was unclear for all four infection‐related outcomes.
4.5. Analysis.
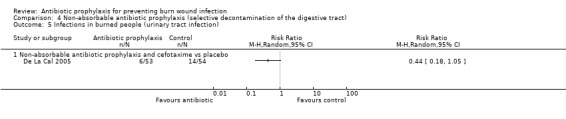
Comparison 4 Non‐absorbable antibiotic prophylaxis (selective decontamination of the digestive tract), Outcome 5 Infections in burned people (urinary tract infection).
Outcome 6: Antibiotic resistance
In De La Cal 2005 the results of colonization by organisms resistant to antibiotics were reported as endogenous secondary infections, which was defined as the colonization caused by micro‐organisms that were not present at admission, but that were acquired during treatment in the intensive care unit.
According to De La Cal 2005, the number of participants who developed an MRSA infection was significantly higher in the non‐absorbable antibiotics and cefotaxime group (24/53) than in the placebo group (11/54). This difference was statistically significant (RR = 2.22; 95% CI: 1.21 to 4.07) (Analysis 4.7). The overall risk of bias was unclear.
4.7. Analysis.
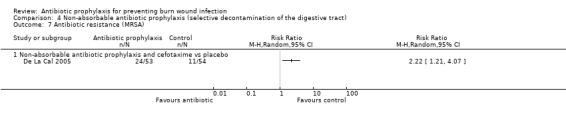
Comparison 4 Non‐absorbable antibiotic prophylaxis (selective decontamination of the digestive tract), Outcome 7 Antibiotic resistance (MRSA).
Outcome 7: All‐cause mortality
In De La Cal 2005, significantly fewer participants in the non‐absorbable antibiotics and cefotaxime group (5/53) died during the study than in the placebo group (15/54) (RR = 0.34; 95% CI: 0.13 to 0.87) (Analysis 4.8). The overall risk of bias for this outcome was unclear in both trials.
Outcome 8: Length of hospital stay
In De La Cal 2005, the mean LOS was 50.6 days in the non‐absorbable antibiotics and cefotaxime group and 52.3 days in the placebo group; this difference was not statistically significant (MD = ‐1.70 days; 95% CI: ‐15.82 to 12.42) (Analysis 4.9). The overall risk of bias for this outcome was unclear.
5. Local antibiotic prophylaxis (airway)
Only one trial (30 participants) evaluated local antibiotics (gentamicin 80 mg in 2 ml of diluent) administered by airway compared with placebo (2 ml of saline solution) (Levine 1978). burns of all participants included in the study were treated with SSD cream (Silvadene) or mafenide acetate (Sulfamylon).
Outcome 2: Infections in burned people
There was no statistically significant difference in incidence of sepsis between the gentamicin and placebo group (RR = 1.04; 95% CI: 0.67 to 1.60) (Analysis 5.1). There was an overall high risk of bias.
5.1. Analysis.
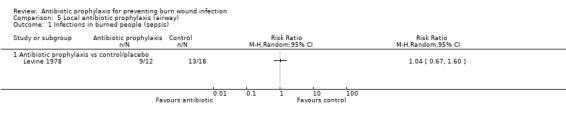
Comparison 5 Local antibiotic prophylaxis (airway), Outcome 1 Infections in burned people (sepsis).
Outcome 7: All‐cause mortality
There was no statistically significant difference between the antibiotic and placebo groups for all‐cause mortality (RR = 0.75; 95% CI: 0.39 to 1.44) (Analysis 5.2). The overall risk of bias for this outcome was high. A significant number of participants who died in the placebo group had more than 60% of their total body surface area burned.
5.2. Analysis.
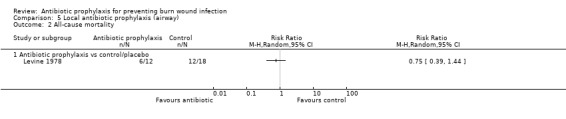
Comparison 5 Local antibiotic prophylaxis (airway), Outcome 2 All‐cause mortality.
6. Antibiotic prophylaxis compared with inactive control (no intervention or placebo)
We now present a summary of the results of studies comparing any antibiotic with an inactive control (i.e. no intervention or placebo) (Alexander 1982; Barret 2001; De La Cal 2005; Durtschi 1982; Fisher 1968; Kimura 1998; Levine 1978; Livingston 1990; Munster 1986; Rodgers 1997) (Analysis 6.1 to Analysis 6.10). In this section, we just describe the results concerning the primary outcome variables of the review.
6.1. Analysis.
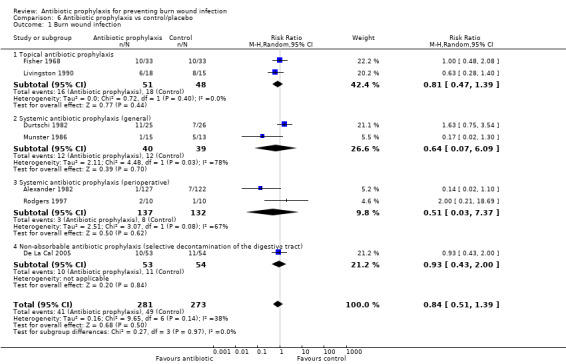
Comparison 6 Antibiotic prophylaxis vs control/placebo, Outcome 1 Burn wound infection.
6.10. Analysis.
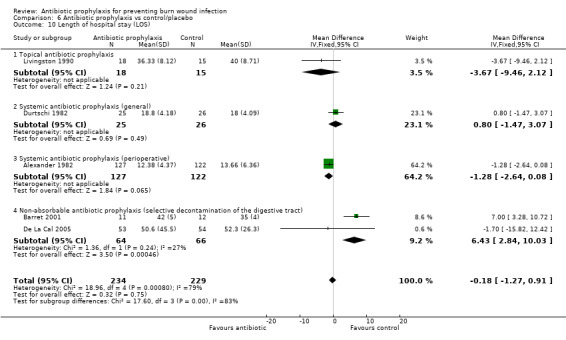
Comparison 6 Antibiotic prophylaxis vs control/placebo, Outcome 10 Length of hospital stay (LOS).
Outcome 1: Burn wound infection
Pooling seven trials (554 participants) revealed no statistically significant difference between treatments in the number of participants with burn wound infection (RR = 0.84; 95% CI: 0.51 to 1.39) (Alexander 1982; De La Cal 2005; Durtschi 1982; Fisher 1968; Livingston 1990; Munster 1986; Rodgers 1997), with moderate statistical heterogeneity (I2 = 38%) (Analysis 6.1).
Outcome 2: Infections in burned people
Meta‐analysis of six trials (231 participants) showed no statistically significant difference between treatments in the number of participants who developed sepsis (RR = 1.06; 95% CI: 0.54 to 2.10) (Barret 2001; Durtschi 1982; Fisher 1968; Levine 1978; Livingston 1990; Munster 1986), without relevant statistical heterogeneity (I2 = 25%) (Analysis 6.2).
6.2. Analysis.
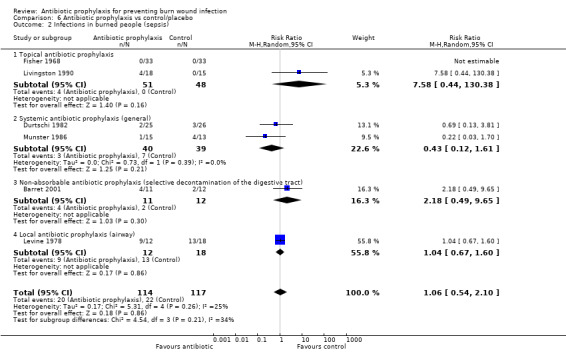
Comparison 6 Antibiotic prophylaxis vs control/placebo, Outcome 2 Infections in burned people (sepsis).
Pooling of five trials (313 participants) (Alexander 1982; De La Cal 2005; Durtschi 1982; Fisher 1968; Rodgers 1997), showed no statistically significant difference between treatments for the number of participants who developed bacteraemia (RR = 1.08; 95% CI: 0.67 to 1.72), without statistical heterogeneity (I2 = 0%) (Analysis 6.3).
6.3. Analysis.
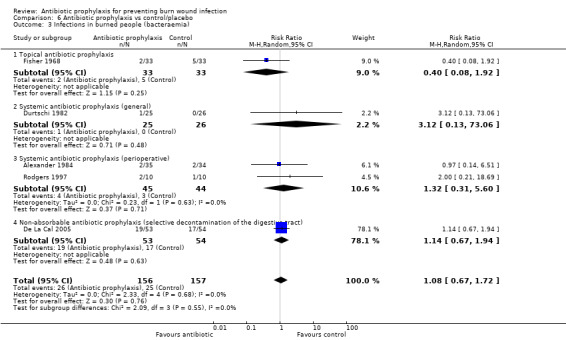
Comparison 6 Antibiotic prophylaxis vs control/placebo, Outcome 3 Infections in burned people (bacteraemia).
Pooling of four trials (203 participants) suggested that the number of participants with pneumonia was lower in the antibiotic prophylaxis group (Barret 2001; De La Cal 2005; Kimura 1998; Livingston 1990), however, given that the meta‐analysis had substantial heterogeneity (I² = 56%), its results are not presented (Analysis 6.4).
6.4. Analysis.
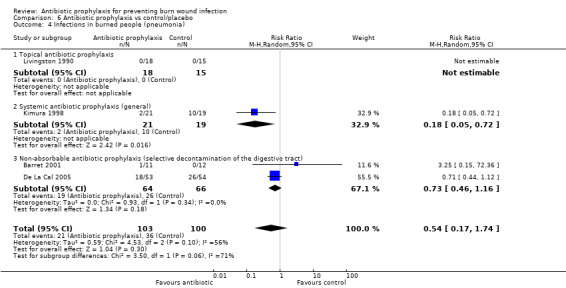
Comparison 6 Antibiotic prophylaxis vs control/placebo, Outcome 4 Infections in burned people (pneumonia).
Pooling of two trials (158 participants) revealed no statistically significant difference between treatments for the number of participants who developed UTI (RR = 0.43; 95% CI: 0.18 to 1.00) (De La Cal 2005; Durtschi 1982), without statistical heterogeneity (I2 = 0%) (Analysis 6.5).
6.5. Analysis.
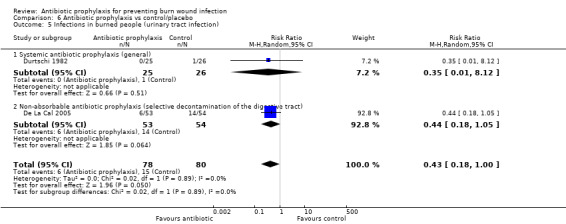
Comparison 6 Antibiotic prophylaxis vs control/placebo, Outcome 5 Infections in burned people (urinary tract infection).
Outcome 3: Infection‐related mortality
Pooling of two trials (79 participants) revealed no statistically significant differences between treatments for the number of participants who died of infection (RR = 0.27; 95% CI: 0.05 to 1.58) (Durtschi 1982; Munster 1986), with no statistical heterogeneity (I2 = 0%) (Analysis 6.6).
6.6. Analysis.
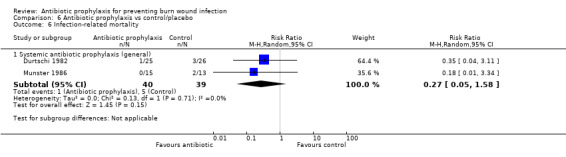
Comparison 6 Antibiotic prophylaxis vs control/placebo, Outcome 6 Infection‐related mortality.
Outcome 4: Adverse events
Meta‐analysis of four trials (340 participants) showed that the number of participants who developed at least one adverse event was statistically higher in the antibiotic groups than in the control groups (RR = 3.12; 95% CI: 1.22 to 7.97) (Alexander 1982; Barret 2001; Kimura 1998; Munster 1986), with no statistical heterogeneity (I2 = 0%) (Analysis 6.7).
6.7. Analysis.
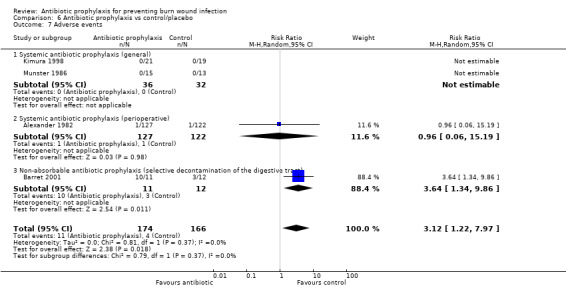
Comparison 6 Antibiotic prophylaxis vs control/placebo, Outcome 7 Adverse events.
Outcome 6: Antibiotic resistance
Pooling three trials (180 participants) showed no statistically significant difference between treatment groups for the number of participants who developed MRSA infection (De La Cal 2005; Kimura 1998; Livingston 1990), however, given that the meta‐analysis had high heterogeneity (I² = 79%), its results are not presented (Analysis 6.8).
6.8. Analysis.
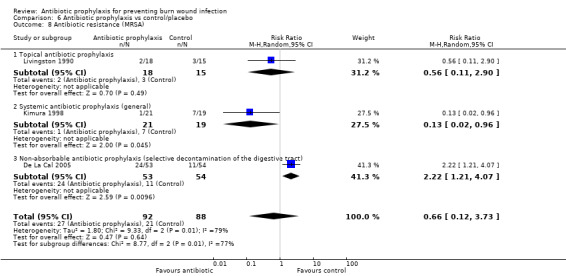
Comparison 6 Antibiotic prophylaxis vs control/placebo, Outcome 8 Antibiotic resistance (MRSA).
Outcome 7: All‐cause mortality
Pooling seven trials (348 participants) showed that a statistically significantly greater number of people died in the control group (RR = 0.62; 95% CI: 0.39 to 0.99) (Alexander 1982; Barret 2001; De La Cal 2005; Durtschi 1982; Kimura 1998; Levine 1978; Munster 1986), with low statistical heterogeneity (I2 = 9%) (Analysis 6.9).
6.9. Analysis.
Comparison 6 Antibiotic prophylaxis vs control/placebo, Outcome 9 All‐cause mortality.
Outcome 8: Length of hospital stay
Pooling five trials (463 participants) showed that there was no statistically significant difference between treatment groups for LOS (Alexander 1982; Barret 2001; De La Cal 2005; Durtschi 1982; Livingston 1990). However, given that the meta‐analysis had high heterogeneity (I² = 79%), its results are not presented (Analysis 6.10).
Subgroup analysis
We did not perform subgroup analyses considering the factors specified in the protocol (participants’ age and severity of the burn) due to a lack of data in the included studies; even though 15 of the 36 included studies included children and adults, none presented the results separately according to age group.
Sensitivity analysis
Analysis with 'cluster designs':
No cluster RCTs were identified.
Risk of bias
Only one study was classified as ‘low risk of bias’ (Barret 2001), therefore, it was not possible to conduct the corresponding sensitivity analysis.
Levels of missing data
Most of the included trials had low levels of missing data (less than 20%); one trial had 21% of data missing (Noordenbos 1999), and three had more than 40% (Durtschi 1982; Livingston 1990; Rodgers 1997). We performed a sensitivity analysis to explore the impact of the levels of missing data on the overall treatment effect for the main variable of burn wound infection.
When comparing analyses of silver sulfadiazine versus dressings or skin substitutes, there was no significant difference between the analysis that included all studies (RR = 1.74; 95% CI: 1.08 to 2.79; I2 = 0%) (Analysis 1.1), and the analysis that included only studies with less than 20% of data missing (RR= 1.64; 95% CI: 1.02 to 2.65; I2 = 0%).
In the comparison of antibiotic prophylaxis versus other treatments, no significant difference was found when the analysis that included all studies (RR = 1.39; 95% CI: 1.11 to 1.75; I2 = 0%) (Analysis 1.1) was compared with the analysis that included only studies with less than 20% of data missing (RR = 1.37; 95% CI: 1.07 to 1.74; I2 = 0%), however, it was not possible to compare the results to the remaining comparisons because it was not possible to estimate the effect from studies with less than 20% of data missing.
Worst case scenario analysis (considering dichotomous missing data as negative events)
The worst‐case scenario included 10 trials with incomplete outcome data (Bugmann 1998; Caruso 2006; Durtschi 1982; Gerding 1990; Hosseini 2009; Livingston 1990; Miller 1987; Noordenbos 1999; Rodgers 1997; Soroff 1994) and found no significant differences with respect to the main analysis strategy.
Statistical model for meta‐analysis
We performed a sensitivity analysis using a fixed‐effect model. Overall, there were no significant differences in the results for any outcome with respect to the analysis under the random‐effects model. As expected, the confidence intervals tended to be narrower when applying the fixed‐effect model, particularly for moderate and highly heterogeneous comparisons (i.e. I2 = more than 50%) when no explanation for heterogeneity was found (e.g. clinical or pharmacological intervention, or population differences among trials).
Analyses restricted to studies including participants with specific co‐morbidities
It was not possible to perform an analysis restricted to studies including participants with specific co‐morbidities because the vast majority of included trials did not report the co‐morbidities. Only two trials reported that eligible participants did not have any co‐morbidity (Kimura 1998; Mohammadi 2009), and eight trials reported that they had included participants with co‐morbidities such as diabetes, kidney disease, liver dysfunction, immunodeficiency, massive obesity, or severe malnutrition (De La Cal 2005; Durtschi 1982; Gong 2009; Gotschall 1998; Khorasani 2009; Miller 1990; Munster 1986; Silver 2007).
Reporting bias
We were able to evaluate the possibility of publication bias in only one comparison, topical antibiotic prophylaxis. We produced a funnel plot for burn wound infection including twenty‐six trials with five comparisons. We found symmetry in this plot and, therefore, we did not detect evidence of publication bias.
Discussion
This systematic review summarizes the best available evidence on the effects of antibiotic prophylaxis in people with burn wounds. Thirty‐six randomised controlled trials with a total of 2117 participants met the eligibility criteria for the review. The results were analysed according to the following groups: 1) topical antibiotic prophylaxis; 2) systemic antibiotic prophylaxis (general); 3) systemic antibiotic prophylaxis (perioperative); 4) non‐absorbable antibiotics (selective decontamination of the digestive tract); 5) local antibiotic prophylaxis (administered by airway); and 6) any antibiotic prophylaxis versus control.
Summary of main results
Efficacy of antibiotic prophylaxis
1. Topical antibiotic prophylaxis
Overall there is no evidence that the use of prophylactic, topical antibiotics (compared with other topical preparations, dressings, placebo or no treatment_ reduces the risk of burn wound infection, invasive infections (pneumonia, bacteraemia, sepsis or UTI), or mortality associated with infection. Meta‐analysis of data from 11 RCTs indicates that participants treated with topical silver sulfadiazine (SSD) have a higher risk of burn wound infection than those treated with dressings/skin substitutes, although the trials included in this analysis had either a high or an unclear risk of bias.
There is no evidence either that topical antibiotics have an influence on the secondary outcomes of this review. Generally time to wound healing was poorly analysed (as a continuous outcome rather than a time to event outcome) in the trials identified so it is difficult to judge the impact of the interventions on burn healing time. The average length of hospital stay (LOS) was significantly longer in participants whose burns were treated with SSD compared with dressings or skin substitute.
2. Systemic antibiotic prophylaxis in the non‐surgical patient
There is no evidence that general systemic antibiotic prophylaxis compared with placebo or no active treatment has an influence on any of the primary outcome variables assessed (burn wound infection, sepsis, bacteraemia, UTI, or death associated with infection). The only clear benefit was a reduction in the incidence of pneumonia with TMP‐SMX compared with placebo, however, this was obtained from a small trial (40 participants) with an uncertain risk of bias (Kimura 1998). Additionally, it was supported by Shionogi Pharmaceutical Company, which provided technical assistance in the measurement of TMP‐SMX concentrations. There is no evidence that systemic antibiotic prophylaxis has an effect on the secondary outcomes of this review.
3. Perioperative systemic antibiotic prophylaxis
There is no evidence that perioperative systemic antibiotic prophylaxis compared with placebo or another antibiotic influences any of the outcome variables of this review (primary or secondary).
4. Selective decontamination of the digestive tract (SDD)
There is no evidence that selective digestive tract decontamination (SDD) influences the frequency of burn wound infection, sepsis, or bacteraemia. Evidence indicates, however, that people in the SDD group developed more adverse events (diarrhoea) compared with those receiving placebo (Barret 2001). With regard to secondary outcomes, one study suggested that the number of participants who developed MRSA infection was higher in the SDD group than in the placebo group and LOS stay was greater in participants treated with SDD than in those who received placebo (Barret 2001).
5. Local antibiotic prophylaxis (administered by airway)
There is no evidence that gentamicin administered by airway influences on the frequency of sepsis or total mortality when compared to placebo.
Safety of antibiotic prophylaxis
In general, it could not be demonstrated that antibiotic prophylaxis is associated with an increase in adverse events in any of the comparisons, except for one study that suggested an increased frequency in people receiving SDD compared to those receiving placebo (Barret 2001).It should be noted that adverse events were poorly reported.
Overall completeness and applicability of evidence
Despite the fact that during recent years several measures to improve care of burn patients have been implemented, treating burn wounds continues to be a complex process. With the establishment of early excision of damaged tissue, skin grafts, and the strict implementation of infection‐control measures in burn‐care centres, it has been possible to reduce bacterial resistance, though not the incidence of infections. Topic application of SSD has shown a higher risk of burn wound infection and an increased length of hospital stay.
The results given in this review are still limited; few data could be pooled in most comparisons. Outcome measures and follow‐up times were heterogeneous, or not even defined, which made it difficult to interpret the results of the review and to determine their applicability. These results, however, will undoubtedly evolve with the establishment of new strategies and the standardization of care for burn wounds. As a result of these factors, it was not possible to identify or generate definitive evidence on the effects of antibiotic prophylaxis in people with burn wounds.
Quality of the evidence
Overall, the risk of bias of the studies was high or uncertain, and many had small sample sizes. Consequently, there is little evidence about the effects of antibiotic prophylaxis in people with burn wounds. Many of the key methodological aspects considered in the risk of bias tool were not described in the reports of the studies (or were not presented with sufficient information to allow their evaluation). Most of the studies did not follow the recommendations of the CONSORT statement (Moher 2001), even when they were published after 2001 ‐ the year in which the CONSORT statement was published.
Only one study presented a low risk of selection bias (i.e. used adequate methods to generate a random allocation sequence and to conceal this sequence) (Barret 2001), however, this study had only 23 participants. Only seven studies adequately described the methods used to generate the random sequence (and were deemed appropriate), and only four studies adequately described the methods used to conceal the sequence (and were deemed appropriate). Often, there was not enough information to assess the blinding of the study, or whether participants had been blinded. A key methodological point in this kind of study is blinding the person in charge of measuring outcomes, but most of the included studies did not report whether this was done. Loss of participants in the included studies was generally low, although the sample sizes for many of them were small. Most studies did not explain if, or how, the sample size was predetermined. Another key methodological aspect of some of the included studies was using more than one burn wound per participant (Gerding 1988; Gerding 1990; Hauser 2007; Khorasani 2009; Noordenbos 1999; Soroff 1994). In these cases, the strategies of analysis implemented were inadequate, because they did not take into account the methodological peculiarities of such designs.
There was a high degree of heterogeneity between studies in terms of interventions evaluated, types of burn, and outcomes assessed. This made it difficult to determine the effectiveness of antibiotic prophylaxis.
We evaluated the possibility of publication bias for one of the comparisons and one of the outcome measures, namely topical antibiotic prophylaxis and burn wound infection, respectively (Figure 4). The figure included twenty‐five trials with five comparisons. Given that this graph displayed symmetry, we did not detect evidence that suggested publication bias.
Potential biases in the review process
Publication bias is a major threat to the validity of systematic reviews. To minimize the risk of publication bias, we conducted an exhaustive search across numerous clinical trial databases. Nonetheless, as for any systematic review, we cannot rule out distortion of the results by publication bias.
Some studies reported the mean time to healing in each group, but did not provided complete data for time‐to‐event analysis. Only one trial reported outcome measures with hazard ratio (HR), therefore, we could not pool the trial data to estimate the hazard ratio. Also, the lack of information regarding the standard deviation around the mean in several trials, did not allow us to perform a pooled estimation of mean difference. This may have hindered the consideration of all relevant information available for the outcome of 'time to wound healing'.
Agreements and disagreements with other studies or reviews
There have been a number of other systematic reviews in the field although none precisely overlaps with ours in focus.
The review by Avni 2010 also evaluated the effect of antibiotic prophylaxis in people with burn wounds. This review differed from ours with respect to the inclusion criteria for the studies, and to the methodology. One of the main differences was that Avni 2010 considered mortality from any cause as the main outcome, and bacteraemia, pneumonia, and burn wound infection as secondary outcomes. Avni 2010 agreed with the results of our review when suggesting that systemic antibiotic prophylaxis (general or perioperative) may reduce the incidence of pneumonia (RR = 0.55; 95% CI: 0.36 to 0.84, three trials) and all‐cause mortality (RR = 0.54; 95% CI: 0.34 to 0.87, five trials). This review suggests that non‐absorbable antibiotics do not significantly affect mortality, however, Avni 2010 concluded that systemic antibiotic prophylaxis applied perioperatively may have a beneficial effect in reducing burn wound infection, which disagrees with the results of our review. Avni 2010 found the risk of bias of trials was high.
The systematic review by Lee 2009, suggests that the available evidence does not justify the use of general systemic antibiotic prophylaxis in the management of burn wounds in children, we cannot confirm this as we did not have data on children. Lee 2009, however, mentions that currently there are topical antimicrobial agents that guarantee lower rates of colonization and infection ‐ a claim that does not correspond the results of our review.
The review by Wasiak 2008 studied the effects of wound dressings rather than antibiotics and was focused only on superficial and partial thickness burns, whereas our review included studies of people with burns of any severity. Another key difference between Wasiak 2008 and our review is our primary focus on infection, mortality and adverse events as primary outcomes rather than wound healing. Nevertheless we reached the same conclusion as
Wasiak 2008 regarding SSD; they concluded that dressings impregnated with SSD decrease the healing of burn wounds; these results were confirmed by our review, which is based on a greater number of studies.
Hoogewerf 2013 assessed the effects of topical interventions for wound healing on facial burns and therefore the eligibility criteria defined are somewhat different from ours. Hoogewerf 2013 concluded that “there is insufficient reliable evidence as to whether topical treatments improve outcomes for people with facial burns including improved wound healing, rates of infection, the need for surgery...". Our findings echo this; there is a relative lack of evidence for the effects of the prophylactic use of topical antibiotics in people with burns (as compared to other topical treatments, placebo or no treatment), due to the volume and quality of the existing research.
The systematic review by Rosanova 2012compared different topical agents for preventing burn wound infections. Rosanova 2012 included both randomised and quasi‐randomised trials, and considered infections and sepsis as primary outcomes. This review concluded that there is no evidence to prove the superiority of any one topical agent to reduce infection or sepsis in the burn patient. Our review supports these conclusions.
Finally our review is in broad agreement with recommendations made by recent clinical practice guidelines on the management of burn wounds, which do not recommend antibiotic prophylaxis for the prevention of infection in the burned person (Alsbjörn 2007; Brychta 2011;Hospenthal 2011; NSW Severe Burn Injury Service 2008; WLDI 2008). In contrast the New Zealand Guidelines Group (NZGG 2007) recommended the use of products with antimicrobial action (such as silver sulphadiazine cream) on all burns for the first 72 hours (three days) after burn injury. However the New Zealand guideline clearly stated that there was little evidence supporting the use of silver sulphadiazine for non‐infected burns, and that the recommendation for its routine use during the first three days was supported only by clinical experience in New Zealand populations, specifically by the high incidence of community‐acquired Staphylococcus aureus sepsis (Miles 2005).
Authors' conclusions
Implications for practice.
The available evidence is limited and, in general, does not demonstrate that antibiotic prophylaxis reduces the risk of burn wound infection, invasive infections, or mortality associated with infection.
The use of topical antibiotics in burn wounds needs to be reconsidered,and specifically the use of SSD, since the available evidence suggests that patients treated with topical silver sulfadiazine have a higher risk of burn wound infection and longer length of hospital stay than those treated with dressings. The evidence concerning the safety of antibiotic prophylaxis is limited, and it is not possible to generate conclusions about it, although one study suggested that patients treated with selective digestive tract decontamination had a higher frequency of adverse events than those treated with placebo.
Implications for research.
The results of this review suggest that the effects of antibiotic prophylaxis in burn patients have not been studied sufficiently. Clinical trials with adequate statistical power are required to evaluate the effects of the different modalities of antibiotic prophylaxis (topical, general systemic, perioperative systemic, selective digestive decontamination, and delivered by airway), compared with placebo or standard treatment on the prevention of burn wound infection (burn wound infection), other infections, or mortality associated with infection. The safety of these interventions is to be determined. Additionally, an economic evaluation of such interventions is warranted.
