

Abstract
Mutations of ankyrin-1 are the most frequent cause of the inherited hemolytic anemia, hereditary spherocytosis (HS), in people of European ancestry. Ankyrin-1, which provides the primary linkage between the erythrocyte membrane skeleton and the plasma membrane, has numerous isoforms generated by alternative splicing, alternate polyadenylation, use of tissue-specific promoters, and alternate NH2 or COOH-termini. Mutation detection in erythrocyte membrane protein genes, including ankyrin, has been a challenge, primarily due to the large size of these genes, and the apparent frequent occurrence of HS-associated null alleles. Using denaturing high-performance liquid chromatography (DHPLC), we screened the ankyrin gene of the proband of a large, three generation African–American kindred with ankyrin-deficient HS. DHPLC yielded an abnormal chromatogram for exon 1. Examination of the corresponding exon 1 sequence in genomic DNA from the proband revealed heterozygosity for a mutation of the initiator methionine (ATG to ATA Met 1 Ile). Coupled in vitro transcription/translation studies with rabbit reticulocyte lysates demonstrated that the wild-type ankyrin erythroid cDNA initiates only from the known initiator methionine, indicating that the use of alternate initiator methionine is not a mechanism of isoform diversity in erythroid cells. The mutant ankyrin allele, unlike some initiator methionine mutations that utilize downstream codons for translation initiation, was associated with a null allele. This is the first report describing ankyrin-linked HS in an African–American kindred.
Introduction
Hereditary spherocytosis (HS) is a common congenital hemolytic anemia characterized by the presence of spherical-shaped erythrocytes on peripheral blood smear. HS affects patients in all ethnic groups worldwide and it is the most common inherited anemia affecting the people of European ancestry [1]. The primary biochemical defects in HS leading to membrane loss are in the proteins of the erythrocyte membrane, particularly those proteins involved in the interactions between the membrane skeleton and the lipid bilayer, including ankyrin, band 3, α-spectrin, β-spectrin, and protein 4.2.
Knowledge of the primary structure of these proteins has allowed determination of the precise genetic defects in many cases of HS [1,2]. Analyses of these mutations will provide additional insights into the pathogenesis of HS, facilitate the understanding of the genetic bases of HS in various populations, and allow for rapid, specific DNA diagnosis, particularly important for transfusion-dependent patients, where transfused red cells hinder standard diagnostic testing, and for kindreds who have produced affected fetuses with severe anemia and its often fatal complications. These studies will also provide important information on the structure and function of these proteins both in the red cell, and in other cell types, where mutation of these and homologous proteins has been associated with abnormalities in nonerythroid cells [2].
Ankyrin deficiency is the most common biochemical abnormality observed in the erythrocyte membranes of HS patients [3–13]. In the erythrocyte, ankyrin provides the primary linkage between the spectrin-based membrane skeleton and the plasma membrane. Ankyrin-1, initially discovered in preparations of erythrocyte membranes, is the prototype of a homologous family of multifunctional proteins involved in the local segregation of integral membrane proteins within functional domains on the plasma membrane [2]. This important cellular localization of membrane proteins is provided by the relative affinities of the many different tissue-specific and developmentally regulated isoforms of ankyrin for target proteins. The composition and specialized functions of cellular membranes differ in erythroid and nonerythroid cells. This specialization appears to have evolved from the tissue-specific, developmentally regulated expression of multiple protein isoforms. Ankyrin isoform diversity arises from different gene products, from differential, alternative splicing of the same gene product, from alternate polyadenylation, from the use of tissue-specific promoters, and/or the use of alternate NH2 or COOH-termini [14–29]. Although numerous ankyrin-1 isoforms are visualized on Western blots of erythrocyte membranes [16], it is unknown if isoforms of ankyrin-1 are generated in erythroid cells by use of alternate translation initiation codons.
Mutation screening has identified a number of ankyrin gene mutations in HS individuals, primarily point mutations in the coding region that lead to unstable mRNA and ankyrin deficiency [30,31]. Detection of HS-associated ankyrin mutations has been difficult. The ankyrin gene is encoded by 42 exons spread over ~200 kb of genomic DNA [32]. In most cases, ankyrin gene mutations are not only unique to individual kindreds (private mutations) but are also located throughout the coding exons and promoter region of the ankyrin gene. Biochemical studies typically only reveal ankyrin deficiency and do not provide clues to where causative mutations are located. Finally, decreased ankyrin mRNA has been observed in up to 30% of HS-associated ankyrin gene mutations, making cDNA-based mutation detection strategies problematic [33].
We recently developed a denaturing high-performance liquid chromatography (DHPLC)-based screening method for rapid and sensitive detection of HS-associated ankyrin gene mutations in genomic DNA [34]. DHPLC is a high-throughput, economical, and highly sensitive technique based on the detection of heteroduplexes in PCR amplicons by ion-pair reverse phase HPLC under partially denaturing conditions. Using this technique, we studied a large, three generation African–American kindred with typical, dominant, ankyrin-deficient HS, where other screening methods had failed to detect any ankyrin gene abnormalities, and identified a mutation in the initiator methionine, ankyrinNew Haven, the first of this type described in the ankyrin gene. Coupled in vitro transcription/translation studies with rabbit reticulocyte lysates demonstrated that wild-type ankyrin utilizes a single translation initiation codon, indicating that the use of alternate initiator methionine is not a mechanism of isoform diversity in erythroid cells. The mutant ankyrin allele, unlike some initiator methionine mutations that utilize downstream codons for translation initiation, was associated with a null allele. This is the first report describing ankyrin-linked HS in an African–American kindred.
Results
Patients
We studied a three-generation African–American kindred with typical, autosomal dominant HS. At presentation, the proband was investigated for unexplained splenomegaly, diagnosed while being evaluated for adenocarcinoma of the sigmoid colon. The patient had no history of hematologic disease. The patient’s brother had also suffered from splenomegaly and his sister had undergone cholecystectomy for cholelithiasis. Other than the splenomegaly, the only other physical finding was scleral icterus. The patient’s blood count was hemoglobin 11.8 g/dL, hematocrit 36.9%, mean corpuscular volume 96 fL, and mean corpuscular hemoglobin concentration 36.6%. Reticulocyte count was 5.6%. Peripheral blood films showed polychromasia and spherocytosis (see Fig. 1). The total bilirubin was 5.5 mg/dL with an indirect fraction of 4.6 mg/dL. His erythrocytes demonstrated marked increase in osmotic fragility after incubation.
Figure 1.
