

Abstract
Fentanyl, and the numerous drugs derived from it, are contributing to the opioid overdose epidemic currently underway in the United States. To identify human exposure to these growing public health threats, an LC-MS/MS method for 5 μL dried blood spots was developed. This method was developed to detect exposure to 3-methylfentanyl, alfentanil, α-methylfentanyl, carfentanil, fentanyl, lofentanil, sufentanil, norcarfentanil, norfentanyl, norlofentanil, norsufentanil, and using a separate LC-MS/MS injection, cyclopropylfentanyl, acrylfentanyl, 2-furanylfentanyl, isobutyrylfentanyl, ocfentanil, and methoxyacetylfentanyl. Preparation of materials into groups of compounds was used to accommodate an ever increasing need to incorporate newly identified fentanyls. This protocol was validated within a linear range of 1.00–100 ng/mL, with precision ≤12% CV and accuracy ≥93%, as reported for the pooled blood quality control samples, and limits of detection as low as 0.10 ng/mL. The use of dried blood spots to assess fentanyl analogue exposures can facilitate rapid sample collection, transport, and preparation for analysis that could enhance surveillance and response efforts in the ongoing opioid overdose epidemic.
Introduction
Fentanyl was the first in a family of synthetic narcotic analgesics. Modification of the 4-N-anilinopiperidine core has yielded many novel analogues that retain or improve upon the original potency and rapid onset of fentanyl (1). To date over 30 analogues have been produced, with some used in surgical anesthesia, palliative care, and chronic pain management. Fentanyl derived compounds, both produced illicitly and obtained diversionally, have also been used as street drugs and chemical weapons both in the United States and Europe (2–4). Due to high potency and potential for addiction, a wide array of fentanyl-derived compounds have been mixed with heroin (5) and cocaine (6) or sold as counterfeit medications (7, 8). This distribution has resulted in an alarming 426% increase in drug products containing fentanyl as determined by US law enforcement and a 79% increase in synthetic opioid overdose deaths from 2013 to 2014 (4).
To better respond to the ongoing opioid overdose epidemic, identification of fentanyl-related exposures, including overdoses and deaths, is needed for surveillance to inform prevention and response efforts. This need was recently highlighted in recent exposures to novel fentanyl analogues (9, 10). Confirmation of exposure is best provided through the analysis of both fentanyl-derived compounds and their corresponding metabolites. Measurement of metabolites can extend the time window for detection, up to 96 hours post exposure, and often increases positive laboratory identification of genuine exposures (11, 12). However, exclusively monitoring fentanyl metabolites does not conclusively determine the causative parent drug, as many fentanyl analogues share the same metabolite (e.g. sufentanil and alfentanil). In whole blood, fentanyl concentrations have been detected from 0.04–383 ng/mL resulting from misuse, fatal overdose, and therapeutic drug monitoring (TDM) (13–16). Other illicit fentanyl analogues have been detected in whole blood from 0.01–79 ng/mL (17–28). Additionally, the N-dealkylated metabolite of fentanyl, norfentanyl, has been detected in blood from 1.4–8.9 ng/mL in cases of fentanyl overdose (29, 30). Metabolites of other analogues (i.e. norsufentanil, norcarfentanil, and norlofentanil) have been confirmed in both plasma and urine (31, 32).
Fentanyl compounds and their metabolites have been measured to support TDM in clinical matrices. Initially, these methods utilized high-pressure liquid chromatography ultraviolet detection (HPLC/UV) and gas chromatography mass spectrometry (GC/MS) (17, 33–35). When coupled to liquid-liquid extraction or other sample preparation, these methods were able to detect fentanyl compounds as low as 1 ng/mL using 1 mL of blood. Immunoassays have also been developed for the detection of fentanyl compounds; however, select fentanyl analogues may not be identified due to cross-reactivity of the antibodies (36–38). Modern high-pressure liquid chromatography tandem mass spectrometry (LC-MS/MS) methods have detected sub-nanogram amounts of fentanyl. Many methods have focused on the detection of a specific analogue for the purpose of TDM (39, 40) or included only fentanyl as a part of a wide-ranging pain panel (41–45). Consequently, few LC-MS/MS methods detect the wide array of illicit and prescribed fentanyl analogues that are now contributing to the ongoing opioid overdose epidemic (46–50).
Sample preparation of serum, plasma, and whole blood is routine, but typically requires lengthy or expensive sample clean-up. Using dried blood spots (DBS) can simplify the analysis of whole blood, and may even eliminate the need for additional sample processing such as solid phase extraction (51). Furthermore, DBS allow for small sample volume, deceased risk of infection from bloodborne pathogens, increased analyte stability, and simple storage and shipping requirements (52), which reduces costs in preparation, shipping, and storage. Previously, fentanyl has been quantitated from DBS using automated flow through desorption (53, 54) as well as manual extraction (45, 55, 56). The inclusion of additional fentanyl analogues and metabolites would improve these methods as diagnostic tools for exposure identification to support surveillance, prevention, and response efforts for the opioid overdose epidemic.
This paper describes the development and validation of a method applied for the quantitation of thirteen fentanyl analogues and four corresponding metabolites in DBS using solvent extraction followed by LC-MS/MS. With five microliters of dried whole blood, the method accurately quantitates analogues from 1.00 to 100 ng/mL. Due to the reduced complexity of sample preparation, this method is easily transferrable to additional laboratories and readily adaptable for new fentanyl analogues.
Methods
Appropriate safety control measures, including engineering, administrative, and personal protective equipment, were used for all procedures based on a site-specific risk assessment that identified physical, health, and procedural hazards.
Materials
Fentanyl, norfentanyl, 2H5-fentanyl, and 2H5-norfentanyl were purchased from Cerilliant (Round Rock, TX). Norlofentanil and 2H3-norlofentanil were purchased from Toronto Research Chemicals (Toronto, Canada). Cyclopropylfentanyl, 2H5-cyclopropylfentanyl, 2-furanylfentanyl, 2H5-furanylfentanyl (3-furancarboxamide isomer), acrylfentanyl, 2H5-acrylfentanyl, isobutyrylfentanyl, 2H5-isobutyrylfentanyl, ocfentanil, 2H5-ocfentanil, methoxyacetylfentanyl, and 2H5-methoxyacetylfentanyl were purchased from Cayman Chemical (Ann Arbor, MI). Carfentanil, 2H5-carfentanil, norcarfentanil, 2H5-norcarfentanil, sufentanil, 2H5-sufentanil, norsufentanil, 2H5-norsufentanil, and 13C6-alfentanil were custom synthesized by Battelle (Columbus, OH). Lofentanil, alfentanil, cis/trans 3-methylfentanyl mixture, and α-methylfentanyl were generous gifts from a variety of sources as listed in the Acknowledgements. Structures of all target analytes are shown in Figure 1. High pressure liquid chromatography (HPLC) grade acetonitrile was purchased from The Lab Depot (Dawsonville, GA). HPLC grade formic acid and methanol was purchased from Fisher (Hampton, NH). Deionized (DI) water (>18 MΩ·cm) was prepared on-site using an installed water purification system (Aqua Solutions Inc., Jasper, GA). Indicating desiccant used for storage was purchased from Caulfield Industrial Ltd. (Galway, IR). Pooled whole blood and a convenience set of individual whole blood samples were purchased from Tennessee Blood Services (Memphis, TN), with K2EDTA as an additive. Postmortem blood was harvested from a single individual by Golden West Biological (Temecula, CA) within 25 hours following death. The method used blood products acquired from commercial sources, and the work did not meet the definition of human subjects as specified in 45 CFR 46.102 (f).
Figure 1-.
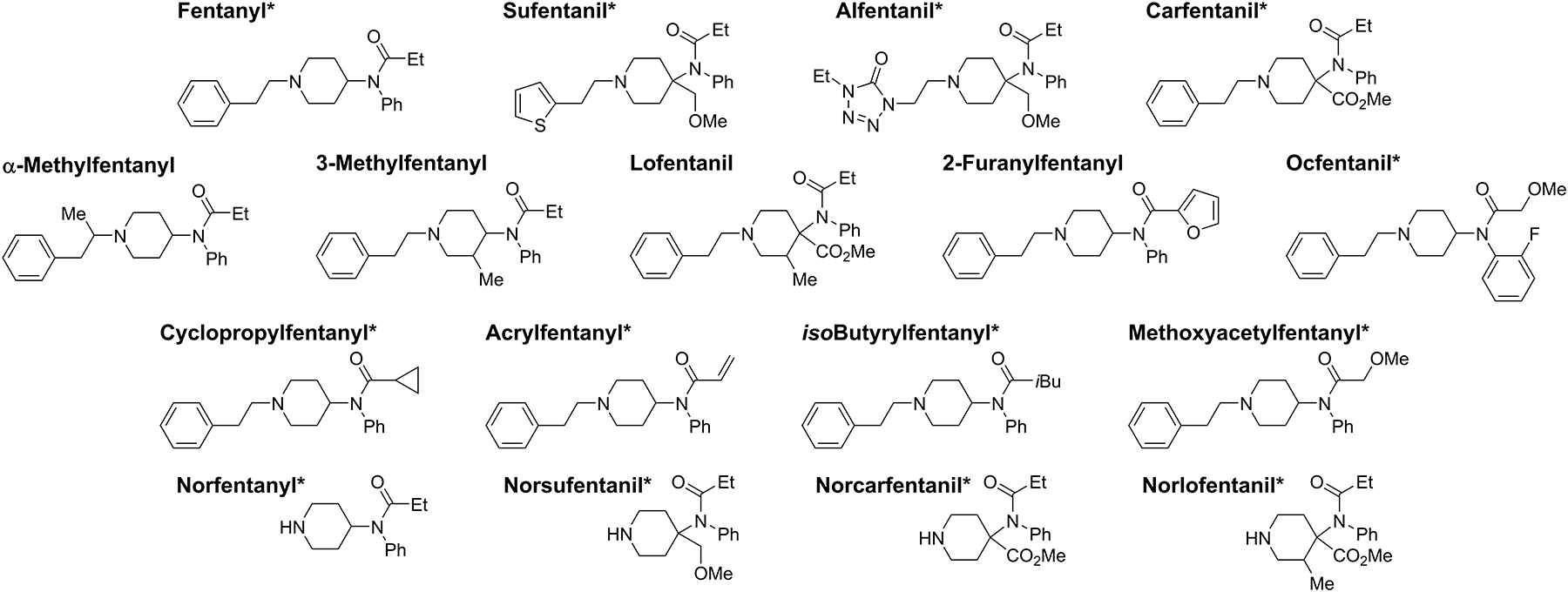
Chemical structures of fentanyl analogues and metabolites. *Represents analogues with isotopically labeled matched internal standards.