Future randomised trials should be designed and conducted rigorously. The design and implementation of future studies must guarantee adequate generation and concealment of the randomisation sequence, as well as blinding of participants and evaluators of outcomes. In addition, researchers must ensure proper monitoring of participants, minimize losses, and handle losses in agreement with sound statistical analysis. With specific reference to trials that randomise burns on the same person to different interventions, methodological characteristics inherent to this type of design must be taken into account (Louis 1984; Mills 2009). Many of these characteristics are present in self‐controlled trials, and should be considered when pre‐determining the sample size and analysing the data, ideally with the advice of a statistician. The corresponding reports should present relevant information in a clear manner, and allow critical appraisal of their methodology, results and applicability. It is recommended that they abide by the guidelines of the CONSORT declaration for clinical trials (Moher 2001; Schulz 2010), or any of its extensions, when pertinent (Boutron 2008).
As for the participants, it is necessary to define the degree of burns clearly; describing depth and total body surface burned to allow assessment of the applicability of the interventions tested. There should be a protocol for management and burn care that must be applied consistently across study arms, so that the effect of antibiotic prophylaxis can be determined. In addition, consensus is needed amongst researchers and clinicians regarding valid and reproducible criteria for diagnosis of infection of the burn and a consistent and standardised approach to outcome reporting.
Notes
Leticia A Barajas‐Nava is a doctoral candidate in Public Health and Methodology of Biomedical Research, at the Pediatria, d'Obstetrícia i Ginecologia i de Medicina Preventiva Department, Universitat Autònoma de Barcelona, Barcelona, Spain.
Acknowledgements
The authors would like to acknowledge the Iberoamerican Cochrane Center and the Hospital de la Santa Creu i Sant Pau (Barcelona, Spain) for lending their facilities to conduct this review, as well as the Consejo Nacional de Ciencia y Tecnología (CONACYT) of Mexico for its support.
The authors would also like to thank Carlos Jiménez Gutiérrez for his contribution with data extraction; Beatriz Galván Guijo for her comments, and expert opinions during discussion of review results; Ma Ximena Rojas for her methodological advice and Héctor Pardo for translating the final Spanish version of the manuscript into English.
The authors would like to acknowledge the contribution of the referees (Richard Kirubakaran, Roy Buffery, Rachel Richardson and Mary Mondozzi) and Wounds Group Editors (Joan Webster and Susan O'Meara) and copy editor Elizabeth Royle.
Appendices
Appendix 1. Search strategies for Ovid Medline, Ovid Embase & EBSCO CINAHL
Ovid Medline
1 exp Burns/ (17746) 2 (burn or burns or burned or scald*).tw. (20006) 3 (thermal adj injur*).tw. (1989) 4 or/1‐3 (25841) 5 exp Anti‐Bacterial Agents/ (140513) 6 exp Anti‐Infective Agents, Local/ (7169) 7 (antibiotic* or amoxicillin or ampicillin* or bacitracin or cephalothin or cefazolin or cefotaxime or cefoperazone or ceftazidime or ceftriaxone or cefuroxime or chloramphenicol or ciprofloxacin or clarithromycin or clindamycin or cloxacillin or colistin or colymycin or erythromycin or flucloxacillin or furazolidone or fusidic acid or gentamicin or gramicidin or imipenem or mafenide acetate or mupirocin or natamycin or neomycin or nitrofurazone or oxacillin or penicillin or piperacillin or polymyxin or rifam* or silver nitrate or silver sulfadiazine or sulfacetamide sodium or tobramycin or amphotericin or tazocin or teicoplanin or tetracylcin or (trimethopri* adj sulfamethoxazole) or vancomycin).tw. (172269) 8 or/5‐7 (243914) 9 4 and 8 (1662) 10 randomized controlled trial.pt. (241879) 11 controlled clinical trial.pt. (39610) 12 randomized.ab. (196839) 13 placebo.ab. (91804) 14 clinical trials as topic.sh. (79729) 15 randomly.ab. (135266) 16 trial.ti. (73044) 17 or/10‐16 (546261) 18 (animals not (humans and animals)).sh. (1619207) 19 17 not 18 (497352) 20 9 and 19 (136)
Ovid Embase
1 exp burn/ (26490) 2 (burn or burns or burned or scald*).tw. (28523) 3 (thermal adj injur*).tw. (2628) 4 or/1‐3 (39099) 5 exp antibiotic agent/ (539517) 6 (antibiotic* or amoxicillin or ampicillin* or bacitracin or cephalothin or cefazolin or cefotaxime or cefoperazone or ceftazidime or ceftriaxone or cefuroxime or chloramphenicol or ciprofloxacin or clarithromycin or clindamycin or cloxacillin or colistin or colymycin or erythromycin or flucloxacillin or furazolidone or fusidic acid or gentamicin or gramicidin or imipenem or mafenide acetate or mupirocin or natamycin or neomycin or nitrofurazone or oxacillin or penicillin or piperacillin or polymyxin or rifam* or silver nitrate or silver sulfadiazine or sulfacetamide sodium or tobramycin or amphotericin or tazocin or teicoplanin or tetracylcin or (trimethopri* adj sulfamethoxazole) or vancomycin).tw. (252042) 7 or/5‐6 (623723) 8 4 and 7 (3669) 9 Clinical trial/ (714073) 10 Randomized controlled trials/ (25328) 11 Random Allocation/ (50422) 12 Single‐Blind Method/ (15458) 13 Double‐Blind Method/ (85567) 14 Cross‐Over Studies/ (31669) 15 Placebos/ (164396) 16 Randomi?ed controlled trial$.tw. (79498) 17 RCT.tw. (10411) 18 Random allocation.tw. (900) 19 Randomly allocated.tw. (14141) 20 Allocated randomly.tw. (1207) 21 (allocated adj2 random).tw. (264) 22 Single blind$.tw. (9611) 23 Double blind$.tw. (89754) 24 ((treble or triple) adj blind$).tw. (233) 25 Placebo$.tw. (136413) 26 Prospective Studies/ (198323) 27 or/9‐26 (1055024) 28 Case study/ (15609) 29 Case report.tw. (165315) 30 Abstract report/ or letter/ (508867) 31 or/28‐30 (685575) 32 27 not 31 (1026480) 33 animal/ (721470) 34 human/ (8575374) 35 33 not 34 (482095) 36 32 not 35 (1004334) 37 8 and 36 (535)
EBSCO CINAHL
S10 S4 and S9 S9 S5 or S6 or S7 or S8 S8 AB antibiotic* or amoxicillin or ampicillin* or bacitracin or cephalothin or cefazolin or cefotaxime or cefoperazone or ceftazidime or ceftriaxone or cefuroxime or chloramphenicol or ciprofloxacin or clarithromycin or clindamycin or cloxacillin or colistin or colymycin or erythromycin or flucloxacillin or furazolidone or fusidic acid or gentamicin or gramicidin or imipenem or mafenide acetate or mupirocin or natamycin or neomycin or nitrofurazone or oxacillin or penicillin or piperacillin or polymyxin or rifam* or silver nitrate or silver sulfadiazine or sulfacetamide sodium or tobramycin or amphotericin or tazocin or teicoplanin or tetracylcin or trimethopri* or vancomycin S7 TI antibiotic* or amoxicillin or ampicillin* or bacitracin or cephalothin or cefazolin or cefotaxime or cefoperazone or ceftazidime or ceftriaxone or cefuroxime or chloramphenicol or ciprofloxacin or clarithromycin or clindamycin or cloxacillin or colistin or colymycin or erythromycin or flucloxacillin or furazolidone or fusidic acid or gentamicin or gramicidin or imipenem or mafenide acetate or mupirocin or natamycin or neomycin or nitrofurazone or oxacillin or penicillin or piperacillin or polymyxin or rifam* or silver nitrate or silver sulfadiazine or sulfacetamide sodium or tobramycin or amphotericin or tazocin or teicoplanin or tetracylcin or trimethopri* or vancomycin S6 (MH "Antiinfective Agents, Local+") S5 (MH "Antibiotics+") S4 S1 or S2 or S3 S3 TI thermal* injur* or AB thermal* injur* S2 TI ( burn or burns or burned or scald* ) or AB ( burn or burns or burned or scald* ) S1 MH "Burns+")
Appendix 2. Risk of bias judgement criteria for RCT studies
1. Random sequence generation (selection bias)
Low risk of bias
The investigators describe a random component in the sequence generation process such as: referring to a random number table; using a computer random number generator; coin tossing; shuffling cards or envelopes; throwing dice; drawing of lots.
High risk of bias
The investigators describe a non‐random component in the sequence generation process. Usually, the description would involve some systematic, non‐random approach, for example: sequence generated by odd or even date of birth; or by some rule based on date (or day) of admission; or some rule based on hospital or clinic record number.
Unclear risk
Insufficient information available about the sequence generation process to permit a judgement of ‘low risk’ or ‘high risk’ to be made.
2. Allocation concealment (selection bias)
Low risk of bias
Participants and investigators enrolling participants could not foresee assignment because one of the following methods, or an equivalent, was used to conceal allocation: central allocation (including telephone, web‐based and pharmacy‐controlled randomisation); sequentially‐numbered drug containers of identical appearance; sequentially‐numbered, opaque, sealed envelopes.
High risk of bias
Participants or investigators enrolling participants could possibly foresee assignments ‐ due to allocation based on: an open, random allocation schedule (e.g. a list of random numbers); assignment envelopes without appropriate safeguards (e.g. envelopes that were unsealed, or nonopaque, or not sequentially numbered); alternation or rotation; date of birth; case record number; or any other explicitly unconcealed procedure ‐ and thus introduce selection bias.
Unclear risk
Insufficient information available to permit judgement of ‘low risk’ or ‘high risk’. This is usually the case if the method of concealment is not described, or not described in sufficient detail to allow a definite judgement to be made – for example if the use of assignment envelopes is described, but it is not clear whether envelopes were sequentially numbered, opaque and sealed.
3. Blinding for participants and personnel (performance bias)
Low risk of bias
No blinding or incomplete blinding, but the review authors judge that the outcome is not likely to be influenced by lack of blinding;
Blinding of participants and key study personnel ensured, and unlikely that the blinding could have been broken.
High risk of bias
Either of the following:
No blinding or incomplete blinding, and the outcome is likely to be influenced by lack of blinding;
Blinding of key study participants and personnel attempted, but likely that the blinding could have been broken, and the outcome is likely to be influenced by lack of blinding.
Unclear risk
Insufficient information available to permit judgement of ‘low risk’ or ‘high risk’;
The study did not address this outcome.
4. Blinding of outcome assessment (detection bias)
Low risk of bias
Either of the following:
No blinding of outcome assessment, but the review authors judge that the outcome measurement is not likely to be influenced by lack of blinding;
Blinding of outcome assessment ensured, and unlikely that the blinding could have been broken.
High risk of bias
Either of the following:
No blinding of outcome assessment, and the outcome measurement is likely to be influenced by lack of blinding;
Blinding of outcome assessment, but likely that the blinding could have been broken, and the outcome measurement is likely to be influenced by lack of blinding.
Unclear risk
Either of the following:
Insufficient information available to permit judgement of ‘low risk’ or ‘high risk’;
The study did not address this outcome.
5. Incomplete outcome data (attrition bias)
Low risk of bias
Any one of the following:
No missing outcome data.
Reasons for missing outcome data unlikely to be related to true outcome (for survival data, censoring unlikely to be introducing bias).
Missing outcome data balanced in numbers across intervention groups, with similar reasons for missing data across groups.
For dichotomous outcome data, the proportion of missing outcomes compared with the observed event risk was not enough to have a clinically‐relevant impact on the intervention effect estimate.
For continuous outcome data, plausible effect size (difference in means or standardised difference in means) among missing outcomes not enough to have a clinically‐relevant impact on observed effect size.
Missing data have been imputed using appropriate methods.
High risk of bias
Any one of the following:
Reason for missing outcome data likely to be related to true outcome, with either imbalance in numbers, or reasons for missing data, across intervention groups.
For dichotomous outcome data, the proportion of missing outcomes compared with the observed event risk was enough to induce clinically‐relevant bias in the intervention effect estimate.
For continuous outcome data, plausible effect size (difference in means or standardized difference in means) among missing outcomes enough to induce clinically‐relevant bias in observed effect size.
‘As‐treated’ analysis done with substantial departure of the intervention received from that assigned at randomisation.
Potentially inappropriate application of simple imputation.
Unclear risk
Either of the following:
Insufficient reporting of attrition/exclusions to permit judgement of ‘low risk’ or ‘high risk’ to be made (e.g. number randomized not stated, no reasons for missing data provided).
The study did not address this outcome.
6. Selective reporting (reporting bias)
Low risk of bias
Either of the following:
The study protocol is available and all of the study’s pre‐specified (primary and secondary) outcomes that are of interest in the review have been reported in the pre‐specified way.
The study protocol is not available but it is clear that the published reports include all expected outcomes, including those that were pre‐specified (convincing text of this nature may be uncommon)
High risk of bias
Any one of the following:
Not all of the study’s pre‐specified primary outcomes have been reported.
One or more primary outcomes is reported using measurements, analysis methods or subsets of the data (e.g. subscales) that were not pre‐specified.
One or more reported primary outcomes were not pre‐specified (unless clear justification for their reporting is provided, such as an unexpected adverse effect).
One or more outcomes of interest in the review are reported incompletely so that they cannot be entered in a meta‐analysis.
The study report fails to include results for a key outcome that would be expected to have been reported for such a study.
Unclear risk
Insufficient information available to permit judgement of ‘low risk’ or ‘high risk’ to be made. It is likely that the majority of studies will fall into this category.
7. Other bias
Low risk of bias
The study appears to be free of other sources of bias:
For cluster‐randomised trials: there is a balance in baseline characteristics in either clusters or individuals (patient).
For trials where the unit of randomisation is the patient: the baseline characteristics of the patients are similar in both groups
High risk of bias
There is at least one important risk of bias:
Extreme baseline imbalance (recruitment bias).
In cluster‐randomised trials: recruitment bias; baseline imbalance in either clusters or individuals (patient); loss of clusters and incorrect analysis.
For trials where the unit of randomisation is the patient: baseline characteristics vary significantly between groups.
Unclear risk
There may be a risk of bias, but there is either:
Insufficient information to assess whether an important risk of bias exists; or
Insufficient rationale or evidence that an identified problem will introduce bias.
Data and analyses
Comparison 1. Topical antibiotic prophylaxis.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Burn wound infection | 25 | Odds Ratio (Random, 95% CI) | 1.37 [1.02, 1.82] | |
| 1.1 Neomycin, bacitracin and polymyxin B vs control/placebo | 2 | Odds Ratio (Random, 95% CI) | 0.75 [0.32, 1.73] | |
| 1.2 Silver sulfadiazine vs dressings or skin substitute | 11 | Odds Ratio (Random, 95% CI) | 1.87 [1.09, 3.19] | |
| 1.3 Silver sulfadiazine vs traditional medicine | 4 | Odds Ratio (Random, 95% CI) | 1.05 [0.54, 2.06] | |
| 1.4 Other topical antibiotics vs dressings or skin substitute | 3 | Odds Ratio (Random, 95% CI) | 1.03 [0.19, 5.48] | |
| 1.5 Antibiotic prophylaxis vs other treatments | 7 | Odds Ratio (Random, 95% CI) | 1.51 [0.94, 2.42] | |
| 2 Infections in the burned people (sepsis) | 3 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 Neomycin, bacitracin and polymyxin B vs control/placebo | 2 | 99 | Risk Ratio (M‐H, Random, 95% CI) | 7.58 [0.44, 130.38] |
| 2.2 Antibiotic prophylaxis vs other treatments | 3 | 227 | Risk Ratio (M‐H, Random, 95% CI) | 4.31 [1.61, 11.49] |
| 3 Infections in burned people (bacteraemia) | 3 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 3.1 Neomycin, bacitracin and polymyxin B vs control/placebo | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.2 Silver sulfadiazine vs dressings or skin substitute | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.3 Silver sulfadiazine vs traditional medicine | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.4 Antibiotic prophylaxis vs other treatments | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 4 Infections in burned people (pneumonia) | 2 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 4.1 Neomycin, bacitracin and polymyxin B vs control/placebo | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.2 Silver sulfadiazine vs traditional medicine | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.3 Antibiotic prophylaxis vs other treatments | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 5 Infections in burned people (urinary tract infection) | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 5.1 Silver sulfadiazine vs traditional medicine | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 6 Adverse events | 7 | Odds Ratio (Random, 95% CI) | Subtotals only | |
| 6.1 Silver sulfadiazine vs polymyxin B/bacitracin | 1 | Odds Ratio (Random, 95% CI) | 0.20 [0.02, 2.16] | |
| 6.2 Silver sulfadiazine vs dressings or skin substitute | 4 | Odds Ratio (Random, 95% CI) | 1.00 [0.47, 2.14] | |
| 6.3 Other topical antibiotics vs dressings or skin substitute | 1 | Odds Ratio (Random, 95% CI) | 1.0 [0.05, 18.57] | |
| 6.4 Antibiotic prophylaxis vs other treatments | 1 | Odds Ratio (Random, 95% CI) | 1.0 [0.06, 18.08] | |
| 7 Infection‐related mortality | 2 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 7.1 Neomycin, bacitracin and polymyxin B vs control/placebo | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 7.2 Silver sulfadiazine vs traditional medicine | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 7.3 Antibiotic prophylaxis vs other treatments | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 8 Antibiotic resistance (MRSA) | 3 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 8.1 Neomycin, bacitracin and polymyxin B vs control/placebo | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 8.2 Silver sulfadiazine vs dressings or skin substitute | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 8.3 Silver sulfadiazine vs traditional medicine | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 8.4 Antibiotic prophylaxis vs other treatments | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 9 All‐cause mortality | 5 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 9.1 Silver sulfadiazine vs dressings or skin substitute | 2 | 132 | Risk Ratio (M‐H, Random, 95% CI) | 0.35 [0.01, 8.34] |
| 9.2 Silver sulfadiazine vs traditional medicine | 1 | 112 | Risk Ratio (M‐H, Random, 95% CI) | 1.86 [0.17, 19.95] |
| 9.3 Antibiotic prophylaxis vs other treatments | 2 | 161 | Risk Ratio (M‐H, Random, 95% CI) | 5.95 [1.10, 32.33] |
| 10 Length of hospital stay (LOS) | 8 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 10.1 Neomycin, bacitracin and polymyxin B vs control/placebo | 1 | 33 | Mean Difference (IV, Fixed, 95% CI) | ‐3.67 [‐9.46, 2.12] |
| 10.2 Silver sulfadiazine vs dressings or skin substitute | 4 | 196 | Mean Difference (IV, Fixed, 95% CI) | 2.11 [1.93, 2.28] |
| 10.3 Antibiotic prophylaxis vs other treatments | 4 | 216 | Mean Difference (IV, Fixed, 95% CI) | 3.26 [1.45, 5.07] |
Comparison 2. Systemic antibiotic prophylaxis (general).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Burn wound infection | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 Antibiotic prophylaxis vs control/placebo | 2 | 79 | Risk Ratio (M‐H, Random, 95% CI) | 0.64 [0.07, 6.09] |
| 2 Infections in burned people (sepsis) | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 Antibiotic prophylaxis vs control/placebo | 2 | 79 | Risk Ratio (M‐H, Random, 95% CI) | 0.43 [0.12, 1.61] |
| 3 Infections in burned people (bacteraemia) | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 3.1 Antibiotic prophylaxis vs control/placebo | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 4 Infections in burned people (pneumonia) | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 4.1 Antibiotic prophylaxis vs control/placebo | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 5 Infections in burned people (urinary tract infection) | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 5.1 Antibiotic prophylaxis vs control/placebo | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 6 Infection‐related mortality | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 6.1 Antibiotic prophylaxis vs control/placebo | 2 | 79 | Risk Ratio (M‐H, Random, 95% CI) | 0.27 [0.05, 1.58] |
| 7 Antibiotic resistance (MRSA) | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 7.1 Antibiotic prophylaxis vs control/placebo | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 8 All‐cause mortality | 3 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 8.1 Antibiotic prophylaxis vs control/placebo | 3 | 119 | Risk Ratio (M‐H, Random, 95% CI) | 0.41 [0.17, 1.02] |
| 9 Length of hospital stay (LOS) | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 9.1 Antibiotic prophylaxis vs control/placebo | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
Comparison 3. Systemic antibiotic prophylaxis (perioperative).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Burn wound infection | 3 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 Antibiotic prophylaxis vs control/placebo | 2 | 269 | Risk Ratio (M‐H, Random, 95% CI) | 0.51 [0.03, 7.37] |
| 1.2 Cephazolin vs other antibiotic | 2 | 51 | Risk Ratio (M‐H, Random, 95% CI) | 0.99 [0.49, 2.01] |
| 2 Infections in burned people (bacteraemia) | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 Antibiotic prophylaxis vs control/placebo | 2 | 89 | Risk Ratio (M‐H, Random, 95% CI) | 1.32 [0.31, 5.60] |
| 2.2 Cephazolin vs other antibiotic | 1 | 4 | Risk Ratio (M‐H, Random, 95% CI) | 0.83 [0.28, 2.51] |
| 3 Infections in burned people (pneumonia) | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 3.1 Cephazolin vs other antibiotic | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 4 Infections in burned people (urinary tract infection) | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 4.1 Cephazolin vs other antibiotic | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 5 Adverse events | 2 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 5.1 Antibiotic prophylaxis vs control/placebo | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 5.2 Cephazolin vs other antibiotic | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 6 All‐cause mortality | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 6.1 Antibiotic prophylaxis vs control/placebo | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 7 Length of hospital stay (LOS) | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 7.1 Antibiotic prophylaxis vs control/placebo | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
Comparison 4. Non‐absorbable antibiotic prophylaxis (selective decontamination of the digestive tract).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Burn wound infection | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 1.1 Non‐absorbable antibiotic prophylaxis and cefotaxime vs placebo | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2 Infections in burned people (sepsis) | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2.1 Non‐absorbable antibiotic prophylaxis vs placebo | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 3 Infections in burned people (bacteraemia) | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 3.1 Non‐absorbable antibiotic prophylaxis and cefotaxime vs placebo | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 4 Infections in burned people (pneumonia) | 2 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 4.1 Non‐absorbable antibiotic prophylaxis vs placebo | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.2 Non‐absorbable antibiotic prophylaxis and cefotaxime vs placebo | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 5 Infections in burned people (urinary tract infection) | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 5.1 Non‐absorbable antibiotic prophylaxis and cefotaxime vs placebo | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 6 Adverse events | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 6.1 Non‐absorbable antibiotic prophylaxis vs placebo | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 7 Antibiotic resistance (MRSA) | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 7.1 Non‐absorbable antibiotic prophylaxis and cefotaxime vs placebo | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 8 All‐cause mortality | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 8.1 Non‐absorbable antibiotic prophylaxis vs placebo | 1 | 23 | Risk Ratio (M‐H, Random, 95% CI) | 2.18 [0.23, 20.84] |
| 8.2 Non‐absorbable antibiotic prophylaxis and cefotaxime vs placebo | 1 | 107 | Risk Ratio (M‐H, Random, 95% CI) | 0.34 [0.13, 0.87] |
| 9 Length of hospital stay (LOS) | 2 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 9.1 Non‐absorbable antibiotic prophylaxis vs placebo | 1 | 23 | Mean Difference (IV, Fixed, 95% CI) | 7.0 [3.28, 10.72] |
| 9.2 Non‐absorbable antibiotic prophylaxis and cefotaxime vs placebo | 1 | 107 | Mean Difference (IV, Fixed, 95% CI) | ‐1.70 [‐15.82, 12.42] |
Comparison 5. Local antibiotic prophylaxis (airway).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Infections in burned people (sepsis) | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 1.1 Antibiotic prophylaxis vs control/placebo | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2 All‐cause mortality | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2.1 Antibiotic prophylaxis vs control/placebo | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
Comparison 6. Antibiotic prophylaxis vs control/placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Burn wound infection | 7 | 554 | Risk Ratio (M‐H, Random, 95% CI) | 0.84 [0.51, 1.39] |
| 1.1 Topical antibiotic prophylaxis | 2 | 99 | Risk Ratio (M‐H, Random, 95% CI) | 0.81 [0.47, 1.39] |
| 1.2 Systemic antibiotic prophylaxis (general) | 2 | 79 | Risk Ratio (M‐H, Random, 95% CI) | 0.64 [0.07, 6.09] |
| 1.3 Systemic antibiotic prophylaxis (perioperative) | 2 | 269 | Risk Ratio (M‐H, Random, 95% CI) | 0.51 [0.03, 7.37] |
| 1.4 Non‐absorbable antibiotic prophylaxis (selective decontamination of the digestive tract) | 1 | 107 | Risk Ratio (M‐H, Random, 95% CI) | 0.93 [0.43, 2.00] |
| 2 Infections in burned people (sepsis) | 6 | 231 | Risk Ratio (M‐H, Random, 95% CI) | 1.06 [0.54, 2.10] |
| 2.1 Topical antibiotic prophylaxis | 2 | 99 | Risk Ratio (M‐H, Random, 95% CI) | 7.58 [0.44, 130.38] |
| 2.2 Systemic antibiotic prophylaxis (general) | 2 | 79 | Risk Ratio (M‐H, Random, 95% CI) | 0.43 [0.12, 1.61] |
| 2.3 Non‐absorbable antibiotic prophylaxis (selective decontamination of the digestive tract) | 1 | 23 | Risk Ratio (M‐H, Random, 95% CI) | 2.18 [0.49, 9.65] |
| 2.4 Local antibiotic prophylaxis (airway) | 1 | 30 | Risk Ratio (M‐H, Random, 95% CI) | 1.04 [0.67, 1.60] |
| 3 Infections in burned people (bacteraemia) | 5 | 313 | Risk Ratio (M‐H, Random, 95% CI) | 1.08 [0.67, 1.72] |
| 3.1 Topical antibiotic prophylaxis | 1 | 66 | Risk Ratio (M‐H, Random, 95% CI) | 0.4 [0.08, 1.92] |
| 3.2 Systemic antibiotic prophylaxis (general) | 1 | 51 | Risk Ratio (M‐H, Random, 95% CI) | 3.12 [0.13, 73.06] |
| 3.3 Systemic antibiotic prophylaxis (perioperative) | 2 | 89 | Risk Ratio (M‐H, Random, 95% CI) | 1.32 [0.31, 5.60] |
| 3.4 Non‐absorbable antibiotic prophylaxis (selective decontamination of the digestive tract) | 1 | 107 | Risk Ratio (M‐H, Random, 95% CI) | 1.14 [0.67, 1.94] |
| 4 Infections in burned people (pneumonia) | 4 | 203 | Risk Ratio (M‐H, Random, 95% CI) | 0.54 [0.17, 1.74] |
| 4.1 Topical antibiotic prophylaxis | 1 | 33 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 4.2 Systemic antibiotic prophylaxis (general) | 1 | 40 | Risk Ratio (M‐H, Random, 95% CI) | 0.18 [0.05, 0.72] |
| 4.3 Non‐absorbable antibiotic prophylaxis (selective decontamination of the digestive tract) | 2 | 130 | Risk Ratio (M‐H, Random, 95% CI) | 0.73 [0.46, 1.16] |
| 5 Infections in burned people (urinary tract infection) | 2 | 158 | Risk Ratio (M‐H, Random, 95% CI) | 0.43 [0.18, 1.00] |
| 5.1 Systemic antibiotic prophylaxis (general) | 1 | 51 | Risk Ratio (M‐H, Random, 95% CI) | 0.35 [0.01, 8.12] |
| 5.2 Non‐absorbable antibiotic prophylaxis (selective decontamination of the digestive tract) | 1 | 107 | Risk Ratio (M‐H, Random, 95% CI) | 0.44 [0.18, 1.05] |
| 6 Infection‐related mortality | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 6.1 Systemic antibiotic prophylaxis (general) | 2 | 79 | Risk Ratio (M‐H, Random, 95% CI) | 0.27 [0.05, 1.58] |
| 7 Adverse events | 4 | 340 | Risk Ratio (M‐H, Random, 95% CI) | 3.12 [1.22, 7.97] |
| 7.1 Systemic antibiotic prophylaxis (general) | 2 | 68 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 7.2 Systemic antibiotic prophylaxis (perioperative) | 1 | 249 | Risk Ratio (M‐H, Random, 95% CI) | 0.96 [0.06, 15.19] |
| 7.3 Non‐absorbable antibiotic prophylaxis (selective decontamination of the digestive tract) | 1 | 23 | Risk Ratio (M‐H, Random, 95% CI) | 3.64 [1.34, 9.86] |
| 8 Antibiotic resistance (MRSA) | 3 | 180 | Risk Ratio (M‐H, Random, 95% CI) | 0.66 [0.12, 3.73] |
| 8.1 Topical antibiotic prophylaxis | 1 | 33 | Risk Ratio (M‐H, Random, 95% CI) | 0.56 [0.11, 2.90] |
| 8.2 Systemic antibiotic prophylaxis (general) | 1 | 40 | Risk Ratio (M‐H, Random, 95% CI) | 0.13 [0.02, 0.96] |
| 8.3 Non‐absorbable antibiotic prophylaxis (selective decontamination of the digestive tract) | 1 | 107 | Risk Ratio (M‐H, Random, 95% CI) | 2.22 [1.21, 4.07] |
| 9 All‐cause mortality | 7 | 348 | Risk Ratio (M‐H, Random, 95% CI) | 0.62 [0.39, 0.99] |
| 9.1 Systemic antibiotic prophylaxis (general) | 3 | 119 | Risk Ratio (M‐H, Random, 95% CI) | 0.41 [0.17, 1.02] |
| 9.2 Systemic antibiotic prophylaxis (perioperative) | 1 | 69 | Risk Ratio (M‐H, Random, 95% CI) | 1.62 [0.42, 6.25] |
| 9.3 Non‐absorbable antibiotic prophylaxis (selective decontamination of the digestive tract) | 2 | 130 | Risk Ratio (M‐H, Random, 95% CI) | 0.64 [0.11, 3.61] |
| 9.4 Local antibiotic prophylaxis (airway) | 1 | 30 | Risk Ratio (M‐H, Random, 95% CI) | 0.75 [0.39, 1.44] |
| 10 Length of hospital stay (LOS) | 5 | 463 | Mean Difference (IV, Fixed, 95% CI) | ‐0.18 [‐1.27, 0.91] |
| 10.1 Topical antibiotic prophylaxis | 1 | 33 | Mean Difference (IV, Fixed, 95% CI) | ‐3.67 [‐9.46, 2.12] |
| 10.2 Systemic antibiotic prophylaxis (general) | 1 | 51 | Mean Difference (IV, Fixed, 95% CI) | 0.80 [‐1.47, 3.07] |
| 10.3 Systemic antibiotic prophylaxis (perioperative) | 1 | 249 | Mean Difference (IV, Fixed, 95% CI) | ‐1.28 [‐2.64, 0.08] |
| 10.4 Non‐absorbable antibiotic prophylaxis (selective decontamination of the digestive tract) | 2 | 130 | Mean Difference (IV, Fixed, 95% CI) | 6.43 [2.84, 10.03] |
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Alexander 1982.