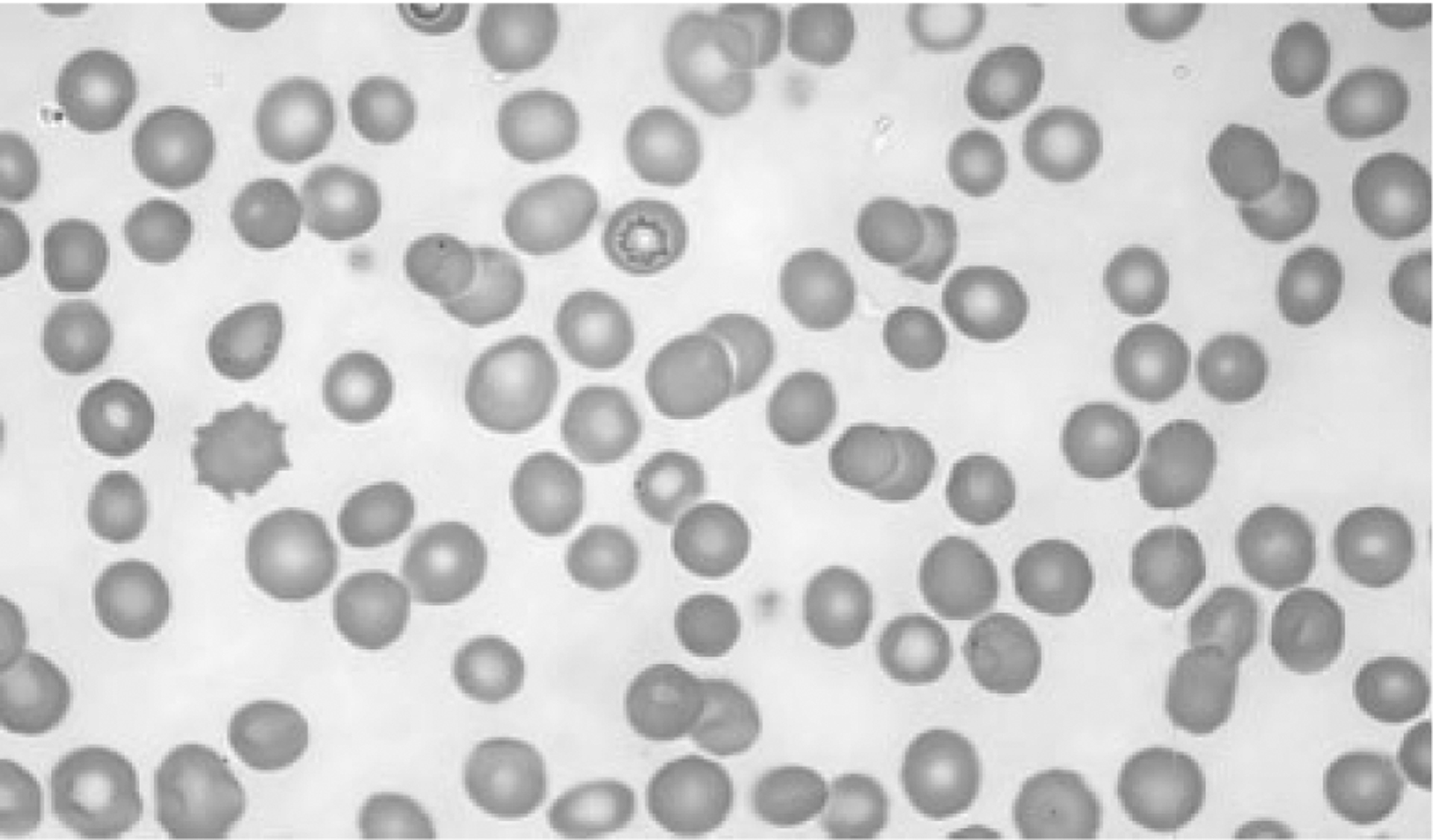
Peripheral blood smear. Wright-Giemsa stained peripheral blood smear from the proband reveals polychromasia and spherocytosis.
Further investigation revealed that numerous family members, including a sister and brother, as well as all three of the affected brother’s children, and one grandchild were affected with HS.
Qualitative and quantitative analyses of erythrocyte membrane proteins
One-dimensional SDS-PAGE analyses of erythrocyte membranes from the proband were qualitatively normal and no abnormal isoforms of any major membrane proteins were observed (data not shown). Quantitative analyses of spectrin and ankyrin content, as measured by the ratios of spectrin/band 3 and ankyrin/band 3, respectively, are shown in Table I. The erythrocyte membranes of the proband demonstrated combined ankyrin and spectrin deficiency at a level typical for other cases of ankyrin-linked, mild-to-moderate HS, ~70–90%.
TABLE I.
Biochemical Studies
| Spectrin/band 3 ratio | Ankyrin/band 3 ratio | |
|---|---|---|
| Proband | 0.78 ± 0.08 | 0.14 ± 0.01 |
| Controla | 1.0 ± 0.10 | 0.20 ± 0.04 |
Control values are mean ± 2 SD.
Mutation screening
To search for HS mutations, the ankyrin gene was initially screened by single-stranded conformational polymorphism (SSCP) and HA of amplified genomic DNA from the HS proband. No abnormal patterns were detected by SSCP or HA in the proband. DHPLC identified several variant chromatogram patterns. Nucleotide sequence analyses of the corresponding regions identified several sequence variations (Table II). One variant chromatogram (Fig. 2A) corresponded to exon 1 and flanking sequences. Examination of the exon 1 sequence in amplified genomic DNA from the proband revealed heterozygosity for a mutation of the initiator methionine, ATG to ATA, Met 1 Ile (Fig. 2B).
TABLE II.
Ankyrin Gene Variants Detected by Denaturing High-Performance Liquid Chromatography and Nucleotide Sequencing
| Location | Variant | Name |
|---|---|---|
| Exon 1 | 5′UTR + 115 G insertion | |
| Exon 1 | Codon 1 ATG-ATA | AnkyrinNew Haven |
| Intron 25 | T-G 50 bp before exon 26 | |
| Intron 26 | C-T 46 bp after end exon 26 |
Figure 2.