Working Solutions
Stock solutions of the individual 13 fentanyl analogues and four metabolites were prepared volumetrically at 10 μg/mL in DI water with the exception of norlofentanil and methoxyacetylfentanyl, which were prepared at 20 μg/mL in methanol and 10.5 μg/mL in water, respectively. A working solution of 3-methylfentanyl, alfentanil, α-methylfentanyl, carfentanil, fentanyl, lofentanil, sufentanil, norcarfentanil, norfentanyl, norlofentanil, and norsufentanil was prepared from the individual stock solutions at 500 ng/mL in DI water (Stock Solution I for Group I compounds). A separate working solution of cyclopropylfentanyl, 2-furanylfentanyl, acrylfentanyl, isobutyrylfentanyl, ocfentanil, and methoxyacetylfentanyl was prepared from the individual stock solutions at 500 ng/mL in DI water (Stock Solution II for Group II). 2H5-Fentanyl, 2H5-norfentanyl, 2H5-carfentanil, 2H5-sufentanil, 2H5-norsufentanil, and 2H5-norcarfentanil stock solutions were prepared at 100 μg/mL in methanol. 13C6-Alfentanil and 2H3-norlofentanil stock solutions were prepared at 20 μg/mL in methanol. 2H5-cyclopropylfentanyl, 2H5-furanylfentanyl (3-furancarboxamide isomer), 2H5-acrylfentanyl, 2H5-isobutyrylfentanyl, 2H5-ocfentanil, 2H5-methoxyacetylfentanyl stock solutions were prepared at 10 μg/mL in DI water. Group I internal standard solution was prepared as a mixture of 13C6-alfentanil, 2H5-carfentanil, 2H5-norcarfentanil, 2H3-norlofentanil and 2H5-norsufentanil at 25 ng/mL and 2H5-Fentanyl, 2H5-norfentanyl, 2H5-sufentanil at 50 ng/mL. Group II internal standard solution was prepared as a mixture of 2H5-cyclopropylfentanyl at 25 ng/mL and 2H5-furanylfentanyl (3-furancarboxamide isomer), 2H5-acrylfentanyl, 2H5-isobutyrylfentanyl, 2H5-ocfentanil, 2H5-methoxyacetylfentanyl at 50 ng/mL. All solutions were stored at −20 °C.
Materials Preparation
Calibrators were prepared volumetrically in pooled whole antemortem human blood using Stock Solution I for Group I and Stock Solution II for Group II to achieve final concentrations of 1.00, 2.50, 5.00, 10.0, 25.0, 50.0, 80.0, and 100 ng/mL. Group I and Group II quality control (QC) samples were prepared in pooled whole blood in the same manner as the calibrators at low (QL) and high (QH) concentrations of 7.50 and 75.0 ng/mL, respectively. Using Stock Solution I and II, six separate individual blood samples were fortified at 6.00 ng/mL and at 60.0 ng/mL in the same manner as the calibrators. Pooled whole antemortem human blood was used for matrix effect experiments. Total recovery experiments pooled whole antemortem human blood in the same manner as the calibrators at concentrations of 5.00 and 50.0 ng/mL. Using Stock Solution I and II, postmortem blood was spiked via pipette dilution at 7.50 and 75.0 ng/mL; a set of pooled whole blood controls at the same concentrations were prepared at the same time. Spiked samples at 0.10, 0.30, 0.50, and 0.80 ng/mL were prepared volumetrically in pooled human blood using diluted Stock Solution I and II.
Adjusted hematocrit blood was prepared at 17%, 25%, 58% and 65% via the addition of pooled plasma to washed red blood cells until the desired hematocrit was achieved. Washed red blood cells were prepared by separating pooled whole blood in a centrifuge (Eppendorf 5810 Hauppauge, NY) at 3,500 rpm (2,465 g) for 15 min, the plasma was removed and the remaining red blood cells were washed three times with twice the volume of 1x PBS buffer. The adjusted hematocrit blood was spiked via pipette dilution with either Stock Solution I or II at 7.50 and 75.0 ng/mL for each hematocrit level. To measure hematocrit, adjusted hematocrit bloods and individual whole blood samples were centrifuged in a 75 mm hematocrit capillary tube (Drummond, Broomall, PA) at 12,000 rpm using a M24 Hematocrit Centrifuge (LW Scientific, Lawrenceville, GA) in triplicate and read with the attached Micro-Capillary Reader.
All calibrators, QC samples and samples were spotted at 5.00 μL onto Whatman 903 protein saver cards (Eastern Business Forms, Inc., Atlanta, GA) using a Rainin E4–10XLS+ pipette (Mettler-Toledo, Columbus, OH). The cards were allowed to dry at ambient temperature in a biological safety cabinet overnight (~18 hours) before analysis or storage in a resealable plastic bag with desiccant at −20°C.
Sample Preparation
Dried blood spot calibrators, QCs, and samples were punched into a 2.0-mL 96 DeepWell plate (ThemoFisher Scientific, Waltham, MA) using a 0.25 inch hole punch (S.P. Richards Co., Smyrna, GA). Extraction solvent (1.0 mL of 50:50 methanol:acetonitrile) followed by 25.0 μL of the internal standard solution was added to each well containing a punch. The DeepWell plate was sealed with adhesive foil and mixed at 1,000 rpm for 10 min (Eppendorf MixMate, Hauppauge, NY). The solvent was then transferred to a second 96 DeepWell plate, leaving the extracted spot behind, and dried under a stream of 60 °C nitrogen using a Porvair TurboVap (Ashland, VA). Dried extracts were reconstituted with 100 μL of a 90:10 water:acetonitrile solution containing with 0.1% formic acid. The plate was again sealed with adhesive foil and mixed at 1,000 rpm for 5 min. Finally, the reconstituted extracts were transferred to a 96-well PCR plate, heat sealed, and loaded into the instrument for analysis.
LC-MS/MS conditions
LC separation was performed on an Agilent Technologies Liquid Chromatography 1290 Infinity I using a 3 μm 2.0 × 50 mm Pursuit pentafluorophenyl (PFP) column from Agilent (Santa Clara, CA). Target analytes were separated using a flow rate of 450 μL/min. The mobile phases used were aqueous 0.1% formic acid (Solvent A) and acetonitrile with 0.1% formic acid (Solvent B). The gradient program was as follows: an initial flow of 90% A was held for 0.5 min and then reduced to 80% A over 0.3 min; at 1.5 min 80% A was reduced to 50% A over 2.3 min; at 4.1 min 50% A was reduced to 5% A over 0.3 min; and at 7.4 min 5% A was increased to 90% A over 0.6 min and held for 2 min for a total run time of 10 min. A 20 μL injection was used for separate analysis of Group I and Group II compounds. A needle wash of acetonitrile with 0.1% formic acid was performed 10 seconds before each injection.
Analytes were detected in positive mode ESI on a Sciex 6500 Triple Quadrupole Mass Spectrometer (Foster City, CA). Two transitions were monitored per analyte and one transition was monitored for each internal standard (Table 1), with Group I and Group II analytes monitored in separate analytical runs. Analyte-specific parameters were optimized for maximum signal (Table S1). Additional parameters used during analysis include the following values: curtain gas, 35 psi; collision gas, 7 psi; IonSpray voltage, 5500 volts; source temperature, 550 °C; ion source gas 1, 70 psi; ion source gas 2, 60 psi; entrance potential, 10 volts. A dwell time of 20 msec was used for all transitions except for the confirmation transition of sufentanil which was set to 40 msec. The instrument was controlled using Analyst software (Sciex, Version 1.6).
Table 1-.
