

Abstract
Background
Neddylation (NAE) inhibition, affecting posttranslational protein function and turnover, is a promising therapeutic approach to cancer. We report the cytotoxic vulnerability to NAE inhibitors in a subset of glioblastoma (GBM) preclinical models and identify genetic alterations and biological processes underlying differential response.
Methods
GBM DNA sequencing and transcriptomic data were queried for genes associated with response to NAE inhibition; candidates were validated by molecular techniques. Multi-omics and functional assays revealed processes implicated in NAE inhibition response.
Results
Transcriptomics and shotgun proteomics depict PTEN signaling, DNA replication, and DNA repair pathways as significant differentiators between sensitive and resistant models. Vulnerability to MLN4924, a NAE inhibitor, is associated with elevated S-phase populations, DNA re-replication, and DNA damage. In a panel of GBM models, loss of WT PTEN is associated with resistance to different NAE inhibitors. A NAE inhibition response gene set could segregate the GBM cell lines that are most resistant to MLN4924.
Conclusions
Loss of WT PTEN is associated with non-sensitivity to 3 different compounds that inhibit NAE in GBM. A NAE inhibition response gene set largely consisting of DNA replication genes could segregate GBM cell lines most resistant to NAEi and may be the basis for future development of NAE inhibition signatures of vulnerability and clinical trial enrollment within a precision medicine paradigm.
Keywords: glioblastoma, DNA replication, multi-omics, Neddylation, PTEN
Key Points.
GBM cell lines sensitive to MLN4924 have increased baseline S-phase and post-treatment > 4N populations and are enriched in transcripts for processes related to DNA replication and the cell cycle.
Loss of functional PTEN confers resistance to neddylation inhibition in GBM.
The development of Neddylation Inhibition Response Gene Set that can segregate and identify the GBM models most resistant to MLN4924, potentially setting the groundwork for the development of a signature of resistance.
Importance of the Study.
As the most common malignant and most lethal primary CNS tumor, glioblastoma (GBM) calls for novel therapeutic development. Neddylation inhibition has shown promising antitumor activity in GBM models but sufficient knowledge of response status across tumors is urgently needed to support the design of successful clinical trials. This study demonstrates that WT PTEN and dysregulation of the cell cycle and DNA replication are associated with enhanced response to NEDD8 activating enzyme (NAE) inhibition-induced cell death and provides a gene set––largely consisting of genes involved in DNA replication––that may predict high de novo resistance to NAE inhibitor response.
Glioblastoma (GBM) is the most common, primary malignant brain tumor in adults, with a 5-year survival rate of 7.2%.1 The heterogeneity and complexity of GBM molecular aberrations not only confounds diagnostics,2 but leads to variability in therapeutic response.3 There is an unmet need to define molecular aberrations predictive of therapeutic response for the deployment of current and new agents.
Neddylation (NAE) is a posttranslational modification pathway that conjugates NEDD8 to target protein substrates, affecting protein stability and function.4 Analogous to ubiquitination, NEDD8 is activated by its E1 ligase NEDD8 activating enzyme (NAE), then transferred to its E2 ligase UBC12, and, via NEDD8 E3 ligases, conjugated to target substrate(s).4,5 Turnover of known cancer-related proteins is mediated, in part, through the neddylation of cullin proteins.6,7 Numerous solid tumors demonstrate increased activation of the NAE pathway.8 Hua et al. showed elevated NAE pathway activation in GBM tumor tissues, as well as a significant correlation between pathway activation and tumor grade, recurrence, and shorter survival.9
The role of NAE in tumorigenesis led to development of MLN4924 (pevonedistat), a small molecule inhibitor that binds to NAE creating a covalent NEDD8-MLN4924 adduct, blocking subsequent NAE activity.10 MLN4924 demonstrated antitumor effects alone and in combination with DNA-damaging agents across tumor types which led to more than thirty clinical trials.7,11 In GBM, MLN4924 demonstrated antitumor effects in vitro and in vivo.9,12,13 MLN4924 stabilizes CDT1, a DNA licensing factor, which induces re-replication, DNA damage, and cell death.4,14–16 Garcia et al. demonstrated that depletion of DNA damage repair pathways, which largely overlap with DNA replication pathways, reduced efficacy of MLN4924.17,18 Since the development of MLN4924, more compounds have been developed or found to possess anti-neddylation properties, such as the highly potent selective NAE inhibitor TAS4464,19,20 or the clinically ubiquitous antihypertensive agent, candesartan cilexetic (CDC).21
The tumor suppressor gene PTEN is mutated or deleted across cancer types, including 40% of GBM.17 The loss of PTEN allows run-away PI3K/AKT signaling in the cytoplasmic compartment, driving cancer cell proliferation, survival, and cell migration,22 as well as resistance to GBM therapeutics.23 Nuclear localization of PTEN can impact genome integrity, cell cycle control, and DNA damage response.24,25 Phosphorylation at PTEN Y240––a site critical for chromatin binding, interaction with Ki67 and RAD51, and DNA damage repair––is associated with resistance to radiation treatment in GBM.26 While the status and localization of PTEN impact therapeutics currently employed against GBM, the role of PTEN in response to MLN4924 is unknown.
In this study, we employed GBM models with different vulnerabilities to MLN4924 to characterize molecular mechanisms underlying therapeutic sensitivity. We showed that the efficacy of MLN4924 is dependent on the PTEN tumor suppressor gene and pathways related to its nuclear functions such as DNA replication and the cell cycle. Furthermore, we established a NAEi Response Gene set using a broader panel of GBM PDX models and inhibitors that could separate the GBM cell lines most resistant to MLN4924 in PCA space. Our results reveal that molecular subsets of GBM patients may be identified and benefit from treatment with NAE inhibitors.
Materials and Methods
Cell Lines, Cultures, and Reagents
LN18 and M059K were purchased from the American Type Culture Collection. GB1 and SNU1105 were obtained from the Japanese Collection of Research Bioresources Cell Bank and Korean Cell Line Bank, respectively. Immortalized human astrocytes were provided by the Pieper lab.27 Established human glioma cell lines were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM, Hyclone) containing 10% fetal bovine serum, FBS (Biochrom AG), and 1% penicillin-streptomycin at 37 °C with 5% CO2. Patient-derived xenograft (PDX) models (Mayo Clinic Hospital),28,29 and isogenic PTEN glioma stem cell (GSC) lines were cultured in DMEM/F12 (Hyclone), 20 ng/mL EGF, 20ng/ml FGF, 2% B12 supplement, 1% N2 supplement, and 1% gentamicin solution at 37°C with 5% CO2.23 Isogenic lines HCT116 PTEN (+/+) and (−/−) were cultured in McCoy’s 5a (Modified) Medium, 10% FBS (Biochrom AG) and 1% penicillin-streptomycin.30
MLN4924 and TAS4464 were purchased from Selleckchem (Cat# S7109 and S8849). CDC was purchased from Millipore Sigma (Cat# SML0245). Bortezomib, was from Sigma-Aldrich (Cat# 179324-69-7 and M8699).
Colony Formation Assay
Cells were seeded into 6-well plates in triplicate (300-500 cells per well). 24 hours later, the media was replaced with new media with or without MLN4924, and cells were incubated at 37°C for 2 weeks. Cultures were fixed with 4% paraformaldehyde and stained with crystal violet, and colonies (> 50 cells) were counted.
Cell Cycle Analysis
Cells treated with DMSO or MLN4924 (0.1, 0.25, or 0.5 μM) for 24 hours were harvested, fixed in 70% ethanol − 20°C overnight, stained with FxCycle TM propidium iodide/RNAse staining solution (ThermoFisher Scientific, Cat # F10797) at 37°C for 30 minutes, and then analyzed for cell-cycle profile by flow cytometry (FACSCalibur, BD Bioscience). Data were analyzed with FlowJo software (Becton, Dickinson & Co, 2019).