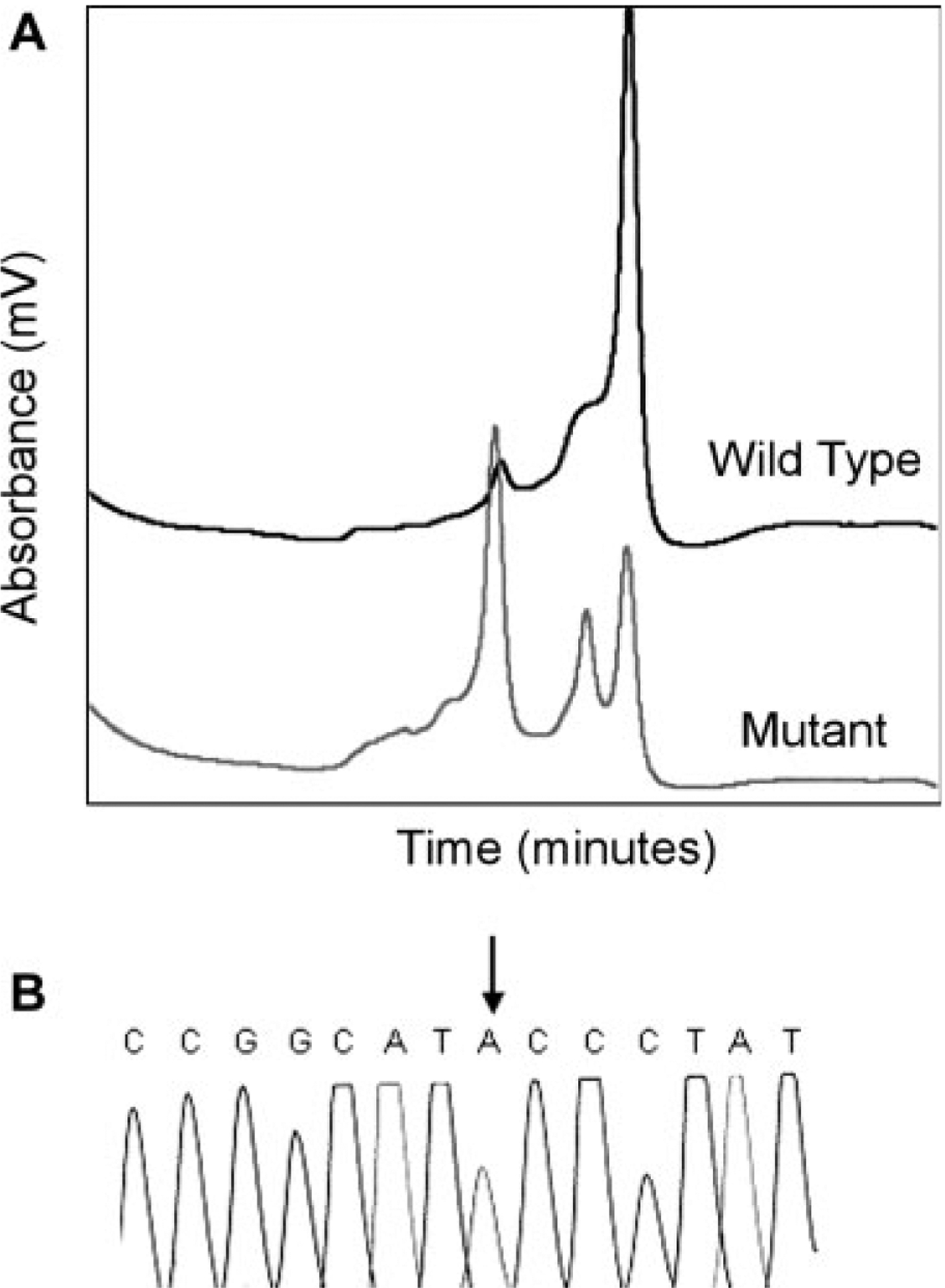
Detection of the ankyrinNew Haven mutation. A: Chromatograms corresponding to exon 1 of the ankyrin gene. Compared to wild-type (black), the proband (gray) exhibits a variant chromatogram. B: Partial nucleotide sequence of exon 1 of the ankyrin gene from a PCR-amplified, subcloned fragment from the spherocytosis proband. The wild-type sequence ATG encoding codon 1 is mutated to ATA, changing the initiator methionine to isoleucine.
Molecular genetic analyses
The mutation abolishes a recognition site for the enzyme SphI, GCATGC, in exon 1, allowing for rapid PCR-based detection in genomic DNA of family members. Genomic DNA corresponding to exon 1 and flanking DNA of the ankyrin gene was amplified by PCR. Amplification products were digested with SphI, fractionated by agarose gel electrophoresis, and visualized by ethidium bromide staining (see Fig. 3). Amplification products from a normal control yielded a 232-bp fragment which after SphI digestion cleaves into fragments of 155 and 77 bp. Amplification products from unaffected family members demonstrated patterns of SphI digestion similar to control. Digestion of amplification products from the proband and other affected HS family members yielded fragments of 155 and 77 bp, as well as an undigested fragment of 232 bp, indicating heterozygosity for the ankyrinNew Haven allele.
Figure 3.
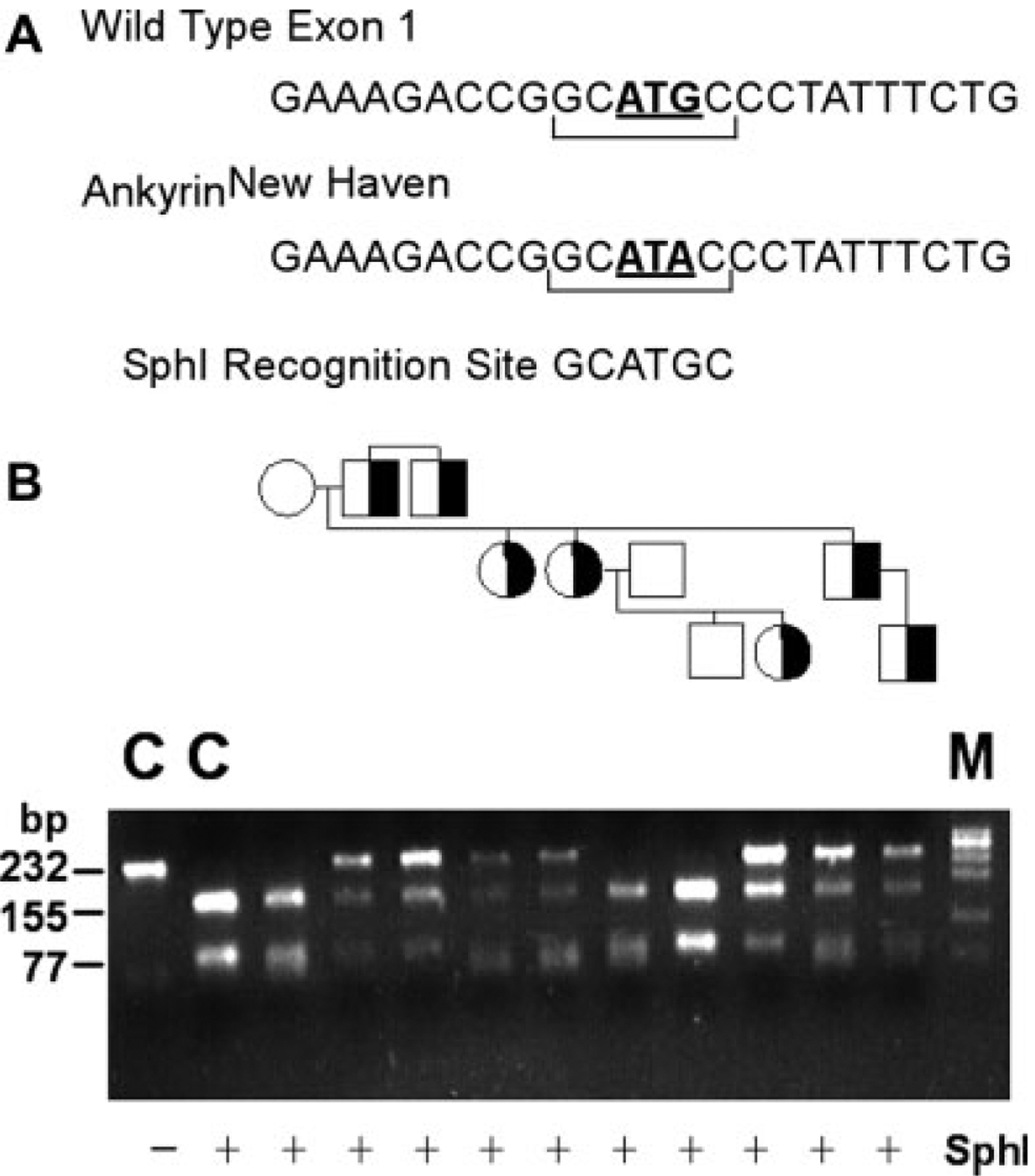
Analysis of the ankyrinNew Haven mutation in a three generation, African–American kindred. A: The ankyrinNew Haven mutation abolishes a SphI restriction site, allowing discrimination of wild-type and mutant alleles. B: The region of the ankyrin gene containing the ankyrinNew Haven mutation was amplified by PCR from genomic DNA of the proband, his family members, and a normal control (C), amplification products digested with SphI, fractionated by agarose gel electrophoresis, and visualized by ethidium bromide staining. The control PCR product yields a 232 bp fragment (Lane 1) which after SphI digestion cleaves into fragments of 155 and 77 bp (Lane 2). Amplification products from unaffected patients demonstrate patterns of SphI digestion similar to control. Digestion of amplification products from spherocytosis patients yields fragments of 155 and 77 bp, as well as an undigested fragment of 232 bp, indicating heterozygosity for the ankyrinNew Haven allele. Half-filled symbols indicate hereditary spherocytosis individuals. M indicates marker.