| Methods |
Study design: randomised, prospective, double‐blind trial. Setting/location: hospital (Shriners Burns Institute Cincinnati Unit, Ohio). Country: USA. Period of study: 2.1 years. Unit of randomisation: patient. Unit of analysis: patient. Sample size calculation: no. Use of ITT analysis?: yes. |
|
| Participants |
Inclusion criteria: 1. Patients scheduled for clean reconstructive surgery involving skin grafts. Exclusion criteria: 1. Patients who had grafts immediately adjacent to the mouth or anus. 2. Another indication for the administration of an antibiotic during the preoperative period of hospitalisation. 3. Presence of open areas in the anatomic preparation area of either the donor site or reconstructive site. 4. Known or suspected allergy to penicillin or to cephalosporins. Randomised: 249 patients (Intervention group: n = 127, Control group: n = 122). Withdrawals: Intervention group: 1 (2.5%) Reasons: adverse reaction. Patients assessed: 249 (100%). Age (years): (mean): Intervention group: 10.5 ± 0.4, Control group: 10.8 ± 0.4. Burned surface (% TBSA): not described. Inhalation injury: not stated. Time post‐burn (h): (mean): Intervention group: 72.5 ± 0.4, Control group: 71.0 ± 4.4. Burn type: Intervention group: thermal (100%), Control group: thermal (100%). Wounds infected at baseline?: no. Co‐morbidity: not described. |
|
| Interventions |
Type of antibiotic prophylaxis: systemic antibiotic prophylaxis (perioperative). Type of interventions: cephalothin vs placebo. Intervention group: iv cephalothin (Keflin, Eli Lilly, Indianapolis) 15 mg/kg in 50 ml of 5% dextrose in water. Control group: placebo (identical volume of 50 ml of 5% dextrose in water). 1st dose given with preoperative medications, 2nd dose at start of skin incision, 3rd dose 4 h later during the recovery phase. Duration of intervention: perioperative (1 day). Co‐interventions: not described. |
|
| Outcomes | Incidence of infection graft (infection was defined as discharge of pus from the graft site associated with graft loss). Adverse effects. LOS (days). |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "After informed consent . . . patients were randomised by draw of a card in a sealed envelope according to the anatomic site of the operation" (Page 687). Comment: insufficient information to make a judgement. |
| Allocation concealment (selection bias) | Unclear risk | Quote: "After informed consent . . . patients were randomised by draw of a card in a sealed envelope according to the anatomic site of the operation" (Page 687). Quote: "The sealed envelope designating whether or not the patient would receive the prophylactic antibiotic was identified on the outside by a research nurse with the patient's name, hospital number and weight in kilograms and given to the hospital pharmacist" (Page 687 trial report). Comment: insufficient information to permit judgement of ‘low risk’ or ‘high risk’. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "The sealed envelope designating whether or not the patient would receive the prophylactic antibiotic was identified on the outside by a research nurse with the patient's name, hospital number and weight in kilograms and given to the hospital pharmacist. On the morning of operation, the pharmacist dispensed three doses of either the antibiotic or placebo . . . All of the doses of antibiotic or placebo were given intravenously by 'piggy‐back' infusion, using identical infusion sets." (Page 687 trial report). "Only the pharmacist knew whether the patient received the antibiotic or a placebo until the end of the study" (page 688). Comment: patients and key study personnel were probably blinded. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "Only the pharmacist knew whether the patient received the antibiotic or a placebo until the end of the study" (Page 688 trial report). Comment: the outcome assessment was probably blinded. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Comment: no missing outcome data. All patients who were randomised were included in the final analysis. ITT analysis was conducted. |
| Selective reporting (reporting bias) | Low risk | No protocol provided, but given the outcomes listed in the methods section, all pre‐specified outcomes were reported. |
| Other bias | Low risk | The study appears to have been free of other sources of bias. |
Alexander 1984.
| Methods |
Study design: prospective, randomised trial. Setting/location: hospital (Shriners Burns Institute Cincinnati Unit, Ohio). Country: USA. Period of study: not stated (published in 1984). Unit of randomisation: patient. Unit of analysis: patient and procedure. Sample size calculation: no. Use of ITT analysis?: yes. |
|
| Participants |
Inclusion criteria: 1. Patients admitted to the Burns Center (acute care). 2. Burns of ≥ 20% of TBSA. Exclusion criteria: 1. History of sensitivity to multiple antibiotics. Randomised: 69 patients (Intervention group: n = 35, Control group: n = 34). Withdrawals: not stated. Burned surface (% TBSA): 20‐50% TBSA: Intervention group: 19 (54.3%), Control group: 21 (62%). ≥ 50% TBSA: Intervention group: 16 (45.7%), Control group: 13 (38%). Inhalation injury: not stated. Time post‐burn (h): not stated. Burn type: not stated. Wounds infected at baseline?: no. Co‐morbidity: not described. |
|
| Interventions |
Type of antibiotic prophylaxis: systemic antibiotic prophylaxis (perioperative). Type of interventions: prophylactic antibiotics vs no prophylactic antibiotics. Intervention group: prophylactic antibiotics during the perioperative period for debridement and skin grafting. Selection of the antibiotic(s) for use was based upon antibiotic sensitivity of the dominant organism and most recent wound cultures. Antibiotics were not given at other times except for specific medical indications. Control group: no prophylactic antibiotics. Duration of intervention: perioperative (1 day). Co‐interventions: not described. |
|
| Outcomes | Infection (total number of bacteraemic episodes/days at risk). Postoperative blood cultures. Mortality. |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "Patients were randomised to receive prophylactic antibiotics or no prophylactic antibiotics within the size ranges of 20‐50 % and greater than 50% to assure equal distribution. Randomization was done at the time of admission and an attempt was made to place all control patients on one ward and all treatment patients on another ward" (Page 20 trial report). Comment: insufficient information to make a judgement. |
| Allocation concealment (selection bias) | Unclear risk | No information provided. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | No information provided. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No information provided. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Did not report number of withdrawals. Comment: denominator values suggested complete follow‐up. |
| Selective reporting (reporting bias) | High risk | No protocol provided. Not all of the outcomes reported were mentioned in the methods section of the paper (for example, mortality was not specified). |
| Other bias | Unclear risk | Although mentioned that the groups were homogeneous, no data on age, sex, or comorbidity in each comparison group were presented. |
Ang 2001.
| Methods |
Study design: prospective, randomised, controlled clinical trial. Setting/location: hospital (Singapore National Burns Center, Singapore General Hospital). Country: Singapore. Period of study: 1 April 1997‐24 October 1998 (1.6 years). Unit of randomisation: patient. Unit of analysis: patient. Sample size calculation: no. Use of ITT analysis?: yes. Quote: "All analyses were made using intention‐to‐treat" (Page 95). |
|
| Participants |
Inclusion criteria: 1. Age: ≥ 6 and < 80 years. 2. Partial‐thickness burns. Exclusion criteria: 1. Age: < 6 and > 80 years. 2. Electrical or chemical burns. 3. Burns > 40% TBSA. Randomised: 115 patients (Group 1: n = 58, Group 2: n = 57). Excluded (post‐randomisation): Group 2: 3 (5.3%). Reason for exclusion: Quote: "In the MEBO group, data were not obtained from 3 patients (1 who was inadvertently randomised with BSA 68 %, technically violating the inclusion criteria, 1 illegal immigrant who was repatriated to his country of origin following first aid care, and 1 who withdrew consent immediately after randomisation for no specific reason)" (Page 95). Withdrawals: Group 1: 2 (3.4%), Group 2: 1 (2%). Reasons: death. Patients assessed: 112 (97.4%) Group 1: 58, Group 2: 54. Age (years): (mean, range): Group 1: 33.7 (11‐68), Group 2: 38.2 (7‐68). Gender (male: female): Group 1: 43 (74%): 15 (26%), Group 2: 40 (74%): 14 (26%). Burned surface (% TBSA): (mean, range): Group 1: 8.7% (1.5‐32), Group 2: 10.5% (1.5‐37.5). Inhalation injury: not stated. Time post‐burn (h): not stated. Burn type: Group 1: fire (flame) 28 (48%), scald (hot liquid or steam) 22 (38%), other (several agents, oil scald) 8 (14%); Group 2: fire 29 (54%), scald 20 (37%), other 5 (9%). Wounds infected at baseline?: no. Co‐morbidity: not described. |
|
| Interventions |
Type of antibiotic prophylaxis: topical antibiotic prophylaxis. Type of interventions: SSD (conventional management) vs MEBO (traditional medicine). Group 1: topical SSD twice daily. Group 2: 4‐hourly MEBO ointment (oil‐based ointment containing sesame oil, beta‐sitosterol, berberine, and other small quantities of plant ingredients). Duration of intervention: 14 days. Co‐interventions: in the SSD group (designated "C" by the trialists), burns were cleansed with plain chlorhexidine 0.05 % and de blistered where necessary. Areas of superficial burns were covered with paraffin‐impregnated gauze (Jelonet, Smith & Nephew Inc, Largo, Florida) or polyurethane dressing (Opsite, Smith & Nephew). In limited areas, not amenable to surgery with slough persisting beyond 14 days, chemical debridement using Elase (fibrinolysin and desoxyribonuclease) (Warner‐Lambert, Parke‐Davis) was used. In the MEBO group, the wounds were cleansed with normal saline gauze. In both groups, the excision and skin grafting were carried out on deep dermal wounds that showed minimal signs of healing after 14 days. Antibiotics were given only for clinically septic patients. |
|
| Outcomes | Wound healing rate. Bacterial infection rate. Burn wound infection (a clinical assessment was made daily for the presence of fever and/or reddening of the wound to indicate infection). |
|
| Notes | Conflict of interest: Quote: "None of the investigators maintain any financial interest in the company manufacturing MEBO" (Page 95 trial report). | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "After the initial assessment, patients were randomly assigned to C or MEBO . . . Randomly alternating permuted sub‐blocks of sizes 4 and 6, with equal numbers per treatment within each sub‐block, were used to obtain an overall block size of 10" (Page 93 trial report). Comment: insufficient information to make a judgement. |
| Allocation concealment (selection bias) | Low risk | Quote: "After the initial assessment, patients were randomly assigned to C or MEBO either by telephone to the National Medical Research Council Clinical Trials & Epidemiology Research Unit, Singapore (trial office), or by sealed envelopes. Envelopes were provided for patients requiring treatment allocation outside "office hours." These were numbered sequentially and a list was provided with the envelopes and completed with the trial number, allocated treatment, and patient name. The date the envelope was opened (i.e. the date of randomisation) was added. Notification of this procedure was sent to the trial office by facsimile". (Page 93 trial report). |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Comment: there was no information about blinding of participants and personnel. Blinding can be difficult for this study due to the different nature of the interventions being evaluated (topical interventions with different characteristics are easily noticed). The antibiotic was applied topically, and, with different time points for applications, was obviously different to the intervention administered in the other (MEBO) group; we assumed that the participants, personnel or outcome assessors were not blinded. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No information provided. Comment: there was no information about blinding of outcome assessment. Blinding can be difficult for this study due to the different nature of the interventions being evaluated (topical interventions applied at different time points ‐ easily noticeable). |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Losses and reasons for dropping out of the study were reported. 58/58 and 54/57 patients in the SSD and the MEBO groups, respectively, were included in the final analysis. ITT analysis was conducted. |
| Selective reporting (reporting bias) | Low risk | No protocol provided, but given the outcomes nominated in the methods section, all pre‐specified outcomes were reported. |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
Barret 2000.
| Methods |
Study design: prospective, randomised study. Setting/location: hospital (Shriners Burns Hospital and the University of Texas Medical Branch, Galveston). Country: USA. Period of study: not stated (published in 2000). Unit of randomisation: patient. Unit of analysis: patient. Sample size calculation: yes. Quote: "Based on previous studies published in the literature with similar populations,1–3 sample size was estimated at 20 patients (10 patients per group), taking a power of 0.80 and an alpha level of 0.05" (Page 62). Use of ITT analysis?: yes. |
|
| Participants |
Inclusion criteria: 1. Age: 0‐17 years. 2. Partial‐thickness burns. 3. Burn type: thermal, flame or scald. 4. 2‐ 29% TBSA. 5. Admitted within 24 h of injury. 6. Clean wound, uninfected (diagnosed by the attending physician). Exclusion criteria: 1. Age: > 17 years. 2. Full‐thickness burns. 3. Admitted > 24 h after injury. 4. Other types of burn injuries (chemical, electrical, or contact). 5. Evidence of contaminated or infected wounds. Randomised: 20 paediatric patients (Group 1: n = 10, Group 2: n = 10). Patients assessed: 20 (100%). Withdrawals: not stated. Age (years): (mean): Group 1: 3.7 ± 0.6, Group 2: 3.1 ± 0.5. Gender (male:female): Group 1: 8 (80%):2 (20%), Group 2: 7 (70%):3 (30%). Burned surface (% TBSA): (mean): Group 1: 7.8% ± 0.9, Group 2: 8.9% ± 4.9. Inhalation injury: not stated. Time post‐burn (h): < 24 h after injury (both groups). Burn type: Group 1: fire (flame) 3 (30%), scald (hot liquid or steam) 7 (70%); Group 2: fire 2 (20%), scald 8 (80%). Wounds infected at baseline?: no. Co‐morbidity: not described. |
|
| Interventions |
Type of antibiotic prophylaxis: topical antibiotic prophylaxis. Type of interventions: SSD vs biosynthetic dressing. Group 1: topical SSD 1% (Silvadene) twice a day. Group 2: biosynthetic dressing (Biobrane ‐ skin substitute). Application of a temporary cover to all open wounds. Wounds were inspected within 24 h, and patients discharged when parents were ready to assume wound care. Patients included in SSD group received wound care until wounds were healed. Patients dressed with Biobrane received no other treatment until wounds were considered to be healed. Duration of intervention: until wounds healed. Co‐interventions: before wound debridement, all patients were sedated with ketamine (1 mg/kg iv or 4 mg/kg intramuscularly). Pain medication regimen included 0.3 mg/kg/dose morphine by mouth for procedural pain and acetaminophen 15 mg/kg/dose by mouth every 4 h for background pain. The anxiolytic regimen included 4‐hourly Lorazepam, 0.03 mg/kg/dose by mouth. |
|
| Outcomes | Wound healing time (days): wounds were considered healed when all areas affected in the initial injury were closed. LOS (days). Infection. Wound infection. |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "After obtaining informed consent as mandated by the University of Texas Medical Branch Institutional Review Board, patients included in the study were randomised into two groups" (Page 63 trial report). Comment: insufficient information to make a judgement. |
| Allocation concealment (selection bias) | Unclear risk | No information provided. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | It was not reported whether participants and personnel were blinded but, due to the different nature of the interventions (SSD versus biosynthetic dressing), they were probably not blinded. |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Quote: "Pain assessment was not blinded due to the nature of the study" (Page 63)" Comment: outcome assessment was not blinded. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Did not report number of withdrawals. Comment: all patients who were randomised were included in the final analysis. ITT analysis was conducted. |
| Selective reporting (reporting bias) | High risk | No protocol provided, and some of the outcomes reported were specified in the methods section of this paper. |
| Other bias | Low risk | Quote: "Patients included in both groups were comparable, and all data followed normal distribution (Kolmogorov‐ Smirnov normality test)" (Page 63 trial report). |
Barret 2001.
| Methods |
Study design: prospective, randomised, double‐blinded study. Setting/location: hospital (Shriners Burns Hospital Galveston and The University of Texas Medical Branch). Country: USA. Period of study: 9 months. Unit of randomisation: patient. Unit of analysis: patient. Sample size calculation: yes. Use of ITT analysis?: yes. |
|
| Participants |
Inclusion criteria: 1. Age: 0‐18 years. 2. Full‐thickness burns. 3. Burns ≥ 30% TBSA 4. Admission within 5 days of injury. 5. No evidence of sepsis or organ failure. Exclusion criteria: 1. Evidence of sepsis or organ failure. Randomised: 23 patients (Intervention group: n = 11, Control group: n = 12). Age (years): (mean): Intervention group: 8 ± 1, Control group: 9.4 ± 2. Burned surface (% TBSA): (mean, SEM): Intervention group: 67% ± 6, Control group: 58% ± 6. TBSA full thickness burns: (mean, SEM): Intervention group: 59% ± 6, Control group: 54% ± 6. Inhalation injury: Intervention group: 9/12 (75%), Control group: 7/11 (64%). Time post‐burn: < 5 days from injury. Burn type: not stated. Wounds infected at baseline?: no. Co‐morbidity: not stated. |
|
| Interventions |
Type of antibiotic prophylaxis: non‐absorbable antibiotic prophylaxis (selective digestive decontamination). Type of interventions: selective digestive decontamination vs placebo. Intervention group: suspension of polymyxin E (100 mg), tobramycin (100 mg), and amphotericin B (500 mg) given by nasogastric tube, 4 times/day. Control group: isotonic physiologic solution (Ringer's lactate). Duration of intervention: until open burn wound area < 10% TBSA (48 days). Co‐interventions: resuscitation was given immediately after burn wound (lactated Ringer’s solution). Within 24 h of admission, patients underwent total burn wound excision of all full‐thickness burns, and coverage with autografts and homografts. Systemic antibiotics (vancomycin, amikacin, and piperacillin) were given preoperatively before 1st operative session in order to prevent postoperative sepsis due to perioperative bacteriaemia. All patients received nasoduodenal feedings with Vivonex TEN (Sandoz Nutrition, Minneapolis, MN), an elemental formula containing 82.3% carbohydrate, 3% fat (linoleic acid), and 14.7% protein. Oral nystatin in the form of ‘swish‐and‐swallow’ was used to prevent oral and oesophageal candidiasis. |
|
| Outcomes | Episodes of pneumonia (with positive bacteria and white cells on a class III, or sputum specimen). Episodes of sepsis (positive blood culture). Episodes of diarrhoea (culture results from faeces). Episodes of UTI (with 105 organisms/ml urine). Wound infection (biopsy with more than 105 organisms/g tissue and/or histologic evidence of viable tissue invasion). Time until wound closure (days). Miscellaneous complications. LOS (days). Mortality. |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Randomization was performed with a random‐number chart (Page 440), "Randomization was stratified for age, time from burn to admission, burn size, and presence of inhalation injury and ventilatory support" (Page 441 trial report). |
| Allocation concealment (selection bias) | Low risk | Central allocation at pharmacy. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "The study drug was prepared by the pharmacy and all patients, physicians, microbiologists, nursing staff, dieticians and laboratory personnel were blinded" (Page 440 trial report). Comment: participants and personnel were probably blinded. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "The study drug was prepared by the pharmacy and all patients, physicians, microbiologists, nursing staff, dieticians and laboratory personnel were blinded" (Page 440 trial report). Comment: outcome assessment was probably blinded. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Number of withdrawals not reported. Comment: all patients who were randomised were included in the final analysis. ITT analysis was conducted. |
| Selective reporting (reporting bias) | Low risk | No protocol provided, but given the outcomes listed in the methods section, all pre‐specified outcomes were reported. |
| Other bias | Low risk | Data analysis showed no significant differences between groups. |
Bugmann 1998.
| Methods |
Study design: prospective, randomised trial. Setting/location: hospital (Service de Chirurgie Pédiatrique, Hôpital des enfant, Geneva). Country: Switzerland. Period of study: 1995‐1996 (1 year). Unit of randomisation: patient. Unit of analysis: patient. Sample size calculation: no. Use of ITT analysis?: no. |
|
| Participants |
Inclusion criteria: 1. Burned paediatric patients. 2. Emergency admissions. Exclusion criteria: 1. Patients with facial burns or associated lesions. 2. > 24 h since burn. 3. Patients treated elsewhere prior to admission. Randomised: 76 paediatric patients (Group 1: n = 35, Group 2: n = 41). Excluded (post‐randomisation): Group 1: 5 (14.3%), Group 2: 5 (12.2%) Reasons: participants underwent tangential skin excision and skin graft. Patients assessed: 66 (87%) Group 1: 30 (86%), Group 2: 36 (88%). Age (years): (mean, SD): Group 1: 3.43 ± 3.7, Group 2: 3.29 ± 3.09. Gender (male: female): Group 1: 20 (57%): 15 (43%), Group 2: 22 (54%): 19 (46%). Burned surface (% TBSA): Group 1: 1.92% ± 2.05, Group 2: 2.29% ± 1.96. Inhalation injury: not stated. Time post‐burn (h): < 24 h. Burn type: Group 1: scald (hot liquid or steam) 21 (60%), contact (hot solids) 9 (26%), fire (flame) 4 (11%), electrical 1 (3%); Group 2: scald 28 (68%), contact 11 (27%), fire 2 (5%), electrical 0. Wounds infected at baseline?: no. Co‐morbidity: not described. |
|
| Interventions |
Type of antibiotic prophylaxis: topical antibiotic prophylaxis. Type of interventions: SSD vs silicone‐coated nylon dressing. Group 1: SSD (Flamazine, Duphar) topically every 2‐3 days. Group 2: silicone‐coated nylon dressing (Mepitel; Mölnlycke, Sweden) every 2‐3 days. Duration of intervention: until complete healing. Co‐interventions: initial debridement and disinfection under sedation or general anaesthesia was performed in the same manner in the two groups. Disinfection and cleaning of the wound done with chlorhexidine. |
|
| Outcomes | Epithelialization time (days). Wound infection. Adverse events (allergy). |
|
| Notes | Conflict of interest: the study was not sponsored by the manufacturer Mölnlycke. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "After written consent was obtained, we randomly assigned the patients to treatment with Mepitel (group M) or Flamazine (group F), our standard silver sulfadiazine burn dressing" (Page 609‐10 trial report). Comment: insufficient information to make a judgement. |
| Allocation concealment (selection bias) | Unclear risk | No information provided. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | No information provided. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No information provided. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 30/35 (86%) and 36/41 patients (88%) in the SSD and the silicone‐coated nylon dressing group respectively were included in the final analysis. |
| Selective reporting (reporting bias) | Low risk | No protocol provided, but given the outcomes listed in the methods section, all pre‐specified outcomes were reported. |
| Other bias | Unclear risk | The study appeared to be free of other sources of bias. |
Caruso 2006.
| Methods |
Study design: prospective, randomised study. Setting/location: hospital (8 burn centres ‐ see notes for details). Country: USA. Period of study: January 2003‐September 2004 (1.8 years). Unit of randomisation: patient. Unit of analysis: patient. Sample size calculation: yes. Quote: "A sample size of at least 82 patients was selected to obtain a minimum of 64 evaluable patients" (Page 301 trial report). Use of ITT analysis?: no, Quote: "In the AQUACEL® Ag dressing group, all 42 patients were included in the safety and intent‐to‐treat analyses. In the silver sulfadiazine group, 40 of 42 patients were included in the safety and intent‐to‐treat analyses because 2 patients did not receive study treatment" (Page 301 trial report). |
|
| Participants |
Inclusion criteria: 1. Patients with burn injuries acquired within 36 h preceding enrolment. 2. Age: ≥ 2 months. 3. Partial‐thickness burns, superficial burns, mid‐dermal, or mixed partial‐thickness burns. 4. 5%‐40% TBSA Exclusion criteria: 1. Electrical or chemical burns, or burns caused by frostbite. 2. Antibiotic taken during 2 days preceding burn injury. 3. Evidence of inhalation injury. 4. Fractures and/or neurological injury. 5. Treatment of the burn with an active agent (e.g. SSD) before study entry. 6. Pregnant. 7. Deep‐partial burns or full‐thickness burns (i.e. areas likely to require excision and grafting). Randomised: 84 patients (Group 1: n = 42, Group 2: n = 42). Excluded (post‐randomisation): Group 1: 2 (5%). Reason for exclusion: these 2 patients did not receive study treatment. Patients assessed: 82 patients (97.6%) (Group 1: n = 40 (95%) Group 2: n = 42 (100%)). Age (years): (mean, range): Group 1: 24 (0.5‐76.5), Group 2: 29.4 (0.8‐80.6). Gender (male: female): Group 1: 30 (75%): 10 (25%), Group 2: 27 (64%): 15 (36%). Burned surface (% TBSA): (mean, range): Group 1: 10.8% (5.0‐27.5%), Group 2: 12% (5.0‐35.0%). Inhalation injury: none. Time post‐burn (h): (mean, range): Group 1: 5.5 (0.0‐18.7), Group 2: 7.2 (1.0‐49.5). Burn type: Group 1: scald (hot liquid or steam) 18 (45%), fire (flame) 8 (20%), contact (hot solids) 1 (2.5%), other (several agents) 13 (32.5%); Group 2: scald 27 (64.3%), fire 4 (9.5%), other 11 (26.19%). Wounds infected at baseline?: no. Co‐morbidity: not described. |
|
| Interventions |
Type of antibiotic prophylaxis: topical antibiotic prophylaxis. Type of interventions: SSD vs Ag dressing. Group 1: SSD 1% cream topically once daily. Group 2: AQUACEL® Ag dressing (ConvaTec, a Bristol‐Myers Squibb company, Skillman, NJ) topically every 2‐3 days. Duration of intervention: 21 days, or until complete re‐epithelialization. Co‐interventions: not stated. |
|
| Outcomes | Rate of full re‐epithelialization (healing was defined as either 100% re‐epithelialization, or within 21 days). Time to complete wound healing (days). Adverse event (defined as any untoward medical occurrence that was new, or worsened during the study). Infection. Wound infection. Mortality. |
|
| Notes |
Sources of support: Quote: "This study was supported by a grant from ConvaTec, a Bristol‐ Myers Squibb company" (Page 298). "Study centers were compensated for performing the study, and ConvaTec provided AQUACEL® Ag dressing and SSD. Patients were not compensated for their participation. ConvaTec supervised the design of the study, the data analyses, and the development of the manuscript" (Page 309 trial report). Note on Methods section (above): Arizona Burn Center, Phoenix, Arizona; Lehigh Valley Hospital, Allentown, Pennsylvania; Hennepin County Medical Center, Minneapolis, Minnesota; Shriners Burns Hospital‐Galveston, Galveston, Texas; University of South Alabama Medical Center, Mobile, Alabama; Integris Baptist Medical Center, Oklahoma City, Oklahoma; Los Angeles County and University of Southern California Medical Center, Los Angeles, California; New York‐Presbyterian Hospital, New York. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "Patients were assigned randomly to a protocol of care that included either AQUACEL® Ag dressing or silver sulfadiazine. The randomisation schedule was stratified by extent of burns (5% to 20% or > 20% to 40% of TBSA) and age (0–3 years or 4 years and older)" (Page 299 trial report). Quote: "Baseline characteristics were comparable between treatment groups (Table 1)" (Page 301 trial report). Comment: insufficient information to make a judgement. |
| Allocation concealment (selection bias) | Unclear risk | No information provided. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Quote: "Study treatment was not blinded" (Page 299 trial report). |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | No blinding. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 42/42 (100%) and 40/42 patients (95%) in the AQUACEL Ag dressing and in the SSD group, respectively, were included in the final analysis. Although a per protocol analysis was performed (2 participants from the SSD group were excluded due to not having received study treatment), it probably did not bias the results of the study. |
| Selective reporting (reporting bias) | Low risk | No protocol provided, but given the outcomes listed in the methods section, all pre‐specified outcomes were reported. |
| Other bias | Unclear risk | Insufficient information to assess whether an important risk of bias existed. |
De La Cal 2005.