Caspase-Glo 3/7 Assay
The Caspase-Glo 3/7 Assay (Promega, Madison, WI) was performed according to the manufacturer’s protocol. GB1 and M059K cells were seeded in a 384-well plate (1250 cells/well) and treated with MLN4924. After 48- and 72-hour drug treatment, 25 uL of Caspase-Glo 3/7 reagent was added to wells, mixed with 50 uL of medium, incubated in the dark at room temperature for 30 minutes then read using the EnVision multimode plate reader (PerkinElmer Life and Analytical Sciences, Waltham, MA). Levels of caspase-3/7 were normalized to those of the DMSO controls.
siRNA Knockdown Assay
Qiagen FlexiTube siRNA was obtained for PTEN (GS5728). Cells were seeded in a 6-well plate (150 000-300 000/well), then reverse-transfected with siRNA prepared in lipofectamine RNAiMax reagent (Cat# 13778150) in Opti-Mem Reduced Serum Media (Cat# 31985062). After 48 hours, cells were lysed for immunoblot analysis. Transcriptomics and Proteomics Analyses are described in Supplementary Methods.
Data and Materials Availability
RNA-Seq accession number PRJNA1023909 (https://www.ncbi.nlm.nih.gov/bioproject/PRJNA1023909) and PRIDE repository ID # (pending) for the proteomics data.
Results
Response Status to MLN4924 is Independent of Differences in Canonical CRL Substrate Accumulation in a Subset of Glioma Cell Models
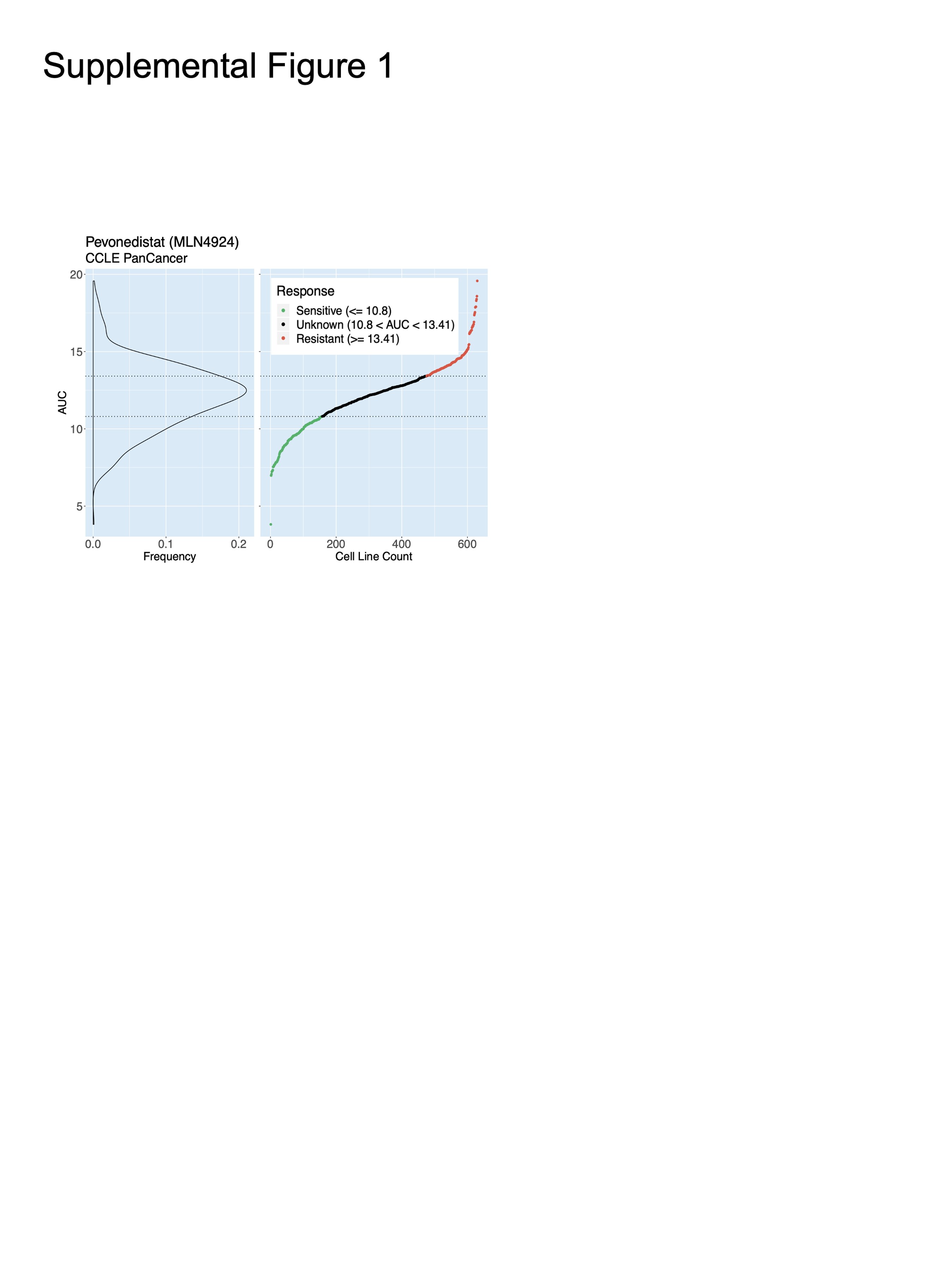
Across 860 cancer cell lines in The cancer therapeutics response portal,31 MLN4924 demonstrated a broad range of efficacy (Supplementary Figure 1). We selected 2 sensitive (LN18, GB1) and 2 non-sensitive (M059K, SNU1105; hereafter referred to as “resistant”) glioma cell lines for further investigation. Viability assay using serial dilution of MLN4924 validated LN18 and GB1 as sensitive (IC50 = 0.195 µM and IC50 = 0.279 µM, respectively) and M059K and SNU1105 as resistant (IC50 = 5.50 µM and IC50 = 20.9 µM, respectively; Figure 1A). Differential vulnerability to MLN4924 was further substantiated by significant differences between the ability of LN18 and SNU1105 to form colonies in the presence of 0.1 and 0.5 µM MLN4924 (Figure 1B). Differential sensitivity was not observed with bortezomib, a proteasome inhibitor, in these models (Figure 1C) indicating distinct therapeutic vulnerabilities between these 2 drugs that target protein turnover. Furthermore, bortezomib killed TERT-immortalized normal human astrocytes (IC50 = 5.81 nM) whereas MLN4924 was nontoxic at tested doses (Figure 1D).
Figure 1.

Differential response of glioma cell lines to MLN4924 is independent of NEDD8 activating enzyme (NAE) targeted inhibition. (A) Viability of LN18, GB1, M059K, and SNU1105 cell lines 72 hours after MLN4924 treatment was measured. Data are normalized to DMSO (5 replicates). P < .001 by non-linear regression fit comparison. (B) Colony formation by LN18 and SNU1105 cell lines 2 weeks after MLN4924 treatment (24 hours; 5 replicates). Colonies (> 50 cells) were counted and reported as surviving fractions (mean ± SD, *** P < .001 by Student’s t-test). Drug dose–response assay of Bortezomib was conducted on glioma models (C) and TERT-immortalized normal human astrocytes (D). (E) Indicated cell lines were treated with increasing concentrations of MLN4924 for 8 hours; isolated proteins were subjected to Immunoblot (IB) using antibodies (Ab) against Nedd8-cullin or NAE1 of proteins from glioma cells after MLN4924 treatment (alpha-tubulin is loading control). (F) IB analysis of selected CRL substrates and DNA damage proteins from GB1 and M059K exposed to 500 nM MLN4924 for indicated times, (GAPDH is loading control).
Cullin neddylation was suppressed in each glioma cell line (Figure 1E), with concomitant accumulation of CRL substrates such as CDT1, P27, and P21 (Figure 1F). Other than a slightly earlier accumulation of CDT1 and a more pronounced accumulation of P27, there were no clear differences in CRL substrate accumulation explaining MLN4924 differential sensitivity.