In vitro transcription/translation
Some proteins utilize alternate translation initiation codons as a mechanism of generating isoform diversity. Moreover, some disease-associated initiator methionine mutations lead to translation initiation from a downstream methionine or other amino acid, producing a truncated protein that retains partial or full function. To address these possibilities, in vitro transcription/translation studies were performed with full-length wild-type or ankyrinNew Haven plasmids, rabbit reticulocyte lysate, and antibodies corresponding to either the NH2-terminus or full length erythrocyte ankyrin.
To validate the NH2-terminal anti-peptide ankyrin antibody, immunoblots using human erythrocyte membrane ghosts were performed. The affinity-purified NH2-terminal anti-peptide antibody raised against NH2-terminal sequences of ankyrin-1 detected bands of 200, 205, and 210 kDa, appropriate for erythrocyte ankyrin. A well-characterized polyclonal antibody raised to full length ankyrin detected comparable bands of 200, 205, and 210 kDa in erythrocyte ghosts [20,29], validating the anti-peptide antibody for use in immunoblotting ankyrin (see Fig. 4).
Figure 4.
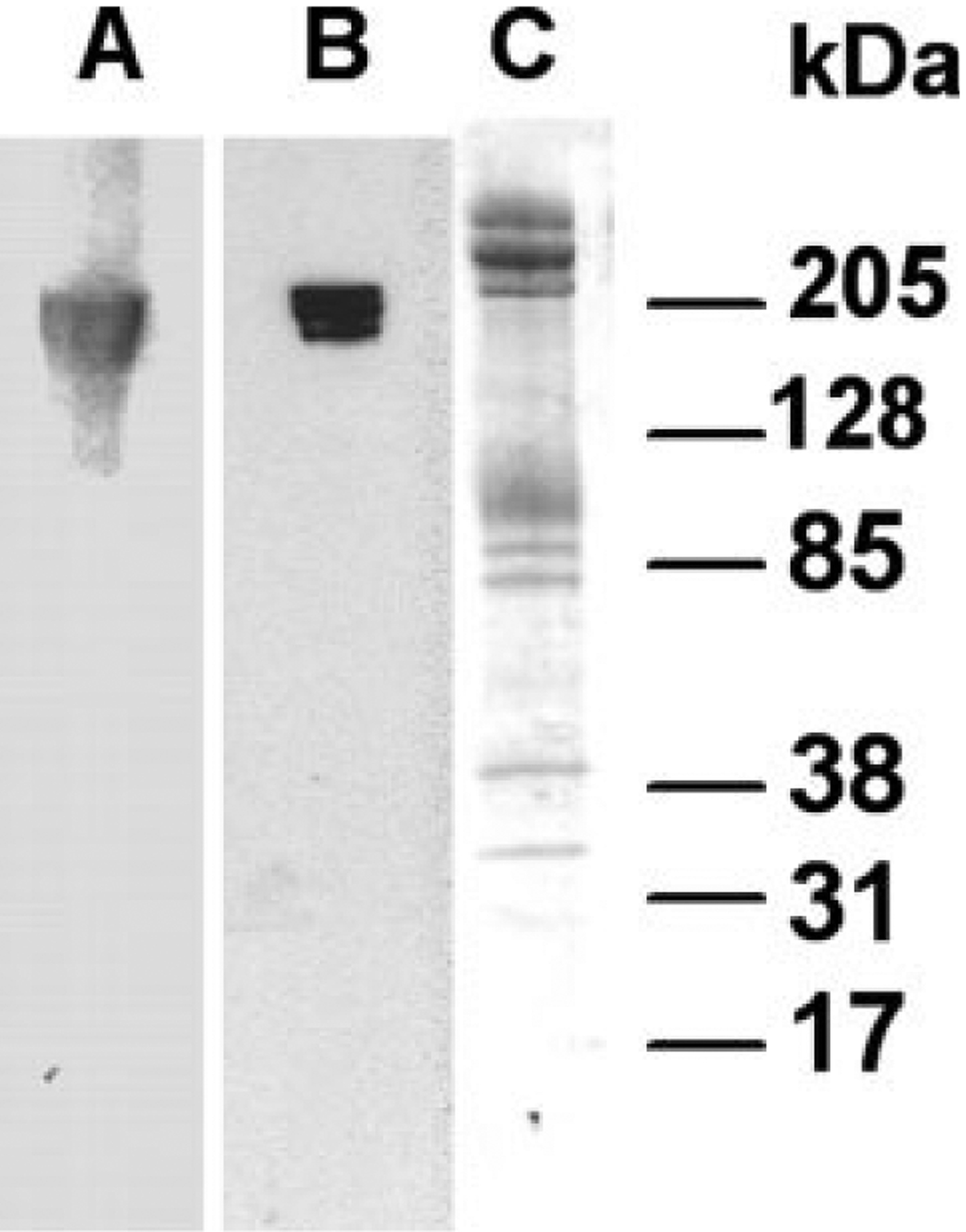
Immunoblotting with ankyrin-1 specific antibodies. Human erythrocyte membrane ghosts were separated by SDS-PAGE in a 4–20% gel. The gel was cut and part transferred to an Immobilon-P. A: Incubation of a strip of this membrane to a polyclonal antibody raised to full length erythrocyte ankyrin detected bands of 200, 205, and 210 kDa in erythrocyte membranes. B: Incubation of a separate strip of this membrane with an anti-peptide antibody raised to ankyrin sequences corresponding to codons 1–13 identified bands of 200, 205, and 210 kDa in erythrocyte membranes. C: Strip of the same gel stained with Coomassie Blue.