| Methods |
Study design: prospective, randomised, double blind, placebo‐controlled trial. Setting/location: hospital (burn ICU of a tertiary hospital, Getafe). Country: Spain. Period of study: 1 May 1997‐31 January 2000 (2.6 years). Unit of randomisation: patient. Unit of analysis: patient, event. Sample size calculation: yes. Use of ITT analysis?: yes. Quote:"The analysis was considered to be by intention to treat because all 10 excluded patients did not fulfil the inclusion criteria of the trial" (Page 426 trial report). |
|
| Participants |
Inclusion criteria: 1. Age: ≥ 14 years. 2. Burns of ≥ 20% TBSA. 3. Suspected, or evidence of, inhalation injury. 4. Interval between injury and admission to burn ICU ≤ 3 days. Exclusion criteria: 1. < 3 day stay in burn ICU. 2. Withdrawal of treatment within 3 days. 3. Immunosuppression or pregnancy. 4. Inhalation injury not requiring mechanical ventilation within first 3 days. Randomised: 117 patients (Intervention group: n = 58, Control group: n = 59). Excluded (post‐randomisation): 10 (8.5%) (Intervention group: 5 (8.6%), Control group: 5 (8.5%)). Reason for exclusion: Age < 14 years: 1 (Control group); length of stay < 72 h (Intervention group: 5, Control group: 3); treatment withdrawal: 1 (Control group). Withdrawals: Control group: 1 (0,85%). Reasons: treatment was withdrawn. Patients assessed: 107 (91.4%) (Intervention group: 53 (91.4%), Control group: 54 (91.5%)). Age (years): (mean, SD): Intervention group: 41.4 ± 17.7, Control group: 48.1 ± 18.5. Gender (male: female): Intervention group: 44 (83%): 9 (17%), Control group: 40 (74%): 14 (26%). Burned surface (% TBSA): (mean, SD): Intervention group: 34.0% ± 21.4, Control group: 37.7% ± 21.1. TBSA full thickness burns: (mean, SD): Intervention group:19.3 ± 15.3, Control group: 19.0 ± 18.8. Inhalation injury: Intervention group: 34 (64%), Control group: 37 (68%). Ventilator support: Intervention group: 39 (74%), Control group: 43 (80%). Time post‐burn: ≤ 3 days (in both groups). Burn type: not stated. Wounds infected at baseline?: no. Co‐morbidity: not stated. |
|
| Interventions |
Type of antibiotic prophylaxis: non‐absorbable antibiotic prophylaxis (selective digestive decontamination). Type of interventions: selective digestive decontamination (SDD) and cefotaxime vs placebo. Intervention group: (1) 100 mg polymyxin E, 100 mg tobramycin, and 500 mg amphotericin B given by digestive tract, 4 times/day; (2) cefotaxime 1 g iv 8‐hourly for 4 days; (3) non absorbable polymyxin E, tobramycin and amphotericin B 0.5 g of a 2% paste, topical application in the oropharynx 4 times/day. Control group: (1) placebo solution, given via digestive tract; (2) placebo solution, isotonic 0.9% saline iv; (3) placebo paste, topical application in the oropharynx. Duration of intervention: total duration of treatment in the burn ICU. Other co‐interventions: . Enteral nutrition: all patients received a diet supplemented with ω‐3‐acids, nucleotides and arginine, (Perative, Abbott). Protein support was between 1.5‐2.0 g/kg/d. Burn wounds were treated with closed dressings and daily application of SSD or iodine‐povidone ointment. Systemic antibiotics, such as vancomycin, ceftazidime and aminoglycoside, were administered empirically when clinical signs of infection developed and were adjusted according to the microbiologic results. |
|
| Outcomes | Mortality. Endogenous pneumonia (defined as the presence of new (or progressive) pulmonary infiltrates persisting for more than 48 h on chest X‐ray, in addition to at least 2 of the following criteria: (1) fever ≥ 38.5°C or hypothermia < 35.0°C; (2) leukocytosis 10,000/mm or leukopenia 3000/mm; (3) isolation of potential pathogens in high concentration of ≥4+ [107 colony forming units/ml] using semi‐quantitative culture, from unprotected purulent tracheal aspirates). UTI. Bloodstream infections (bacteraemia). Burn wound infection. |
|
| Notes | Sources of support: Quote: "This study has been partially supported by two grants from Fondo de Investigación Sanitaria: FIS 02/1883 and Respira C 03/11" (Page 424 trial report). | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "The patients were stratified according to the suspicion of inhalation injury" (Page 425 trial report). Comment: insufficient information to make a judgement. |
| Allocation concealment (selection bias) | Low risk | Central allocation at pharmacy, Quote: "The result of randomisation was introduced in a sealed envelope that was kept in the Department of Pharmacy" (Page 425 trial report). |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "Both control and test medication was prepared in the Department of Pharmacy of the hospital . . . The hospital pharmacist was the only person to be informed about the identity of the study medication" (Page 425 trial report). Comment: participants and personnel were probably blinded. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "Both control and test medication was prepared in the Department of Pharmacy of the hospital . . . The hospital pharmacist was the only person to be informed about the identity of the study medication" (Page 425). Comment: the outcome assessor was probably blinded. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Quote: "The analysis was considered to be by intention to treat because all 10 excluded patients did not fulfil the inclusion criteria of the trial" (Page 426 trial report). Comment: 53/58 (91.3%) and 54/59 patients (91.5%) in the intervention and in the placebo group, respectively, were included in the final analysis. |
| Selective reporting (reporting bias) | Low risk | No protocol provided, but the published report includes all expected outcomes. |
| Other bias | Low risk | The 2 groups were similar with respect to sex, age, total burn area, full‐thickness burn area, and inhalation injury. |
Demling 1999.
| Methods |
Study design: randomised, prospective study. Setting/location: hospital (Trauma and Burn Center, Brigham and Women's Hospital, Boston). Country: USA. Period of study: not stated (published in 1999). Unit of randomisation: patient. Unit of analysis: patient. Sample size calculation: no. Use of ITT analysis?: yes. |
|
| Participants |
Inclusion criteria: 1. Age: ≥ 18 years. 2. Partial‐thickness burns of the face (mid‐dermal). 3. Burns < 50% of the facial surface. 4. Burns produced by flash flames or flame exposure. Exclusion criteria: not stated. Randomised: 21 patients (Group 1: n = 11, Group 2: n = 10). Withdrawals: not stated. Age (years): (mean, SD): Group 1: 34.5 ± 7.5, Group 2: 37.5 ± 9. Burned surface (% TBSA): (mean, SD): Group 1: 18.5% ± 5, Group 2: 21% ± 6. Inhalation injury: not stated. Time post‐burn (h): not stated. Burn type: Group 1: fire (flame) 28 (48%); Group 2: fire 29 (54%). Wounds infected at baseline?: no. Co‐morbidity: not described. |
|
| Interventions |
Type of antibiotic prophylaxis: topical antibiotic prophylaxis. Type of interventions: bacitracin or SSD vs skin substitute. Group 1: bacitracin ointment (mid‐dermal areas) or SSD (deeper areas), topical 2 to 3 times a day. Group 2: skin substitute coated with fibronectin (TransCyte, Advanced Tissue Sciences, La Jolla, CA). Duration of intervention: until re‐epithelialization. Co‐interventions: cleaning dermal surface before treatment. All patients underwent complete debridement of non‐viable epidermis and upper dermis using blunt debridement (moist gauze) using systemic and topical analgesia. No tangential excision was performed. |
|
| Outcomes | Healing time (defined as ≥ 90% re‐epithelialization). Wound infection (diagnosed if local wound demonstrated increased exudate and surrounding cellulitis). LOS. |
|
| Notes | Sources of support: supported in part by The Heather Foundation. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "Once categorized into major and minor burns, patients were randomised into one of the treatment modalities" (Page 257 trial report). Comment: insufficient information to make a judgement. |
| Allocation concealment (selection bias) | Unclear risk | No information provided. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Blinding of participants and key study personnel not reported but probably not done due to the different nature of the interventions evaluated (ointment versus skin substitute). |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Blinding of outcome assessment not reported but probably not done due to the different nature of the interventions evaluated (ointment versus skin substitute). |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Did not report number of withdrawals. Comment: denominator values suggested complete follow‐up. |
| Selective reporting (reporting bias) | Low risk | Study protocol was not available, but it was clear that the published reports include all expected outcomes. |
| Other bias | Unclear risk | Insufficient information to assess whether an important risk of bias existed. |
Demling 2003.
| Methods |
Study design: randomised clinical trial. Setting/location: hospital. Country: not stated. Period of study: 1999‐2001 (2 years). Unit of randomisation: patient. Unit of analysis: patient. Sample size calculation: no. Use of ITT analysis?: yes. |
|
| Participants |
Inclusion criteria: 1. Adult patients. 2. Partial‐thickness burns. 3. Burns ≥ 50% of surface area of the foot. 4. Burns ≥ 10% of TBSA. Exclusion criteria: not stated. Randomised: 44 patients: Group 1: n = 20 (13 = 1 foot, 7 = both feet); Group 2: n = 24 (16 = 1 foot, 8 = both feet). Withdrawals: not stated. Age (years): (mean, SD): Group 1: 1 foot: 39 ± 8, both feet: 29 ± 10; Group 2: 1 foot: 41 ± 9, both feet: 32 ± 11. Burned surface (% TBSA): (mean, SD): Group 1: 1 foot: 3% ± 2, both feet: 5% ± 3; Group 2: 1 foot: 3% ± 1, both feet: 6% ± 2. Inhalation injury: not stated. Time post‐burn (h): not stated. Burn type: scald (hot liquid or steam) 26 (60%), chemical 9 (20%), other (several agents) 9 (20%). Wounds infected at baseline?: no. Co‐morbidity: not described. |
|
| Interventions |
Type of antibiotic prophylaxis: topical antibiotic prophylaxis. Type of interventions: bacitracin vs skin substitute. Group 1: bacitracin ointment, xeroform gauze, and a soft gauze topical dressing daily. Group 2: skin substitute (TransCyte® Smith & Nephew, Inc. Largo, Florida). Duration of intervention: until healed. Co‐interventions: after initial assessment, wounds were debrided of necrotic debris. Narcotics and nonsteroidal analgesics were used before, during, and after dressing changes. |
|
| Outcomes | Time to re‐epithelialization (95% of total). Burn wound infection (defined using a quantitative swab culture method). |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "After initial assessment . . . wounds were debrided of necrotic debris and dirt then randomised into the standard of care, which included bacitracin ointment . . . or placement of the skin substitute TransCyte® . . ." (Page 2/3 trial report). Comment: insufficient information to make a judgement. |
| Allocation concealment (selection bias) | Unclear risk | No information provided. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Blinding of participants and key study personnel not reported, but probably not done due to the different nature of the interventions evaluated (ointment versus skin substitute). |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Blinding of outcome assessment not reported, but probably not done due to the different nature of the interventions evaluated (ointment versus skin substitute). |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Did not report number of withdrawals. Comment: denominator values suggested complete follow‐ up. |
| Selective reporting (reporting bias) | Low risk | No protocol provided, but, given the outcomes listed in the methods section, all pre‐specified outcomes were reported. |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
Desai 1991.
| Methods |
Study design: randomised clinical trial. Setting/location: hospital (Shriners Burns Institute Galveston and the University of Texas). Country: USA. Period of study: not stated (published in 1991). Unit of randomisation: patient. Unit of analysis: patient. Sample size calculation: no. Use of ITT analysis?: yes. |
|
| Participants |
Inclusion criteria: 1. Age: ≥ 5 years (able to respond to tactile stimulation and to communicate feelings of discomfort verbally). 2. Patients with burns resulting from traffic accidents. 3. Admitted within 72 h of burn injury. Exclusion criteria: not stated. Randomised: 15 patients (Group 1: n = 7, Group 2: n = 8). Patients assessed: 15 (100%). Withdrawals: not stated. Age (years): (mean): Group 1: 11.4 ± 1.2, Group 2: 9.5 ± 1.6. Burned surface (% TBSA): (mean): Group 1: 35% ± 7, Group 2: 50% ± 6. TBSA full thickness burns: (mean): Group 1: 20% ± 9, Group 2: 32% ± 7. Inhalation injury: not stated. Ventilator support: not stated. Time post‐burn (h): ≤ 72 h. Burn type: Group 1: fire (flame) 100%, Group 2: fire 100%. Wounds infected at baseline?: no. Co‐morbidity: not stated. |
|
| Interventions |
Type of antibiotic prophylaxis: topical antibiotic prophylaxis. Type of interventions: gentamicin iontophoresis vs routine care. Group 1: gentamicin 1% cream (tube 30 g) layered over the ear, which was covered with the iontophoresis (treatment electrode) for 15‐20 minutes, 2 times/day. Group 2: routine care (6‐hourly ear cleaning and dressing changes). Duration of intervention: until final closure of the ear wound. Co‐interventions: all patients bathed once a day and had their ears cleaned and dressed with mafenide acetate cream 6‐hourly. |
|
| Outcomes | Wound infection (defined as chondritis, destruction of unburned cartilage, and ear deformities). Resistant organisms (qualitative cultures, quantitative cultures). LOS (days). |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "Subjects were randomly assigned to receive gentamicin iontophoresis . . . or to receive routine care alone" (Page 522 trial report). Comment: insufficient information to make a judgement. |
| Allocation concealment (selection bias) | Unclear risk | No information provided. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Blinding of participants and study personnel not reported, but probably not done due to the different nature of the interventions evaluated (cream given by iontophoresis versus routine care). |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Blinding of outcome assessment not reported, but probably not done due to the different nature of the interventions evaluated (cream given by iontophoresis versus routine care). |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Did not report number of withdrawals. Comment: denominator values suggested complete follow‐up. |
| Selective reporting (reporting bias) | Low risk | No protocol provided, but given the outcomes listed in the methods section, all pre‐specified outcomes were reported. |
| Other bias | Unclear risk | Insufficient information to assess whether an important risk of bias existed. |
Durtschi 1982.
| Methods |
Study design: prospective, randomised, double‐blind clinical trial. Setting/location: hospital (Regional Burn Center at the University of Washington). Country: USA. Period of study: 1 September 1978‐1 February 1980 (17 months). Unit of randomisation: patient. Unit of analysis: patient. Sample size calculation: no. Use of ITT analysis?: no. |
|
| Participants |
Inclusion criteria: 1. Age: ≥ 18 years. 2. Burns ≥ 20% TBSA. 3. All patients hospitalised in Regional Burn Center, University of Washington. Exclusion criteria: 1. Electrical burns. 2. Admission > 48 h after burn injury. 3. Allergy to penicillin. 4. Received antibiotics in previous 30 days. 5. Infection, or suspected infection, of the burn at admission. 6. If burn previously treated with biological dressings. 7. Insulin‐dependent diabetes, a disease requiring steroids or immunosuppressive therapy, massive obesity, severe malnutrition, or malignant disease Randomised: 97 patients. Excluded (post‐randomisation): 46 (47.4%) (reported as withdrawn). Reason for exclusion: Patient discharged before completion of 5‐day course of penicillin or placebo. Additional antibiotics begun for undocumented reason. Inappropriate entry into the study. Additional antibiotics given before excision and grafting. Patients assessed: 51 (52.6%) (Intervention group: 25 (25.7%), Control group: 26 (26.8%)). Age (years): (mean, range): Intervention group: 31.1 (18‐77), Control group: 36.8 (18‐66). Gender (male: female): Intervention group: 20 (80%): 5 (20%), Control group: 24 (92%): 2 (8%). Burned surface (% TBSA): (mean, range): Intervention group: 14.9% (1‐70%), Control group: 20% (1‐91%) TBSA full thickness burns: (mean, SD): not stated. Inhalation injury: not stated. Time post‐burn (h): ≤ 48 h. Burn type: Intervention group: thermal (100%), Control group: thermal (100%). Wounds infected at baseline?: no. Co‐morbidity: not reported. |
|
| Interventions |
Type of antibiotic prophylaxis: systemic antibiotic prophylaxis (general). Type of interventions: penicillin vs placebo. Intervention group: penicillin V potassium (250 mg), orally 6‐hourly for 5 days, or aqueous penicillin 1.2 million units iv 12‐hourly for 5 days. Control group: oral administration of placebo 6‐hourly for 5 days. The majority received medication or placebo by the oral route. Duration of intervention: 5 days. Co‐interventions: wound cleansing (2 times/day) and topical application of SSD. Patients received no additional antibiotics during initial 5 days of study period. Early tangential excision and grafting were performed when deemed appropriate by attending physician. All patients received clinical care according to the standards of the Burn Center. |
|
| Outcomes | Burn wound sepsis: syndrome resulting from presence of > 100,000 organisms/g biopsied wound tissue, associated with variable temperature and leucocyte count, blood chemistry abnormalities, and occasionally accompanied by positive blood cultures. Cellulitis: an area of warm, spreading, cutaneous erythema, accompanied by local pain and fever. Documented infection in lungs or urinary tract. LOS (days). |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "After obtaining informed consent, patients were randomised to receive either penicillin or an identical‐appearing placebo" (Page 12 trial report). Comment: insufficient information to make a judgement. |
| Allocation concealment (selection bias) | Unclear risk | No information provided. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: " . . . penicillin or an identical‐appearing placebo beginning on the day of admission" (Page 12 trial report). Comment: blinding of participants and personnel probably done. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "Because the diagnosis of cellulitis is primarily based on clinical criteria, a placebo group was essential for the study design. The physicians responsible for diagnosing cellulitis knew only that a patient was receiving either penicillin or placebo, but were unaware of the patient assortment". Comment: blinding of outcome assessment probably done. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Only 51/97 randomised patients included in the analysis. |
| Selective reporting (reporting bias) | Low risk | No protocol provided, but given the outcomes listed in the methods section, all pre‐specified outcomes were reported. |
| Other bias | Unclear risk | Insufficient information to assess whether an important risk of bias existed. |
Fisher 1968.
| Methods |
Study design: randomised clinical trial. Setting/location: hospital (Red Cross War Memorial Children's Hospital). Country: South Africa. Period of study: not stated (published in 1968). Unit of randomisation: patient. Unit of analysis: patient. Sample size calculation: no. Use of ITT analysis?: yes. |
|
| Participants |
Inclusion criteria: 1. Age: ≤ 12 years. 2. Burns ≥ 5% TBSA. 3. Burns sustained up to 10 h before admission. Exclusion criteria: not described. Randomised: 99 children (Group 1: n = 33, Group 2: n = 33, Group 3: n = 33). Burned surface (% TBSA): 22 patients < 10% TBSA burn, 10 patients 10‐20% TBSA burn, 1 patient > 20% TBSA burn (same percentages for each group). Inhalation injury: not described. Time post‐burn (h): ≤ 10 h. Burn type: Group 1: scald (hot liquid or steam) 30 (91%), fire (flame) 3 (9%); Group 2: scald 30 (91%), fire 3 (9%); Group 3: scald 30 (91%), fire 3 (9%). Wounds infected at baseline?: no. Co‐morbidity: not described. |
|
| Interventions |
Type of antibiotic prophylaxis: topical antibiotic prophylaxis. Type of interventions: polybactrin vs Dermoplast vs control. Group 1: polybactrin spray (mixture of zinc bacitracin, neomycin and polymyxin B), administered topically. Group 2: Dermoplast spray (benzocaine 4.5%, benzethonium chloride1.1%, menthol 0.5%, methyl paraben 2% and 8‐hydroxyquinoline 0.83 %), administered topically. Group 3: (Control): no spray. Sprays applied in 5‐second bursts 24 inches (61 cm) from the burn, 4‐hourly throughout exposure treatment. Duration of intervention: until final healing of burn wound. Co‐interventions: morphine 1.0 mg/12 lb (5.44 kg) body‐weight, or pethidine 0.5 mg/lb (0.45 kg) body‐weight, was given on admission, with further doses as necessary. No excision or debridement of burn surface done, home remedies not removed and blisters not opened. If infection supervened, instituted closed treatment with framycetin sulphate antibiotic cream. Systemic antibiotics administered only in presence of systemic illness or if beta‐haemolytic streptococcus was isolated. After preparation by regular dressing changes, deep burns were grafted as soon as possible, usually between days 17‐25, using large sheets of split‐thickness skin placed edge to edge. |
|
| Outcomes | Infection (bacteraemia, sepsis). Wound infection (bacterial culture swabs). Healing time (days). |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "To meet the likelihood that inherent differences in the size and depth of burns would cause sufficient variation in healing to conceal differences between treatments, a random block' experimental design was utilized . . . After assignment to a block, treatment was randomly allocated . . . " (Page 903 trial report). Comment: insufficient information to make a judgement. |
| Allocation concealment (selection bias) | Unclear risk | No information provided. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "A sealed box at each patient's bedside contained the appropriate spray, or none, and medical staff responsible for clinical management were unaware of the local treatment" (Page 903 trial report). Comment: participants and personnel were probably blinded. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No information provided. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Did not report number of withdrawals. Comment: denominator values suggested that all patients who were randomised were included in the final analysis. |
| Selective reporting (reporting bias) | High risk | The study protocol was not available and the methods section did not pre‐specify outcomes to be reported. |
| Other bias | Low risk | Quote: "In this study the problem of such bias was met by means of a blind trial, while a random block experimental design has permitted analysis of the effects of treatment over a wide range of burn size and severity. Significant differences between blocks in the time required for healing have been found, vindicating this method of analysis" (Page 904 trial report). |
Gerding 1988.
| Methods |
Study design: randomised prospective study. Setting/location: hospital (Burn Center, Cleveland Metropolitan General Hospital, Cleveland). Country: USA. Period of study: not stated (published in 1988). Unit of randomisation: burn wound. Unit of analysis: burn wound. Sample size calculation: no. Use of ITT analysis?: unclear. |
|
| Participants |
Inclusion criteria: 1. Patients admitted to the Burn Center. 2. Age: > 2 months. 3. Acute partial‐thickness burns (with a moist, sensate surface and prompt capillary refill). 4. Thermal burns. Exclusion criteria: 1. Grossly contaminated wounds. 2. Wounds > 6 h old. 3. Wounds previously treated by topical creams or other agents before admission. 4. Chemical and electrical burns. Randomised: 50 partial‐thickness burns (in 43 patients) (Group 1: n = 27, Group 2: n = 23) (paired controls). Excluded (before randomisation): 4 patients (8%). Reason for exclusion: 1 died of puImonary embolism, 1 transferred to a home‐state hospital before completion of protocol, 1 due to protocol violation, and 1 because of skin infection which antedated the burn wound. Assessed: 50 burn wounds (100%). Withdrawals: not stated. Age (years): (mean, range): Group 1: 17.6 (6 months‐71), Group 2: 22 (6 months‐71). Gender (male: female): 34 (79%): 9 (21%). Burned surface (% TBSA): (mean ± SEM, range): Group 1: 6.1% ± 0.9 (1.5‐26%), Group 2: 6.5% ± 0.1 (1.5‐12%). Inhalation injury: not stated. Time post‐burn (h): < 6 h. Burn type: scald (hot liquid or steam) 29 (67%), fire (flame) 12 (28%), contact (hot solids) 2 (5%). Wounds infected at baseline?: no. Co‐morbidity: not stated. |
|
| Interventions |
Type of antibiotic prophylaxis: topical antibiotic prophylaxis. Type of interventions: biosynthetic skin dressing vs SSD. Group 1: biosynthetic dressing (Biobrane, Woodroof Laboratories, Winthrop, Santa Ana, CA), daily application. Group 2: SSD 1% cream (Marion Laboratories, Kansas City, MO) twice daily. Wounds in both groups then dressed with gauze bandage and elastic outer wraps. Duration of intervention: 21 days. Co‐interventions: initial therapy of study wounds consisted of complete debridement of blisters and loose tissue, and cleansing with sterile saline. Detergents and antiseptic solutions were not utilised. |
|
| Outcomes | Healing time (days) (wounds considered healed when completely re‐epithelialized). Infection rate (wound infections diagnosed on clinical grounds in conjunction with semi‐quantitative surface swab cultures). |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Randomization was achieved by computer‐generated codes within sealed, numbered envelopes that were opened sequentially . . . Twenty‐seven burn wounds were randomised for treatment with Biobrane (Group I) and 23 wounds (Group II) randomised for treatment with 1% silver sulfadiazine cream (Marion Laboratories, Kansas City, MO). Seven patients who presented with anatomically separate but similar burn wounds were chosen to serve as matched controls by randomising the paired wounds to treatment by opposite modalities" (Page 1265 trial report). The 2 groups were similar with respect to sex, race, and burn agent (Table I). |
| Allocation concealment (selection bias) | Unclear risk | Quote: "Randomization was achieved by computer‐generated codes within sealed, numbered envelopes that were opened sequentially" (Page 1265 trial report). Comment: the authors did not describe whether the envelopes were opaque. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Blinding of participants and key study personnel not reported, however, the study was probably not blinded (as for other similar studies by the same author (Gerding 1990)). |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Blinding of outcome assessors not reported, however, it was probably not blinded (as for other similar studies by the same author (Gerding 1990)). |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | It is not clear when the exclusions occurred (before or after randomisation), so it is not possible to make a judgement about its impact in the study results. |
| Selective reporting (reporting bias) | Low risk | No protocol provided, but given the outcomes listed in the methods section, all pre‐specified outcomes were reported. |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
Gerding 1990.
| Methods |
Study design: randomised, prospective study. Setting/location: hospital (Emergency Department and Burn Clinic of the Cleveland Metropolitan General Hospital, Cleveland). Country: USA. Period of study: not stated (published in 1990). Unit of randomisation: burn wound. Unit of analysis: burn wound. Sample size calculation: no. Use of ITT analysis?: no. |
|
| Participants |
Inclusion criteria: 1. Age: ≥ 2 months. 2. Acute partial‐thickness burns. 3. No history of sulphonamide sensitivity. 4. Wounds with moist, sensate surface and prompt capillary refill. Exclusion criteria: 1. Chemical or electrical burns. 2. Grossly contaminated wounds. 3. Wounds > 24 h old. 4. Wounds treated by any topical agent before admission. 5. Pregnancy. Randomised: 64 patients. Excluded (post‐randomisation): Group 1: 7 patients (21%), Group 2: 5 patients (16%). Reason for exclusion: Group 1: 4 protocol violations by non‐investigators, 2 lost to follow‐up, 1 found to be suffering from scarlet fever (without evidence of wound infection); Group 2: 1 had protocol violations by non‐investigators, 4 lost to follow‐up. Withdrawals: Group 1: 2 patients (6%), Group 2: 4 patients (13%). Reasons: lost to follow‐up. Patients assessed: 52 patients (81%) (56 burn wounds: Group 1: 30, Group 2: 26). Age (years): (mean ± SEM, range): Group 1: 18.3 ± 2.6 (10 months‐55), Group 2: 22.1 ± 3.5 (8 months‐79). Gender (male: female): Group 1: 19 (74%): 11 (26%), Group 2: 18 (69%): 8 (31%). Burned surface (% TBSA): (mean ± SEM, range): Group 1: 2.0% ± 0.3 (0.5‐5.0), Group 2: 2.4% ± 0.5 (0.5‐10.0). Inhalation injury: not stated. Time post‐burn (h): not stated. Burn type: aqueous scald, grease, contact, other. Aqueous scald burns were the most common and were attributable to hot water, coffee, tea, soup, or steam. Wounds infected at baseline?: no. Co‐morbidity: not described. |
|
| Interventions |
Type of antibiotic prophylaxis: topical antibiotic prophylaxis. Type of interventions: biosynthetic dressing vs SSD. Group 1: biosynthetic dressing (Biobrane). Group 2: SSD 1% topically twice daily. Duration of intervention: until complete healing. Co‐interventions: all study wounds were completely debrided of blisters and loose tissue and cleansed with sterile saline before randomisation. Wounds in both groups were covered with dry gauze and elastic wraps. Adult patients were given prescriptions for acetaminophen with codeine, and children were treated with acetaminophen alone. Wounds that developed eschar were treated with SSD 1% or surgically excised. |
|
| Outcomes | Healing time (defined as time required to re‐epithelialize the burn surface fully). Burn wound infection (infected and skin grafted wounds were considered failures of therapy and excluded from healing time analysis). |
|
| Notes | Quote: "The groups also were well matched by mechanisms of injury (Figure 1)" (Page 122 trial report). | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Randomization was achieved by computer‐generated codes within sealed, numbered envelopes that were opened in sequential fashion" (Page 122 trial report). Quote: "There were no significant differences in age, race, or gender distribution between the two groups (Table)" (Page 122). |
| Allocation concealment (selection bias) | Unclear risk | Quote: "Randomization was achieved by computer‐generated codes within sealed, numbered envelopes that were opened in sequential fashion" (Page 122). Comment: the authors do not describe if the envelopes were opaque. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Quote: "Although this study was randomised, it is limited by the fact that it was not blinded. Neíther the treating physicians nor those who judged healing times were or could have been blinded to the treatment type" (Page 124 trial report). |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Quote: "Although this study was randomised, it is limited by the fact that it was not blinded. Neíther the treating physicians nor those who judged healing times were or could have been blinded to the treatment type" (Page 124 trial report). |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Only 52/64 randomised patients included in the analysis. Comment: the magnitude of losses during the study was > 20%. |
| Selective reporting (reporting bias) | Low risk | No protocol provided, but given the outcomes listed in the methods section, all pre‐specified outcomes were reported. |
| Other bias | Unclear risk | Insufficient information to assess whether an important risk of bias existed. |
Glat 2009.