Monitored Transitions.
| Analyte | Group | Precursor Ion (m/z) | Product Ion (m/z) |
|---|---|---|---|
| 3-Methylfentanyl# | I | 351.3 | 105.1†, 202.1‡ |
| Alfentanil | I | 417.2 | 197.1†,165.1‡ |
| 13C6-Alfentanil | I | 423.0 | 268.0 |
| α-Methylfentanyl# | I | 351.3 | 119.1†, 202.1‡ |
| Carfentanil | I | 395.4 | 335.2†, 363.2‡ |
| 2H5-Carfentanil | I | 400.3 | 340.3 |
| Fentanyl | I | 337.3 | 188.3†, 105.0‡ |
| 2H5-Fentanyl | I | 342.1 | 188.3 |
| Lofentanil# | I | 409.3 | 349.2†, 134.2‡ |
| Sufentanil | I | 387.4 | 238.3†, 111.2‡ |
| 2H5-Sufentanil | I | 392.1 | 111.0 |
| Norcarfentanil | I | 291.3 | 231.2†,113.0‡ |
| 2H5-Norcarfentanil | I | 296.1 | 142.1 |
| Norfentanyl | I | 233.3 | 84.1†,56.1‡ |
| 2H5-Norfentanyl | I | 238.5 | 182.2 |
| Norlofentanil | I | 305.2 | 273.1†,156.1‡ |
| 2H3-Norlofentanil | I | 309.1 | 249.0 |
| Norsufentanil | I | 277.2 | 128.0†,245.3‡ |
| 2H5-Norsufentanil | I | 282.4 | 128.2 |
| Cyclopropylfentanyl | II | 348.9 | 188.1†, 105.0‡ |
| 2H5-Cyclopropylfentanyl | II | 353.9 | 188.1 |
| 2-Furanylfentanyl | II | 374.8 | 187.9†, 104.9‡ |
| 2H5-Furanylfentanyl (3-furancarboxamide isomer) | II | 379.8 | 188.1 |
| Acrylfentanyl | II | 334.9 | 188.1†,105.0‡ |
| 2H5-Acrylfentanyl | II | 339.9 | 188.1 |
| isoButyrylfentanyl | II | 350.9 | 188.0†, 104.8‡ |
| 2H5-isoButyrylfentanyl | II | 355.9 | 188.1 |
| Ocfentanil | II | 370.9 | 188.1†, 105.0‡ |
| 2H5-Ocfentanil | II | 375.9 | 193.2 |
| Methoxyacetylfentanyl | II | 352.9 | 188.1†, 105.0‡ |
| 2H5-Methoxyacetylfentanyl | II | 357.8 | 188.1 |
Quantitation ion,
Confirmation Ion,
Uses 2H5-Carfentanil as an internal standard.
Data analysis was performed using MultiQuant software (Sciex, Version 2.1). Linear regression analysis of the calibrator concentration versus the ratio of the quantitation ion area to the internal standard ion area with 1/x weighting was used for quantitation. A sum-of-least-squares calculation was performed to determine the best weighting factor. Isotopically-labeled analytes were used as internal standards for corresponding analogues where available. Isotopically-labeled carfentanil was used as internal standard for 3-methylfentanyl, α-methylfentanyl, and lofentanil; isotopically-labeled furanylfentanyl (3-furancarboxamide isomer) was used as internal standard for 2-furanylfentanyl. All calibration curves met the correlation of determination (R2) requirement of 0.980 or greater and were accepted for use.
Method Validation
This method was optimized for Group I compounds but validated for both Group I and II compounds to confirm the quantitation of emerging fentanyls. The validation included determination of accuracy, precision, reportable range, selectivity, limits of detection, matrix effects, and total recovery. Stability and storage were assessed for Group I analytes.
Data from 20 replicate calibration curves and QC samples were evaluated to assess accuracy and precision. These replicates were completed by three analysts with a maximum of two calibration curves per day over 16 weeks for Group I and over 12 weeks for Group II (Table 1). Potential matrix interferences affecting analyte transitions (selectivity) were evaluated by analyzing 100 unidentified patient samples from individuals with no anticipated exposure to fentanyls. Six individual whole blood samples were also fortified at both 6.00 and 60.0 ng/mL and analyzed in triplicate for all analytes to examine variation between individual samples.
Matrix Effects and Total Recovery
Matrix effects were evaluated by comparing a post extraction spiked sample to a solvent spiked sample at the same concentration. A blank DBS was extracted, dried, and then fortified with 5 μL of either a 5.00 or 50.0 ng/mL working solution and 25 μL of the internal standard solution during reconstitution. A matrix free spike was prepared by adding 5 μL of either a 5.00 or 50.0 ng/mL working solution and 25 μL of the internal standard solution to 70 μL of solvent such that the solvent composition was identical to reconstituted samples. The average area counts (n=5) was used to calculate matrix effects for each compound using the following equation:
Total recovery was evaluated by comparing an extracted DBS to a solvent spike at the same concentration. A 5 μL dried blood spot at either 5.00 or 50.0 ng/mL was extracted with internal standard as described previously, dried, and reconstituted. A solvent spike was prepared by adding 5 μL of either a 5.00 or 50.0 ng/mL working solution and 25 μL of the internal standard solution to 70 μL of solvent such that the solvent composition was identical to reconstituted samples. The average area counts (n=5) was used to calculate total recovery for each compound using the following equation:
Humidity Exposure and Benchtop DBS Storage
For humidity exposure experiments, a DBS card was placed in a resealable plastic tub filled with water to maintain an approximate humidity of 85% (measured using an uncalibrated analog hygrometer).The effect of humidity was evaluated at 5.00 ng/mL by determining the average deviation between experimental DBS and a set of control DBS maintained at −20 °C with desiccant.
For benchtop storage experiments, DBS were stored at room temperature in resealable plastic bags with and without desiccant. The experiment was run in triplicate for humidity tests and in quintuplicate for benchtop storage.
Autosampler Stability
Extracted DBS samples were also evaluated for processed sample stability at 4 °C (autosampler temperature) by running a calibration curve, and three replicates at both 7.5 and 75.0 ng/mL. The curve and samples were run and then run a second time after 24 hours had elapsed.
Results and Discussion
An analytical method was developed to identify and quantify human exposure to fentanyl, thirteen analogues, and four metabolites using an extraction of the DBS followed by LC-MS/MS analysis. Extraction parameters, chromatographic conditions, internal standard addition, DBS drying time, and storage conditions were optimized for Group I compounds. Method performance was evaluated using simulated patient and postmortem samples prepared from unexposed blood samples fortified at levels typical of abuse.
Method Optimization (Solvent Selection and Quantity, Chromatography, Internal Standard Addition)
The extraction of fentanyl compounds from DBS was optimized for several solvent conditions including composition, volume, and acidity. Methanol, acetonitrile, and varying ratios of those solvents were investigated. Aqueous-organic mixtures were briefly examined but additional matrix was extracted by the aqueous component from the card, noted by the pink extract color. A 50:50 ratio of methanol and acetonitrile was the most effective extraction solvent when examining total analyte response. Formic acid, documented to release protein bound fentanyls, was added to the extraction solution (57) however, the recovery of lofentanil was reduced by 84% following this 1% addition of acid and was not included in the final solvent mixture. Extraction solvent volumes were also examined with no response increase from 0.50 to 1.25 mL; therefore, 1.0 mL of extraction solvent was used for ease of manipulation.
Although C18 reversed phase chromatography has typically been used for fentanyl analogue separation, these compounds readily separated using a PFP column, which contains a polar modified sorbent designed for selectivity towards structurally similar aromatic compounds (58). The metabolites and fentanyl analogues of Group I were separated as noted in Figure 2, with the constitutional isomers α-methylfentanyl and 3-methylfentanyl resolved from one another (inset Figure 2). Adjacent matrix peaks were separated from the analytes of interest from all ions monitored. When the chromatographic conditions were applied to the Group II fentanyl analogues in a separate injection, no adjustments were necessary to separate the analytes from any detectable matrix interferences. 3-Methylfentanyl and isobutyrylfentanyl had similar retention times and would coelute if run together, however while they share the 350 > 105 transition they both have a unique transition which can aid in differentiation. It is likely that butyrylfentanyl and isobutyrylfentanyl would also coelute; work is currently underway on a chromatographic method that can better resolve constitutional isomers commonly encountered. No carryover was observed in a water injection after the injection of a 100 ng/mL calibrator.
Figure 2-.