After transcription/translation of wild-type and ankyrinNew Haven plasmids, products were separated on polyacrylamide gels, transferred to Immobilon-P membranes, and immunoblotted with the antibodies described earlier. The wild-type plasmid yielded a single 210-kDa protein detected by the full-length and NH2-terminal ankyrin antibodies, demonstrating that the wild-type ankyrin erythroid cDNA initiates only from the known initiator methionine. This indicates that use of an alternate initiator methionine is not a mechanism of ankyrin isoform diversity in erythroid cells. The ankyrinNew Haven plasmid did not yield any functional protein detectable by either antibody (see Fig. 5). This indicates that unlike some initiator methionine mutations that utilize downstream codons for translation initiation, the ankyrinNew Haven initiator methionine mutation is associated with a null ankyrin allele.
Figure 5.
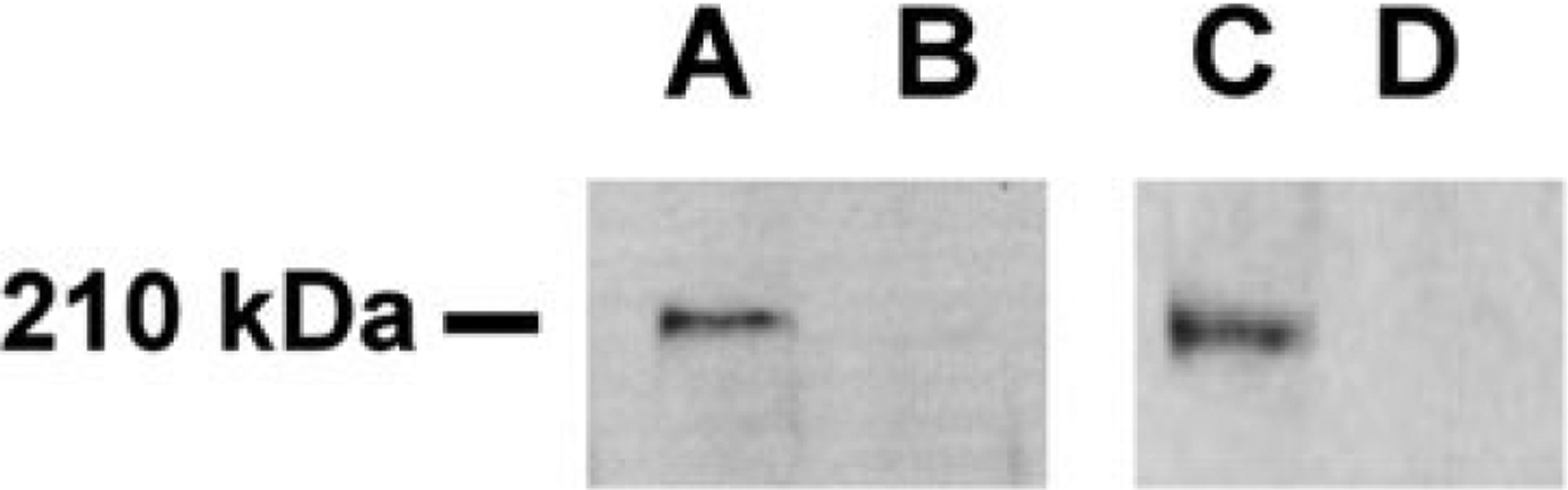
Immunoblotting of in vitro transcription/translation products using ankyrin antibodies. Products of in vitro transcription/translation experiments were analyzed by SDS-PAGE on 4–20% gels and transferred to Immobilon-P membranes. A: Incubation of a strip of this membrane containing products of wild-type plasmids with a NH2-terminal ankyrin antibody recognized a band of ~210 kDa. B: Incubation of a strip of immobilon-P membrane containing products of AnkyrinNew Haven plasmids with a NH2-terminal ankyrin antibody did not recognize any bands. C: Incubation of a strip of Immobilon-P membrane containing products of wild-type plasmids with a full length ankyrin antibody recognized a band of ~210 kDa. D: Incubation of a strip of Immobilon-P membrane containing products of AnkyrinNew Haven plasmids with full length ankyrin antibody did not recognize any bands.
Discussion
Deficiency of ankyrin is the most common biochemical abnormality found in the erythrocyte membranes of patients with HS, often with concomitant spectrin deficiency [3–13]. Decreased ankyrin synthesis or accumulation on the erythrocyte membrane leads to decreased assembly of spectrin on the membrane because the spectrin binding sites in ankyrin may be decreased, absent, or defective. HS gene mutation detection has primarily been carried out in Europeans, where, not unexpectedly, mutations of the ankyrin gene are the most common abnormality detected [1,31]. These are typically point mutations associated with decreased or absent expression of the mutant allele, including frame shift, nonsense, and splicing mutations. This is the first report of an initiation codon mutation of the ankyrin gene associated with HS that, too, is associated with a null ankyrin allele.
HS is found worldwide, but its incidence and prevalence in non-European populations are not clearly established. Similarly, the genetic bases of HS in non-European populations have not been defined. The exceptions are Japan and Brazil, where mutation screening has been performed [35,36]. These studies found that ankyrin gene mutations were less common than in Europeans. In Japan, mutations in the protein 4.2 gene were frequently found, an uncommon finding in European HS patients [35].
Cases of HS in Africans and African–Americans have been described, but molecular studies have not been performed [37,38]. This is the first report describing ankyrin-linked HS in an African–American HS kindred. Study of the additional affected patients of African descent will shed light on the predominant etiology of HS in this population.
Utilization of alternate translation initiation codons was initially thought of as an uncommon biologic process, limited to yeast, plants, and lower organisms. However, there has been growing recognition of the use of alternate initiation codons in mammalian cells, often producing proteins with very different functions [39–41], as well as recognition of new mechanisms of translational control of protein structure and function [42,43]. Initiator codon mutations are uncommon, but typically they are associated with null alleles as no functional protein is produced [44–47]. However, in a few cases, translation of the mutant allele initiates from a downstream methionine or other amino acid, producing a truncated protein that retains partial or full function [48–50]. The in vitro transcription/translation experiments indicated that the wild-type ankyrin cDNA initiates only from the known initiator methionine and that the ankyrinNew Haven cDNA does not utilize an alternate translation initiation codon.
Ankyrin is of critical importance in both erythroid and nonerythroid cells. However, the large size of the ankyrin molecule has complicated its study at the genetic, biochemical, and structural levels. The identification and characterization of mutations in patients with defined phenotypes greatly clarifies and facilitates the focus on critical regions of ankyrin and permits the in vitro and in vivo testing of fundamental questions in membrane and cellular biology. A DHPLC-based screening strategy identified the mutation in this ankyrin-deficient kindred, even when other techniques (SSCP, HA) failed.