| Methods |
Study design: prospective, randomised study. Setting/location: hospital (Burn Unit, St Christopher’s Hospital for Children, Pennsylvania). Country: USA. Period of study: not stated (published in 1988). Unit of randomisation: patient. Unit of analysis: patient. Sample size calculation: no. Use of ITT analysis?: yes. |
|
| Participants |
Inclusion criteria: 1. Paediatric patients (2 months‐18 years). 2. Partial‐thickness burns (superficial and mid‐dermal wounds). 3. Burn wounds acquired within previous 36 h. 4. Burns > 1% to < 40% TBSA. 5. Patients or their parents able to consent to both inclusion in the study and treatment until wounds were completely healed. Exclusion criteria: 1. Deep burns or full‐thickness burns. 2. Burn associated with electrical or chemical injury. 3. Patient not expected to survive for duration of study. 4. Burn site previously treated with an antimicrobial agent or debrided with an enzymatic agent. 5. Previous participation in a similar study. 6. Pregnancy. Randomised: 24 patients (Group 1: n = 12, Group 2: n = 12). Patients assessed: 24 (100%). Age (years): (mean ± SD, range): Group 1: 43 months ± 29.10 (9 months‐9 years), Group 2: 22.78 months ± 13.51, (13 months‐5 years). Burned surface (% TBSA): TBSA for the wound injury site was comparable for both study arms and ranged from 1‐10%. Inhalation injury: not stated. Time post‐burn (h): ≤ 36 h (in both groups). Burn type: not reported. Wounds infected at baseline?: no. Co‐morbidity: not stated. |
|
| Interventions |
Type of antibiotic prophylaxis: topical antibiotic prophylaxis. Type of interventions: biocompatible hydrogel (silver ions) vs SSD. Group 1: SilvaSorb Gel (silver ions) amorphous, biocompatible hydrogel, applied topically every 2‐3 days. Group 2: SSD cream (Silvadene) applied topically every 2‐3 days. Outpatients and/or their guardians were allowed to change their own dressing and were provided with general practice instructions by the burn centre and on the use of the treatments. Duration of intervention: until complete healing. Co‐interventions: not stated. |
|
| Outcomes | Time to full re‐epithelialization (days). Adverse events. Wound infection. |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "Patients were randomly assigned to a protocol of care that included either SSD cream or SilvaSorb Gel" (Page 263 trial report). Quote: "Baseline characteristics were comparable between the treatment and control arms of the study with the exception of patient age" (Page 264). Comment: insufficient information to make a judgement. |
| Allocation concealment (selection bias) | Unclear risk | No information provided. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Quote: ". . . without blinding of the physician investigator or other medical personnel to the type of treatment" (Page 263 trial report). Comment: participants and personnel were probably not blinded. |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Quote: " . . . without blinding of the physician investigator or other medical personnel to the type of treatment" (Page 263 trial report). Comment: outcome assessors were probably not blinded. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Number of withdrawals not reported. Comment: denominator values suggested that all randomised patients were included in the final analysis. |
| Selective reporting (reporting bias) | Low risk | No protocol provided, but given the outcomes listed in the methods section, all pre‐specified outcomes were reported. |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
Gong 2009.
| Methods |
Study design: randomised controlled trial. Setting/location: hospital (First People's Hospital of Nantong, Jiangsu). Country: China. Period of study: May 2007‐May 2009 (2 years). Unit of randomisation: patient. Unit of analysis: patient. Sample size calculation: no. Use of ITT analysis?: yes. |
|
| Participants |
Inclusion criteria: 1. Age: 20‐40 years. 2. 2nd‐degree burns. 3. Burns < 10% TBSA. 4. Burns thermal flame or scald (hot fluids). 5. No infection on wound surface. Exclusion criteria: 1. Serious dysfunction of liver or serious renal dysfunction. 2. Chronic consumption. 3. Allergy to silver dressing and hydrogel. 4. Cephalofacial and cervicalis wound surface. 5. Patient and family wanted surgery. Randomised: 104 patients (Group 1: n = 52, Group 2: n = 52). Patients assessed: 104 (100%). Withdrawals: none withdrew or were lost to follow‐up. Age (years): (mean, range): Superficial degree II: Group 1: 27.3 ± 3.8, Group 2: 27.6 ± 3.4; Deep degree II: Group 1: 29.2 ± 4.7, Group 2: 28.6 ± 3.7. Gender (male:female): 62 (60%): 42 (40%). Burned surface (% TBSA): (mean, range): Superficial degree II: Group 1: 7.4 ± 1.6, Group 2: 7.1 ± 1.5; Deep degree II: Group 1: 7.7 ± 1.4, Group 2: 7.3 ± 1.3. Inhalation injury: not stated. Time post‐burn (h): not stated. Burn type: Superficial degree II: Group 1: scald (hot liquid or steam) 15 (29%), fire (flame) 13 (25%); Group 2: scald 15 (29%), fire 13 (25%); Deep degree II: Group 1: scald 13 (25%), fire 11 (21%); Group 2: scald 12 (23%), fire 12 (23%). Wounds infected at baseline?: no. Co‐morbidity: not described. |
|
| Interventions |
Type of antibiotic prophylaxis: topical antibiotic prophylaxis. Type of interventions: ionic silver dressing/hydrogel vs SSD. Group 1: ionic silver pressure dressing once daily for 7 days; then hydrogel was used. Group 2: SSD 1% topically once daily. Duration of intervention: 21 days. Co‐interventions: routine antiinflammatory treatment, treatment to activate blood circulation, and provision of nutritional support. |
|
| Outcomes | Burn wound infection. Wound healing time. Adverse events. |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Participants were allocated to interventions on the basis of a sequence generated by random‐number tables. |
| Allocation concealment (selection bias) | Unclear risk | No information provided. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | The trial was not blinded. |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | The trial was not blinded. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No missing outcome data. Denominator values suggested that all patients who were randomised were included in the final analysis. |
| Selective reporting (reporting bias) | Low risk | No protocol provided, but given the outcomes listed in the methods section, all pre‐specified outcomes were reported. |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
Gotschall 1998.
| Methods |
Study design: prospective, randomised, controlled clinical trial. Setting/location: hospital (Children's National Medical Center, Washington). Country: USA. Period of study: 1 November 1993‐31 December 1996 (3.1 years). Unit of randomisation: patient. Unit of analysis: patient. Sample size calculation: no. Use of ITT analysis?: unclear. |
|
| Participants |
Inclusion criteria: 1. Age: ≤ 12 years. 2. Admitted to the regional paediatric burn centre. 3. Partial‐thickness burns. 4. Scald burns (defined as burns resulting from hot non‐viscous fluids and did not include substances such as oatmeal or mashed potato). 5. Burns ≤ 15% TBSA 6. When the burns affected only were flat body surfaces. Exclusion criteria: 1. Children suspected to be victims of child abuse. 2. History of allergy to silicone. 3. Chronic diseases that might affect the healing process (e.g. white blood cell deficiency). Randomised: 63 children (Group 1: n = 33, Group 2: n = 30). Withdrawals: children whose wounds converted to full‐thickness were withdrawn from the study. Burned surface (% TBSA): (mean, SD):Group 1: 6.8% ± 3.4%, Group 2: 5.1% ± 2.2%. Inhalation injury: not stated. Time post‐burn (h): not stated. Burn type: Group 1: scald (hot liquid or steam) 33 (100%); Group 2: scald (hot liquid or steam) 30 (100%). Wounds infected at baseline?: no. Co‐morbidity: not described. |
|
| Interventions |
Type of antibiotic prophylaxis: topical antibiotic prophylaxis. Type of interventions: silicone mesh dressing vs SSD. Group 1: silicon‐coated nylon dressing (Mepitel, Mölnlycke Health Care, USA). Group 2: SSD cream. Gauzes wet and dry were applied under cotton gauze dressing over both treatment arms. Duration of intervention: until complete healing. Co‐interventions: not stated. |
|
| Outcomes | Wound healing. Healing time (measured by number of days until wounds were 25%, 50%, 75% and 100% epithelialized). Burn wound infection (clinical data and swab culture). |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "Treatment was assigned randomly" (Page 280 trial report). Quote: "There were no significant differences between the two groups with respect to age, sex, or race" (Page 280 trial report). Comment: insufficient information to make a judgement. |
| Allocation concealment (selection bias) | Unclear risk | No information provided. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | It was not reported whether participants and personnel were blinded, but they were probably not, due to the different nature of the interventions. |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Quote: "The principal Iimitation of this investigation is the lack of "blinding" to treatment assignment by the people assessing the wounds at the dressing changes" (Page 283 trial report). |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Did not report number of withdrawals. Quote: ". . . children whose wounds had converted to full‐thickness were withdrawn from the study" (Page 280 trial report). Comment: it is not clear whether the outcome data were incomplete. |
| Selective reporting (reporting bias) | Low risk | No protocol provided, but given the outcomes listed in the methods section, all pre‐specified outcomes were reported. |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
Hauser 2007.
| Methods |
Study design: a randomised, controlled, intra‐individual comparative study. Setting/location: hospital. Country: Germany. Period of study: not stated (published in 2007). Unit of randomisation: burn wound. Unit of analysis: burn wound. Sample size calculation: no. Use of ITT analysis?: yes (per protocol cohort). |
|
| Participants |
Inclusion criteria: 1. 2nd‐degree burns (partial‐thickness burns). 2. 2 wounds of comparable size and location (noncontiguous). 3. Wounds without clinical data suggesting infection. 4. Burns of ≤ 50% TBSA. 5. ≤ 3 h post‐burn. Exclusion criteria: 1. Drug and alcohol abuse. 2. Pregnant or breastfeeding women. 3. Contraindications to SSD or hydrosome gel. Randomised: 47 patients (94 burn wounds). Patients assessed: 43 (91.5%) 86 burn wounds, Group 1: 43 burn wounds, Group 2: 43 burn wounds. Withdrawals: not stated. Age (years): (mean, range): 37.2 (3‐78). Gender (male: female): 34 (72%): 13 (28%). Burned surface (% TBSA): (mean): 11.1%. Inhalation injury: not stated. Time post‐burn (h): ≤ 3 h. Burn type: not stated. Wounds infected at baseline?: no. Co‐morbidity: 15 patients were smokers, 31 non‐smokers and former smokers. |
|
| Interventions |
Type of antibiotic prophylaxis: topical antibiotic prophylaxis. Type of interventions: hydrosome gel vs SSD. Group 1: SSD cream (Flammazine®, Solvay Arzneimittel GmbH, Hannover, Deutschland) topically once a day. Group 2: hydrosome wound gel (Repithel®, Mundipharma GmbH, Limburg/Lahn, Deutschland) topically once a day. Duration of intervention: until complete healing (21 days). Co‐interventions: not stated. |
|
| Outcomes | Time to complete wound healing (days). Wound infection (clinical evaluation and analysis by microbiological laboratory). |
|
| Notes |
Conflict of interest: the research activities of Prof Steinau are funded on the basis of contracts between the BG‐Kliniken Bergmannsheil Bochum and the company Mundipharma. The corresponding author argues that, despite the potential conflict of interest, they conducted the study in an independent manner. Quote: "Interessenkonflikt. Die Forschungstätigkeit von Prof. Steinau wird auf der Grundlage von Verträgen zwischen den BG‐Kliniken Bergmannsheil Bochum und der Firma Mundipharma gefördert. Der korrespondierende Autor versichert, dass trotz des möglichen Interessenkonflikts der Beitrag unabhängig und produktneutral ist" (Page 994 trial report). |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Both wounds were randomly assigned for each treatment. Randomization was performed using the computer program "Rancode 3.6". |
| Allocation concealment (selection bias) | Unclear risk | No information provided. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Quote: "Eine Verblindung der Studie war aufgrund der präparatspezifischen Farbe des HW bedingt durch die PVP‐Iod‐ Komponente nicht möglich" (Page 990 trial report). Comment: blinding of participants and personnel was not possible due to the different colours of the topical interventions evaluated. |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Not done, because of the different colours of the topical interventions evaluated. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 43/47 patients included in the final analysis; although 4 randomised participants were not included in the analysis, this probably did not bias the results of the study. |
| Selective reporting (reporting bias) | Low risk | No protocol provided, but given the outcomes listed in the methods section, all pre‐specified outcomes were reported. |
| Other bias | Unclear risk | Insufficient information available to assess whether an important risk of bias existed. |
Hosseini 2009.
| Methods |
Study design: prospective, randomised clinical trial. Setting/location: hospital (Shafieeh Hospital in Zanjan). Country: Iran. Period of study: March 2006‐November 2007 (1.8 years). Unit of randomisation: patient. Unit of analysis: patient. Sample size calculation: no. Use of ITT analysis?: no. |
|
| Participants |
Inclusion criteria: 1. Burns patients. 2. Partial‐thickness burns (2nd‐degree burns). 3. Burns 10–60% TBSA. 4. Scald or flames. Exclusion criteria: 1. 3rd‐degree burns. 2. Contact burns and others. 3. Infection. 4. Contaminated wounds (with chemical or faecal material, or soil). 5. Comorbid diseases. 6. Fractures. 7. Neurological injury. 8. Pregnancy. Randomised: 78 patients (Group 1: n = 39, Group 2: n = 39). Excluded (post‐randomisation): Group 1: 2 (5%). Reason for exclusion: left the hospital 2 days after admission. Patients assessed: 76 (97.4%) Group 1: 37, Group 2: 39. Withdrawals: not stated. Age (years): (mean, range): Group 1: 24.9 (1‐67), Group 2: 18.9 (1‐74). Gender (male: female): Group 1: 24 (64.9%): 13 (35.1%), Group 2: 26 (66.7%): 13 (33.3%). Burned surface (% TBSA): (mean, range): Group 1: 16.4% (10‐54%), Group 2: 17.6% (10‐45%). TBSA full thickness burns: Group 1: 9 (24.3%), Group 2: 12 (30.8%). Inhalation injury: Group 1: 6 (16.2%), Group 2: 11 (28.2%). Time post‐burn: participants admitted on day on which they were burned: Group 1: 30 (81.1%), Group 2: 37 (94.9%). Burn type: Group 1: scald (hot liquid or steam) 15 (40.5%), fire (flame) 22 (59.5%); Group 2: scald 20 (51.3%), fire 19 (48.7%). Wounds infected at baseline?: no. Co‐morbidity: no comorbidity. |
|
| Interventions |
Type of antibiotic prophylaxis: topical antibiotic prophylaxis. Type of interventions: SSD vs skin substitute. Group 1: SSD 1% daily topical application, and the wound was treated daily with washing and cleaning. Group 2: lyophilised porcine skin (Xenoderm, Medical Biomaterial Products, Berlin, Germany). After tangential excision or dermabrasion of the burned area with a dermatome, and rinsing the wound with normal saline, Xenoderm was placed on the wound by the surgeon and fixed in place using a suture, dressing, or bandage. Duration of intervention: 2‐5 weeks. Co‐interventions: In the Xenoderm group, all patients received cefazolin during the surgery (tangential escisión or dermabrasion), before the application of Xenoderm. The burned region was immobilised by a splint if necessary. |
|
| Outcomes | Wound infection (secretion of pus). Length of hospital stay (days). |
|
| Notes | Sources of support: quote. "This study was supported by a grant from the Deputy for Research of Zanjan University of Medical Sciences" (Page 239 trial report). | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "The patients were randomly divided into two groups consecutively" (Page 235 trial report). Quote: "There were no significant differences between the two groups with respect to age, gender, % TBSA, cause of burn, burn thickness or burn site (Table 1)" (Page 236 trial report). Comment: insufficient information to make a judgement. |
| Allocation concealment (selection bias) | Unclear risk | No information provided. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | It was not reported whether participants and personnel were blinded, but they were probably not, because of the different nature of the interventions (topical SSD vs porcine skin). |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | It was not reported whether outcome assessors were blinded, but they were probably not, because of the different nature of the interventions (topical SSD vs porcine skin). |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 37/39 (95 %) and 39/39 patients (100%) in the SSD and lyophilised porcine skin groups, respectively, were included in the final analysis. |
| Selective reporting (reporting bias) | Low risk | No protocol was provided, but given the outcomes listed in the methods section, all pre‐specified outcomes were reported. |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
Khorasani 2009.
| Methods |
Study design: randomised controlled study. Setting/location: hospital (Zare’s Burn Hospital, Sari). Country: Iran. Period of study: not stated (published in 2009). Unit of randomisation: site burn. Unit of analysis: site burn. Sample size calculation: no. Use of ITT analysis?: yes. |
|
| Participants |
Inclusion criteria: 1. Burns acquired within 24 h preceding initiation of treatment. 2. 2 same site burns (such as on the feet or hands) on each participant. 3. Partial‐thickness burns (2nd‐degree burns with respect to depth and similar surface areas in 2 different parts of the body). 4. Burns ≤ 40% TBSA. Exclusion criteria: 1. Diabetes. 2. Immunodeficiency. 3. Pregnancy. 4. Kidney diseases. 5. Electrical and chemical burns. Randomised: 30 patients (60 burn wounds: Group 1: n = 30, Group 2: n = 30). Age (years): (mean, SD): 33 ±11. Gender (male: female): 25 (83%): 5 (17%). Burned surface (% TBSA): (mean, SD, range): 19.8 ± 7.9 (10‐40). Inhalation injury: not stated. Time post‐burn (h): < 1 h: 15 patients (50%); 1‐3 h: 12 patients (40%); > 3 h: 3 patients (10%). Burn type: not stated. Wounds infected at baseline?: no. Co‐morbidity: not described. |
|
| Interventions |
Type of antibiotic prophylaxis: topical antibiotic prophylaxis. Type of interventions: SSD vs Aloe vera. Group 1: SSD topically twice daily. Group 2:Aloe vera cream (Aloe vera powder (Zarband Phytopharmaceutical, Tehran, Iran), white paraffin 2 g, sterile alcohol 7.5 g, cetyl alcohol 7.5 g, solid white paraffin 3 g, and propylene paraben 0.015 g) topically twice daily. Duration of intervention: until burns were fully healed and epithelialized. Co‐interventions: all patients were treated with fluid resuscitation, daily dressings, and other treatment protocols during their hospitalisation. After admission, the wounds were cleaned with water or normal saline solution and the topical agent. All patients were given oral nutrition with occasional iv support in the form of amino acid infusion and blood products during their hospital stay. |
|
| Outcomes | Burn wound infection (wound observed clinically for signs of infection). Healing time. |
|
| Notes | Sources of support: quote: "This work was supported by a grant from Mazandaran University of Medical Sciences, Sari, Iran" (Page 590 trial report). | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No information provided. |
| Allocation concealment (selection bias) | Unclear risk | No information provided. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "Patients and nursing staff were blinded to the procedure" (Page 588 trial report). |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No information provided to permit a judgement to be made. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Did not report number of withdrawals. Comment: denominator values suggested complete follow‐ up |
| Selective reporting (reporting bias) | Low risk | No protocol provided, but given the outcomes listed in the methods section, all pre‐specified outcomes were reported. |
| Other bias | Unclear risk | Insufficient information provided to assess whether an important risk of bias existed. |
Kimura 1998.
| Methods |
Study design: prospective, randomised, placebo controlled study. Setting/location: hospital (Emergency and Critical Care Center of Nippon Medical School Hospital). Country: Japan. Period of study: April 1994‐September 1996 (2.4 years). Unit of randomisation: patient. Unit of analysis: patient. Sample size calculation: no. Use of ITT analysis?: yes. |
|
| Participants |
Inclusion criteria: 1. Age: ≥ 10 years. 2. Burns of ≥ 20% TBSA. 3. Requirement for ventilator support. Exclusion criteria: 1. Children (< 10 years old). 2. Pregnant women. 3. Patients with severe underlying diseases such as renal failure, hepatic failure, and leukopenia. Randomised: 40 participants (Intervention group: n = 21, Control group: n = 19). Age (years): (mean, range): Intervention group: 44 (10‐91), Control group: 48 (12‐85). Burned surface (% TBSA): (mean, range): Intervention group: 49% (22‐87%), Control group: 43% (20‐80%). Inhalation injury: Intervention group: 11 (52.4%), Control group: 12 (63.2%). Ventilator support: Intervention group: 21 (100%), Control group: 19 (100%). Time post‐burn (days): 4‐6 days. Burn type: Intervention group: fire (flame) 19 (90%), scald (hot liquid or steam) 2 (10%); Control group: fire 16 (84%), scald 3 (16%). Wounds infected at baseline?: no. Co‐morbidity: none. |
|
| Interventions |
Type of antibiotic prophylaxis: systemic antibiotic prophylaxis (general). Type of interventions: TMP‐SMX vs placebo. Intervention group: TMP‐SMX (1.0 g) containing 400 mg SMX and 80 mg TMP. Control group: placebo (lactose 1.0 g). Both TMP‐SMX and placebo were administered orally or by means of a nasogastric tube 3 times daily. Other antibacterial therapies (ampicillin; cefazolin; cefamandole; cefmetazole; flomoxef) were used with TMP‐SMX or placebo when deemed necessary by the treating physicians. Duration of intervention: 10 days. Co‐interventions: not described. |
|
| Outcomes | Incidence of pneumonia (defined when all the following criteria present: (1) infiltration of lung fields on chest X‐ray films, (2) fever (> 38°C) for at least 3 consecutive days, (3) peripheral white blood cell count > 104/mm3, (4) pathogenic bacteria (> 103 colony forming units/ml) detected in airway secretions). Incidence of MRSA pneumonia. Mortality. Airway flora. Side effects. |
|
| Notes | Sources of support: Quote: "We are indebted to MI. Yasuji Aoto, laboratory technician at Nippon Medieal Sehool Hospital, for collecting bacterial strains, and Shionogi Pharmaceutical Company for technical assistance in measuring TMP‐SMX concentrations" (Page 386). | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "The subjects were divided into TMP‐SMX and placebo groups in a randomised manner by the attending pharmacist" (Page 384 trial report). Comment: insufficient information provided to permit a judgement to be made. |
| Allocation concealment (selection bias) | Low risk | Central pharmacy. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: " . . with both the patient and the attending physician being blinded to the treatment protocol" (Page 384 trial report). Comment: participants and personnel were probably blinded. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | It was not reported whether the outcome assessors were blinded (but they probably were, if the attending physician was the person who assessed the outcomes). |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Did not report number of withdrawals Comment: denominator values suggested that all patients randomised were included in the final analysis. |
| Selective reporting (reporting bias) | Low risk | No protocol provided, but given the outcomes listed in the methods section, all pre‐specified outcomes were reported. |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
Levine 1978.
| Methods |
Study design: prospective, randomised trial. Setting/location: hospital (US Army Institute of Surgical Research, Houston, Texas). Country: USA. Period of study: not stated (published in 1978). Unit of randomisation: patient. Unit of analysis: patient. Sample size calculation: no. Use of ITT analysis?: yes. |
|
| Participants |
Inclusion criteria: 1. Age: ≥ 16 years. 2. Suspected, or evidence of, inhalation injury (with positive bronchoscopic findings of carbonaceous material, or of tracheobronchial mucosal edema, erythema, haemorrhage, or ulceration). 3. Patient admitted within 72 h of acquiring burn injury. Exclusion criteria: not described. Randomised: 30 patients (Intervention group: n = 12, Control group: n = 18). Age (years): (mean): Intervention group: 34.3, Control group: 28.1. Burned surface (% TBSA): (mean): Intervention group: 57.6%, Control group: 53.8%. Inhalation injury: Intervention group: 12 (100%), Control group: 18 (100%). Time post‐burn (h): ≤ 72 h (in both groups). Burn type: Intervention group: thermal (100%), Control group: thermal (100%). Wounds infected at baseline?: no. Co‐morbidity: not described. |
|
| Interventions |
Type of antibiotic prophylaxis: local antibiotic prophylaxis (airway). Type of interventions: gentamicin vs placebo. Intervention group: aerosolized gentamicin, 80 mg in 2 ml of diluent. Control group: placebo (2 ml of aerosolized normal saline). Both groups received treatment 3 times daily, administered by airway. Duration of intervention: 10 days. Co‐interventions: All burn wounds were cleaned and debrided at time of admission and treated with either SSD (Silvadene) or mafenide acetate (Sulfamylon) creams. Escharotomies were performed only as necessary on all circumferentially burned extremities and the thorax. All patients in both trials were resuscitated over the first 24 h with Ringer's lactate solution. Humidified oxygen was given by face mask. Endotracheal intubation with controlled mechanical ventilation was instituted on the basis of recognized criteria (hypoxia, hypercarbia, and markedly increased respiratory rate). |
|
| Outcomes | Mortality. Sepsis. Pulmonary complications attributable to inhalation injury (e.g. pneumonitis, bronchitis, severe atelectasis, and lobar collapse) |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "Thirty patients with proven inhalation injuries were allocated in a prospective randomised manner into either a gentamicin or placebo‐treated group" (Page 189 trial report). Comment: insufficient information provided to permit a judgement to be made |
| Allocation concealment (selection bias) | Unclear risk | No information provided. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | No information provided. Placebo inhalations used, so likely that participants and personnel were blinded. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Insufficient information provided. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Did not report number of withdrawals. Comment: denominator values suggested that all patients randomised were included in the final analysis. |
| Selective reporting (reporting bias) | High risk | The study protocol was not available, and the methods section did not pre‐specify the outcomes to be reported. |
| Other bias | Unclear risk | Insufficient information to assess whether an important risk of bias existed. |
Livingston 1990.
| Methods |
Study design: prospective randomised study. Setting/location: hospital (Adult Burn Unit, University of Louisville, Kentucky). Country: USA. Period of study: January 1987‐January 1988 (12 months). Unit of randomisation: patient. Unit of analysis: patient. Sample size calculation: no. Use of ITT analysis?: no. |
|
| Participants |
Inclusion criteria: 1. Patients admitted to the Adult Burn Unit. 2. Thermal burns requiring skin grafting. Exclusion criteria: 1. Use of non‐meshed grafts (hand and face grafts). 2. Allergy to any of the test agents (neomycin, bacitracin, silver nitrate). Enrolled: 90 patients. Randomised: unclear. Active participants: 52 patients (57.7%) (Group 1: n = 15, Group 2: n = 18 , Group 3: n = 19). Withdrawals: not described. Patients assessed: 52 patients. Age (years): (mean): Group 1: < 20% TBSA: 46 ± 22, 20‐40% TBSA: 27 ± 5, > 40% TBSA: 49 ± 10. Group 2: < 20% TBSA: 48 ± 24, 20‐40% TBSA: 38 ± 22, > 40% TBSA: 52 ± 13. Group 3: < 20% TBSA: 43 ± 27, 20‐40% TBSA: 34 ± 20, > 40% TBSA: 43 ± 19. Burned surface (% TBSA): (mean): Group 1: < 20% TBSA: 14% ± 5, 20‐40% TBSA: 29% ± 7, >40% TBSA: 47% ± 6. Group 2: < 20% TBSA: 11% ± 3, 20‐40% TBSA: 28% ± 6, >40% TBSA: 53% ± 16. Group 3: < 20% TBSA: 13% ± 5, 20‐40% TBSA: 30% ± 6, >40% TBSA: 52% ± 11. Inhalation injury: Group 1: 5 (33.3%), Group 2: 4 (22.2%), Group 3: 2 (10.5%). Time post‐burn: not stated. Burn type: Group 1: thermal (100%), Group 2: thermal (100%), Group 3: thermal (100%). Wounds infected at baseline?: no. Co‐morbidity: not described. |
|
| Interventions |
Type of antibiotic prophylaxis: topical antibiotic prophylaxis. Type of interventions: placebo vs neomycin plus bacitracin vs silver nitrate. Group 1: placebo (Ringer's lactate (RL)). Group 2: neomycin (1 g/litre) plus bacitracin (50,000 units/litre) (NB). Group 3: silver nitrate 0.5% (Ag). The solutions were applied topically with gauze soaked in the operating room. The solutions were reapplied as necessary to keep the grafted areas moist (every 2‐6 h). Duration of intervention: until grafts were healed, or there was evidence of graft loss or infection. Co‐interventions: patients received standardized resuscitation with Ringer's lactate solution. Enteral or parenteral nutrition was started as soon as the patient could tolerate it, and full nutritional support was applied 5 days postburn. All patients received cefazolin perioperatively (during excision and grafting, ¯before randomization). Escharotomies were performed when clinically indicated in patients with circumferential burns. The catheters (arterial, central venous, pulmonary artery) were placed when clinically indicated. Tangential excision and split‐thickness skin grafting were performed as soon as the clinical condition of the patient permitted (2‐5 days postburn). |
|
| Outcomes | Burn wound infection (defined as > 105 organisms/g of tissue in both the nonadherent graft and recipient site). Hospital stay (days). Antibiotic‐resistant organisms. |
|
| Notes | Quote: "The study was originally designed to include 90 patients; however, intermittent analysis showed that poor results occurred in two of the three groups (NB and RL), and therefore, data were evaluated after 45 patients had completed the study" (Page 1060 trial report). | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Cards shuffled at assignment. Quote: "Patients were selected at random to receive one of the three treatments . . . Randomization was achieved by labelling 10 cards for each topical agent in each of three percentage total body surface area (TBSA) burn categories, specifically, less than 20 percent, 20 to 40 percent, and more than 40 percent" (Page 1060 trial report). |
| Allocation concealment (selection bias) | Unclear risk | Quote: "The cards were shuffled and drawn in blinded fashion when the patient was entered into the study" (Page 1060 trial report). Comment: did not provide sufficient information to permit a judgement to be made. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | No information provided. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No information provided. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Only 52/90 randomised patients included in the analysis. |
| Selective reporting (reporting bias) | Low risk | No protocol provided, but given the outcomes listed in the methods section, all pre‐specified outcomes were reported. |
| Other bias | High risk | Quote: "The study was originally designed to include 90 patients; however, intermittent analysis showed that poor results occurred in two of the three groups (NB and RL), and therefore, data were evaluated after 45 patients had completed the study" (Page 1060 trial report). |
Maya 1986.