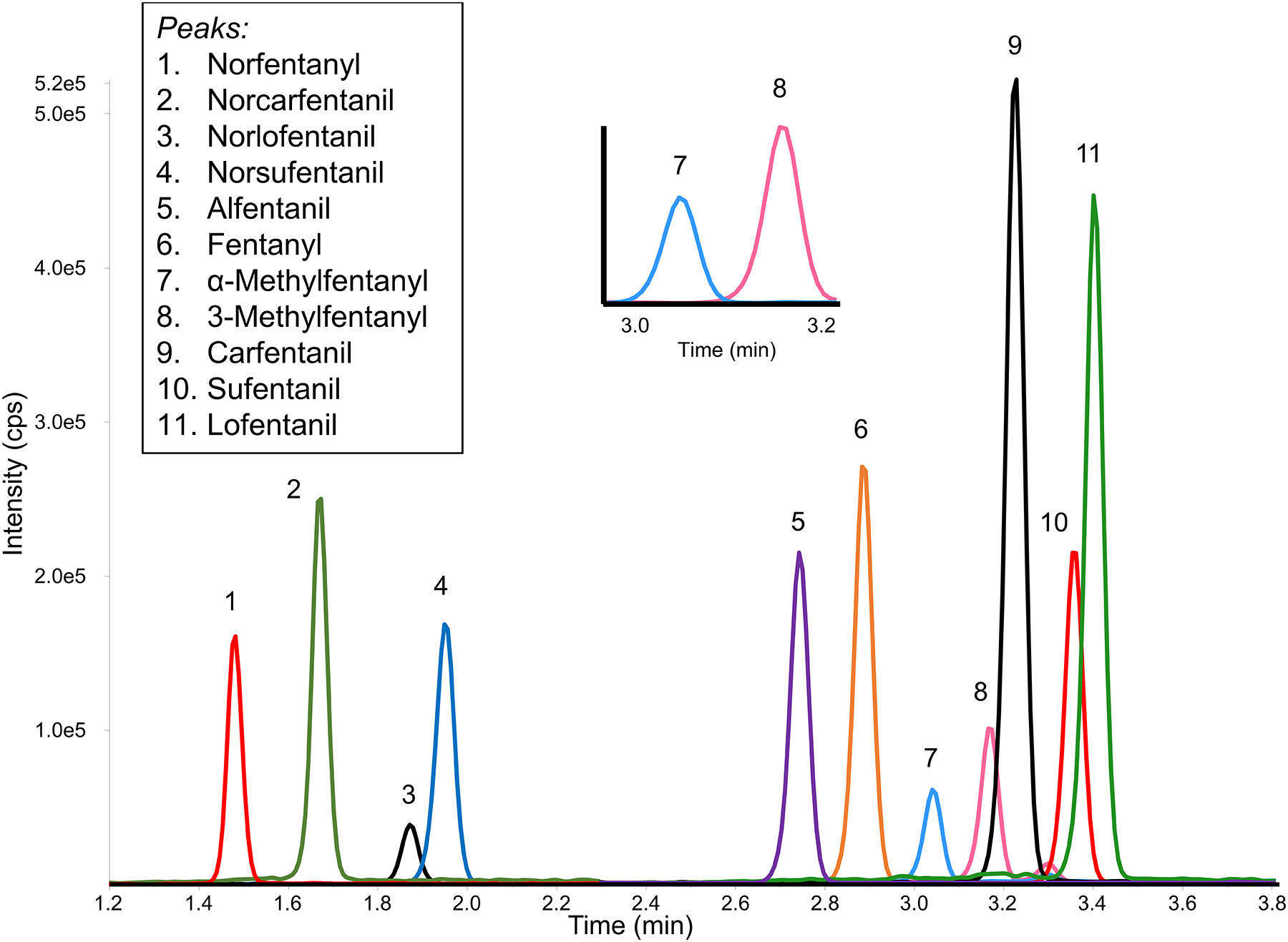
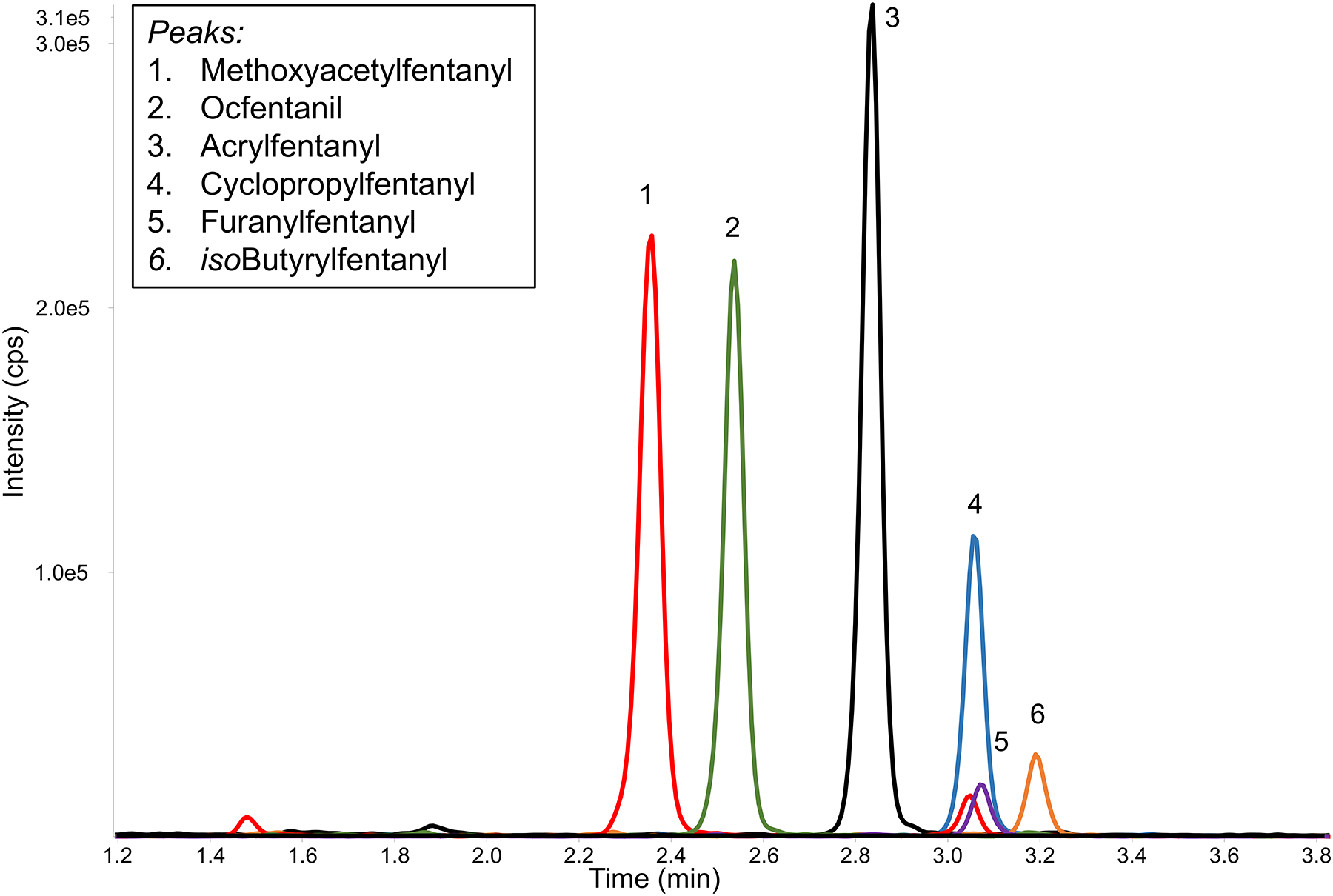
Chromatographic resolution of Group I and II compounds in two injections. PFP separation results in the resolution of key pairs (inset: resolution of constitutional isomers α-methylfentanyl and 3-methylfentanyl). 20μL injection of a 5 ng/mL dried blood spot.
MS parameters and transitions were selected to maximize signal and minimize matrix interferences. Two transitions were monitored for each analyte, with the second transition used solely for confirmatory purposes. While common fragmentations were noted between analogues, only 3-methylfentanyl and α-methylfentanyl share a transition, but were chromatographically resolved.
Corresponding isotopically labeled internal standards were used where available. For analytes without matching internal standards, a surrogate internal standard was selected based on proximity to retention time and similarity of structure and molecular weight.
With the analytical parameters established, the addition of internal standard (IS) was examined. To compensate for sample preparation variations, IS should be added as early as possible in the sample preparation process. However, premixing the 5 μL blood sample with IS would require extremely small volumes (<1 μL) to maintain spot size and maximize sample volume, likely resulting in poor accuracy. A larger volume of IS could be mixed with a larger aliquot of whole blood prior to spotting, but this would negate the advantages of the small sample size and preparation of DBS prior to shipment to the analysis laboratory; therefore, was not further assessed. The addition of the IS directly onto the dried blood sample, onto the card prior to spotting, and concurrent with the extraction solvent were also considered. When the IS was pipetted on top of the DBS, the solution did not adhere and absorbed into the paper surrounding the spot, exceeding the punch size. Another approach investigated was the addition of IS to the card both before sample spotting. A 1.0 μL volume was required to maintain the IS spot within a 0.25 inch punch area. However, once dried, the IS spot was difficult to identify on the card, resulting in inconsistent coverage of the IS spot with the whole blood sample. Another approach could be to impregnate the card with IS prior to spotting or spray the IS on the card after spotting, which has been used in other DBS studies (59). However, the extreme potency of fentanyl compounds would result in the creation an undesired controlled substance. Finally, the addition of the IS to the DBS with the extraction solution was evaluated; the resulting high reproducibility between samples confirmed this approach to be effective for the analysis of DBS for fentanyls.