Mutation detection in very large genes with mutations spread across the entire gene has been a challenge, as conventional methods for large-scale are expensive, technically demanding, and time-consuming. Similarly, mutation detection in the erythrocyte membrane protein genes associated with inherited hemolytic anemia, including alpha spectrin (53 exons), ankyrin (42 exons), beta spectrin (32 exons), band 3 (20 exons), and protein 4.2 (13 exons), has been prohibitively expensive and laborious. DHPLC offers a high-capacity low-cost mutation detection method that is highly sensitive, high-capacity, semiautomated, low-cost method using ion-pairing reverse phase chromatography (IPRPC) columns for the screening of gene mutations and polymorphisms [51,52]. This technique of mutation detection has been validated for a number of genes, including several very large genes such as dystrophin (79 exons) and fibrillin-1 (65 exons) [53,54]. It is anticipated that either massively parallel sequencing of large number of exons or whole genome sequencing techniques will evolve to allow rapid, DNA-based diagnosis in many genetic disorders [55–59]. Until such technology becomes widely accessible and affordable, DHPLC offers a viable strategy for mutation screening.
Methods
Erythrocyte membrane preparation and quantitation of protein content
Erythrocyte membranes were prepared from peripheral blood as previously described [60,61]. Membrane proteins were analyzed by electrophoresis in SDS-PAGE either with a 5–15% polyacrylamide gradient as described by Laemmli [62] or using a 3.5% polyacrylamide gel as described by Fairbanks et al. [60]. To estimate spectrin/band-3 and ankyrin/band ratios, SDS polyacrylamide slab gels were scanned after Coomassie blue staining using a DU8 spectrophotometer (Beckman Instruments, Fullerton, CA) at 550 nm.
Mutation detection
Genomic DNA from the proband was amplified by PCR using pairs of primers flanking the 42 coding exons and promoter region of the ankyrin-1 gene. Amplification products were subjected to SSCP according to Eber et al. [30]. The same PCR amplification products were also subjected to HA in mutation detection enhancement gels (MDE, Cambrex Bioproducts) as described [63].
DHPLC was performed on a WAVE DNA fragment analysis system (Transgenomic, Omaha, Nebraska) as described. Primer pairs were designed using Primer 3 and WAVEmaker software version 4.1.40 (Transgenomic) to optimize the conditions for both PCR amplification and DHPLC separation. Primers were typically ~40 nucleotides away from the intron-exon boundary to facilitate the detection of splice junction mutations. PCR amplification products were analyzed by ethidium bromide staining agarose gel electrophoresis to ensure robust amplification. Patient and wild-type amplification products were mixed in a 1:1 ratio before DHPLC analysis. Each sample was subjected to denaturation (5 min at 94°C) followed by reannealing, with the sample temperature gradually decreased from 94°C to the desired temperature. Amplification products were separated on a DNASep column (Transgenomics) by ion-pair reversed-phase liquid chromatography through a 2% linear acetonitrile gradient under partially denaturing conditions at the desired temperature. Temperatures for DHPLC were determined by melting domain profile analysis of each amplicon to produce a ≥90% double helical fraction of wild-type DNA. When two or more distinct melting domains were predicted, DHPLC analysis was performed at two or more different temperatures. DHPLC chromatograms were compared with wild-type for analysis.
SSCP, HA, or DHPLC was repeated on samples with abnormal patterns (SSCP, HA) or chromatograms (DHPLC) on initial screening. Genomic DNA fragments demonstrating a consistently abnormal pattern were amplified in a second PCR reaction, subcloned and subjected to nucleotide sequence analysis, or directly sequenced.
Molecular genetic analyses
The mutant ankyrinNew Haven allele creates an SphI recognition site, GCATGC, allowing discrimination of wild-type and mutant alleles by SphI restriction enzyme digestion. Primers flanking the mutation, 5′-CCCGGCCCGACAGCAAGCGCCTCTG-3′ (sense) and 5′-GTGGCCCCCTCCTGACATCTCCCCG-3′ (antisense) were used to amplify genomic DNA from the proband, his family members, and a control. Amplification products were digested with SphI, fractionated by electrophoresis in a 3% agarose gel, and visualized by ethidium bromide staining.
In vitro transcription/translation assays
Plasmids.
The coding region of the human ankyrin-1 cDNA was cloned into pGEM7 (Promega). The initiator methionine mutation was introduced using PCR mutagenesis followed by DpnI digestion, religation, and plasmid isolation. The nucleotide sequence of both plasmids was verified by DNA sequencing.
Antibodies.
Polyclonal antibodies raised to full length 2.1 erythrocyte ankyrin [20,29] or a peptide representing the NH2-terminus of the ankyrin protein were utilized. This anti-peptide antibody was produced by immunizing the rabbits with the peptide MPYSVGFREANAA, conjugated to keyhole limpet hemocyanin. This peptide corresponds to codons 1–13 and is located at the NH2-terminal of the second methionine in ankyrin at codon 60.
In vitro transcription/translation.
The rabbit reticulocyte coupled transcription/translation system (TNT, Promega) was utilized for expression of ankyrin proteins in vitro. TnT reactions containing the pGEM7 ankyrin cDNA plasmid constructs, T7 RNA polymerase, rabbit reticulocyte lysate, and [35S]methionine were carried out at 30°C for 90 min as described by the manufacturer. Approximately 1 μg of plasmid DNA and 3 μL [35S]methionine were used in 50 μL assays.
Western blotting.
From the reaction mixture, 5 μL aliquots were added to Laemmli sample buffer. Reaction mixture and human ghost proteins were analyzed using 4–20% gradient minigels and transferred to Immobilon-P membranes overnight at 30 V. Membranes were incubated with either the full length or NH2-terminal ankyrin antibodies for 1 hr at 21°C. After washing, membranes were incubated with anti-rabbit IgG horseradish peroxidase secondary antibody for 30 min at 21°C. Detection of bands was achieved using Western Lightning chemiluminescence reagent (Perkin Elmer LAS, Boston, MA).
Contract grant numbers:
NICHD T32 HD07094 to JS, NIDDK RO1DK19482 to BGF, and NIDDK RO1DK62039 to PGG.
Footnotes
Conflict of Interest: Nothing to Report.