| Methods |
Study design: randomised controlled trial. Setting/location: hospital (Hospital Infantil de Tacubaya de los Servicios Médicos del D.D.F.). Country: Mexico. Period of study: not stated (published in 1986). Unit of randomisation: patient. Unit of analysis: patient. Sample size calculation: no. Use of ITT analysis?: yes. |
|
| Participants |
Inclusion criteria: 1. Paediatric patients. 2. 2nd‐degree burns. 3. Patients admitted to the hospital. Exclusion criteria: not described. Randomised: 40 patients (Intervention group: n = 20, Control group: n = 20). Patients assessed: 40 patients (100%). Age (months): (mean, SD): Intervention group: 45.65 (26.87), Control group: 35.45 (26.44). Gender (male: female): Intervention group: 12 (60%): 8 (40%), Control group: 11 (55%): 9 (45%). Burned surface (% TBSA): (mean, SD): Intervention group: 12.25% (7.8), Control group: 12.65% (8). TBSA full thickness burns: Intervention group: 3 (15%), Control group: 2 (10%). Inhalation injury: not stated. Time post‐burn (h): (mean): Intervention group: 11.75 h, Control group: 14.87 h. Burn type: Intervention group: scald (hot liquid or steam) 17 (85%), fire (flame) 3 (15%); Control group: scald 16 (80%), fire 2 (10%), chemical 2 (10%). Wounds infected at baseline?: no. Co‐morbidity: not described. |
|
| Interventions |
Type of antibiotic prophylaxis: topical antibiotic prophylaxis. Type of interventions: rifamycin and amniotic membranes vs amniotic membranes. Intervention group: amniotic membranes and rifamycin. Rifamycin was sprayed over the amniotic membranes. Membranes were kept dry, and sprayed again with rifamycin in case of apparent discharge. Control group: amniotic membranes. Amniotic membranes were separated from the chorion. Then they were washed in 250 ml 0.9% saline solution plus 1 g of neomycin. Membranes were stored at 4ºC for 14 days. Cultures of control were conducted every five days to verify the absence of bacterial contamination. Afterwards, they were used for treatment. In both groups, once the membranes had been placed, they were stretched over the burn area and dried with a hair dryer. In case of slippage or infection, the amniotic membranes were changed. Duration of intervention: until wound healing (16 days). Co‐interventions: mechanical wash with Isodine and debridement of necrotic tissue, including blisters. Fluid replacement. |
|
| Outcomes | LOS (days) Wound infection Time to re‐epithelialization (days) |
|
| Notes | We tried to contact authors to obtain data on time to re‐epithelialization, given that the study did not provide enough information on this matter, but It was not possible to obtain data that could be used for our review. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "Forty patients with second‐degree burns were treated with amniotic membranes and rifamycin. They were randomly assigned to two treatment groups " (Page 73 trial report). Comment: insufficient information to make a judgement. |
| Allocation concealment (selection bias) | Unclear risk | No information provided. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | No blinding described (therefore, probably open). |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Insufficient information provided. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Did not report number of withdrawals. Comment: denominator values suggested complete follow‐ up. |
| Selective reporting (reporting bias) | High risk | No protocol provided, and none of the outcomes reported were nominated in the methods section of this paper. |
| Other bias | Unclear risk | Insufficient information to assess whether an important risk of bias existed. |
Miller 1987.
| Methods |
Study design: randomised clinical trial. Setting/location: hospital (Regional Burn Center, San Diego, California). Country: USA. Period of study: not stated (published in 1987). Unit of randomisation: patient. Unit of analysis: patient. Sample size calculation: no. Use of ITT analysis?: no. |
|
| Participants |
Inclusion criteria: 1. Age: 18‐67 years. 2. Gender: men and women. 3. Full‐tickness burn injury. 4. Admitted within 72 h of burn injury. 5. Requirement for surgical excision and autografting. Exclusion criteria: 1. Pregnant or breastfeeding women. 2. Impaired renal function (serum creatinine ≥ 1.5 mg%). 3. History of hypersensitivity to penicillin and cephalosporin. 4. Those who received antimicrobial drug therapy within 72 h preceding administration of preoperative study drug, or who were likely to receive concomitant antibiotics. Randomised: 48 patients (Group 1: n = 24, Group 2: n = 24). Excluded (post‐randomisation): 7 (14.5%) (Group 1: 2 (8.3%), Group 2: 5 (20.8%)). Reason for exclusion: Did not receive ceforanide: 1 (Group 1). Received additional antibiotic therapy after surgery: 1 (Group 1), topical antimicrobial: 1 (Group 2). Premature hospital discharge: 1 (Group 2). Late administration of the first dose of study drug: 1 (Group 2). Concurrent administration of other antibiotics during the study period: 1 (Group 2). Graft loss, due to inadequate operative debridement: 1 (Group 2). Patients assessed: 47 patients (97.9%) (Group 1: 23, Group 2: 24). Number evaluable: 41 patients (87.2%), Group 1: 22 (95.6%), Group 2: 19 (79.1%). Age (years): (mean, range): Group 1: 34 (18‐67), Group 2: 35 (20‐65). Burned surface (% TBSA): (mean, range): Group 1: 7.9% (2‐22%), Group 2: 6% (2‐15%). Inhalation injury: not stated. Time post‐burn (h): ≤ 72 h (in both groups). Burn type: not stated. Wounds infected at baseline?: no. Co‐morbidity: not described. |
|
| Interventions |
Type of antibiotic prophylaxis: systemic antibiotic prophylaxis (perioperative). Type of interventions: ceforanide vs cefazolin. Group 1: ceforanide (1 g iv within 1 h prior to surgery). Group 2: cefazolin (3 x 1 g iv doses, 1st within 1 hour of surgery, then doses at 6 h and 12 h after 1st dose) Duration of intervention: during perioperative period (1 day). Co‐interventions: not described. |
|
| Outcomes | Infection. Burn wound infection (bacterial culture). Cultures and sensitivities. Overall prophylactic response (freedom of infection and successful graft take). Side effects. |
|
| Notes | Sources of support: quote: "This study was supported in part by an educational graft from Bristol Myers, lnc." (Page 951 trial report). | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "After randomisation, patients received either ceforanide 1 gm intravenously within one hour prior to surgery or three 1 gm doses of cefazolin, given intravenously within one hour of surgery . . . " (Page 948 trial report). Comment: insufficient information to make a judgement. |
| Allocation concealment (selection bias) | Unclear risk | No information provided. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Quote: "An open‐labelled, randomised, single‐centre study" (Page 946). |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Quote: "An open‐labelled, randomised, single‐centre study" (Page 946 trial report). |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 22/24 (92 %) and 19/24 patients (79%) in the ceforanide and in the cefazolin groups, respectively, were included in the final analysis. Comment: not all randomised patients were included in the final analysis. |
| Selective reporting (reporting bias) | Low risk | No protocol provided, but given the outcomes listed in the methods section, all pre‐specified outcomes were reported. |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
Mohammadi 2009.
| Methods |
Study design: randomised clinical trial. Setting/location: hospital (Ghotbeddin Burn Hospital, Shiraz). Country: Iran. Period of study: October 2005‐February 2007 (16 months). Unit of randomisation: patient. Unit of analysis: patient. Sample size calculation: no. Use of ITT analysis?: unclear. |
|
| Participants |
Inclusion criteria: 1. Patients with deep burns (2nd‐ and 3rd‐degree). 2. Burns over 20‐50% TBSA. 3. Patients admitted to burn centre. Exclusion criteria: 1. Age: > 60 years. 2. History of cardiac disease, renal failure, diabetes mellitus or any other severe metabolic disorder. Randomised: 124 patients (Group 1: n = 63, Group 2: n = 61). Excluded (post‐randomisation): Group 1 (control group): 1 (1.6%). Reasons for exclusion: Quote: "... an 18‐year‐old female, a case of suicide with 40% burn, who underwent amniotic membrane dressing. On the 5th day, the patient had high grade fever due to which her dressing was changed to regular antibiotic and gauze dressing. She was expired on the 14th day of hospitalization. This was the only case whose treatment policy was changed during hospitalization and so she was excluded from the survey". Withdrawals: Group 2: 5 (8.6%) (2 males and 3 females). Reasons: died during the study. Age (years): (mean): Group 1: 23.31 ± 14.53, Group 2: 25.3 ± 11.81. Gender (male: female): Group 1: 35 (55.5%): 28 (44.4%), Group 2: 35 (57.3%): 26 (42.6%). Burned surface (% TBSA): (mean): Group 1: 32.4% ± 8.9%, Group 2: 31.2% ± 8.3%. TBSA full thickness burns: Group 1: 63 (100%), Group 2: 61 (100%). Inhalation injury: not stated. Time post‐burn (h): not stated. Burn type: in both groups most common mechanism was flame followed by flash. Wounds infected at baseline?: no. Co‐morbidity: no comorbidity. |
|
| Interventions |
Type of antibiotic prophylaxis: topical antibiotic prophylaxis. Type of interventions: amniotic membrane vs SSD. Group 1: amniotic membrane. Wounds were washed with normal saline and diluted Betadine and then covered with a layer of amniotic membrane, then a layer of Vaseline gauze, and a dressing with gauze and band. Placentas were acquired from elective caesarean sections. The amniotic membrane was separated from chorion and placenta and washed thoroughly with normal saline, kept in a sterile pot of normal saline plus 80 mg gentamycin, and stored in refrigerator at 4°C. A blood sample drawn from the umbilical cord was checked for syphilis (VDRL test), HIV, HCV, and HBS, and, only if all these tests were negative, was the amniotic membrane used. Dressings were changed every 3‐4 days. Group 2: SSD or mafenide acetate. Wounds were irrigated twice daily with normal saline and diluted Betadine and then covered with SSD, or in some cases mafenide acetate dressing. Duration of intervention: before skin graft (26 days). Co‐interventions: not described. |
|
| Outcomes | Wound infection. Sepsis (suspected as present with the following symptoms: signs of hypothermia, hypotension, abrupt hyperglycaemia, decreased urine output, thrombocytopenia and diet intolerance, including blood culture and urine culture). LOS (days). Mortality. |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: " . . . were admitted in our center were randomly divided into two groups, using random allocation (regardless of the depth of the burn)" (Page 67 trial report). Comment: insufficient information to make a judgement. |
| Allocation concealment (selection bias) | Unclear risk | No information provided. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "In this double‐blinded randomised clinical trial". Comment: it is not clear who was blinded. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "In this double‐blinded randomised clinical trial" Comment: it is not clear who was blinded. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Comment: it is not clear how the authors considered the post randomisation exclusions and withdrawals in the analysis, however, due to their low number (6/124, i.e. 5%) they may have not distorted the study results. |
| Selective reporting (reporting bias) | High risk | No protocol provided, and some of the outcomes reported were listed in the methods section of the trial report. |
| Other bias | Unclear risk | Insufficient information to assess whether an important risk of bias existed. |
Moharamzad 2010.
| Methods |
Study design: double‐blinded, randomised clinical trial. Setting/location: hospital. Country: Iran. Period of study: not stated (published in 2010). Unit of randomisation: patient. Unit of analysis: patient. Sample size calculation: no. Use of ITT analysis?: unclear. |
|
| Participants |
Inclusion criteria: 1. Burn patients. 2. Admitted to hospital < 24 h after burn injury. 3. 2nd‐degree burns (partial‐thickness burns). 4. Burns over ≤ 5% TBSA. Exclusion criteria: not stated. Randomised: 111 patients (Group 1: n = 55, Group 2: n = 56). Withdrawals: not stated. Burned surface (% TBSA): ≤ 5%. Inhalation injury: not stated. Time post‐burn (h): ≤ 24 h. Burn type: not stated. Wounds infected at baseline?: no. Co‐morbidity: not described. |
|
| Interventions |
Type of antibiotic prophylaxis: topical antibiotic prophylaxis. Type of interventions: SSD vs herbal cream. Group 1: SSD topically once daily. Group 2: herbal cream (Aloe vera, Geranium robertianum, and Lavandula stoechas) topically once daily. Duration of intervention: 14 days. Co‐interventions: debridement and cleaning the wound. |
|
| Outcomes | Duration of wound healing. Wound infection. | |
| Notes | We only had data published in an abstract. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "Patients: In this double‐blinded randomised clinical trial". Comment: insufficient information to make a judgement. |
| Allocation concealment (selection bias) | Unclear risk | No information provided. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "In this double‐blinded randomised clinical trial" |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "In this double‐blinded randomised clinical trial" Comment: probably done. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Insufficient information to permit judgement of ‘low risk’ or ‘high risk’. |
| Selective reporting (reporting bias) | Unclear risk | Insufficient information in poster report. |
| Other bias | Unclear risk | Insufficient information to assess whether an important risk of bias existed. |
Muangman 2006.
| Methods |
Study design: randomised controlled trial. Setting/location: hospital (Burn Unit, Siriraj Hospital). Country: Thailand. Period of study: May 2002‐September 2005 (3.4 years). Unit of randomisation: patient. Unit of analysis: patient. Sample size calculation: no. Use of ITT analysis?: yes. |
|
| Participants |
Inclusion criteria: 1. Partial‐thickness burn wounds. 2. Burns < 25% TBSA. 3. Patients admitted to Burn Unit. Exclusion criteria: not described. Randomised: 50 patients (Group 1: n = 25, Group 2: n = 25). Patients assessed: 50 (100%). Age (years): (mean, SD): Group 1: 38 ± 25, Group 2: 26 ± 27. Burned surface (% TBSA): (mean, SD): Group 1: 15 ± 7, Group 2: 15 ± 5. Inhalation injury: not stated. Time post‐burn (h): not stated. Burn type: Group 1: fire (flame) 14 (56%), scald (hot liquid or steam) 9 (36%), electrical 1 (4%), chemical 1 (4%); Group 2: fire 12 (48%), scald 12 (48%), chemical 2 (10%), electrical 1 (4%). Wounds infected at baseline?: no. Co‐morbidity: not described. |
|
| Interventions |
Type of antibiotic prophylaxis: topical antibiotic prophylaxis. Type of interventions: silver‐coated dressing vs SSD. Group 1: Acticoat (Smith & Nephew USA, Largo, FL), a non‐adherent nanocrystalline silver‐coated dressing material. Acticoat, moistened in sterile water, was applied, then a dry dressing. The inner gauze was moistened twice a day with sterile water and the Acticoat was changed every 3 days. Group 2: SSD 1% and dry gauze dressings twice daily. Duration of intervention: until burn wound closure. Co‐interventions: all patients were routinely given 2 x 500 mg tablets of acetaminophen (paracetamol) before dressing changes. |
|
| Outcomes | LOS (days). Days until burn wound closure. Type of cultured organisms. Wound colonization (bacterial culture and signs of infection such as erythema, induration, purulent discharge and malodour). Infection. Mortality. |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "Fifty patients were identified and randomised into 2 groups and given burn wound treatment . . . " (Page 954 trial report). No significant differences in age, TBSA (%), type of burn, length of hospital stay, between the two groups. Comment: insufficient information to make a judgement. |
| Allocation concealment (selection bias) | Unclear risk | No information provided. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | No information provided. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No information provided. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Did not report number of withdrawals. Comment: denominator values suggested complete follow‐up. |
| Selective reporting (reporting bias) | Low risk | No protocol provided, but given the outcomes listed in the methods section, all pre‐specified outcomes were reported. |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
Munster 1986.
| Methods |
Study design: randomised, prospective study. Setting/location: hospital (Baltimore Regional Burn Center, Maryland). Country: USA. Period of study: not stated (published in 1986). Unit of randomisation: patient. Unit of analysis: patient. Sample size calculation: no. Use of ITT analysis?: yes. |
|
| Participants |
Inclusion criteria: 1. Age: 18‐50 years. 2. Burns 20%‐70% TBSA. 3. Patients admitted to Burn Center. Exclusion criteria: 1. Tests of hepatic and renal function with abnormal values (post‐randomisation). Participants: 28 patients (Intervention group: n = 15, Control group: n = 13). Withdrawals: no patient had to be discontinued from the study because of adverse side effects, or adverse effects on renal or hepatic function. Age (years): (mean, SD): Intervention group: 36.9 ± 12, Control group: 40 ± 15.9. Burned surface (% TBSA): (mean, SD): Intervention group: 30.8 ± 8.5, Control group: 38.4 ± 17.3. Inhalation injury: not stated. Time post‐burn (h): not stated. Burn type: Intervention group: thermal (100%), Control group: thermal (100%). Wounds infected at baseline?: no. Co‐morbidity: not described. |
|
| Interventions |
Type of antibiotic prophylaxis: systemic antibiotic prophylaxis (general). Type of interventions: polymixin B vs control. Intervention group: polymixin B 5000 units/kg iv on 1st day of study, reducing by increments of 500 units/kg/day to 1500 units on last day. Control group: untreated patients. Duration of intervention: 7 days. Duration of follow‐up: 2 weeks. Co‐interventions: not described. |
|
| Outcomes | Burn wound sepsis (determined by presence of ≥ 105 organisms/g tissue). Sepsis (determined by either: (1) presence of a positive blood culture; or, (2) presence of a quantitative biopsy on one or more occasion of ≥ 105 organisms/g tissue, coupled with any one of the following clinical parameters: hypothermia, disorientation and paralytic ileus). Mortality. |
|
| Notes | Sources of support: quote: "Supported in part by NIH Grant GM 26235 and by the Baltimore Metropolitan Firefighters Unions" (Page 995 trial report). | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "All patients were admitted . . . following which they were randomised to polymyxin or control group" (Page 995 trial report). Comment: insufficient information to make a judgement. |
| Allocation concealment (selection bias) | Unclear risk | No information provided. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Blinding of participants and study personnel not reported, but probably not done due to the different nature of the interventions (polymixin iv vs no treatment). |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Blinding of outcome assessors not reported. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | The authors stated that, "A total of 28 patients completed the study, 15 in the polymixin and 13 in the control group". Quote: "No patient had to be discontinued from the study because of adverse side effects or adverse effects on renal or hepatic function" (Page 996 trial report). |
| Selective reporting (reporting bias) | Low risk | No protocol provided, but given the outcomes nominated in the methods section, all pre‐specified outcomes were reported. |
| Other bias | Unclear risk | Insufficient information provided to enable an assessment of whether an important risk of bias existed. |
Noordenbos 1999.
| Methods |
Study design: prospective, randomised, paired‐site study. Setting/location: hospital (San Diego Medical Center, California). Country: USA. Period of study: 1 year. Unit of randomisation: wound site. Unit of analysis: wound site. Sample size calculation: no. Use of ITT analysis?: no. |
|
| Participants |
Inclusion criteria: 1. Age: 1‐70 years. 2. Partial‐thickness burns (moderate to deep partial‐thickness in depth, i.e. wounds that it was estimated would require 7‐21 days to heal). 3. Burns 2%‐30% TBSA. Exclusion criteria: 1. Wounds of the hands, face, buttocks, feet, and genitalia. Randomised: 14 patients, 28 wound sites: Group 1: n = 14, Group 2: n = 14. Patients assessed: 11 patients (78.6%). Age (years): (mean, range): 23.4 (1.1‐52), SD: 19.4. Burned surface (% TBSA): (mean, range): 13.3% (4‐30), SD: 7.2. Inhalation injury: not stated. Time post‐burn (h): unclear, quote: "Attempts were made to enrol patients into the study as soon as possible after injury" (Page 276). Burn type: not stated. Wounds infected at baseline?: no. Co‐morbidity: not described. |
|
| Interventions |
Type of antibiotic prophylaxis: topical antibiotic prophylaxis. Type of interventions: biosynthetic dressing (TransCyte) vs SSD. Group 1: biosynthetic dressing (TransCyte (Advanced Tissue Sciences, La Jolla, California, USA), twice‐daily. TransCyte was never applied more than 24 h postburn. Group 2: SSD (BASF Inc, Mt Olive, New Jersey, USA) applied topically twice daily. Duration of intervention: until wound was clean of necrotic tissue and debris. Co‐interventions: wound debridement according to standard protocol of the burn centre. All cases of cellulitis were solved with short cycles of intravenous antibiotics. |
|
| Outcomes | Time until 90% healing (number of days until epithelial closure of at least 90% of the study site wound). Burn wound infection. |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "After patients were enrolled into the study, the 2 wound sites were chosen randomly to received either topical therapy with SSD (BASF Inc, Mt Olive, NJ) and twice‐daily dressing changes or TransCyte" (Page 276 trial report). Comment: insufficient information to make a judgement. |
| Allocation concealment (selection bias) | Unclear risk | No information provided. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Blinding of participants and study personnel not reported. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Blinding of outcome assessors not reported. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Not all patients who were randomised were included in the final analysis, and this may cause attrition bias. Comment: the magnitude of losses during the study was > 20%. |
| Selective reporting (reporting bias) | High risk | No protocol provided, and some of the outcomes reported were listed in the methods section of the trial report. |
| Other bias | Unclear risk | Insufficient information to assess whether an important risk of bias existed. |
Rodgers 1997.
| Methods |
Study design: randomised, prospective, partially‐blinded study. Setting/location: hospital (St Christopher's Hospital for Children, Pennsylvania). Country: USA. Period of study: October 1993‐September 1994 (11 months). Unit of randomisation: patient. Unit of analysis: patient. Sample size calculation: no. Use of ITT analysis?: no. |
|
| Participants |
Inclusion criteria: 1. Patients admitted for care of burns. 2. Burns < 35% TBSA. 3. Requirement for surgical debridement and grafting. Exclusion criteria: 1. Clinical evidence of infection at the time of debridement. 2. Received antibiotic therapy before debridement. 3. History of allergy to penicillin or cephalosporins. Randomised: 48 patients (42 patients < 35% TBSA; 6 patients > 35% TBSA). One patient with ≥ 35% TBSA burns was re‐entered into the randomisation for a 2nd debridement procedure. Excluded (before randomisation): < 35% TBSA burned: 21 patients (50%); > 35% TBSA burned: 3 patients (50%). Reason for exclusion: Failure to consent: 10 patients < 35% TBSA burned. Receipt of antibiotics before surgical debridement: < 35% TBSA burned: 10 patients, > 35% TBSA burned: 3 patients. Allergy to penicillin: < 35% TBSA burned: 1 patient. Withdrawals: < 35% TBSA burned: Intervention group: 1 (2%). Reasons: became hypotensive and hypothermic during surgery and was not responsive to volume resuscitation. Patients assessed: 24 patients (50%). < 35% TBSA burned: 20 patients (Intervention group: n = 10, Control group: n = 10). > 35% TBSA burned: 4 patients (Intervention group: n = 3, Control group: n = 1). Age (years): (mean): < 35% TBSA: Intervention group: 1.5, Control group: 1.9. > 35% TBSA: Intervention group: 5.4, Control group: 8. Gender (male: female): <35% TBSA: Intervention group: 6 (60%): 4 (40%), Control group: 4 (40%): 6 (60%). >35% TBSA: Intervention group: 3 (100%): 0, Control group: 1 (100%): 0. Burned surface (% TBSA): (mean): <35% TBSA: Intervention group: 10%, Control group: 11%. >35% TBSA: Intervention group: 45%, Control group: 55%. TBSA full thickness burns: > 35% TBSA: Intervention group: 3 (100%), Control group: 1 (100%). Inhalation injury: not stated. Time post‐burn (h): not stated. Burn type: < 35% TBSA: Intervention group: scald (hot liquid or steam) 9 (90%), fire (flame) 1 (10%); Control group: scald 6 (60%), fire 2 (20%), electrical 1 (10%), contact (hot solids) 1 (10%) > 35% TBSA: Intervention group: fire 3 (100%), Control group: fire 1 (100%) Wounds infected at baseline?: no. Co‐morbidity: not described. |
|
| Interventions |
Type of antibiotic prophylaxis: systemic antibiotic prophylaxis (perioperative). Type of interventions: cefazolin vs placebo, cefazolin vs targeted antibiotics. < 35% TBSA Intervention group: iv cefazolin 25 mg/kg 6‐hourly for 24 h. Administered in the operating room after a blood culture. Control group: placebo (normal saline in the volume corresponding to the cefazolin dose). > 35% TBSA Intervention group: iv cefazolin 25 mg/kg 6‐hourly for 24 h. Control group: targeted antibiotics in the volume corresponding to the cefazolin dose (selected by the infectious diseases consultant based on the results of the latest surveillance cultures). Duration of intervention: perioperative (1 day). Co‐interventions: surgical debridement of the burn wound . |
|
| Outcomes | Infection (bacteraemia). Organisms isolated. Burn wound infection (quantitative tissue biopsy cultures). |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "After categorization of TBSA burn, patients were randomised to treatment groups with use of a standard random number table" (Page 343 trial report). |
| Allocation concealment (selection bias) | Unclear risk | Comment: no information provided |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Quote: "All antibiotics and placebo were prepared by the hospital pharmacy and administered by one unblinded investigator (GR)" (Page 343). There was not enough information about blinding of participants, although it was reported that the professional who administered the intervention was not blinded. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "All grafts were inspected by one of two attending plastic surgeons (who were blinded to study treatment assignment) . . . " (Page 343 trial report). |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 20/21 (95 %) and 4/4 (100%) in the < 35% TBSA group and in the ≥ 35% TBSA group, respectively, were included in the final analysis. |
| Selective reporting (reporting bias) | Low risk | No protocol provided, but given the outcomes listed in the methods section, all pre‐specified outcomes were reported. |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
Silver 2007.
| Methods |
Study design: open, prospective, single‐centred, parallel group. Setting/location: hospital (Loyola University Medical Center, Chicago). Country: USA. Period of study: December 2003‐January 2005 (1.1 years). Unit of randomisation: patient. Unit of analysis: patient. Sample size calculation: no. Use of ITT analysis?: yes. |
|
| Participants |
Inclusion criteria: 1. Age: ≥ 13 years. 2. Burn requiring grafting with meshed autografts. 3. Consent according to institutional research board guidelines. Exclusion criteria: 1. Clinically significant history of major system disorder. 2. Active immunosuppressive therapy. 3. Pregnant or breastfeeding. 4. Chemotherapy or radiation therapy. 5. Immunocompromised state. 6. Sensitivity to silver or sulphonamide. 7. Clinical wound infection before enrolment. Randomised: 20 patients (Group 1: n = 10, Group 2: n = 10). Patients assessed: 20 patients (100%). Withdrawals: not stated. Age (years): (mean, range): Group 1: 41.4 ± 15.8, Group 2: 44.0 ± 17.9. Gender (male: female): Group 1: 6 (60%): 4 (40%), Group 2: 8 (80%): 2 (20%). Burned surface (% TBSA): (mean, SD): Group 1: 17 ± 9.9, Group 2: 18.7 ± 10.3. Inhalation injury: not stated. Time post‐burn (days): not stated. Burn type: not stated. Wounds infected at baseline?: no Co‐morbidity: not described. |
|
| Interventions |
Type of antibiotic prophylaxis: topical antibiotic prophylaxis. Type of interventions: mafenide acetate vs silver dressing. Group 1: mafenide acetate (Sulfamylon® 5% topical solution) applied through dressing (Exu‐Dry, Smith & Nephew) soaked in the solution. Dressing was left on for 3 days, then changed every 3 days. Group 2: silver dressing (Acticoat, Smith & Nephew Inc. Largo, Florida, USA). The dressing stayed intact for 2 days, and was then changed daily. Duration of intervention: until complete healing, or 2 weeks from initial assessment. Co‐interventions: not described. |
|
| Outcomes | Healing time. Infectious complications. Adverse effects. |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "All wounds were placed in 5% sulfamylon solution except test sites in the group of subjects randomised to Acticoat" (Page 716 trial report). Comment: insufficient information to make a judgement. |
| Allocation concealment (selection bias) | Unclear risk | No information provided. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Quote: "The study was designed as an open, prospective, single center, parallel group, comparative evaluation . . ." (Page 716 trial report). |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Quote: "The study was designed as an open, prospective, single center, parallel group, comparative evaluation . . . " (Page 716 trial report). |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Altough the authors stated that "there were no withdrawals from the study" (Page 718), there was insufficient information to permit judgement of ‘low risk’ or 'high risk’ for incomplete outcome data, as the denominators of the comparisons for each outcome were not reported. |
| Selective reporting (reporting bias) | Low risk | No protocol provided, but given the outcomes listed in the methods section, all pre‐specified outcomes were reported. |
| Other bias | Unclear risk | Insufficient information to assess whether an important risk of bias existed. |
Soroff 1994.