Multi-omics Reveal Enrichment of DNA Replication and Repair and Chromosome Regulation Processes in Models Sensitive to MLN4924
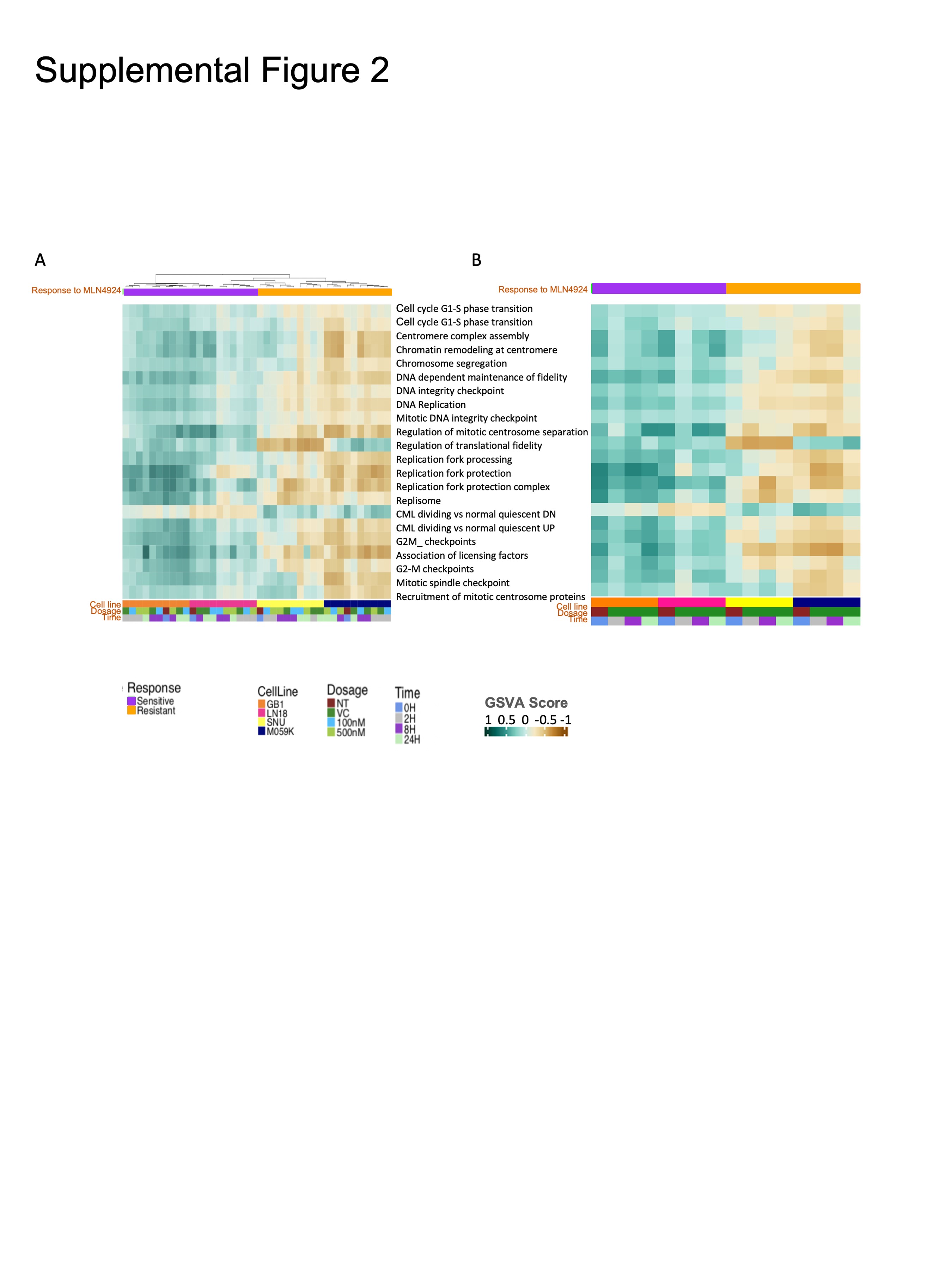
We next performed RNA-seq and shotgun proteomics from sensitive and resistant glioma models ± MLN4924. Gene set variation analysis was done on all samples, and unsupervised hierarchical clustering indicated that differences in transcriptomes were primarily due to cell line heterogeneity rather than changes induced by MLN4924 treatment (Figure 2A and Supplementary Figure 2A). Twenty-two gene sets were significantly different between sensitive and resistant models, and sensitive models were enriched in processes related to DNA replication, chromosome regulation, and the cell cycle and its checkpoints at baseline and through treatment (Supplementary Figure 2A and Figure 2A). Proteomics results validated the discrimination between sensitivity and resistance to MLN4924 by DNA replication pathways: Sensitive cell lines were more enriched in replication-related proteins than resistant cell lines (Figure 2B).
Figure 2.

DNA replication processes and cell cycle dynamics distinguish glioma cells sensitive and resistant to MLN4924. Analytes from sensitive (LN18 and GB1) and resistant (M059K and SNU1105) glioma cells were collected at 0-, 2-, 8-, and 24 hours post-exposure to 0 (vehicle control), 100 or 500 nM MLN4924 and analyzed by next-generation RNA sequencing and mass spectrometry (LN18 and SN1105 only). (A) Supervised gene set enrichment analysis of RNA Seq data was carried out with 22 gene sets that were significantly differentially enriched between sensitive and resistant models (P < .05, t-test with multiple testing corrections) (Gene set names in Supplementary Table. Fig 2). (B) Hierarchical clustering of protein sets that differentiate sensitive and resistant samples highlights replication and repair pathways. (C). Sensitive (LN18 and GB1) and resistant (M059K and SNU1105) cell lines were treated with DMSO (NT) or MLN4924 (100, 250, or 500 nM) for 48 hours in triplicates and analyzed for cell-cycle distribution and percentage. (D) MLN4924 induced apoptosis (Caspase-Glo 3/7 Assay) in GB1 (sensitive) but far less so in M059K (resistant) glioma cells; levels of caspase-3/7 were normalized to those of the DMSO-treated controls.