Method Validation
Monitoring fentanyl analogue use for the Opioid Epidemic is challenging as novel analogues are constantly emerging on the illicit market. For that reason, the ability to quickly incorporate new analytes to the method is highly desired. To assess the adaptability of this method, a variety of emerging fentanyl analogues were selected for evaluation. DBS Group II calibrators and QC materials containing cyclopropylfentanyl, 2-furanylfentanyl, acrylfentanyl, isobutyrylfentanyl, ocfentanil, and methoxyacetylfentanyl were prepared and analyzed in addition to the Group I analytes previously defined in Table 1. While the method was optimized for Group I compounds, a full validation was run separately on Group II compounds using the same conditions.
The analytical method was validated using 20 sets of calibration curves and QC samples at concentrations of 7.50 and 75.0 ng/mL (Table 2). Precision, accuracy, and sensitivity were calculated based on the quantitation ion transition. The precision, defined by the coefficient of variation (CV), ranged from 2.68–12.0% over the two QC concentrations. This variability was reduced for analytes with corresponding isotopically labeled internal standards, resulting in CVs less than 7.35%. Bias for both QC samples were within 6.98% for all Group I and II compounds. Both precision and accuracy for all analytes included in this method met the FDA guidelines for biomedical testing (60) and met or exceeded results obtained by previous methods for the detection of fentanyls in dried blood spots (53, 54). Prepared DBS extracts were assessed for autosampler stability at 4 °C over a 24 hour period. All analytes, at both levels, did not vary more than 7.4% from the initial T0 measurement.
Table 2-.
Accuracy and precision for QC samples (n=20, interday). Percent error is calculated from the averaged QC value.
| QC Low (7.50 ng/mL) | QC High (75.0 ng/mL) | ||||
|---|---|---|---|---|---|
| Analyte | Group | Coefficient of Variation (%) | Percent Error (%) | Coefficient of Variation (%) | Percent Error (%) |
| 3-Methylfentanyl | I | 6.96 | 2.85 | 8.60 | 1.74 |
| Alfentanil* | I | 2.68 | 0.85 | 4.18 | 1.93 |
| α-Methylfentanyl | I | 6.29 | 0.47 | 10.7 | 0.70 |
| Carfentanil* | I | 3.50 | 0.97 | 4.63 | 1.47 |
| Fentanyl* | I | 4.11 | 0.44 | 5.11 | −0.75 |
| Lofentanil | I | 12.0 | 6.98 | 6.53 | 5.07 |
| Sufentanil* | I | 4.16 | 1.51 | 5.62 | 1.79 |
| Norcarfentanil* | I | 5.20 | −0.02 | 4.16 | 4.50 |
| Norfentanyl* | I | 7.35 | 0.28 | 6.51 | 3.55 |
| Norlofentanil* | I | 5.95 | −0.06 | 4.58 | 2.77 |
| Norsufentanil* | I | 3.47 | −2.23 | 3.80 | 2.97 |
| Cyclopropylfentanyl* | II | 3.85 | 4.47 | 4.59 | −0.88 |
| Acrylfentanyl* | II | 5.41 | 1.49 | 4.86 | −3.72 |
| 2-Furanylfentanyl | II | 9.66 | 4.02 | 7.53 | 0.67 |
| isobutyrylfentanyl* | II | 5.47 | 2.61 | 5.75 | −3.50 |
| Ocfentanil* | II | 4.49 | 3.40 | 5.34 | −3.79 |
| Methoxyacetylfentanyl* | II | 5.51 | −2.33 | 7.07 | −3.61 |
Represents analogues with isotopically labeled matched internal standards.
Humidity Exposure and Benchtop DBS Storage
DBS cards were stored at −20 °C in resealable plastic bags with individual desiccant pouches that were replaced as necessary (when desiccant indicator turned pink; typically once every two months) as recommended by Verplaetse, et al. (53). For DBS stored in these conditions, no loss of sensitivity was detected for the lowest calibrator and a negative trend was not detected for any of the QC materials observed over 11 months. To evaluate the impact of humidity, DBS were stored at room temperature (~20 °C) in plastic bags both with desiccant and without desiccant as well as in a sealed chamber with elevated humidity. The DBS stored in plastic bags showed no loss of analyte over 4 weeks for all Group I compounds, including the nor-metabolites. Although previous reports indicated that norfentanyl was not stable in DBS after 5 days at room temperature, this was not observed experimentally here (55). After 7 days in elevated humidity (~85% humidity), the quantitated analytes deviated 32 – 60% from the control DBS for 10 of the 11 Group I analytes evaluated. Alfentanil was the sole exception, which quantitated within 1% of the control samples. After 14 days in the humid environment, the DBS card was consumed by mold. Although the absence of a desiccant pouch did not negatively impact the results, all DBS cards used in this study were stored with desiccant and monitored to ensure minimal exposure to moisture.
Reportable Range and Limit of Detection
The calibration range established from 1.00–100 ng/mL was designed for the detection of the majority of overdose cases and most therapeutic levels. 2-furanylfentanyl was evaluated from 1.00–80.0 ng/mL. A weighting of 1/x was selected following a regression analysis (evaluation of least squares). Each calibration curve met the requirement of r2>0.98. The performance of the lowest calibrator did not exceed a CV of 11.4%, with exception of lofentanil at 22.0%. Percent error ranged 4.20–16.8% for the lowest calibrator for both Group I and II compounds, carfentanil and sufentanil were the only analytes to exceed 15% error. The highest calibrator did not exceed 4.62% CV, confirming the range was appropriate for this protocol.
Theoretical limits of detection (LoD) were calculated and ranging from 0.127–0.704 ng/mL for all analytes. Recent carfentanil exposures have reported blood concentrations <0.10 ng/mL however, the mean and median carfentanil concentration reported in these case studies was greater than 0.10 ng/mL (24–26). For this reason, sensitivity beyond the theoretical LoD was investigated. DBS were prepared at 0.10, 0.30, 0.50, and 0.80 ng/mL for both Group I and II compounds and were examined (n=10). Carfentanil, fentanyl, sufentanil, norcarfentanil, norfentanyl, and norsufentanil were readily observed at 0.10 ng/mL with >3:1 S/N for all replicates (representative traces are presented in the Supplementary Data). Lofentanil, alfentanil, acrylfentanyl, ocfentanil, and methoxyacetylfentanyl were readily observed at 0.30 ng/mL >3:1 S/N for all replicates. Although the 1.00–100 ng/mL calibration range is acceptable for most overdoses, this method has the potential to identify potent analogues at levels below the lowest calibrator.
Selectivity, Matrix Effects and Total Recovery
Specificity of the method was confirmed through the analysis of 100 unidentified patient samples from individuals with no known exposure to fentanyls. No interfering matrix peaks were identified for either the quantitation or confirmation ions for all analytes included in this study.
Matrix effects, investigated at 50.0 ng/mL (Table 3) and at 5.00 ng/mL (Table S2), ranged from −17% to 43%. Matrix effects were reproducible with a CV of 13% or less for all analytes. Positive values for matrix effects indicated ion suppression and negative values indicated ion enhancement. Although the method was optimized for Group I analytes, all Group II compounds maintained matrix effects < 20% at both concentrations evaluated. The matrix effects obtained for alfentanil, fentanyl, sufentanil, and norfentanyl for this method were similar to those previously reported in dried blood spots (56). The results reported here demonstrate an improvement for matrix effects from other biological matrices previously reported up to 100% (48). This is likely due to the small DBS sample size and the fixation of interfering matrix by the DBS collection paper coupled with a fully organic extraction solvent.
Table 3-.
Matrix Effects and Total Recovery at 50.0 ng/mL (n=5). Presented as mean ± the standard deviation.
| Analyte | Matrix Effects (%) | Total Recovery (%) |
|---|---|---|
| 3-Methylfentanyl | 41 ± 6 | 86 ± 24 |
| Alfentanil* | 11 ± 8 | 63 ± 11 |
| α-Methylfentanyl | 43 ± 6 | 107 ± 28 |
| Carfentanil* | 22 ± 13 | 92 ± 16 |
| Fentanyl* | 38 ± 10 | 91 ± 24 |
| Lofentanil | 37 ± 11 | 80 ± 16 |
| Sufentanil* | 41 ± 11 | 81 ± 21 |
| Norcarfentanil* | 14 ± 10 | 79 ± 16 |
| Norfentanyl* | 26 ± 9 | 72 ± 22 |
| Norlofentanil* | 20 ± 8 | 67 ± 14 |
| Norsufentanil* | 24 ± 8 | 70 ± 20 |
| Cyclopropylfentanyl* | 1 ± 10 | 74 ± 26 |
| 2-Furanylfentanyl | −17 ± 13 | 91 ± 27 |
| Acrylfentanyl* | 5 ± 10 | 56 ± 21 |
| isobutyrylfentanyl* | −6 ± 11 | 82 ± 29 |
| Ocfentanil* | 10 ± 14 | 79 ± 24 |
| Methoxyacetylfentanyl* | 15 ± 12 | 77 ± 22 |
Represents analogues with isotopically labeled matched internal standards.
Total recovery, which includes extraction recovery, transfer losses, and matrix effects, were investigated at 50.0 ng/mL (Table 3) and at 5.00 ng/mL (Table S1). Total recovery values at the 50.0 ng/mL level were similar to those at the 5.00 ng/mL with exception to α-methylfentanyl, fentanyl, and norfentanyl whose average recovery differed more than 20% between levels. The majority of the analytes maintained a total recovery above 70%, with the exception of alfentanil, norlofentanil, and acrylfentanyl; however, this did not negatively impact the detection of the lowest calibrator at 1.00 ng/mL.