| Methods |
Study design: pilot study. Setting/location: hospital (Hospital Institutional Review Board, Minnesota). Country: USA. Period of study: not stated (published in 1994). Unit of randomisation: burn wound. Unit of analysis: burn wound. Sample size calculation: no. Use of ITT analysis?: no. |
|
| Participants |
Inclusion criteria: 1. Age: ≥ 18 years. 2. Partial‐thickness burns (defined as partial destruction of the dermal layer with dead tissue adherent to underlying viable dermis). 3. Two wounds of similar size and severity (noncontiguous). Exclusion criteria: 1. Chemical or electrical burns. 2. Burns ≥ 25% TBSA. 3. Known hypersensitivity to collagenase, silver sulfadiazine, polymyxin B sulfate, or bacitracin. 4. Pregnant or breastfeeding women. Randomised: 15 patients (30 burn wounds). Excluded (post‐randomisation): 2 (13.3%). Reason for exclusion: refused treatment (1); had an infection at an unrelated burn site (1). Assessed: 13 patients (86.6%). Age (years): not stated. Gender (male: female): 14 (93.3%): 1 (6.6%). Burned surface (% TBSA): (mean, SD): 11.7 ± 9.7 (range 2%‐34%). TBSA full thickness burns: > 25% TBSA: 1 (34%), 1 (30%). Inhalation injury: not stated. Time post‐burn (h): not stated. Burn type: scald (hot liquid or steam) 5 (33.3%), fire (flame) 5 (33.3%), other agents (hot ashes, flash, combined flame/flash) 5 (33.3%). Wounds infected at baseline?: no. Co‐morbidity: not described. |
|
| Interventions |
Type of antibiotic prophylaxis: topical antibiotic prophylaxis. Type of interventions: polymyxin B sulfate/bacitracin vs SSD. Group 1 (1st burn site): polymyxin B sulfate/bacitracin spray, covered with collagenase ointment, topically administered twice daily. Group 2 (2nd burn site): SSD cream, topically administered twice daily . Before treatment wound cultures were taken, and burns cleansed with normal saline solution. Duration of intervention: until wound bed was clean (disappearance of injured dermis). Co‐interventions: not described. |
|
| Outcomes | Time to wound healing. Adverse events. |
|
| Notes | In total, only 15 patients for both treatment groups were treated for different, non‐contiguous wounds. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "Two noncontiguous burns of similar size and severity were treated according to a randomisation schedule" (Page 13 trial report). Comment: insufficient information provided to make a judgement. |
| Allocation concealment (selection bias) | Unclear risk | No information provided. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | No information provided (probably an open trial due to the different natures of the interventions (spray + ointment versus cream). |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Blinding of outcome assessors not reported, and probably not done, because of the different natures of the interventions (spray + ointment versus cream). |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 13/15 (87%) and 13/15 burn wounds (87%) in the collagenase and in the control groups, respectively, were included in the final analysis. Incomplete outcome data probably did not distort the study results. |
| Selective reporting (reporting bias) | High risk | No protocol provided, and none of the outcomes reported were listed in the methods section of the trial report. |
| Other bias | Low risk | The basal characteristics of participants did not present significant differences between comparison groups. |
Subrahmanyam 1998.
| Methods |
Study design: prospective randomised trial. Setting/location: hospital (Department of Surgery, Dr Vaishampayan Memorial Medical College, Maharashtra). Country: India. Period of study: June 1995‐December 1996 (18 months). Unit of randomisation: patient. Unit of analysis: patient. Sample size calculation: no. Use of ITT analysis?: yes. |
|
| Participants |
Inclusion criteria: 1. Superficial burns. 2. Burns ≥ 40% TBSA. 3. Treated within 6 h of burn injury. Exclusion criteria: not stated. Randomised: 50 patients (Group 1: n = 25, Group 2: n = 25). Withdrawals: not stated. Age (years): (mean, range): Group 1: 25.2 (3‐58), Group 2: 26.4 (5‐60). Gender (male:female): Group 1: 14 (56%):11 (44%), Group 2: 13 (52%):12 (48%). Burned surface (% TBSA): (mean, range): Group 1: 14.5% (10‐38), Group 2: 15.6% (10.5‐40). Inhalation injury: not stated. Time post‐burn (h): ≤ 6 h. Burn type: Group 1: fire (flame) 23 (92%), scald (hot liquid or steam) 2 (8%), Group 2: fire 22 (88%), scald 3 (12%). Wounds infected at baseline?: no. Co‐morbidity: not described. |
|
| Interventions |
Type of antibiotic prophylaxis: topical antibiotic prophylaxis. Type of interventions: honey vs SSD. Group 1: honey (pure, unprocessed, undiluted, obtained from hives), applied topically daily and at the time of dressing. Group 2: SSD applied topically daily. Burns were washed with normal saline prior to the intervention. Duration of intervention: until wounds healed. Co‐interventions: not described. |
|
| Outcomes | Rates of wound healing (assessed clinically and histologically on days 7 and 21). Wound infection (bacterial cultures, biopsies and clinical assessment). Proportion of participants with completely healed wounds. |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "After the initial management, patients were allocated at random to two groups" (Page 157 trial report). Comment: insufficient information to make a judgement. |
| Allocation concealment (selection bias) | Unclear risk | No information provided. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | No information provided (probably an open trial due to the different nature of the interventions (honey versus SSD). The antibiotic was applied topically, and was obviously different to the intervention administered in the control group, so we assumed that the participants, personnel and outcome assessors were not blinded. |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Blinding of outcome assessors not reported, but probably not done because of the different nature of the interventions (honey versus SSD). |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Did not report number of withdrawals Comment: denominator values suggested complete follow‐ up. |
| Selective reporting (reporting bias) | Low risk | No protocol provided, but given the outcomes listed in the methods section, all pre‐specified outcomes were reported. |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
Tayade 2006.
| Methods |
Study design: prospective controlled trial. Setting/location: hospital and outpatient (Department of Surgery, Grant Medical College and Sir J.J. Group of Hospitals, Mumbai). Country: India Period of study: February 2002‐August 2004 (2.6 years). Unit of randomisation: patient. Unit of analysis: patient. Sample size calculation: no. Use of ITT analysis?: yes. |
|
| Participants |
Inclusion criteria: 1. Age: patients of any age. 2. Partial‐thickness burns. 3. Burns ≤ 15% TBSA. 4. Thermal burns or scalds. 5. Burns sustained up to 24 h prior to treatment. 6. Superficial burns not requiring any kind of graft. Exclusion criteria: not described. Randomised: 50 patients (Group 1: n = 25, Group 2: n = 25). Patients assessed: 50 patients (100%). Age (years): (range): 11‐30. Burned surface (% TBSA): < 10% (both groups). Inhalation injury: not stated. Time post‐burn (h): < 24 h (both groups). Burn type: Group 1: scald (hot liquid or steam) 9 (36%), fire (flame)16 (64%), Group 2: scald 10 (40%), fire 15 (60%). Wounds infected at baseline?: no. Co‐morbidity: not described. |
|
| Interventions |
Type of antibiotic prophylaxis: topical antibiotic prophylaxis. Type of interventions: SSD vs collagen sheet. Group 1: SSD 1%. Group 2: collagen sheet (Kollagen, enzymatically prepared from cattle skin), membrane was applied to the wound after thorough cleansing with chlorhexidine solution and thorough debridement of blisters. Both treatments were topically administered daily. Duration of intervention: until complete epithelization (mean:15.54 days). Co‐interventions: NSAIDs used as first line analgesics with intramuscular (im) pentazocine as second line. |
|
| Outcomes | Healing time (days). Burn wound infection (presence of pus and conversion to full‐thickness wounds). Adverse events (allergic or hypersensitivity reactions). |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "Each patient was then randomly allocated to either of the groups with subsequent application of collagen sheet or silver sulphadiazine respectively", "Hence the two groups were compatible with each other in respect of age, sex ratio and type of burns (Table 1)" (Page 2 trial report). Comment: insufficient information to make a judgement. |
| Allocation concealment (selection bias) | Unclear risk | No information provided. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Blinding of participants and key study personnel not reported, but probably not done due to the different nature of the interventions (SSD vs collagen sheet). |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Blinding of outcome assessors not reported, but probably not done due to the different nature of the interventions (SSD vs collagen sheet). |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Did not report number of withdrawals. Comment: denominator values suggested complete follow‐up. |
| Selective reporting (reporting bias) | Low risk | No protocol provided, but given the outcomes listed in the methods section, all pre‐specified outcomes were reported. |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
Abbreviations
< = less than ≤ = less than or equal to > = more than ≥ = more than or equal to Ag = silver h = hour(s) HBS = sickle cell anaemia HCV = hepatitis C virus HIV = human immunodeficiency virus ICU = intensive care unit im = intramuscular ITT = intention‐to‐treat analysis iv = intravascular LOS = length of hospital stay NSAIDs = non‐steroidal antiinflammatory drugs SMX = sulfamethoxazole SSD = silver sulphadiazine TBSA = total surface body area TMP = trimethoprim TMP‐SMX = trimethoprim‐sulfamethoxazole vs = versus
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Abdel‐Razek 2000 | Inappropriate comparison. All patients received the same antibiotic (used the same treatment with selective gastrointestinal decontamination in two arms). |
| Afilalo 1992 | Relevant data were not reported or available from the authors. |
| Ahuja 2009 | None of the review outcomes was assessed. |
| Baghel 2009 | Wounds were already infected before treatment. The study reported data on patients with positive cultures in the wound at the beginning of the study. |
| Branski 2008 | Inappropriate comparison (used the same antibiotic in both arms). |
| Carneiro 2002 | Wounds were already infected before treatment. The study reported data on patients with positive cultures in the wound at the beginning of the study. |
| Cason 1966 | Quasi‐randomised. |
| De Gracia 2001 | Inappropriate comparison (used the same antibiotic in both arms). |
| Deutsch 1990 | Quasi‐randomised. |
| Donati 1994 | Inappropriate intervention. All patients received the same antibiotic. An immunological treatment was the only one randomised. |
| Fang 1987 | Inappropriate comparison (used the same antibiotic in both arms). |
| Grippaudo 2010 | Even though the authors had mentioned in the abstract that the outcome of burn wound infection had been assessed, the study report did not include the data. We tried to contact the authors, but it was not possible to obtain data that could be used in our review |
| Huang 2006 | Preliminary report of Huang 2007 (see below). |
| Huang 2007 | Study assessed management of residual wounds postburn ‐ wounds were infected before treatment. |
| Hunter 1976 | Quasi‐randomised. |
| Inman 1984 | Inappropriate comparison (used the same antibiotic in both arms). |
| Li XL 2006 | Study assessed management of residual burn wounds. |
| Lowbury 1968 | Quasi‐randomised. |
| Malik 2010 | Quasi‐randomised. |
| Manuskiatti 1999 | Quasi‐randomised. |
| Mashhood 2006 | None of the review outcomes was assessed. |
| Miller 1990 | Inadequate comparison (used the same antibiotic in both arms). |
| Munster 1989 | Quasi‐randomised. |
| Oen 2012 | Inappropriate comparison (used the same antibiotic in both arms). |
| Ostlie 2012 | Inappropriate intervention. All patients received the same antibiotic (SSD) at the beginning of the study. Quote: "After the initial debridement on admission, all patients were dressed with SSD, which was used for the first 2 days of daily debridement. After 2 days, patients were then randomized to continue daily debridement with either SSD or CO for up to10 days" (Page 1205). |
| Piel 1985 | Even though the authors mentioned in the abstract that the outcome of burn wound infection had been assessed, the study report did not include the data. We tried to contact the authors, but it was not possible to obtain data that could be used in our review |
| Proctor 1971 | Quasi‐randomised. |
| Ramos 2008 | Wounds were already infected before treatment. The study reported data on patients with positive cultures in the wound at the beginning of the study. Quote: "The antibiotic regimen was chosen in accordance with the antibiogram of the bacteria isolated from the surveillance wound cultures done once a week. The group of patients with less than 4 days of hospital admission did not have surveillance wound cultures and Cephalothin iv was prescribed" (Page 918). |
| Steer 1997 | The analysis and reported data were not clear; authors reported 134 patients were randomised, but 86 patients who had been re‐intervened or who had undergone change of dressing were included in several analyses. The trial was excluded because of lack of independent information for those patients receiving antibiotic prophylaxis as the first intention treatment. We tried unsuccessfully to contact the study authors to obtain data on these results. |
| Subrahmanyam 1991 | The wounds were already infected before treatment. |
| Ugburo 2004 | The study did not provide information that could be used for our review. |
| Varas 2005 | None of the review outcomes was assessed. |
| Waffle 1988 | Quasi‐randomised. |
Characteristics of studies awaiting assessment [ordered by study ID]
Maghsoudi 2011.
| Methods |
Study design: prospective randomised trial. Setting/location: hospital (Sina Hospital, University of Medical Sciences of Tabriz). Country: Iran. Period of study: 20 March 2010‐20 March 2011 (1 year). Unit of randomisation: patient. Unit of analysis: patient. Sample size calculation: no. Use of ITT analysis?: yes. |
| Participants |
Inclusion criteria: 1. Patients admitted to the hospital. 2. Superficial thermal burns. 3. Burns ≤ 40% TBSA. Exclusion criteria: not stated. Randomised: 100 patients (Group 1: n = 50, Group 2: n = 50). Patients assessed: 100 (100%). Withdrawals: not stated. Age (years): (mean, range): Group 1: 26.4 (5‐70), Group 2: 25.2 (3‐68). Gender (male: female): Group 1: 25 (50%): 25 (50%), Group 2: 23 (46%): 27 (54%). Burned surface (% TBSA): (mean, range): Group 1: 15.6% (10.5‐40), Group 2: 14.5% (10‐40). Inhalation injury: not stated. Time post‐burn (h): not stated. Burn type: Group 1: fire (flame) 39 (78%), scald (hot liquid or steam) 11 (22%), Group 2: fire 43 (86%), scald 7 (14%). Wounds infected at baseline?: no. Co‐morbidity: not described. |
| Interventions |
Type of antibiotic prophylaxis: topical antibiotic prophylaxis. Type of interventions: mafenide acetate dressing vs honey dressing. Group 1: after being washed with normal saline, wounds were covered with gauze impregnated with mafenide acetate. These dressings were replaced daily. Group 2: honey was applied on alternate days and at the time of dressing. After spreading the honey, the wound was covered with dry, sterile gauze, and bandaged. Duration of intervention: until healing. Co‐interventions: not stated. |
| Outcomes | Wound infection (the definitive diagnosis of burn wound infection was made with biopsy of > 105 organisms/g of tissue). Time required for healing (days). Clinical evidence of wound healing. The organisms isolated in positive swab cultures. Side effects. LOS. |
| Notes | Awaiting classification while authors of the trial report respond to a request for further information from the review authors. |
Panahi 2012.
| Methods |
Study design: randomized and double‐blinded clinical trial. Setting/location: hospital (Baqiyatallah University of Medical Sciences of Tehran). Country: Iran. Period of study: not stated. Unit of randomisation: patient. Unit of analysis: patient. Sample size calculation: no. Use of ITT analysis?: yes. |
| Participants |
Inclusion criteria: 1. Second‐degree burns. 2. Burn type: thermal 3. Burns ≤ 5% TBSA. 4. Occurrence of burn in the preceding 48 hours. 5. Without presence of other injuries. 6. Good general physical and mental health. Exclusion criteria: 1. Presence of any renal, hepatic, endocrine, cardiovascular or cerebrovascular disease. 2. Pregnancy. 3. History of drug or alcohol abuse. 4. Use (oral or topical) of antibiotics, steroids or immunosuppressive drugs. Randomised: 120 patients (Group 1: n = 60, Group 2: n = 60). Excluded (post‐randomisation): Group 1: 5 (8.3%), Group 2: 4 (6.6%). Reason for exclusion: Quote: "From the initial 120 patients with superficial second‐degree burn that were recruited into the study, nine were excluded due to study protocol violation." (Page 274). Patients assessed: 111 (92.5%). Withdrawals: not stated. Age (years): (mean, range): Group 1: 37.4 (±12.7), Group 2: 33.6 (±13.4). Gender (male: female): Group 1: 30 (54.5%): 25 (45.5%), Group 2: 35 (62.5%): 21 (37.5%). Burned surface (% TBSA): (mean, range): Group 1: 2.38% (±1.42), Group 2: 2.48% (±1.45). Inhalation injury: not stated. Time post‐burn (h): < 48 h after injury (both groups). Burn agent: Group 1: fire (flame) 18 (32.7%), scald (hot liquid or steam) 33 (60%), contact (hot solids) 3 (5.5%), other (chemical substance) 3 (5.4%); Group 2: fire 22 (39.3%), scald (hot liquid or steam) 29 (51.8%), contact (hot solids) 2 (3.6%), other (chemical substance) 3 (5.4%). Wounds infected at baseline?: no. Co‐morbidity: no chemical substance. |
| Interventions |
Type of antibiotic prophylaxis: topical antibiotic prophylaxis. Type of interventions: SSD vs herbal cream. Group 1: topical SSD 1% cream once a day. Group 2: herbal cream once a day. The constituents of herbal cream were Aloe vera gel and essential oils from Lavandula stoechas and Pelargonium roseum. In both groups, following cleansing and debridement of burn wounds with antimicrobial solution, cream (5 g for each 10 cm2 of burn area) was applied on wounds using a sterile spatula. After application of the creams, sterile gauze was applied and wounds bandaged. Duration of intervention: until recovery. Co‐interventions: not stated. |
| Outcomes | Severity of pain. Frequency of skin dryness. Infection. |
| Notes | Awaiting classification while authors of the trial report respond to a request for further information from the review authors. |
Differences between protocol and review
In the protocol we defined the outcome 'antibiotic resistance' as "the clinical infection or colonization caused by bacteria resistant to one or more of the antibiotics included in the prophylactic regimen (proportion or rate of isolates of a specific pathogen)", however, due to the paucity of studies fulfilling the former definition, for the review we decided to widen the definition for this outcome to "the clinical infection or colonization caused by bacteria resistant to one or more antibiotics".
The ‘risk of bias’ tool was changed during preparation of the review to reflect the changes suggested in Chapter 8 of The Cochrane Handbook version 5.1.0 (Higgins 2011a).
In the protocol we planned to carry out analyses on an intention‐to‐treat basis (i.e. to include all participants randomised to each group in the analyses, irrespective of what happened subsequently), however, there were some studies that included participants whose outcomes were unknown. For these outcomes, we performed an ‘available case analysis’ as the main analysis, that is, data were analysed for every participant for whom the outcome was obtained. We explored the impact of this assumption by performing a worst case scenario sensitivity analysis (we assumed that missing participants experienced a negative outcome).
In the protocol we stated that: “cluster‐randomised trials will be combined with individually randomised trials in the same meta‐analysis only if unit of analysis errors are not detected. If the data analysis is determined to have been performed incorrectly and sufficient information is available, an ’approximately correct analysis’ will be performed for each cluster‐RCT. If it is not possible, the results of the study will be reported as point estimates of the intervention effect without presentation of any statistical analysis (P values) or confidence intervals and they will not be included in the meta‐analysis (Higgins 2011b).”
Contributions of authors
Leticia Barajas: conceived, designed, drafted and wrote the review; identified references for the review background; organised retrieval of papers; performed data extraction and management, statistical analysis and interpretation of results. Provided a methodological, clinical and policy perspective to the manuscript. Approved the final draft document. Jesús López Alcalde: provided support in designing, drafting and writing the review, organising retrieval of papers, and data extraction. Provided a methodological, clinical and policy perspective to the manuscript. Approved the final draft document. Ivan Solá: provided methodological support and comments to the manuscript, and approved the final document. Marta Roqué: provided methodological and statistical support to the review, helped interpret and write the results, commented on the document. Approved the final document. Xavier Bonfill: commented critically on the intellectual content of the review. Approved the final document.
Contributions of editorial base:
Nicky Cullum: edited the protocol; advised on methodology, interpretation and protocol content. Approved the final review prior to submission. Sally Bell‐Syer: co‐ordinated the editorial process. Advised on methodology, interpretation and content. Edited the review. Ruth Foxlee: designed the search strategy,edited the search methods section and ran the searches on the electronic databases.
Sources of support
Internal sources
Iberoamerican Cochrane Centre. IIB Sant Pau. Barcelona, Spain.
Department of Pediatrics, Obstetrics and Gynecology and Preventive Medicine. Universitat Autònoma de Barcelona, Spain.
External sources
Agencia de Calidad del Sistema Nacional de Salud. CIBERESP. Ministerio de Sanidad y Política Social, Spain.
Consejo Nacional de Ciencia y Tecnología (CONACYT), Mexico.
NIHR/Department of Health (England), (Cochrane Wounds Group), UK.
Declarations of interest
Leticia Andrea Barajas Nava: None known. Jesús López Alcalde: None known. Ivan Solà Arnau: None known. Marta Roqué: None known. Xavier Bonfill Cosp: None known.
New
References
References to studies included in this review
Alexander 1982 {published data only}
- Alexander JW, MacMillan BG, Law EJ, Krummel R. Prophylactic antibiotics as an adjunct for skin grafting in clean reconstructive surgery following burn injury. Journal of Trauma 1982;22(8):687‐90. [DOI] [PubMed] [Google Scholar]
Alexander 1984 {published data only}
- Alexander JW, MacMillan BG, Law EJ, Kern R. Lack of beneficial effects of restricted prophylactic antibiotics for debridement and/or grafting of seriously burned patients. Bulletin and Clinical Review of Burn Injuries 1984;1:20. [Google Scholar]
Ang 2001 {published data only}
- Ang ES, Lee ST, Gan CS, See P, Chan YH, Ng LH, et al. The role of alternative therapy in the management of partial thickness burns of the face ‐ experience with the use of moist exposed burn ointment (MEBO)compared with silver sulphadiazine. Annals of the Academy of Medicine 2002;29(1):7‐10. [PubMed] [Google Scholar]
- Ang ES, Lee ST, Gan CS, See PG, Chan YH, Ng LH, et al. Evaluating the role of alternative therapy in burn wound management: randomized trial comparing moist exposed burn ointment with conventional methods in the management of patients with second‐degree burns. Medscape General Medicine 2001;3(12):3‐18. [PubMed] [Google Scholar]
Barret 2000 {published data only}
- Barret JP, Dziewulski P, Ramzy PI, Wolf SE, Desai MH, Herndon DN. Biobrane versus 1% silver sulfadiazine in second‐degree pediatric burns. Plastic and Reconstructive Surgery 2000;105(1):62‐5. [DOI] [PubMed] [Google Scholar]
Barret 2001 {published data only}
- Barret JP, Jeschke MG, Herndon DN. Selective decontamination of the digestive tract in severely burned pediatric patients. Burns 2001;27(5):439‐45. [DOI] [PubMed] [Google Scholar]
Bugmann 1998 {published data only}
- Bugmann P, Taylor S, Gyger D, Lironi A, Genin B, Vunda A, et al. A silicone‐coated nylon dressing reduces healing time in burned paediatric patients in comparison with standard sulfadiazine treatment: a prospective randomized trial. Burns 1998;24(7):609‐12. [DOI] [PubMed] [Google Scholar]
Caruso 2006 {published data only}
- Caruso DM, Foster KN, Blome‐Eberwein SA, Twomey JA, Herndon DN, Luterman A, et al. Randomized clinical study of Hydrofiber dressing with silver or silver sulfadiazine in the management of partial‐thickness burns. Journal of Burn Care and Research 2006;27(3):298‐309. [DOI] [PubMed] [Google Scholar]
De La Cal 2005 {published data only}
- Cal MA, Cerdá E, García‐Hierro P, Saene HK, Gómez‐Santos D, Negro E, et al. Survival benefit in critically ill burned patients receiving selective decontamination of the digestive tract: a randomized, placebo‐controlled, double‐blind trial. Annals of Surgery 2005;241(3):424‐30. [DOI] [PMC free article] [PubMed] [Google Scholar]
Demling 1999 {published data only}
- Demling RH, DeSanti L. Management of partial thickness facial burns (comparison of topical antibiotics and bio‐engineered skin substitutes). Burns 1999;25(3):256‐61. [DOI] [PubMed] [Google Scholar]
Demling 2003 {published data only}
- Demling RH, DeSanti L. The potential benefits of a temporary bioactive skin substitute to treat partial‐thickness burns of the feet. Wounds: A Compendium of Clinical Research and Practice 2003;15(1):10‐5. [Google Scholar]
Desai 1991 {published data only}
- Desai MH, Rutan RL, Heggers JP, Alvarado MI, McElroy K, Henrdon DN. The role of gentamicin iontophoresis in the treatment of burned ears. Journal of Burn Care and Rehabilitation 1991;12(6):521‐4. [DOI] [PubMed] [Google Scholar]
Durtschi 1982 {published data only}
- Durtschi MB, Orgain C, Counts GW, Heimbach DM. A prospective study of prophylactic penicillin in acutely burned hospitalized patients. Journal of Trauma 1982;22(1):11‐4. [DOI] [PubMed] [Google Scholar]
Fisher 1968 {published data only}
- Fisher AJ, Morrison G, Riet Rle S. A blind trial of two aerosol sprays in the exposure treatment of burns. South African Medical Journal 1968;42(34):903‐5. [PubMed] [Google Scholar]
Gerding 1988 {published data only}
- Gerding RL, Imbembo AL, Fratianne RB. Biosynthetic skin substitute vs 1% silver sulfadiazine for treatment of inpatient partial‐thickness thermal burns. Journal of Trauma 1988;28(8):1265‐9. [DOI] [PubMed] [Google Scholar]
Gerding 1990 {published data only}
- Gerding RL, Emerman CL, Effron D, Lukens T, Imbembo AL, Fratianne RB. Outpatient management of partial‐thickness burns: Biobrane versus 1% silver sulfadiazine. Annals of Emergency Medicine 1990;19(2):121‐4. [DOI] [PubMed] [Google Scholar]
Glat 2009 {published data only}
- Glat PM, Kubat WD, Hsu JF, Copty T, Burkey BA, Davis W, et al. Randomized clinical study of SilvaSorb gel in comparison to Silvadene silver sulfadiazine cream in the management of partial‐thickness burns. Journal of Burn Care and Research 2009;30(2):262‐67. [DOI] [PubMed] [Google Scholar]
Gong 2009 {published data only}