MLN4924 Induces Re-replication, Polyploidy, and DNA Damage in Sensitive Glioma Cell Lines
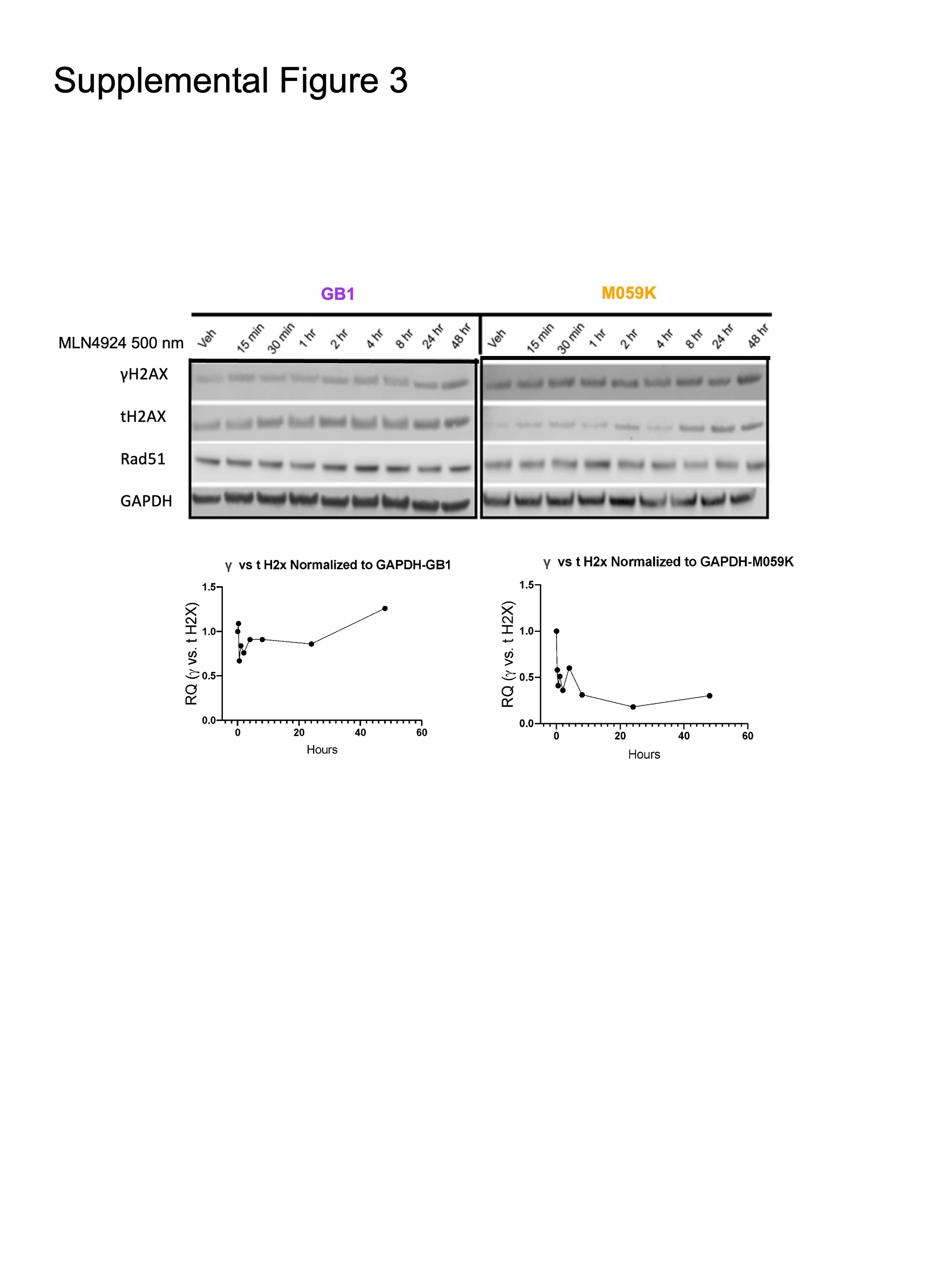
Given these transcriptomic differences in DNA replication, the cell cycle was analyzed across sensitive and resistant glioma lines ± MLN4924. Consistent with previous findings, all cell lines showed dose-dependent increases in G2/M phase after treatment (Figure 2C).4,9,32 In accordance with our -OMICs analyses and MLN4924 response in other cancer types, resistant cell lines showed lower S-phase populations at baseline and through treatment. S-phase cells may be more susceptible to re-replication following MLN4924 treatment, as sensitive lines had higher tetraploid (4N) populations (Figure 2C).16 While the resistant line, M059K, showed a higher baseline level of the DNA damage marker γH2AX, the level did not increase through treatment unlike the sensitive line GB1 (Supplementary Figure 3). Concentration-dependent cell death by apoptosis was evident in GB1 but far less pronounced in M059K (Figure 2D).
Loss of PTEN Drives Resistance to MLN4924 in Glioma Models
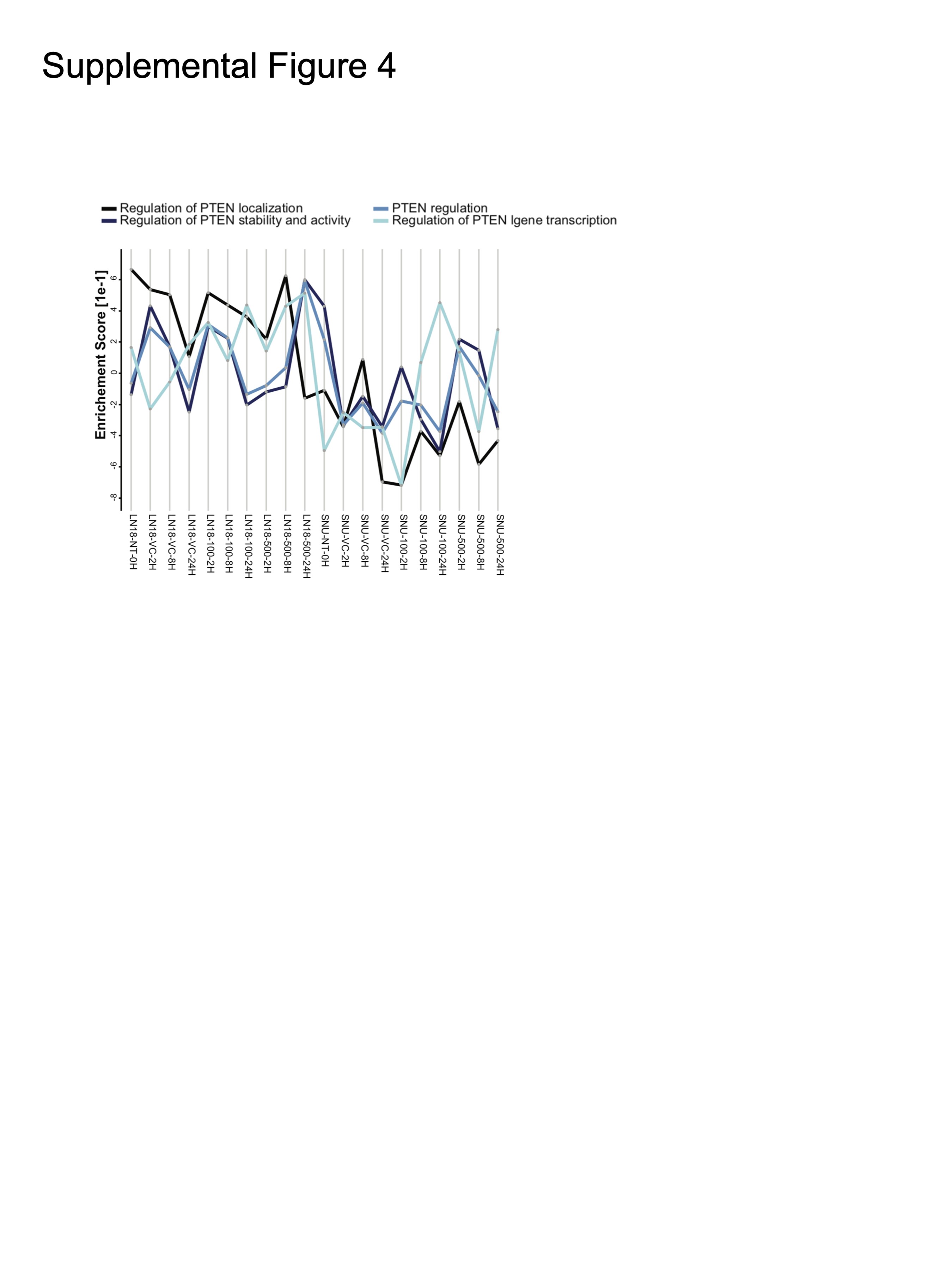
We sought to identify additional features that may discriminate response status to MLN4924. Unbiased Ingenuity Pathway Analysis of signaling networks showed PTEN Signaling to be one of the pathways most strongly associated with sensitivity (Figure 3A, Supplementary Tables 1 and 2). Proteomics also uncovered differences in PTEN signaling, as PTEN gene sets from Reactome showed enrichment in LN18 and a lack thereof in SNU1105 (Supplementary Figure 4) The 2 resistant models harbored nonfunctional PTEN mutations and lacked PTEN protein expression (Supplementary Figure 5A and Figure 3B, respectively). No mutations were found in PIK3CA gene, an additional key mediator of PTEN signaling (Supplementary Figure 5B). Ensuring that this finding was not a feature of the 4 GBM models only, we grouped 21 GBM lines from Cancer Cell Line Encyclopedia (CCLE) and the Genomics of Drug Sensitivity (GDSC) databases (www.broadinstitute.org/ccle and https://www.cancerrxgene.org/) into sensitive and resistant groups based on median AUC values and compared mutation frequencies. For the 6 genes with the largest difference in mutation frequency between sensitive (n = 10) and resistant (n = 11) groups, PTEN mutation status showed the largest delta (P < .05, Fisher’s Exact Test; Figure 3C).