References
- 1.Eber S, Lux SE. Hereditary spherocytosis—Defects in proteins that connect the membrane skeleton to the lipid bilayer. Semin Hematol 2004;41:118–141. [DOI] [PubMed] [Google Scholar]
- 2.Bennett V, Healy J. Organizing the fluid membrane bilayer: Diseases linked to spectrin and ankyrin. Trends Mol Med 2008;14:28–36. [DOI] [PubMed] [Google Scholar]
- 3.Lanciotti M, Perutelli P, Valetto A, et al. Ankyrin deficiency is the most common defect in dominant and non dominant hereditary spherocytosis. Haematologica 1997;82:460–462. [PubMed] [Google Scholar]
- 4.Lee YK, Cho HI, Park SS, et al. Abnormalities of erythrocyte membrane proteins in Korean patients with hereditary spherocytosis. J Korean Med Sci 2000;15:284–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Miraglia del Giudice E, Iolascon A, Pinto L, et al. Erythrocyte membrane protein alterations underlying clinical heterogeneity in hereditary spherocytosis. Br J Haematol 1994;88:52–55. [DOI] [PubMed] [Google Scholar]
- 6.Pekrun A, Eber SW, Kuhlmey A, et al. Combined ankyrin and spectrin deficiency in hereditary spherocytosis. Ann Hematol 1993;67:89–93. [DOI] [PubMed] [Google Scholar]
- 7.Premetis E, Stamoulakatou A, Loukopoulos D. Erythropoiesis: Hereditary spherocytosis in Greece—Collective data on a large number of patients. Hematology 1999;4:361–366. [DOI] [PubMed] [Google Scholar]
- 8.Ricard MP, Gilsanz F, Millan I. Erythroid membrane protein defects in hereditary spherocytosis. A study of 62 Spanish cases. Haematologica 2000;85: 994–995. [PubMed] [Google Scholar]
- 9.Saad ST, Costa FF, Vicentim DL, et al. Red cell membrane protein abnormalities in hereditary spherocytosis in Brazil. Br J Haematol 1994;88:295–299. [DOI] [PubMed] [Google Scholar]
- 10.Savvides P, Shalev O, John KM, et al. Combined spectrin and ankyrin deficiency is common in autosomal dominant hereditary spherocytosis. Blood 1993;82:2953–2960. [PubMed] [Google Scholar]
- 11.Boguslawska DM, Heger E, Chorzalska A, et al. Hereditary spherocytosis: Identification of several HS families with ankyrin and band 3 deficiency in a population of southwestern Poland. Ann Hematol 2004;83:28–33. [DOI] [PubMed] [Google Scholar]
- 12.Rocha S, Rebelo I, Costa E, et al. Protein deficiency balance as a predictor of clinical outcome in hereditary spherocytosis. Eur J Haematol 2005;74:374–380. [DOI] [PubMed] [Google Scholar]
- 13.Sanchez-Lopez JY, Camacho AL, Magana MT, et al. Red cell membrane protein deficiencies in Mexican patients with hereditary spherocytosis. Blood Cells Mol Dis 2003;31:357–359. [DOI] [PubMed] [Google Scholar]
- 14.Bennett V Immunoreactive forms of human erythrocyte ankyrin are present in diverse cells and tissues. Nature 1979;281:597–599. [DOI] [PubMed] [Google Scholar]
- 15.Luna EJ, Kidd GH, Branton D. Identification by peptide analysis of the spectrin-binding protein in human erythrocytes. J Biol Chem 1979;254:2526–2532. [PubMed] [Google Scholar]
- 16.Peters LL, Lux SE. Ankyrins: Structure and function in normal cells and hereditary spherocytes. Semin Hematol 1993;30:85–118. [PubMed] [Google Scholar]
- 17.Birkenmeier CS, Sharp JJ, Gifford EJ, et al. An alternative first exon in the distal end of the erythroid ankyrin gene leads to production of a small isoform containing an NH2-terminal membrane anchor. Genomics 1998;50:79–88. [DOI] [PubMed] [Google Scholar]
- 18.Birkenmeier CS, White RA, Peters LL, et al. Complex patterns of sequence variation and multiple 5′ and 3′ ends are found among transcripts of the erythroid ankyrin gene. J Biol Chem 1993;268:9533–9540. [PubMed] [Google Scholar]
- 19.Doctor RB, Chen J, Peters LL, et al. Distribution of epithelial ankyrin (Ank3) spliceoforms in renal proximal and distal tubules. Am J Physiol 1998;74; F129–F138. [DOI] [PubMed] [Google Scholar]
- 20.Gallagher PG, Forget BG. An alternate promoter directs expression of a truncated, muscle-specific isoform of the human ankyrin 1 gene. J Biol Chem 1998;273:1339–1348. [DOI] [PubMed] [Google Scholar]
- 21.Kordeli E, Bennett V. Distinct ankyrin isoforms at neuron cell bodies and nodes of Ranvier resolved using erythrocyte ankyrin-deficient mice. J Cell Biol 1991;114:1243–1258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kordeli E, Davis J, Trapp B, et al. An isoform of ankyrin is localized at nodes of ranvier in myelinated axons of central and peripheral nerves. J Cell Biol 1990;110:1341–1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kordeli E, Lambert S, Bennett V. AnkyrinG. A new ankyrin gene with neural-specific isoforms localized at the axonal initial segment and node of ranvier. J Biol Chem 1995;270:2352–2359. [DOI] [PubMed] [Google Scholar]
- 24.Lambert S, Yu H, Prchal JT, et al. cDNA sequence for human erythrocyte ankyrin. Proc Natl Acad Sci USA 1990;87:1730–1734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lux SE, John KM, Bennett V. Analysis of cDNA for human erythrocyte ankyrin indicates a repeated structure with homology to tissue-differentiation and cell-cycle control proteins. Nature 1990;344:36–42. [DOI] [PubMed] [Google Scholar]
- 26.Otto E, Kunimoto M, McLaughlin T, Bennett V. Isolation and characterization of cDNAs encoding human brain ankyrins reveal a family of alternatively spliced genes. J Cell Biol 1991;114:241–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Peters B, Kaiser HW, Magin TM. Skin-specific expression of ank-3(93), a novel ankyrin-3 splice variant. J Invest Dermatol 2001;16:216–223. [DOI] [PubMed] [Google Scholar]
- 28.Peters LL, John KM, Lu FM, et al. Ank3 (epithelial ankyrin), a widely distributed new member of the ankyrin gene family and the major ankyrin in kidney, is expressed in alternatively spliced forms, including forms that lack the repeat domain. J Cell Biol 1995;130:313–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Thevananther S, Kolli AH, Devarajan P. Identification of a novel ankyrin isoform (AnkG190) in kidney and lung that associates with the plasma membrane and binds alpha-Na, K-ATPase. J Biol Chem 1998;273:23952–23958. [DOI] [PubMed] [Google Scholar]
- 30.Eber SW, Gonzalez JM, Lux ML, et al. Ankyrin-1 mutations are a major cause of dominant and recessive hereditary spherocytosis. Nat Genet 1996;13:214–218. [DOI] [PubMed] [Google Scholar]
- 31.Gallagher PG. Hematologically important mutations: Ankyrin variants in hereditary spherocytosis. Blood Cells Mol Dis 2005;35:345–347. [DOI] [PubMed] [Google Scholar]