- Gong Z‐H, Yao J, Ji J‐F, Yang J, Xiang T. Effect of ionic silver dressing combined with hydrogel on degree II burn wound healing [in Chinese]. Journal of Clinical Rehabilitative Tissue Engineering Research 2009;13(42):8373‐6. [Google Scholar]
Gotschall 1998 {published data only}
- Gotschall CS, Morrison MIS, Eichelberger MR. Prospective randomized study of the efficacy of Mepitel on children with partial‐thickness scalds. Journal of Burn Care and Rehabilitation 1998;19(4):279‐83. [DOI] [PubMed] [Google Scholar]
Hauser 2007 {published data only}
- Hauser J, Rossbach O, Langer S, Vogt P, Germann G, Steinau HU, et al. Local therapy of grade IIa burns: Efficacy and tolerability of a new hydrosome wound gel for the local treatment of grade IIa burns as compared with silver sulfadiazine ointment [Lokale Behandlung von Grad‐IIa‐Verbrennungen. Wirksamkeit und Verträglichkeit eines neuartigen hydrosomalen Wundgels im Vergleich zu Silbersulfadiazin‐Creme]. Unfallchirurg 2007;110(11):988‐94. [DOI] [PubMed] [Google Scholar]
Hosseini 2009 {published data only}
- Hosseini SN, Karimian A, Mousavinasab SN, Rahmanpour H, Yamini M, Zahmatkesh SH. Xenoderm versus 1% silver sulfadiazine in partial‐thickness burns. Asian Journal of Surgery 2009;32(4):234‐9. [DOI] [PubMed] [Google Scholar]
Khorasani 2009 {published data only}
- Khorasani G, Hosseinimehr SJ, Azadbakht M, Zamani A, Mahdavi MR. Aloe versus silver sulfadiazine creams for second‐degree burns: A randomized controlled study. Surgery Today 2009;39(7):587‐91. [DOI] [PubMed] [Google Scholar]
Kimura 1998 {published data only}
- Kimura A, Mochizuki T, Nishizawa K, Mashiko K, Yamamoto Y, Otsuka T. Trimethoprim‐sulfamethoxazole for the prevention of methicillin‐resistant Staphylococcus aureus pneumonia in severely burned patients. Journal of Trauma 1998;45(2):383‐7. [DOI] [PubMed] [Google Scholar]
Levine 1978 {published data only}
- Levine BA, Petroff PA, Slade CL, Pruitt BA Jr. Prospective trials of dexamethasone and aerosolized gentamicin in the treatment of inhalation injury in the burned patient. Journal of Trauma 1978;18(3):188‐93. [DOI] [PubMed] [Google Scholar]
Livingston 1990 {published data only}
- Livingston DH, Cryer HG, Miller FB, Malangoni MA, Polk HC Jr, Weiner LJ. A randomized prospective study of topical antimicrobial agents on skin grafts after thermal injury. Plastic and Reconstructive Surgery 1990;86(6):1059‐64; discussion 1065. [PubMed] [Google Scholar]
Maya 1986 {published data only}
- Maya BJ, Covarrubias BA, Romero PA, Licona VL, Claesson BE, Filipsson S, et al. Spray rifamycin and amniotic membranes in second degree burns [Rifamicina spray y membranas amnióticas en quemaduras de segundo grado]. Compendium de Investigaciones Clinicas Latinoamericanas 1986;6(2):73‐80. [Google Scholar]
Miller 1987 {published data only}
- Miller LM, Carboll WB, Hansbrough JF. The effect of antimicrobial prophylaxis for burn wound excision: Ceforanide versus cefazolin. Current Therapeutic Research, Clinical and Experimental 1987;41(6):946‐51. [Google Scholar]
Mohammadi 2009 {published data only}
- Mohammadi AA, Riazi H, Hasheminasab MJ, Sabet B, Mohammadi MK, Abbasi S, et al. Amniotic membrane dressing vs conventional topical antibiotic dressing in hospitalized burn patients. Iranian Red Crescent Medical Journal 2009;11(1):66‐70. [Google Scholar]
Moharamzad 2010 {published data only}
- Moharamzad Y, Panahi Y, Beiraghdar F, Feizi I. Focused Conference Group: P16 ‐ Natural products: Past and future? Clinical efficacy of a topical herbal cream (Aloe vera, Geranium robertianum and Lavandula stoechas) in second‐degree burn wounds. Basic and Clinical Pharmacology and Toxicology 2010;107(Suppl. 1):162–69. [Google Scholar]
Muangman 2006 {published data only}
- Muangman P, Chuntrasakul C, Silthram S, Suvanchote S, Benjathanung R, Kittidacha S, et al. Comparison of efficacy of 1% silver sulfadiazine and Acticoat for treatment of partial‐thickness burn wounds. Journal of The Medical Association of Thailand 2006;89(7):953‐8. [PubMed] [Google Scholar]
Munster 1986 {published data only}
- Munster AM, Winchurch RA, Thupari JN, Ernst CB. Reversal of postburn immunosuppression with low‐dose polymyxin B. Journal of Trauma 1986;26(11):995‐8. [DOI] [PubMed] [Google Scholar]
Noordenbos 1999 {published data only}
- Noordenbos J, Dore C, Hansbrough JF. Safety and efficacy of TransCyte for the treatment of partial‐thickness burns. Journal of Burn Care and Rehabilitation 1999;20(4):275‐81. [DOI] [PubMed] [Google Scholar]
Rodgers 1997 {published data only}
- Rodgers GL, Fisher MC, Lo A, Cresswell A, Long SS. Study of antibiotic prophylaxis during burn wound debridement in children. Journal of Burn Care and Rehabilitation 1997;18(4):342‐6. [DOI] [PubMed] [Google Scholar]
Silver 2007 {published data only}
- Silver GM, Robertson SW, Halerz MM, Conrad P, Supple KG, Gamelli RL. A silver‐coated antimicrobial barrier dressing used postoperatively on meshed autografts: a dressing comparison study. Journal of Burn Care and Research 2007;28(5):715‐9. [DOI] [PubMed] [Google Scholar]
Soroff 1994 {published data only}
- Soroff HS, Sasvary DH. Collagenase ointment and polymyxin B sulfate/bacitracin spray versus silver sulfadiazine cream in partial‐thickness burns: a pilot study. Journal of Burn Care and Rehabilitation 1994;15(1):13‐7. [DOI] [PubMed] [Google Scholar]
Subrahmanyam 1998 {published data only}
- Subrahmanyam M. A prospective randomised clinical and histological study of superficial burn wound healing with honey and silver sulfadiazine. Burns 1998;24(2):157‐61. [DOI] [PubMed] [Google Scholar]
Tayade 2006 {published data only}
- Tayade MB, Bakhshi GD, Haobijam N. A comparative study of collagen sheet cover versus 1% silver sulphadiazine in partial thickness burns. Bombay Hospital Journal 2006;48(2):http://www.bhj.org/journal/2006_4802_april/html/org_res_263‐267.html. [Google Scholar]
References to studies excluded from this review
Abdel‐Razek 2000 {published data only}
- Abdel‐Razek SM, Abdel‐Khalek AH, Allam AM, Shalaby H, Mandoor S, Higazi M. Impact of selective gastrointestinal decontamination on mortality and morbidity in severely burned patients. Annals of Burns and Fire Disasters 2000;XIII(4):1‐5. [Google Scholar]
Afilalo 1992 {published data only}
- Afilalo M, Dankoff J, Guttman A, Lloyd J. DuoDERM hydroactive dressing versus silver sulphadiazine/Bactigras in the emergency treatment of partial skin thickness burns. Burns 1992;18(4):313‐6. [DOI] [PubMed] [Google Scholar]
Ahuja 2009 {published data only}
- Ahuja RB, Gupta A, Gur R. A prospective double‐blinded comparative analysis of framycetin and silver sulphadiazine as topical agents for burns: A pilot study. Burns 2009;35(5):672‐6. [DOI] [PubMed] [Google Scholar]
Baghel 2009 {published data only}
- Baghel PS, Shukla S, Mathur RK, Randa R. A comparative study to evaluate the effect of honey dressing and silver sulfadiazene dressing on wound healing in burn patients. Indian Journal of Plastic Surgery 2009;42(2):176‐81. [DOI] [PMC free article] [PubMed] [Google Scholar]
Branski 2008 {published data only}
- Branski LK, Herndon DN, Celis MM, Norbury WB, Masters OE, Jeschke MG. Amnion in the treatment of pediatric partial‐thickness facial burns. Burns 2008;34(3):393‐9. [DOI] [PubMed] [Google Scholar]
Carneiro 2002 {published data only}
- Carneiro PM, Rwanyuma LR, Mkony CA. A comparison of topical Phenytoin with Silverex in the treatment of superficial dermal burn wounds. Central African Journal of Medicine 2002;48(9‐10):105‐8. [PubMed] [Google Scholar]
Cason 1966 {published data only}
- Cason JS, Jackson DM, Lowbury EJL, Ricketts CR. Antiseptic and aseptic prophylaxis for burns: use of silver nitrate and of isolators. British Medical Journal 1966;2(5525):1288‐94. [DOI] [PMC free article] [PubMed] [Google Scholar]
De Gracia 2001 {published data only}
- Gracia CG. An open study comparing topical silver sulfadiazine and topical silver sulfadiazine‐cerium nitrate in the treatment of moderate and severe burns. Burns 2001;27(1):67‐74. [DOI] [PubMed] [Google Scholar]
Deutsch 1990 {published data only}
- Deutsch DH, Miller SF, Finley RK. The use of intestinal antibiotics to delay or prevent infections in patients with burns. Journal of Burn Care and Rehabilitation 1990;11(5):436‐42. [DOI] [PubMed] [Google Scholar]
Donati 1994 {published data only}
- Donati L, Periti P. Antibiotic treatment of burned patients: an Italian multicentre study. Intensive Care Medicine 1994;20(Suppl 4):S30‐4. [DOI] [PubMed] [Google Scholar]
Fang 1987 {published data only}
- Fang CH, Nathan P, Robb EC, Alexander JW, MacMillan BG. Prospective clinical study of Hydron, a synthetic dressing, in delivery of an antimicrobial drug to second‐degree burns. Journal of Burn Care and Rehabilitation 1987;8(3):206‐9. [DOI] [PubMed] [Google Scholar]
Grippaudo 2010 {published data only}
- Grippaudo FR, Carini L, Baldini R. Procutase versus 1% silver sulphadiazine in the treatment of minor burns. Burns 2010;36(6):871‐5. [DOI] [PubMed] [Google Scholar]
Huang 2006 {published data only}
- Huang YS, Li XL, Liao ZJ, Zhang GA, Liu Q, Tang J, et al. Multi‐center clinical study of acticoat (nanocrystalline silver dressing) for the management of residual burn wounds: Summary report. Zhonghua Shao Shang Za Zhi 2006;22(1):15‐8. [PubMed] [Google Scholar]
Huang 2007 {published data only}
- Huang Y, Li X, Liao Z, Zhang G, Liu Q, Tang J, et al. A randomized comparative trial between Acticoat and SD‐Ag in the treatment of residual burn wounds, including safety analysis. Burns 2007;33(2):161‐6. [DOI] [PubMed] [Google Scholar]
Hunter 1976 {published data only}
- Hunter GR, Chang FC. Outpatient burns: a prospective study. Journal of Trauma 1976;16(3):191‐5. [PubMed] [Google Scholar]
Inman 1984 {published data only}
- Inman RJ, Snelling CF, Roberts FJ, Shaw K, Boyle JC. Prospective comparison of silver sulfadiazine 1 per cent plus chlorhexidine digluconate 0.2 per cent (Silvazine) and silver sulfadiazine 1 per cent (Flamazine) as prophylaxis against burn wound infection. Burns, Including Thermal Injury 1984;11(1):35‐40. [DOI] [PubMed] [Google Scholar]
Li XL 2006 {published data only}
- Li XL, Huang YS, Peng YZ, Liao ZJ, Zhang GA, Liu Q, et al. Multi‐center clinical study of acticoat (nanocrystalline silver dressing) for the management of residual burn wounds. Zhonghua Shao Shang Za Zhi 2006;22(1):15‐8. [PubMed] [Google Scholar]
Lowbury 1968 {published data only}
- Lowbury EJ, Jackson DM. Local chemoprophylaxis for burns with gentamicin and other agents. Lancet 1968;1(7544):654‐7. [DOI] [PubMed] [Google Scholar]
Malik 2010 {published data only}
- Malik KI, Malik MA, Aslam A. Honey compared with silver sulphadiazine in the treatment of superficial partial‐thickness burns. International Wound Journal 2010;7(5):413‐7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Manuskiatti 1999 {published data only}
- Manuskiatti W, Fitzpatrick RE, Goldman MP, Krejci‐Papa N. Prophylactic antibiotics in patients undergoing laser resurfacing of the skin. Journal of the American Academy of Dermatology 1999;40(1):77‐84. [DOI] [PubMed] [Google Scholar]
Mashhood 2006 {published data only}
- Mashhood AA, Khan TA, Sami AN. Honey compared with 1% silver sulfadiazine cream in the treatment of superficial and partial thickness burns. Journal of Pakistan Association of Dermatologists 2006;16(1):14‐9. [Google Scholar]
Miller 1990 {published data only}
- Miller L, Hansbrough J, Slater H, Goldfarb IW, Kealey P, Saffle J, et al. Sildimac: a new delivery system for silver sulfadiazine in the treatment of full‐thickness burn injuries. Journal of Burn Care and Rehabilitation 1990;11(1):35‐41. [DOI] [PubMed] [Google Scholar]
Munster 1989 {published data only}
- Munster AM, Xiao GX, Guo Y, Wong LA, Winchurch RA. Control of endotoxemia in burn patients by use of polymyxin B. Journal of Burn Care and Rehabilitation 1989;10(4):327‐30. [DOI] [PubMed] [Google Scholar]
Oen 2012 {published data only}
- Oen IM, van Baar ME, Middelkoop E, Nieuwenhuis MK. Facial Burns Group. Effectiveness of cerium nitrate‐silver sulfadiazine in the treatment of facial burns: a multicenter, randomized,controlled trial. Plastic and Reconstructive Surgery 2012;130(2):274e‐283e. [DOI] [PubMed] [Google Scholar]
Ostlie 2012 {published data only}
- Ostlie DJ, Juang D, Aguayo P, Pettiford‐Cunningham JP, Erkmann EA, Rash DE, et al. Topical silver sulfadiazine vs collagenase ointment for the treatment of partial thickness burns in children: a prospective randomized trial. Journal of Pediatric Surgery 2012;47(6):1204‐7. [DOI] [PubMed] [Google Scholar]
Piel 1985 {published data only}
- Piel P, Scarnati S, Goldfarb IW, Slater H. Antibiotic prophylaxis in patients undergoing burn wound excision. Journal of Burn Care and Rehabilitation 1985;6(5):422‐4. [DOI] [PubMed] [Google Scholar]
Proctor 1971 {published data only}
- Proctor DS. The treatment of burns: a comparative trial of antibiotic dressings. South African Medical Journal 1971;45(2):231‐6. [PubMed] [Google Scholar]
Ramos 2008 {published data only}
- Ramos G, Resta M, Machare Delgado E, Durlach R, Fernandez Canigia L, Benaim F. Systemic perioperative antibiotic prophylaxis may improve skin autograft survival in patients with acute burns. Journal of Burn Care and Research 2008;29(6):917‐23. [DOI] [PubMed] [Google Scholar]
Steer 1997 {published data only}
- Steer JA, Papini RP, Wilson AP, McGrouther DA, Nakhla LS, Parkhouse N. Randomized placebo‐controlled trial of teicoplanin in the antibiotic prophylaxis of infection following manipulation of burn wounds. British Journal of Surgery 1997;84(6):848‐53. [PubMed] [Google Scholar]
Subrahmanyam 1991 {published data only}
- Subrahmanyam M. Topical application of honey in treatment of burns. British Journal of Surgery 1991;78(4):497‐8. [DOI] [PubMed] [Google Scholar]
Ugburo 2004 {published data only}
- Ugburo AO, Atoyebi OA, Oyeneyin JO, Sowemimo GO. An evaluation of the role of systemic antibiotic prophylaxis in the control of burn wound infection at the Lagos University Teaching Hospital. Burns 2004;30(1):43‐8. [DOI] [PubMed] [Google Scholar]
Varas 2005 {published data only}
- Varas RP, O'Keeffe T, Namias N, Pizano LR, Quintana OD, Herrero Tellachea M, et al. A prospective, randomized trial of Acticoat versus silver sulfadiazine in the treatment of partial‐thickness burns: which method is less painful?. Journal of Burn Care and Rehabilitation 2005;26(4):344‐7. [DOI] [PubMed] [Google Scholar]
Waffle 1988 {published data only}
- Waffle C, Simon RR, Joslin C. Moisture‐vapour‐permeable film as an outpatient burn dressing. Burns, Including Thermal Injury 1988;14(1):66‐70. [DOI] [PubMed] [Google Scholar]
References to studies awaiting assessment
Maghsoudi 2011 {published data only}
- Maghsoudi H, Salehi F, Khosrowshahi MK, Baghaei M, Nasirzadeh M, Shams R. Comparison between topical honey and mafenide acetate in treatment of burn wounds. Annals of Burns and Fire Disasters 2011;24(3):132‐7. [PMC free article] [PubMed] [Google Scholar]
Panahi 2012 {published data only}
- Panahi Y, Beiraghdar F, Akbari H, Bekhradi H, Taghizadeh M, Sahebkar A. A herbal cream consisting of Aloe vera, Lavandulastoechas, and Pelargonium roseum as an alternative for silver sulfadiazine in burn management. Asian Biomedicine 2012;6(2):273‐8. [Google Scholar]
Additional references
Alexander 2009
- Alexander JW. When should prophylactic antibiotics be given?. Annals of Surgery 2009;249(4):557‐8. [DOI] [PubMed] [Google Scholar]
Alp 2012
- Alp E, Coruh A, Gunay GK, Yontar Y, Doganay M. Risk factors for nosocomial infection and mortality in burn patients: 10 years of experience at a university hospital. Journal of Burn Care and Research 2012;33(3):379‐85. [DOI] [PubMed] [Google Scholar]
Alsbjörn 2007
- Alsbjörn B, Gilbert P, Hartmann B, Kaźmierski M, Monstrey S, Palao R, et al. Guidelines for the management of partial‐thickness burns in a general hospital or community setting recommendations of a European working party. Burns 2007;33(2):155‐60. [DOI] [PubMed] [Google Scholar]
Altoparlak 2004
- Altoparlak U, Erol S, Akcay MN, Celebi F, Kadanali A. The time‐related changes of antimicrobial resistance patterns and predominant bacterial profiles of burn wounds and body flora of burned patients. Burns 2004;30(7):660‐4. [DOI] [PubMed] [Google Scholar]
Ansermino 2004
- Ansermino M, Hemsley C. Intensive care management and control of infection. BMJ 2004;329(7459):220‐3. [DOI] [PMC free article] [PubMed] [Google Scholar]
Atoyebi 1992
- Atoyebi OA, Sowemimo GO, Odugbemi T. Bacterial flora of burn wounds in Lagos, Nigeria: a prospective study. Burns 1992;18(6):448‐51. [DOI] [PubMed] [Google Scholar]
Avni 2010
- Avni T, Levcovich A, Ad‐El DD, Leibovici L, Paul M. Prophylactic antibiotics for burns patients: systematic review and meta‐analysis. BMJ 2010;340:c241. [DOI] [PMC free article] [PubMed] [Google Scholar]
Bang 2002
- Bang RL, Sharma PN, Sanyal SC, Al Naijadah I. Septicaemia after burn injury: a comparative study. Burns 2002;28(8):746‐51. [DOI] [PubMed] [Google Scholar]
Boutron 2008
- Boutron I, Moher D, Altman DG, Schulz KF, Ravaud P, CONSORT Group. Extending the CONSORT statement to randomized trials of nonpharmacologic treatment: explanation and elaboration. Annals of Internal Medicine 2008;148(4):295‐309. [DOI] [PubMed] [Google Scholar]
Brychta 2011
- Brychta P, Magnette A. . European Practice Guidelines for Burn Care. The Netherlands, 2011.
Church 2006
- Church D, Elsayed S, Reid O, Winston B, Lindsay R. Burn wound infections. Clinical Microbiology Reviews 2006;19(2):403‐34. [DOI] [PMC free article] [PubMed] [Google Scholar]
Curtin 2002
- Curtin F, Elbourne D, Altman DG. Meta‐analysis combining parallel and cross‐over clinical trials. II: Binary outcomes. Statistics in Medicine 2002;21(5):2145‐59. [DOI] [PubMed] [Google Scholar]
De Macedo 2005
- Macedo JL, Santos JB. Bacterial and fungal colonization of burn wounds. Memórias do Instituto Oswaldo Cruz 2005;100(5):535‐9. [DOI] [PubMed] [Google Scholar]
Deeks 2011
- Deeks JJ, Higgins JPT, Altman DG (editors). Chapter 9: Analysing data and undertaking meta‐analyses. In: Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 [updated March 2011]. The Cochrane Collaboration, 2011. Available from www.cochrane‐handbook.org.
DeSanti 2005
- DeSanti L. Pathophysiology and current management of burn injury. Advances in Skin and Wound Care 2005;18(6):323‐32; quiz 332‐4. [DOI] [PubMed] [Google Scholar]
Edwards 2004
- Edwards R, Harding KG. Bacteria and wound healing. Current Opinion in Infectious Diseases 2004;17(2):91‐6. [DOI] [PubMed] [Google Scholar]
Ergün 2004
- Ergün O, Celik A, Ergun G, Ozok G. Prophylactic antibiotic use in pediatric burn units. European Journal of Pediatric Surgery 2004;14(6):422‐6. [DOI] [PubMed] [Google Scholar]
Erol 2004
- Erol S, Altoparlak U, Akcay MN, Celebi F, Parlak M. Changes of microbial flora and wound colonization in burned patients. Burns 2004;30(4):357‐61. [DOI] [PubMed] [Google Scholar]
Haq 1990
- Haq A. Pattern of burn injuries at a Kenyan provincial hospital. Burns 1990;16(3):185‐9. [DOI] [PubMed] [Google Scholar]
Higgins 2003
- Higgins JP, Thompson SG, Deeks JJ, Altman DG. Measuring inconsistency in meta‐analyses. BMJ 2003;327(7414):557‐60. [DOI] [PMC free article] [PubMed] [Google Scholar]
Higgins 2011a
- Higgins JPT, Altman DG, Sterne JAC (editors). Chapter 8: Assessing risk of bias in included studies. In: Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 [updated March 2011]. The Cochrane Collaboration, 2011. Available from www.cochrane‐handbook.org.
Higgins 2011b
- Higgins JPT, Deeks JJ, Altman DG (editors). Chapter 16: Special topics in statistics. In: Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from www.cochrane‐handbook.org.
Hoogewerf 2013
- Hoogewerf CJ, Baar ME, Hop MJ, Nieuwenhuis MK, Oen IMMH, Middelkoop E. Topical treatment for facial burns. Cochrane Database of Systematic Reviews 2013, Issue 1. [DOI: 10.1002/14651858.CD008058.pub2] [DOI] [PubMed] [Google Scholar]
Hospenthal 2011
- Hospenthal DR, Murray CK, Andersen RC, Bell RB, Calhoun JH, Cancio LC, et al. Guidelines for the prevention of infections associated with combat‐related injuries: 2011 update: endorsed by the Infectious Diseases Society of America and the Surgical Infection Society. Journal of Trauma 2011;71(Suppl 2):S210‐34. [DOI] [PubMed] [Google Scholar]
Hyder 2009
- Hyder AA, Peden M, Krug E. Child health must include injury prevention. Lancet 2009;373(9658):102‐3. [DOI] [PubMed] [Google Scholar]
Kumar 2006
- Kumar P. Prophylactic antibiotics in burns. Burns 2006;32(5):655‐6. [DOI] [PubMed] [Google Scholar]
Latarjet 1995
- Latarjet J. A simple guide to burn treatment. International Society for Burn Injuries in collaboration with the World Health Organization. Burns 1995;21(3):221‐5. [DOI] [PubMed] [Google Scholar]
Latenser 2007
- Latenser BA, Miller SF, Bessey PQ, Browning SM, Caruso DM, Gomez M, et al. National Burn Repository 2006: a ten‐year review. Journal of Burn Care and Research 2007;28(5):635‐58. [DOI] [PubMed] [Google Scholar]
Lee 2009
- Lee F, Wong P, Hill F, Burgner D, Taylor R. Evidence behind the WHO guidelines: Hospital care for children: What is the role of prophylactic antibiotics in the management of burns?. Journal of Tropical Pediatrics 2009;55(2):73‐7. [DOI] [PubMed] [Google Scholar]
Lefebvre 2011
- Lefebvre C, Manheimer E, Glanville J. Chapter 6: Searching for studies. In: Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from www.cochrane‐handbook.org.
Louis 1984
- Louis TA, Lavori PW, Bailar JC, Polansky M. Crossover and self‐controlled designs in clinical research. New England Journal of Medicine 1984;310:24‐31. [DOI] [PubMed] [Google Scholar]
Mathers 2003
- Mathers CD, Bernard C, Iburg KM, Inoue M, Ma Fat D, Shibuya K, et al. Global burden of disease in 2002: data sources, methods and results. World Health Organization. Geneva, 2003.
Mayhall 2003
- Mayahall CG. The epidemiology of burn wound infections: then and now. Clinical Infectious Diseases 2003;37(4):543‐50. [DOI] [PubMed] [Google Scholar]
Merriam‐Webster 2012
- Merriam‐Webster's Collegiate® Dictionary, Eleventh Edition. Available from www.merriam‐webster.com/ (accessed 2 May 2012).
Miles 2005
- Miles F, Voss L, Segedin E, Anderson BJ. Review of Staphylococcus aureus infections requiring admission to a paediatric intensive care unit. Archives of Disease in Childhood 2005;90(12):1274‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Mills 2009
- Mills EJ, Chan AW, Wu P, Vail A, Guyatt GH, Altman DG. Design, analysis, and presentation of crossover trials. Trials 2009;10:27. [DOI] [PMC free article] [PubMed] [Google Scholar]
Miniño 2006
- Miniño AM, Anderson RN, Fingerhut LA, Boudreault MA, Warner M. Deaths: injuries, 2002. National Vital Statistics Reports 2006;54(10):1‐124. [PubMed] [Google Scholar]
Mock 2008
- Mock C, Peck M, Peden M, Krug E, Ahuja R, Albertyn H, et al. A WHO plan for burn prevention and care. World Health Organization. Geneva, 2008.
Moher 2001
- Moher D, Schulz KF, Altman D, CONSORT Group (Consolidated Standards of Reporting Trials). The CONSORT statement: revised recommendations for improving the quality of reports of parallel‐group randomized trials. JAMA 2001;285(15):1987‐91. [DOI] [PubMed] [Google Scholar]
Munster 1984
- Munster AM. Immunologic response of trauma and burns. An overview. American Journal of Medicine 1984;76(3A):142‐5. [DOI] [PubMed] [Google Scholar]
Murphy 2003
- Murphy KD, Lee JO, Herndon DN. Current pharmacotherapy for the treatment of severe burns. Expert Opinion on Pharmacotherapy 2003;4(3):369‐84. [DOI] [PubMed] [Google Scholar]
Murray 2008
- Murray C, Hospenthal DR. Burn Wound Infections. eMedicine Infectious Diseases (http://emedicine.medscape.com/article/213595‐overview) 2008.
NSW Severe Burn Injury Service 2008
- NSW Severe Burn Injury Service (SBIS). Clinical Practice Guidelines Burn Wound Management. New South Wales, 2008.
NZGG 2007
- New Zealand Guidelines Group (NZGG). Management of burns and scalds in primary care. Wellington, NZ., 2007.
Onuba 1987
- Onuba O, Udoidiok E. Hospital management of massive burns in the developing countries. Burns, Including Thermal Injury 1987;13(5):386‐90. [DOI] [PubMed] [Google Scholar]
Peden 2002
- Peden M, McGee K, Sharma G. The injury chart book: a graphical overview of the global burden of injuries. World Health Organization, 2002. [Google Scholar]
Peden 2008
- Peden M, Oyegbite K, Ozanne‐Smith J, Hyder AA, Branche C, Fazlur Rahman AKM, et al. World report on child injury prevention. World Health Organization. Geneva, 2008. [PubMed]
Polavarapu 2008
- Polavarapu N, Ogilvie MP, Panthaki ZJ. Microbiology of burn wound infections. Journal of Craniofacial Surgery 2008;19(4):899‐902. [DOI] [PubMed] [Google Scholar]
Pruitt 1998
- Pruitt BA Jr, McManus AT, Kim SH, Goodwin CW. Burn wound infections: current status. World Journal of Surgery 1998;22(2):135‐45. [DOI] [PubMed] [Google Scholar]
RevMan 2011 [Computer program]
- The Nordic Cochrane Centre, The Cochrane Collaboration. Review Manager (RevMan) Version 5.1.. Copenhagen, Denmark: The Nordic Cochrane Centre, The Cochrane Collaboration, 22 March 2011.
Rosanova 2012
- Rosanova MT, Stamboulian D, Lede R. Systematic review: which topical agent is more efficacious in the prevention of infections in burn patients? [Revisión sistemática: ¿Cuál es el agente tópicomás eficaz en la prevención de infeccionesen el paciente quemado?]. Archivos Argentinos de Pediatría 2012;110(4):298‐303. [DOI] [PubMed] [Google Scholar]
Schulz 2010
- Schulz KF, Altman DG, Moher D, CONSORT Group. CONSORT 2010 statement: updated guidelines for reporting parallel group randomized trials. Annals of Internal Medicine 2010;152(11):726‐32. [DOI] [PubMed] [Google Scholar]
SEMPSPH 2008
- Sociedad Española de Medicina Preventiva, Salud Pública e Higiene (SEMPSPH). Estudio de prevalencia de las infecciones nosocomiales en España EPINE 1990‐2007: 18 años. Madrid, 2012:59 p.
Sharma 2007
- Sharma BR. Infection in patients with severe burns: causes and prevention thereof. Infectious Disease Clinics of North America 2007;21(3):745‐59, ix. [DOI] [PubMed] [Google Scholar]
Sheridan 2005
- Sheridan RL. Sepsis in pediatric burn patients. Pediatric Critical Care Medicine 2005;6(3 Suppl):S112‐9. [DOI] [PubMed] [Google Scholar]
SIGN 2011
- Scottish Intercollegiate Guidelines Network (SIGN). Search filters. http://www.sign.ac.uk/methodology/filters.html#random (accessed 8 June 2011).
Singer 2002
- Singer AJ, McClain SA. Persistent wound infection delays epidermal maturation and increases scarring in thermal burns. Wound Repair and Regeneration 2002;10(6):372‐7. [DOI] [PubMed] [Google Scholar]
Soares 2006
- Soares de Macedo JL, Santos JB. Nosocomial infections in a Brazilian burn unit. Burns 2006;32(4):477‐81. [DOI] [PubMed] [Google Scholar]
Sterne 2011
- Sterne JAC, Egger M, Moher D (editors). Chapter 10: Addressing reporting biases. In: Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Intervention. Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from www.cochrane‐handbook.org..
Still 2002
- Still J, Law E, Friedman B, Newton T, Wilson J. Clostridium difficile diarrhea on a burn unit. Burns 2002;28(4):398‐9. [DOI] [PubMed] [Google Scholar]
Wasiak 2008
- Wasiak J, Cleland H, Campbell F. Dressings for superficial and partial thickness burns. Cochrane Database of Systematic Reviews 2008, Issue 4. [DOI: 10.1002/14651858.CD002106.pub3] [DOI] [PubMed] [Google Scholar]
Weber 2002
- Weber JM, Sheridan RL, Schulz JT, Tompkins RG, Ryan CM. Effectiveness of bacteria controlled nursing units in preventing cross‐colonization with resistant bacteria in severely burned children. Infection Control and Hospital Epidemiology 2002;23(9):549‐51. [DOI] [PubMed] [Google Scholar]
Weber 2004
- Weber J, McManus A. Infection control in burn patients. Burns 2004;30(8):A16‐24. [DOI] [PubMed] [Google Scholar]
WHO 2006
- Facts about injuries: burns. World Health Organization and International Society for Burns Injuries. Geneva, 2006.
WLDI 2008
- Work Loss Data Institute (WLDI). Burns. Corpus Christi, Texas, 2008.
Wurtz 1995
- Wurtz R, Karajovic M, Dacumos E, Jovanovic B, Hanumadass M. Nososcomial Infections in a burns intensive care unit. Burns 1995;21(3):181‐4. [DOI] [PubMed] [Google Scholar]