Postmortem Blood
Fentanyl concentrations are routinely investigated in postmortem samples (14, 61–63). To evaluate this application, postmortem blood was spiked at QC levels (7.50 and 75.0 ng/mL), spotted, dried, extracted, and quantitated using whole blood DBS. A positive bias was observed for all analytes in the postmortem DBS. At the 7.50 ng/mL level there was an average 26% difference from nominal values which was reduced to 20% at the 75.0 ng/mL level. Lofentanil, norcarfentanil, norfentanyl, norlofentanil, norsufentanil, cyclopropylfentanyl, isobutyrylfentanyl, ocfentanil, and methoxyacetylfentanyl were within 20% of the nominal value at both levels. Accuracy for all other compounds exceeded 20% bias, indicating this method was not ideal for quantitation from postmortem samples when using whole blood DBS calibrators. However, this method can identify exposure to the selected analogues and metabolites since the transitions for all analytes were free of interfering peaks in postmortem blood.
Individual Spikes and Hematocrit
To test this method, individual blood samples fortified at 6.00 and 60.0 ng/mL were evaluated for accuracy and precision (Figure 3 and Figure S3). Precision for all analytes was <15% CV. Analytes with matched internal standards met the accuracy and precision guidelines set forth by the FDA (60). 3-Methylfentanyl, α-methylfentanyl, and 2-furanylfentanyl were outside of those guidelines.
Figure 3-.
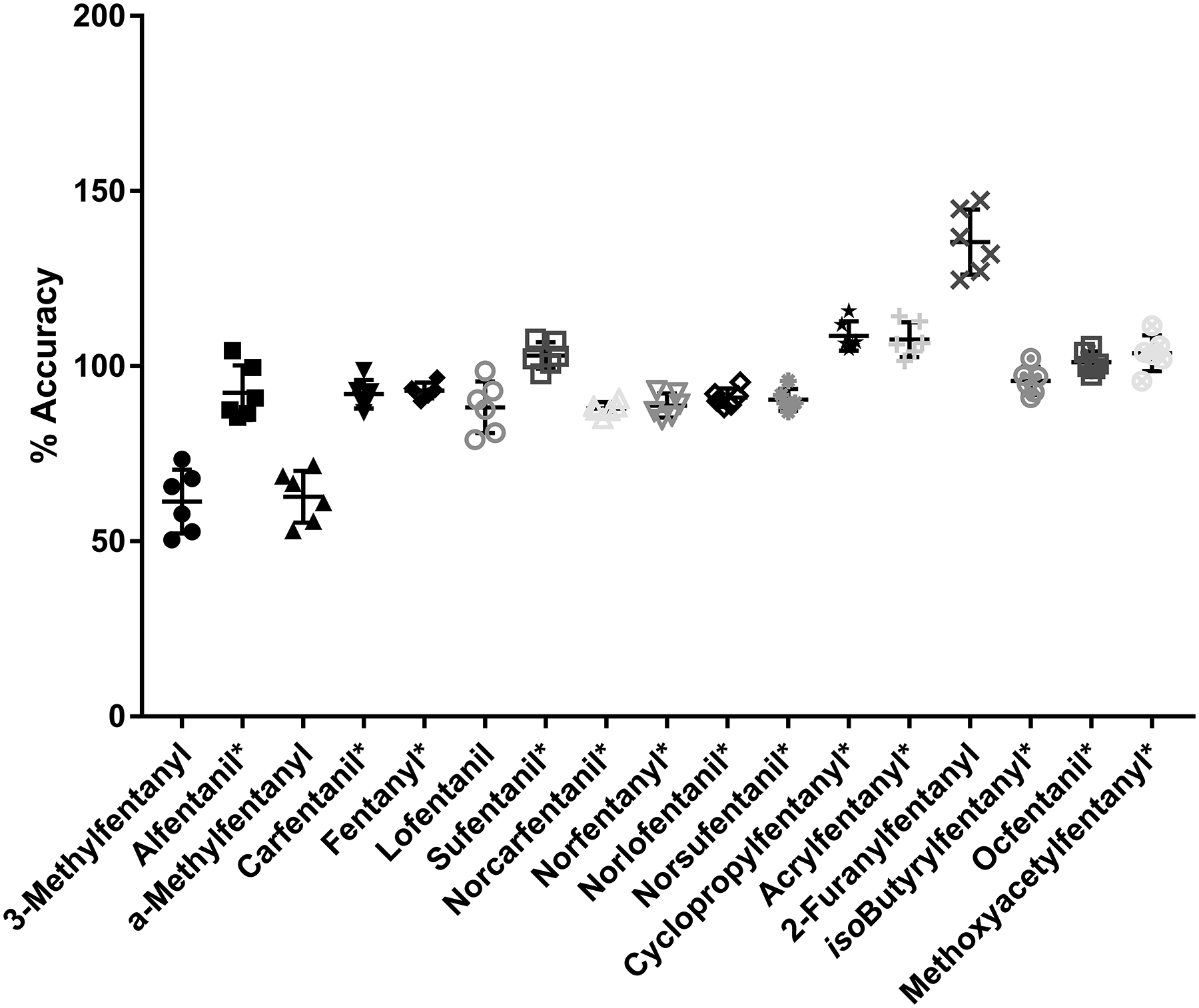
Average (n=6) of individual blood sampled spiked at 6.00 ng/mL. Icons represent individual values. Middle black line represents average. Error bars represent standard deviation. *Represents analogues with isotopically labeled matched internal standards.
For this method, whole spot analysis was selected to more accurately quantitate results and address spot spread caused by varying hematocrit (64). Hematocrit can also impact analyte desorption from the card resulting in bias and increased variability (65). However, further work was required to fully investigate differing analyte desorption at the extreme hematocrit values that are sometimes observed in clinical samples.
To assess any effect atypical hematocrit may have on extraction, spikes at 7.50 and 75.0 ng/mL were made in 17%, 25%, 58%, and 65% hematocrit pooled blood. Percent error was under 15% for all analytes at both levels with the exception to 3-methylfentanyl, alfentanil, α-methylfentanyl, and furanylfentanyl. 3-methylfentanyl and α-methylfentanyl biased low across all hematocrit levels where alfentanil and furanylfentanyl biased high. When examining reproducibility, all analytes at both levels had a value of <15 %CV with exception to 3-methylfentanyl, α-methylfentanyl, and lofentanil. Based on the results observed, a matched internal standard may be necessary when examining a population where large variations in hematocrit are expected. All other analytes did not demonstrate any significant deleterious effect by analyzing samples with vastly different hematocrit to the calibrators.
Method model for expansion
Given that new fentanyls are identified in the opioid overdose epidemic regularly, a model which permits the rapid incorporation of additional compounds to this method is desired. Preparation of materials, including calibrators and controls, into groups of compounds can accommodate this method expansion. As new fentanyls are identified, a third or even fourth group of compounds can easily be prepared, evaluated using the current analytical parameters, and applied to exposure samples for surveillance.
Conclusion
An analytical method was developed to identify human exposure to fentanyl analogues in clinical samples. The LC-MS/MS method quantitated fentanyl compounds over a concentration range of 1.00 to 100 ng/mL extracted from a 5 μL dried blood spot. The use of dried blood spots eliminates the need for additional expensive sample clean-up steps. The method demonstrated versatility by readily incorporating six diverse fentanyl analogues with no change to sample preparation. The assay met FDA accuracy and precision guidelines for the evaluated QC samples and for the individual samples with matched stable isotope labeled internal standard. Identification and quantitation of fentanyl compounds in nonfatal overdose victims will improve awareness of emerging synthetic opioid threats during the current opioid epidemic. The method’s construction allows the rapid incorporation of new fentanyls without revalidation of the entire method and the use of DBS facilitates future surveillance and response efforts.
Supplementary Material
Acknowledgements
For the valuable contribution of analytical standards, we would like to thank Dr. Rita McManamon, Atlanta Fulton County Zoo (Atlanta, GA); the DEA Western Laboratory (San Francisco, CA); Dr. C. Randall Clark, School of Pharmacy, Auburn University (Auburn, AL); Dr Thomas Tobin, Department of Veterinary Science, University of Kentucky (Lexington, KY); Dr. France Varin, Faculty of Pharmacy, University of Montreal (Quebec, Canada); Dr. Jim Baron, Battelle Columbus Laboratories (Columbus, OH); and the NIDA Drug Supply Program.
Footnotes
Supplementary data
Supplementary data are available at Journal of Analytical Toxicology online.