- 32.Gallagher PG, Tse WT, Scarpa AL, et al. Structure and organization of the human ankyrin-1 gene. Basis for complexity of pre-mRNA processing. J Biol Chem 1997;272:19920–19928. [DOI] [PubMed] [Google Scholar]
- 33.Ozcan R, Jarolim P, Lux SE, et al. Simultaneous (AC)n microsatellite polymorphism analysis and single-stranded conformation polymorphism screening is an efficient strategy for detecting ankyrin-1 mutations in dominant hereditary spherocytosis. Br J Haematol 2003;122:669–677. [DOI] [PubMed] [Google Scholar]
- 34.Edelman EJ, Maksimova Y, Duru F, et al. A complex splicing defect associated with homozygous ankyrin-deficient hereditary spherocytosis. Blood 2007;109:5491–5493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yawata Y, Kanzaki A, Yawata S, et al. Characteristic features of the genotype and phenotype of hereditary spherocytosis in the Japanese population. Int J Hematol 2000;71:118–135. [PubMed] [Google Scholar]
- 36.Leite RC, Basseres DS, Ferreira JS, et al. Low frequency of ankyrin mutations in hereditary spherocytosis: Identification of three novel mutations. Hum Mutat 2000;16:529–534. [DOI] [PubMed] [Google Scholar]
- 37.Zerhouni F, Guetarni D, Henni T, et al. Occurrence and characteristics of hereditary spherocytosis in Algeria. Eur J Haematol 1991;47:42–47. [DOI] [PubMed] [Google Scholar]
- 38.Ustun C, Kutlar F, Holley L, et al. Interaction of sickle cell trait with hereditary spherocytosis: Splenic infarcts and sequestration. Acta Haematol 2003;109:46–49. [DOI] [PubMed] [Google Scholar]
- 39.Peabody DS. Translation initiation at non-AUG triplets in mammalian cells. J Biol Chem 1989;264:5031–5035. [PubMed] [Google Scholar]
- 40.Drabkin HJ, RajBhandary UL. Initiation of protein synthesis in mammalian cells with codons other than AUG and amino acids other than methionine. Mol Cell Biol 1998;18:5140–5147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Stoner MA, Auerbach SS, Zamule SM, et al. Transactivation of a DR-1 PPRE by a human constitutive androstane receptor variant expressed from internal protein translation start sites. Nucleic Acids Res 2007;35:2177–2190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kozak M Some thoughts about translational regulation: Forward and backward glances. J Cell Biochem 2007;102:280–290. [DOI] [PubMed] [Google Scholar]
- 43.Sonenberg N, Hinnebusch AG. New modes of translational control in development, behavior, and disease. Mol Cell 2007;28:721–729. [DOI] [PubMed] [Google Scholar]
- 44.Mitchell G, Brody LC, Looney J, et al. An initiator codon mutation in ornithine-delta aminotransferase causing gyrate atrophy of the choroid and retina. J Clin Invest 1988;81:630–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Patten JL, Johns DR, Valle D, et al. Mutation in the gene encoding the stimulatory G protein of adenylate cyclase in Albright’s hereditary osteodystrophy. N Engl J Med 1990;322:1412–1419. [DOI] [PubMed] [Google Scholar]
- 46.Gupta A, Hattori Y, Agarwal S. Initiation codon mutation in an Asian Indian family. Am J Hematol 2002;71:134–136. [DOI] [PubMed] [Google Scholar]
- 47.Quinteiro C, Castro-Feijoo L, Loidi L, et al. Novel mutation involving the translation initiation codon of the growth hormone receptor gene (GHR) in a patient with Laron syndrome. J Pediatr Endocrinol Metab 2002;15:1041–1045. [DOI] [PubMed] [Google Scholar]
- 48.Zoppi S, Wilson CM, Harbison MD, et al. Complete testicular feminization caused by an amino-terminal truncation of the androgen receptor with downstream initiation. J Clin Invest 1993;9:1105–1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.O’Neill GN, Aoki M, Brown RH Jr, et al. ABCD1 translation-initiator mutation demonstrates genotype-phenotype correlation for AMN. Neurology 2001;57: 1956–1962. [DOI] [PubMed] [Google Scholar]
- 50.Nguyen M, He B, Karaplis A. Nuclear forms of parathyroid hormone-related peptide are translated from non-AUG start sites downstream from the initiator methionine. Endocrinology 2001;142:694–703. [DOI] [PubMed] [Google Scholar]
- 51.Lilleberg SL. In-depth mutation and SNP discovery using DHPLC gene scanning. Curr Opin Drug Discov Devel 2003;6:237–252. [PubMed] [Google Scholar]
- 52.Yu B, Sawyer NA, Chiu C, et al. DNA mutation detection using denaturing high-performance liquid chromatography (DHPLC). Curr Protoc Hum Genet 2006;Chapter 7:Unit 7.10. [DOI] [PubMed] [Google Scholar]
- 53.Bennett RR, den Dunnen J, O’Brien KF, et al. Detection of mutations in the dystrophin gene via automated DHPLC screening and direct sequencing. BMC Genet 2001;2:17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Biggin A, Holman K, Brett M, et al. Detection of thirty novel FBN1 mutations in patients with Marfan syndrome or a related fibrillinopathy. Hum Mutat 2004;23:99. [DOI] [PubMed] [Google Scholar]
- 55.Dahl F, Stenberg J, Fredriksson S, et al. Multigene amplification and massively parallel sequencing for cancer mutation discovery. Proc Natl Acad Sci USA 2007;104:9387–9392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Patil N, Berno AJ, Hinds DA, et al. Blocks of limited haplotype diversity revealed by high-resolution scanning of human chromosome 21. Science 2001;294:1719–1723. [DOI] [PubMed] [Google Scholar]
- 57.Chee M, Yang R, Hubbell E, et al. Accessing genetic information with high-density DNA arrays. Science 1996;274:610–614. [DOI] [PubMed] [Google Scholar]
- 58.Margulies M, Egholm M, Altman WE, et al. Genome sequencing in microfabricated high-density picolitre reactors. Nature 2005;437:376–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Porreca GJ, Zhang K, Li JB. Multiplex amplification of large sets of human exons. Nat Methods 2007;4:931–936. [DOI] [PubMed] [Google Scholar]
- 60.Fairbanks G, Steck TL, Wallach DF. Electrophoretic analysis of the major polypeptides of the human erythrocyte membrane. Biochem 1971;10:2606–2617. [DOI] [PubMed] [Google Scholar]
- 61.Marchesi SL, Nowles WJ, Morrow JS, et al. Abnormal spectrin in hereditary elliptocytosis. Blood 1986;67:141–151. [PubMed] [Google Scholar]
- 62.Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 1970;227:680–685. [DOI] [PubMed] [Google Scholar]
- 63.Kambouris M, Jackson CE, Feldman GL. Diagnosis of multiple endocrine neoplasia (MEN) 2A, 2B and familial medullary thyroid cancer (FMTC) by multiplex PCR and heteroduplex analyses of RET proto-oncogene mutations. Hum Mutat 1996;8:64–70. [DOI] [PubMed] [Google Scholar]