Figure 3.

PTEN loss decreases sensitivity to MLN4924. (A) Unbiased ingenuity pathway analysis of RNA Seq data identified the most differentially deregulated pathways following MLN4924 treatment between sensitive and resistant models. (B) Protein expression of PTEN determined by IB analysis. (C) Differential mutation frequencies of selected genes in a glioma cell line panel sensitive (n = 11) and resistant (n = 10) to MLN4924 (21 cell lines total; median analysis). (D) Protein lysates from the treatment naïve cell line (LN18 and GB1) were IB probed for PTEN expression and (E) a PTEN isogenic pair (PTEN WT and PTEN knock-out [PTEN K.O.]) of colorectal cancer cell line HCT116. (F-G) Viability of LN18 and GB1 ± siRNA PTEN knockdown following MLN4924 treatment. (H) Viability of HCT116 (PTEN WT and PTEN knock-out [PTEN K.O.]) after treatment with MLN4924. (I) Viability of a PTEN isogenic pair (PTEN null and PTEN knock-in [PTEN K.I.]) of glioma stem cell line model GSC11 after MLN4924 treatment. F-I Plots show results from 5 replicates and P-values for Nonlinear regression fit comparison of curves are < .001.
PTEN protein levels were suppressed using siRNA in the sensitive LN18 (Figure 3F) and GB1 (Figure 3G) models. We observed a 3- to 4-fold increase in IC50 following PTEN knockdown (Figure 3F, G). This increase in IC50 value was validated in an isogenic colorectal cancer cell line HCT116 ± PTEN knockout (Figure 3E and H). Testing the reciprocal direction, a previously generated PTEN-null GSC model, GSC11, was engineered to re-express PTEN,33 and treated with MLN4924. This resulted in an approximately 4-fold decrease in the IC50 of the PTEN-expressing GSC11 model (Figure 3I).
PTEN-Driven Sensitivity to MLN4924 is Independent of its Phosphatase Activity But Dependent on its Ability to Bind Chromatin
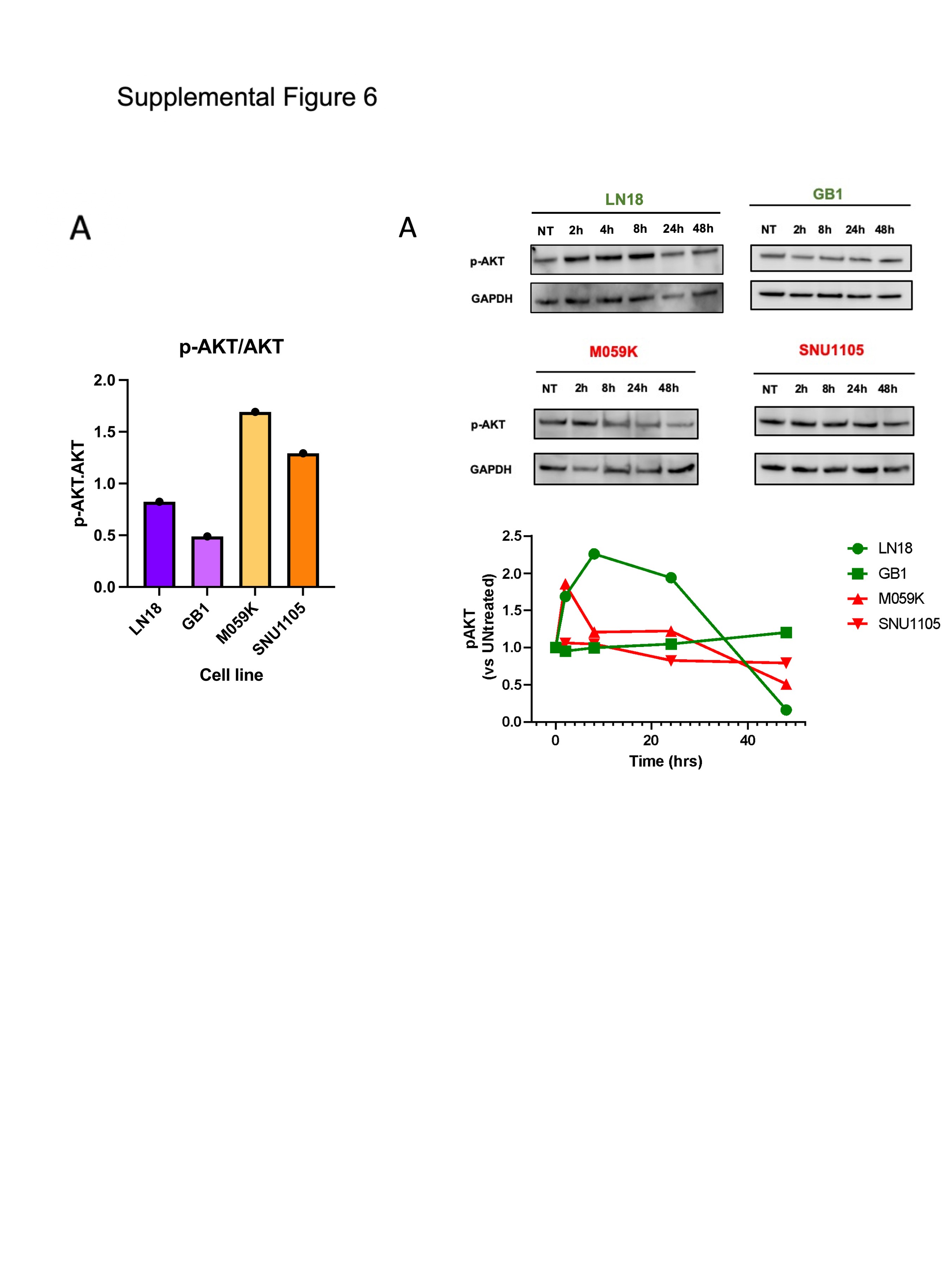
We evaluated whether vulnerability to MLN4924 was dependent on PTEN’s function as a lipid phosphatase and antagonist of PI3K/AKT signaling.22 As expected with PTEN loss, phosphorylated AKT (p-AKT) was elevated in the resistant lines compared to sensitive GBM models (Figure 4A, Supplementary Figure 6A). p-AKT levels showed no consistent change after exposure to MLN4924 (Supplementary Figure 6B). To test whether MLN4924 cytotoxicity is dependent on PTEN’s phosphatase activity, we employed a known PTEN-null GSC model (GSC23), engineered to express WT PTEN or G129R (phosphatase dead).34 Cells with either transgene equally increased sensitivity to MLN4924 (decreased IC50; Figure 4B). Thus, the sensitization effect of PTEN reconstitution is independent of PI3K/AKT axis antagonism in this model.
Figure 4.

Response to MLN4924 is independent of PTEN lipid phosphatase activity but dependent on phosphorylation of PTEN Tyrosine 240. (A) Protein lysates from LN18, GB1, M059K, and SNU1105 were probed for phosphorylated- and total-AKT (pAKT and AKT) by IB analysis, and GAPDH loading control. (B) Viability of Transgene PTEN WT or PTEN G129A (phosphatase-dead) in PTEN-null GSC23 cells after treatment with MLN4924. (C) Viability of PTEN WT or PTEN Y240F mouse embryonic fibroblasts (MEFs) after treatment with MLN4924. B and C are in 5 replicates. (D) Volcano plot of differentially expressed transcripts between isogenic MEFs from Figure 4C (listed in Supplementary Table 3). (E) Waterfall plot (ranked by fold enrichment) of top 100 Reactome pathways from data in Figure 4D. Cell cycle and DNA replication pathways (n = 23) are annotated as "TRUE"; 10 characteristic pathways are labeled for clarity.