References
- (1).Vardanyan RS and Hruby VJ (2014) Fentanyl-related compounds and derivatives: current status and future prospects for pharmaceutical applications. Future Medicinal Chemistry, 6, 385–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Mounteney J, Giraudon I, Denissov G and Griffiths P (2015) Fentanyls: Are we missing the signs? Highly potent and on the rise in Europe. International Journal of Drug Policy, 26, 626–631. [DOI] [PubMed] [Google Scholar]
- (3).Lindsay CD, Riches JR, Roughley N and Timperley CM (2016) Chemical Defense Against Fentanyls. In Worek F, Jenner J and Thiermann H (eds.), Chemical Warfare Toxicology: Volume 2: Management of Poisoning, 1st edition, Chapter 8. The Royal Society of Chemistry, Piccadilly, London, pp. 259–313. [Google Scholar]
- (4).Gladden RM, Martinez P and Seth P (2016) Fentanyl Law Enforcement Submissions and Increases in Synthetic Opioid–Involved Overdose Deaths — 27 States, 2013–2014. Morbidity and Mortality Weekly Report, 65, 837–843. [DOI] [PubMed] [Google Scholar]
- (5).(2016) Influx of Fentanyl-laced Counterfeit Pills and Toxic Fentanyl-related Compounds Further Increases Risk of Fentanyl-related Overdose and Fatalities. Centers for Disease Control and Prevention Health Alert Netwrok. https://emergency.cdc.gov/han/han00395.asp (accessed October 5th, 2016).
- (6).Tomassoni AJ, Hawk KF, Jubanyik K, Nogee DP, Durant T, Lynch KL, et al. (2017) Multiple fentanyl overdoses — New Haven, Connecticut, June 23, 2016. Morbidity and Mortality Weekly Report, 66, 107–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Vo KT, Van Wijk XMR, Lynch KL, Wu AHB and Smollin CG (2016) Counterfeit Norco poisoning outbreak — San Francisco bay area, California, March 25–April 5, 2016. Morbidity and Mortality Weekly Report, 65, 420–423. [DOI] [PubMed] [Google Scholar]
- (8).Arens AM, Van Wijk XMR, Vo KT, Lynch KL, Wu AHB and Smollin CG (2016) Adverse effects from counterfeit alprazolam tablets. JAMA Internal Medicine, 176, 1554–1555. [DOI] [PubMed] [Google Scholar]
- (9).Massey J, Kilkenny M, Batdorf S, Sanders SK, Ellison D, Halpin J, et al. (2017) Opioid overdose outbreak — West Virginia, August 2016. Morbidity and Mortality Weekly Report, 66, 975–980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).O’Donnell J, Gladden RM, Matton CL, and Kariisa M (2018) Overdose Deaths with Carfentanil and Other Fentanyl Analogs Detected — 10 States, July 2016-June 2017. Morbidity and Mortality Weekly Report, 67, 767–768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).De Priest A, Heltsley R, Black DL, Cawthon B, Robert T, Moser F, Caplan YH, et al. (2010) Urine drug testing of chronic pain patients. III. normetabolites as biomarkers of synthetic opioid use. Journal of Analytical Toxicology, 34, 444–449. [DOI] [PubMed] [Google Scholar]
- (12).Silverstein JH, Rieders MF, McMullin M, Schulman S and Zahl K (1993) An analysis of the duration of fentanyl and its metabolites in urine and saliva. Anesthesia and Analgesia, 76, 618–621. [DOI] [PubMed] [Google Scholar]
- (13).Martin TL, Woodall KL and McLellan BA (2006) Fentanyl-Related Deaths in Ontario, Canada: Toxicological Findings and Circumstances of Death in 112 Cases (2002–2004). Journal of Analytical Toxicology, 30, 603–610. [DOI] [PubMed] [Google Scholar]
- (14).McIntyre I and Anderson D (2012) Postmortem fentanyl concentrations: a review. Journal of Forensic Research, 3, 1–10. [Google Scholar]
- (15).Boland JW and Pockley AG (2016) Clinically relevant concentrations of opioids for in vitro studies. Journal of Opioid Management, 12, 313–321. [DOI] [PubMed] [Google Scholar]
- (16).Bista Sudeep R, Haywood A, Hardy J, Norris R and Hennig S (2016) Exposure to Fentanyl After Transdermal Patch Administration for Cancer Pain Management. The Journal of Clinical Pharmacology, 56, 705–713. [DOI] [PubMed] [Google Scholar]
- (17).Henderson GL (1983) Blood concentrations of fentanyl and its analogs in overdose victims. Proceedings of the Western Pharmacology Society, 26, 287–290. [PubMed] [Google Scholar]
- (18).Ojanperä I, Gergov M, Liiv M, Riikoja A and Vuori E (2008) An epidemic of fatal 3-methylfentanyl poisoning in Estonia. International Journal of Legal Medicine, 122, 395–400. [DOI] [PubMed] [Google Scholar]
- (19).Dussy FE, Hangartner S, Hamberg C, Berchtold C, Scherer U, Schlotterbeck G, et al. (2016) An acute ocfentanil fatality: A case report with postmortem concentrations. Journal of Analytical Toxicology, 40, 761–766. [DOI] [PubMed] [Google Scholar]
- (20).Mohr ALA, Friscia M, Papsun D, Kacinko SL, Buzby D and Logan BK (2016) Analysis of Novel Synthetic Opioids U-47700, U-50488 and Furanyl Fentanyl by LC–MS/MS in Postmortem Casework. Journal of Analytical Toxicology, 40, 709–717. [DOI] [PubMed] [Google Scholar]
- (21).Coopman V, Cordonnier J, De Leeuw M and Cirimele V (2016) Ocfentanil overdose fatality in the recreational drug scene. Forensic Science International, 266, 469–473. [DOI] [PubMed] [Google Scholar]
- (22).Guerrieri D, Rapp E, Roman M, Druid H and Kronstrand R (2017) Postmortem and toxicological findings in a series of furanylfentanyl-related deaths. Journal of Analytical Toxicology, 41, 242–249. [DOI] [PubMed] [Google Scholar]
- (23).Sofalvi S, Schueler HE, Lavins ES, Kaspar CK, Brooker IT, Mazzola CD, et al. (2017) An LC–MS-MS Method for the Analysis of Carfentanil, 3-Methylfentanyl, 2-Furanyl Fentanyl, Acetyl Fentanyl, Fentanyl and Norfentanyl in Postmortem and Impaired-Driving Cases. Journal of Analytical Toxicology, 41, 473–483. [DOI] [PubMed] [Google Scholar]
- (24).Shanks KG and Behonick GS (2017) Detection of Carfentanil by LC–MS-MS and Reports of Associated Fatalities in the USA. Journal of Analytical Toxicology, 41, 466–472. [DOI] [PubMed] [Google Scholar]
- (25).Papsun D, Isenschmid D and Logan BK (2017) Observed Carfentanil Concentrations in 355 Blood Specimens from Forensic Investigations. Journal of Analytical Toxicology, 41, 777–778. [DOI] [PubMed] [Google Scholar]
- (26).Müller S, Nussbaumer S, Plitzko G, Ludwig R, Weinmann W, Krähenbühl S, et al. (2017) Recreational use of carfentanil – a case report with laboratory confirmation. Clinical Toxicology, 56, 151–152. [DOI] [PubMed] [Google Scholar]
- (27).Butler DC, Shanks K, Behonick GS, Smith D, Presnell SE and Tormos LM (2018) Three Cases of Fatal Acrylfentanyl Toxicity in the United States and a Review of Literature. Journal of Analytical Toxicology, 42, e6–e11. [DOI] [PubMed] [Google Scholar]
- (28).Hikin L, Smith PR, Ringland E, Hudson S and Morley SR (2018) Multiple fatalities in the North of England associated with synthetic fentanyl analogue exposure: Detection and quantitation a case series from early 2017. Forensic Science International, 282, 179–183. [DOI] [PubMed] [Google Scholar]
- (29).Carson HJ, Knight LD, Dudley MH and Garg U (2010) A fatality involving an unusual route of fentanyl delivery: Chewing and aspirating the transdermal patch. Legal Medicine, 12, 157–159. [DOI] [PubMed] [Google Scholar]
- (30).Teske J, Weller JP, Larsch K, Tröger HD and Karst M (2006) Fatal outcome in a child after ingestion of a transdermal fentanyl patch. International Journal of Legal Medicine, 121, 147–151. [DOI] [PubMed] [Google Scholar]
- (31).Mautz DS, Labroo R and Kharasch ED (1994) Determination of alfentanil and noralfentanil in human plasma by gas chromatography-mass spectrometry. Journal of Chromatography B: Biomedical Sciences and Applications, 658, 149–153. [DOI] [PubMed] [Google Scholar]
- (32).Riches JR, Read RW, Black RM, Cooper NJ and Timperley CM (2012) Analysis of clothing and urine from Moscow theatre siege casualties reveals carfentanil and remifentanil use. Journal of Analytical Toxicology, 36, 647–656. [DOI] [PubMed] [Google Scholar]
- (33).van Rooy HH, Vermeulen NPE and Bovill JG (1981) The assay of fentanyl and its metabolites in plasma of patients using gas chromatography with alkali flame ionisation detection and gas chromatography—mass spectrometry. Journal of Chromatography B: Biomedical Sciences and Applications, 223, 85–93. [DOI] [PubMed] [Google Scholar]
- (34).Gillespie TJ, Gandolfi AJ, Maiorino RM and Vaughan RW (1981) Gas chromatographic determination of fentanyl and its analogues in human plasma. Journal of Analytical Toxicology, 5, 133–137. [DOI] [PubMed] [Google Scholar]
- (35).Kumar K, Ballantyne JA and Baker AB (1996) A sensitive assay for the simultaneous measurement of alfentanil and fentanyl in plasma. Journal of Pharmaceutical and Biomedical Analysis, 14, 667–673. [DOI] [PubMed] [Google Scholar]
- (36).Schuttler J and White PF (1984) Optimization of the radioimmunoassays for measuring fentanyl and alfentanil in human serum. Anesthesiology, 61, 315–320. [DOI] [PubMed] [Google Scholar]
- (37).Ruangyuttikarn W, Law MY, Rollins DEO and Moody DE (1990) Detection of fentanyl and its analogs by enzyme-linked immunosorbent assay. Journal of Analytical Toxicology, 14, 160–164. [DOI] [PubMed] [Google Scholar]
- (38).Wang G, Huynh K, Barhate R, Rodrigues W, Moore C, Coulter C, et al. (2011) Development of a homogeneous immunoassay for the detection of fentanyl in urine. Forensic Science International, 206, 127–131. [DOI] [PubMed] [Google Scholar]
- (39).Shou WZ, Jiang X, Beato BD and Naidong W (2001) A highly automated 96-well solid phase extraction and liquid chromatography/tandem mass spectrometry method for the determination of fentanyl in human plasma. Rapid Communications in Mass Spectrometry, 15, 466–476. [DOI] [PubMed] [Google Scholar]
- (40).Musshoff F, Trafkowski J, Kuepper U and Madea B (2006) An automated and fully validated LC-MS/MS procedure for the simultaneous determination of 11 opioids used in palliative care, with 5 of their metabolites. Journal of Mass Spectrometry, 41, 633–640. [DOI] [PubMed] [Google Scholar]
- (41).Øiestad EL, Johansen U, Øiestad AML and Christophersen AS (2011) Drug screening of whole blood by ultra-performance liquid chromatography-tandem mass spectrometry. Journal of Analytical Toxicology, 35, 280–293. [DOI] [PubMed] [Google Scholar]
- (42).Pedersen AJ, Dalsgaard PW, Rode AJ, Rasmussen BS, Müller IB, Johansen SS, et al. (2013) Screening for illicit and medicinal drugs in whole blood using fully automated SPE and ultra-high-performance liquid chromatography with TOF-MS with data-independent acquisition. Journal of Separation Science, 36, 2081–2089. [DOI] [PubMed] [Google Scholar]
- (43).Remane D, Wetzel D and Peters FT (2014) Development and validation of a liquid chromatography-tandem mass spectrometry (LC-MS/MS) procedure for screening of urine specimens for 100 analytes relevant in drug-facilitated crime (DFC). Analytical and Bioanalytical Chemistry, 406, 4411–4424. [DOI] [PubMed] [Google Scholar]
- (44).Fisichella M, Odoardi S and Strano-Rossi S (2015) High-throughput dispersive liquid/liquid microextraction (DLLME) method for the rapid determination of drugs of abuse, benzodiazepines and other psychotropic medications in blood samples by liquid chromatography-tandem mass spectrometry (LC-MS/MS) and application to forensic cases. Microchemical Journal, 123, 33–41. [Google Scholar]
- (45).Chepyala D, Tsai IL, Liao H-W, Chen G-Y, Chao H-C and Kuo C-H (2017) Sensitive screening of abused drugs in dried blood samples using ultra-high-performance liquid chromatography-ion booster-quadrupole time-of-flight mass spectrometry. Journal of Chromatography A, 1491, 57–66. [DOI] [PubMed] [Google Scholar]
- (46).Wang L and Bernert JT (2006) Analysis of 13 Fentanils, Including Sufentanil and Carfentanil, in Human Urine by Liquid Chromatography-Atmospheric-Pressure Ionization-Tandem Mass Spectrometry. Journal of Analytical Toxicology, 30, 335–341. [DOI] [PubMed] [Google Scholar]
- (47).Gergov M, Nokua P, Vuori E and Ojanperä I (2009) Simultaneous screening and quantification of 25 opioid drugs in post-mortem blood and urine by liquid chromatography-tandem mass spectrometry. Forensic Science International, 186, 36–43. [DOI] [PubMed] [Google Scholar]
- (48).Shaner RL, Kaplan P, Hamelin EI, Bragg WA and Johnson RC (2014) Comparison of two automated solid phase extractions for the detection of ten fentanyl analogs and metabolites in human urine using liquid chromatography tandem mass spectrometry. Journal of Chromatography B: Analytical Technologies in the Biomedical and Life Sciences, 962, 52–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Strayer KE, Antonides HM, Juhascik MP, Daniulaityte R and Sizemore IE (2018) LC-MS/MS-Based Method for the Multiplex Detection of 24 Fentanyl Analogues and Metabolites in Whole Blood at Sub ng mL–1 Concentrations. ACS Omega, 3, 514–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Moody MT, Diaz S, Shah P, Papsun D and Logan BK (2018) Analysis of Fentanyl Analogs and Novel Synthetic Opioids in Blood, Serum/Plasma and Urine in Forensic Casework. Drug Testing and Analysis. doi: 10.1002/dta.2393 [DOI] [PubMed] [Google Scholar]
- (51).Millington DS, Zhang H, Moseley MA, Thompson JW, and Smith P (2014) Direct solvent extraction and analysis of biomarkers in dried blood spots using a flow-through autosampler. In Li W and Lee M (eds.), Dried Blood Spots, John Wiley and Sons, Inc., Hoboken, NJ, pp. 314–324. [Google Scholar]
- (52).Wagner M, Tonoli D, Varesio E and Hopfgartner G (2016) The use of mass spectrometry to analyze dried blood spots. Mass Spectrometry Reviews, 35, 361–438. [DOI] [PubMed] [Google Scholar]
- (53).Verplaetse R and Henion J (2016) Quantitative determination of opioids in whole blood using fully automated dried blood spot desorption coupled to on-line SPE-LC-MS/MS. Drug Testing and Analysis, 8, 30–38. [DOI] [PubMed] [Google Scholar]
- (54).Shaner R, Schulze ND, Seymour C, Hamelin EI, Thomas JD and Johnson RC (2017) Quantitation of fentanyl analogs in dried blood spots by flow-through desorption coupled to online solid phase extraction tandem mass spectrometry. Analytical Methods, 9, 3876–3883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Clavijo CF, Thomas JJ, Cromie M, Schniedewind B, Hoffman KL, Christians U, et al. (2011) A low blood volume LC-MS/MS assay for the quantification of fentanyl and its major metabolites norfentanyl and despropionyl fentanyl in children. Journal of Separation Science, 34, 3568–3577. [DOI] [PubMed] [Google Scholar]
- (56).Odoardi S, Anzillotti L and Strano-Rossi S (2014) Simplifying sample pretreatment: Application of dried blood spot (DBS) method to blood samples, including postmortem, for UHPLC-MS/MS analysis of drugs of abuse. Forensic Science International, 243, 61–67. [DOI] [PubMed] [Google Scholar]
- (57).Meuldermans WEG, Hurkmans RMA and Heykants JJP (1982) Plasma protein binding and distribution of fentanyl, sufentanil, alfentanil and lofentanil in blood. Archives Internationales de Pharmacodynamie et de Therapie, 257, 4–19. [PubMed] [Google Scholar]
- (58).Przybyciel M (2006) Novel Phases for HPLC Separations. LC-GC LC Column Technology Supplement, 24, 49–52. [Google Scholar]
- (59).Mommers J, Mengerink Y, Ritzen E, Weusten J, van der Heijden J and van der Wal S (2013) Quantitative analysis of morphine in dried blood spots by using morphine-d3 pre-impregnated dried blood spot cards. Analytica Chimica Acta, 774, 26–32. [DOI] [PubMed] [Google Scholar]
- (60).US Department of Health and Human Services, Food and Drug Administration. (2016) In Guidance for Industry: Bioanalytical Method Validation. Food and Drug Administration, Washington, DC. [Google Scholar]
- (61).Palmer RB (2010) Fentanyl in postmortem forensic toxicology. Clinical Toxicology, 48, 771–784. [DOI] [PubMed] [Google Scholar]
- (62).Andresen H, Gullans A, Veselinovic M, Anders S, Schmoldt A, Iwersen-Bergmann S, et al. (2012) Fentanyl: Toxic or therapeutic? Postmortem and antemortem blood concentrations after transdermal fentanyl application. Journal of Analytical Toxicology, 36, 182–194. [DOI] [PubMed] [Google Scholar]
- (63).Chatterton CN and Scott-Ham M (2018) The distribution and redistribution of fentanyl and norfentanyl in post mortem samples. Forensic Science International, 284, 146–152. [DOI] [PubMed] [Google Scholar]
- (64).Denniff P and Spooner N (2010) The effect of hematocrit on assay bias when using DBS samples for the quantitative bioanalysis of drugs. Bioanalysis, 2, 1385–1395. [DOI] [PubMed] [Google Scholar]
- (65).Hempen CM, Maarten Koster EH and Ooms JA (2015) Hematocrit-independent recovery of immunosuppressants from DBS using heated flow-through desorption. Bioanalysis, 7, 2019–2029. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.