Nuclear PTEN can be phosphorylated at tyrosine residue-240 (pY240-PTEN), which is essential for PTEN binding to chromatin, recruiting RAD51, and participating in DNA damage repair.26 Using a set of mouse embryonic fibroblasts (MEFs) with transgene WT PTEN and Y240F PTEN (preventing Y240 phosphorylation; Figure 4C), we found that Y240F PTEN increased IC50 of MLN4924 approximately 10-fold (Figure 4C).
We performed RNA sequencing on WT and Y240F PTEN MEFs to determine pathways impacted by the loss of PTEN chromatin binding. The alteration of a single amino acid residue resulted in 4357 significantly differentially expressed genes (Padj < .05 with at least a 2-fold change) with more genes being significantly downregulated (3840 genes) than upregulated (517 genes) in PTEN Y240F cells (Figure 4D). The pathways overrepresented from the significantly downregulated genes in PTEN Y240F MEFs largely consisted of processes related to cell cycle and DNA replication (Figure 4E, Supplementary Table 3). Interestingly, Neddylation (R-MMU-8951664) was the seventh most negatively enriched pathway, as well (Supplementary Table 3).
WT PTEN is Associated With Sensitivity to Neddylation Inhibition Across a Broader Panel of GBM Models and Small Molecules
We sought to determine whether our findings related to PTEN status and MLN4924 response could be extended to other GBM models and small molecules known to inhibit the neddylation pathway.19–21 Eleven PDX GBM models were selected based on a mixed distribution of PTEN status: 4 PTEN WT, 2 PTEN homozygous deletion, and 5 PTEN MUT (Figure 5A-B, Supplementary Figure 7A). The selected GBM models constituted a diverse and authentic representation of molecular and clinical features, such as transcriptional subtype, TP53 status, sex, and PTEN status; however, all models were PIK3CA WT (Supplementary Figure 7B). Given that distinct PTEN mutations differentially impacted MLN4924 response (Figure 4), we ensured the presence of different PTEN mutations (Supplementary Figure 7B).
Figure 5.

Determinants of GBM vulnerability to inhibitors of NEDD8 activating enzyme (NAE). Four PTEN WT (GBM12, 15, 38, and 43), 2 PTEN DEL (GBM120 and 102) and 5 PTEN MUT (GBM10, 39, 76, 116, and 108) lines were treated with MLN4924, TAS4464, or Candesartan Cilexetic (CDC) for 72 hours. (A) Sensitivity ranking of 11 GBM patient-derived xenograft (PDX) cultures to each of 3 different NAE inhibitors. (B) The average ranking of drug response in each group of PTENwt, PTENdel, and PTENmut. Unsupervised hierarchical clustering of (C) Mayo PDX models and (D) 17 Cancer Cell Line Encyclopedia (CCLE) glioma cell lines based on gene sets (Gene set names in Supplementary Table. Fig 5) demonstrated to be significantly differentially expressed between GBM cell lines that were sensitive and resistant to MLN4924 (Figure 2). (E) PCA plot of 17 CCLE lines using the NAEi Response Gene Set.
Drug sensitivity assays validated our previous findings, as PTEN WT PDX models had the lowest EC50s following MLN4924 treatment across all models and compounds (Figure 5A-5B). All EC50 values for the potent TAS4464 were significantly lower than other compounds.20 CDC was the least effective at inducing cell death (Figure 5A–B).21 Despite these differences in potency across all 3 drugs, PTEN WT lines were more sensitive than PTEN altered lines, and accordingly, our previous findings are extendable to NAE inhibition more broadly as opposed to being MLN4924 specific (Figure 5A–B).
GBM PDX Models Sensitive to NAEi are Enriched in DNA Replication and Repair Transcripts
We previously identified a collection of differentially expressed gene sets between GBM cell lines that were sensitive and resistant to MLN4924 (Figure 2A). These gene sets were also differentially expressed between sensitive and resistant Mayo models (Figure 5C) except for the outlier model GBM120, which was enriched in DNA replication and cell cycle transcripts despite being resistant (Figure 5C). Given that these gene sets could broadly segregate sensitive and resistant cell lines across 2 cohorts of GBM cell lines, we hypothesized that these gene sets may be used to predict NAEi response de novo.
For 15 established GBM cell lines, we paired publicly available RNA Seq and MLN4924 EC50 data from the CCLE and the Genomics of Drug Sensitivity (GDSC2) databases as well as including M059K and SNU1105 found to be resistant in our analysis. Unsupervised hierarchical clustering for the 17 CCLE lines revealed that sensitive models tended to be enriched in DNA replication and cell cycle processes (Figure 5D). Despite this trend, some of the resistant cell lines clustered with the sensitive cell lines, including one of the most resistant lines KS1 (Figure 5D). Given these results, we wondered whether we could use the RNA sequencing data from the Mayo cohort to devise a more predictive gene set.
The GBM PDX models were divided into sensitive (ranking 1st-5th on average) and resistant (ranking 6th–11th on average) groups (Figure 5A). Differentially expressed genes between the NAEi-sensitive and NAEi-resistant groups were isolated for DAVID functional analysis (Supplementary Figure 8A). As identified previously, the GBM PDX models that were sensitive to NAE inhibitors were enriched in transcripts related to the cell cycle. More directly gauging the relationship between gene expression and drug sensitivity, we determined the Spearman correlation between gene expression in different GBM PDX models and the NAEi drug EC50 concentrations.35 We isolated genes that strongly correlated with the EC50 of NAE inhibitors in the 11 GBM PDX models (Spearman rho > 0.7 or < 0.7) and conducted DAVID functional analysis (Supplementary Figure 8B). Appreciating the more direct analysis method, we grouped the 40 genes from Figure 5D as the “NAEi Response Gene Set.”
The NAEi Response Gene Set Effectively Distinguishes the Most Resistant Glioma Cell Lines De Novo
For the 17 established GBM cell lines, we first plotted the CCLE cell lines’ MLN4924 EC50 against their expression of genes that were found to be most strongly correlative with NAEi EC50 (Supplementary Figure 8C) and found many to correlate well or trend as expected in CCLE lines (Supplementary Table 4).
We sought to determine whether the combined expression of many NAEi response genes could be used to broadly predict sensitivity to NAE inhibitors. Hierarchical clustering was performed on CCLE lines using the NAEi response gene set (Supplementary Figure 8D). As visualized in a dendrogram annotated by MLN4924 EC50, sensitive cell lines largely segregate from resistant cell lines (Supplementary Figure 8D). However, like the previous analysis (Figure 5D), some resistant lines clustered with sensitive lines using this method. We performed principal component analysis and plotted each cell line across the 2 first principal components. The most resistant cell lines (EC50 > 10 μM) occupied the same PCA space and segregated separately from relatively more sensitive lines (Figure 5E).
Discussion
GBM remains a refractory disease with a high rate of clinical trial failures.36 Neddylation pathway activation has been demonstrated in GBM, along with antitumor efficacy of the NAE inhibitor MLN4924.9 In this work, we employed glioma cell lines and GBM PDX cultures to explore determinants of sensitivity and resistance to MLN4924. Vulnerability to MLN4924 was associated with cell cycle dysregulation, enrichment of DNA replication transcripts, and the chromatin binding functions of PTEN. Loss of WT PTEN correlated with decreased sensitivity to 3 different NAE inhibitors in GBM models. Using genes that correlated strongly with EC50 across all 3 NAE inhibitors, we curated a NAEi response gene set that could segregate the GBM cell lines that were most resistant to MLN4924 in PCA space. These findings highlight the clinical potential of NAE inhibitors to treat molecular subsets of GBM patients.
Knockdown of DNA damage repair and chromatin dynamics pathways has been shown to decrease MLN4924 efficacy.18,37 Garcia et al.,18 proposed that the accumulation of DNA repair machinery at stalled replication forks results in a higher likelihood of fork collisions and DNA damage under the CDT1-driven re-replicative state. While orthogonal evidence, several components of this manuscript align with this model of NAE inhibitors’ mechanism of cell death: namely, in sensitive cell lines there is modestly earlier accumulation of CDT1 (Figure 1F), enrichment of transcripts and proteins broadly related to DNA replication (Figure 2A-B), increased S-phase and > 4N cell populations (Figure 2C), and increased DNA damage and apoptosis (Figure 2D and Supplementary Figure 2). Our interrogation of PTEN and its role in MLN4924 response also provides support for this model on mechanisms of vulnerability.
Our data demonstrate that PTEN mediates vulnerability to NAE inhibitors. Glioma cell lines resistant to MLN4924 were significantly more likely to have PTEN mutations and/or loss (Figure 3B). Knockdown of PTEN in sensitive glioma cells diminished response to MLN4924, while ectopic expression of PTEN-sensitized PTEN-null cells (Figure 3D–H). A phosphatase-dead PTEN construct retained MLN4924 sensitivity (Figure 4B). Instead, we show that the loss of phosphorylation at Y240 drove resistance to MLN4924 (Figure 4C), which has been shown to play a critical role in DNA damage response through chromatin binding and interactions with Ki67 and RAD51.26 Depletion of EP400, a protein that makes chromatin more accessible for DNA damage response proteins like RAD51, eliminated synergy between MLN4924 and mitomycin.18 Although follow-up studies are required, the nuclear functions of PTEN and the processes shown to alter vulnerability to MLN4924 largely overlap. Although not explored in this manuscript, interestingly, PTEN was recently shown to be a substrate for neddylation, impacting its nuclear localization.38
By expanding both the number of NAE inhibitors (3 total) and of GBM models tested, we determined that GBM cell lines with WT PTEN are consistently more sensitive to NAE inhibition (Figure 5A–B). Interestingly, cells with complete loss of PTEN were more sensitive to all compounds than those with PTEN mutations. While our previous results demonstrated that loss of chromatin binding but not its lipid phosphatase function diminished glioma cell sensitivity to MLN4924, we do not believe this exhaustively characterizes the interplay between PTEN and NAEi response (Figure 4). PTEN has many cellular functions, and we propose that retention of some PTEN functions while losing others may be important for response to NAE inhibition.
Despite this complexity, our expanded analysis of NAE inhibitors further reinforced the association between higher activation of DNA replication and cell cycle processes and NAE inhibitor sensitivity, which was observed at every stage of analysis (Figures 2,4, and 5). We curated a NAEi Response Gene Set and found that it could effectively segregate the GBM cell lines most resistant to MLN4924 (Figure 5E). While not in its final form, this gene set provides a starting point for further development of a signature of vulnerability as more clinical sequencing data is produced and other analysis methods improve. Although PTEN alterations occur in at least 50% of GBM cases, which may drive resistance to NAE inhibitors and limit their use, we hope that NAE inhibitors become one of many drugs that can be used for cancer care within a personalized medicine framework.39,40
In summary, our study provides further evidence for the clinical utility of NAE inhibitors in a defined, molecular subset of GBM models. The status and functional state of PTEN and defined genes and pathways related to DNA replication and cell cycle may serve to inform NAEi clinical trial enrollment. As the defined ability of MLN4924 to penetrate the blood-brain barrier makes it an attractive candidate to improve GBM outcomes, better understanding of vulnerability will improve recruitment of patients most likely to benefit and may enhance clinical outcomes.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
Mentorship to trainees in the lab who contributed to this work was provided by Shayesteh Ferdosi, Ph.D. Editorial review of the manuscript by Timothy Whitsett, Ph.D. and Sophia Carvalo is gratefully acknowledged.
Contributor Information
Brett Taylor, Cancer and Cell Biology Division, The Translational Genomics Research Institute, Phoenix, Arizona, USA.
Nanyun Tang, Cancer and Cell Biology Division, The Translational Genomics Research Institute, Phoenix, Arizona, USA.
Yue Hao, Cancer and Cell Biology Division, The Translational Genomics Research Institute, Phoenix, Arizona, USA.
Matthew Lee, Cancer and Cell Biology Division, The Translational Genomics Research Institute, Phoenix, Arizona, USA.
Sen Peng, Cancer and Cell Biology Division, The Translational Genomics Research Institute, Phoenix, Arizona, USA.
Rita Bybee, Cancer and Cell Biology Division, The Translational Genomics Research Institute, Phoenix, Arizona, USA.
Lauren Hartman, Cancer and Cell Biology Division, The Translational Genomics Research Institute, Phoenix, Arizona, USA.
Krystine Garcia-Mansfield, Collaborative Center for Translational Mass Spectrometry, The Translational Genomics Research Institute, Phoenix, Arizona, USA.
Ritin Sharma, Collaborative Center for Translational Mass Spectrometry, The Translational Genomics Research Institute, Phoenix, Arizona, USA.
Patrick Pirrotte, Collaborative Center for Translational Mass Spectrometry, The Translational Genomics Research Institute, Phoenix, Arizona, USA.
Jianhui Ma, Department of Medicine, University of California San Diego, La Jolla, California, USA.
Alison D Parisian, Department of Medicine, University of California San Diego, La Jolla, California, USA.
Frank Furnari, Department of Medicine, University of California San Diego, La Jolla, California, USA.
Harshil D Dhruv, Cancer and Cell Biology Division, The Translational Genomics Research Institute, Phoenix, Arizona, USA.
Michael E Berens, Cancer and Cell Biology Division, The Translational Genomics Research Institute, Phoenix, Arizona, USA.
Funding
National Institutes of Health (U01CA168397 to H.D. and M.B., R01NS080939 to F.F., 5U19CA264512 to M.B.); the Defeat GBM Research Collaborative, a subsidiary of the National Brain Tumor Society (F.F.), and the Ben and Catherine Ivy Foundation to B.T.
Conflict of interest statement
Authors have no financial interests or conflict of interest to disclose.
Authorship statement
Conceptualization: B.T., H.D., and M.B.; Methodology: B.T., H.D., M.B., N.T., Y.H., S.P., and P.P.; Validation: B.T., N.T., Y.H., R.B., L.H., and M.B.; Formal Analysis: Y.H., S.P., M.L., K.G.M., R.S.H., J.M., and A.D.P.; Resources: M.B., P.P., F.F., and H.D.; Data Curation: B.T.; Writing—Original Draft, B.T., Writing—Review & Editing: All the authors; Supervision: M.B. and H.D..
References
- 1. Ostrom QT, Patil N, Cioffi G, Waite K, Kruchko C, Barnholtz-Sloan JS.. CBTRUS statistical report: Primary brain and other central nervous system tumors diagnosed in the United States in 2013-2017. Neuro Oncol. 2020; 22(suppl_1):iv1–iv96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Louis DN, Perry A, Reifenberger G, et al. The 2016 World Health Organization classification of tumors of the central nervous system: A summary. Acta Neuropathol. 2016;131(6):803–820. [DOI] [PubMed] [Google Scholar]
- 3. Stupp R, Mason WP, van den Bent MJ, et al. ; European Organisation for Research and Treatment of Cancer Brain Tumor and Radiotherapy Groups. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352(10):987–996. [DOI] [PubMed] [Google Scholar]
- 4. Soucy TA, Smith PG, Milhollen MA, et al. An inhibitor of NEDD8-activating enzyme as a new approach to treat cancer. Nature. 2009;458(7239):732–736. [DOI] [PubMed] [Google Scholar]
- 5. Enchev RI, Schulman BA, Peter M.. Protein neddylation: beyond cullin-RING ligases. Nat Rev Mol Cell Biol. 2015;16(1):30–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sarikas A, Hartmann T, Pan ZQ.. The cullin protein family. Genome Biol. 2011;12(4):220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Zhou L, Jiang Y, Luo Q, Li L, Jia L.. Neddylation: A novel modulator of the tumor microenvironment. Mol Cancer. 2019;18(1):77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zhou L, Zhang W, Sun Y, Jia L.. Protein neddylation and its alterations in human cancers for targeted therapy. Cell Signal. 2018;44:92–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hua W, Li C, Yang Z, et al. Suppression of glioblastoma by targeting the overactivated protein neddylation pathway. Neuro Oncol. 2015;17(10):1333–1343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Brownell JE, Sintchak MD, Gavin JM, et al. Substrate-assisted inhibition of ubiquitin-like protein-activating enzymes: The NEDD8 E1 inhibitor MLN4924 forms a NEDD8-AMP mimetic in situ. Mol Cell. 2010;37(1):102–111. [DOI] [PubMed] [Google Scholar]
- 11. Sarantopoulos J, Shapiro GI, Cohen RB, et al. Phase I study of the investigational NEDD8-activating enzyme inhibitor pevonedistat (TAK-924/MLN4924) in patients with advanced solid tumors. Clin Cancer Res. 2016;22(4):847–857. [DOI] [PubMed] [Google Scholar]
- 12. Filippova N, Yang X, An Z, Nabors LB, Pereboeva L.. Blocking PD1/PDL1 Interactions Together with MLN4924 therapy is a potential strategy for glioma treatment. J Cancer Sci Ther. 2018;10(8):190–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Smith MA, Maris JM, Gorlick R, et al. Initial testing of the investigational NEDD8-activating enzyme inhibitor MLN4924 by the pediatric preclinical testing program. Pediatr Blood Cancer. 2012;59(2):246–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lin Y, Luo Y, Sun Y, et al. Genomic and transcriptomic alterations associated with drug vulnerabilities and prognosis in adenocarcinoma at the gastroesophageal junction. Nat Commun. 2020;11(1):6091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Nishitani H, Lygerou Z, Nishimoto T, Nurse P.. The Cdt1 protein is required to license DNA for replication in fission yeast. Nature. 2000;404(6778):625–628. [DOI] [PubMed] [Google Scholar]
- 16. Lin JJ, Milhollen MA, Smith PG, Narayanan U, Dutta A.. NEDD8-Targeting Drug MLN4924 Elicits DNA rereplication by stabilizing cdt1 in s phase, triggering checkpoint activation, apoptosis, and senescence in cancer cells. Cancer Research. 2010; 70(24):10310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Cerami E, Gao J, Dogrusoz U, et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2(5):401–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Garcia K, Blank JL, Bouck DC, et al. Nedd8-activating enzyme inhibitor MLN4924 Provides Synergy with Mitomycin C through Interactions with ATR, BRCA1/BRCA2, and Chromatin Dynamics Pathways. Mol Cancer Ther. 2014;13(6):1625–1635. [DOI] [PubMed] [Google Scholar]
- 19. Ochiiwa H, Ailiken G, Yokoyama M, et al. TAS4464, a NEDD8-activating enzyme inhibitor, activates both intrinsic and extrinsic apoptotic pathways via c-Myc-mediated regulation in acute myeloid leukemia. Oncogene. 2021;40(7):1217–1230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yoshimura C, Muraoka H, Ochiiwa H, et al. TAS4464, A highly potent and selective inhibitor of NEDD8-Activating Enzyme, suppresses neddylation and shows antitumor activity in diverse cancer models. Mol Cancer Ther. 2019;18(7):1205–1216. [DOI] [PubMed] [Google Scholar]
- 21. Ni S, Chen X, Yu Q, et al. Discovery of candesartan cilexetic as a novel neddylation inhibitor for suppressing tumor growth. Eur J Med Chem. 2020;185:111848. [DOI] [PubMed] [Google Scholar]
- 22. Milella M, Falcone I, Conciatori F, et al. PTEN: Multiple functions in human malignant tumors. Front Oncol. 2015;5:24:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Benitez JA, Ma J, D’Antonio M, et al. PTEN regulates glioblastoma oncogenesis through chromatin-associated complexes of DAXX and histone H3.3. Nat Commun. 2017:8:15223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Brandmaier A, Hou S-Q, Shen WH.. Cell cycle control by PTEN. J Mol Biol. 2017;429(15):2265–2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hou S-Q, Quyang M, Brandmaier A, Hao H, Shen WH.. PTEN in the maintenance of genome integrity: From DNA replication to chromosome segregation. BioEssays. 2017;39(10):1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ma J, Benitez JA, Li J, et al. Inhibition of nuclear PTEN tyrosine phosphorylation enhances glioma radiation sensitivity through attenuated DNA Repair. Cancer Cell. 2019;35(3):504–518.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sonoda Y, Ozawa T, Hirose Y, et al. Formation of intracranial tumors by genetically modified human astrocytes defines four pathways critical in the development of human anaplastic astrocytoma. Cancer Res. 2001;61(13):4956–4960. [PubMed] [Google Scholar]
- 28. Dhruv HD, Roos A, Tomboc PJ, et al. Propentofylline inhibits glioblastoma cell invasion and survival by targeting the TROY signaling pathway. J Neurooncol. 2016;126(3):397–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Vaubel RA, Tian S, Remonde D, et al. Genomic and phenotypic characterization of a broad panel of patient-derived xenografts reflects the diversity of glioblastoma. Clin Cancer Res. 2020;26(5):1094–1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lee C, Kim J-S, Waldman T.. PTEN gene targeting reveals a radiation-induced size checkpoint in human cancer cells. Cancer Res. 2004;64(19):6906–6914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Jang IS, Neto EC, Guinney J, Friend SH, Margolin AA.. Systematic assessment of analytical methods for drug sensitivity prediction from cancer cell line data. Pac Symp Biocomput. 2014;63–74. [PMC free article] [PubMed] [Google Scholar]
- 32. Jia L, Sun Y.. SCF E3 ubiquitin ligases as anticancer targets. Curr Cancer Drug Targets. 2011;11(3):347–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Benitez JA, Finlay D, Castanza A, et al. PTEN deficiency leads to proteasome addiction: A novel vulnerability in glioblastoma. Neuro Oncol. 2021;23(7):1072–1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Saavedra L, Catarino R, Heinz T, et al. Phosphatase and tensin homolog is a growth repressor of both rhizoid and gametophore development in the moss physcomitrella patens. Plant Physiol. 2015;169(4):2572–2586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Chi C, Ye Y, Chen B, Huang H.. Bipartite graph-based approach for clustering of cell lines by gene expression-drug response associations. Bioinformatics. 2021;37(17):2617–2626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Mandel JJ, Shlomit Y-K, Patel AJ, et al. Inability of positive phase II clinical trials of investigational treatments to subsequently predict positive phase III clinical trials in glioblastoma. Neuro Oncol. 2018;20(1):113–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Blank JL, Liu XJ, Cosmopoulos K, et al. Novel DNA damage checkpoints mediating cell death induced by the NEDD8-Activating Enzyme Inhibitor MLN4924. Cancer Res. 2013;73(1):225–234. [DOI] [PubMed] [Google Scholar]
- 38. Xie P, Peng Z, Chen Y, et al. Neddylation of PTEN regulates its nuclear import and promotes tumor development. Cell Res. 2021;31(3):291–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Adam G, Rampasek L, Safikhani Z, et al. Machine learning approaches to drug response prediction: challenges and recent progress. NJP Precis Oncol. 2020;4:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Partin A, Brettin TS, Zhu Y, et al. Deep learning methods for drug response prediction in cancer: Predominant and emerging trends. Front Med (Lausanne). 2023;10:1086097. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.