

Abstract
Emerging evidence supports the important role of the tumor microbiome in oncogenesis, cancer immune phenotype, cancer progression, and treatment outcomes in many malignancies. In this study, we investigated the metastatic melanoma tumor microbiome and its potential roles in association with clinical outcomes, such as survival, in patients with metastatic disease treated with immune checkpoint inhibitors (ICI). Baseline tumor samples were collected from 71 patients with metastatic melanoma before treatment with ICIs. Bulk RNA sequencing (RNA-seq) was conducted on the formalin-fixed, paraffin-embedded and fresh frozen tumor samples. Durable clinical benefit (primary clinical endpoint) following ICIs was defined as overall survival ≥24 months and no change to the primary drug regimen (responders). We processed RNA-seq reads to carefully identify exogenous sequences using the {exotic} tool. The age of the 71 patients with metastatic melanoma ranged from 24 to 83 years, 59% were male, and 55% survived >24 months following the initiation of ICI treatment. Exogenous taxa were identified in the tumor RNA-seq, including bacteria, fungi, and viruses. We found differences in gene expression and microbe abundances in immunotherapy-responsive versus nonresponsive tumors. Responders showed significant enrichment of bacteriophages in the phylum Uroviricota, and nonresponders showed enrichment of several bacteria, including Campylobacter jejuni. These microbes correlated with immune-related gene expression signatures. Finally, we found that models for predicting prolonged survival with immunotherapy using both microbe abundances and gene expression outperformed models using either dataset alone. Our findings warrant further investigation and potentially support therapeutic strategies to modify the tumor microbiome in order to improve treatment outcomes with ICIs.
Significance:
We analyzed the tumor microbiome and interactions with genes and pathways in metastatic melanoma treated with immunotherapy and identified several microbes associated with immunotherapy response and immune-related gene expression signatures. Machine learning models that combined microbe abundances and gene expression outperformed models using either dataset alone in predicting immunotherapy responses.
Introduction
Advances in immunotherapy, including immune checkpoint inhibitors (ICI), have transformed the standard of care for many types of cancer, including melanoma. Although ICIs have improved outcomes for patients with melanoma, many patients suffer from primary or secondary tumor resistance. For example, at 6.5 years, the overall survival (OS) rates with ipilimumab plus nivolumab, nivolumab, and ipilimumab were 49%, 42%, and 23%, respectively, as reported in the pivotal CheckMate 067 trial (1). Furthermore, mechanisms of resistance to immunotherapy remain poorly understood, and many treatments are associated with immune-mediated toxicities. Therefore, there is an urgent need to develop and improve biomarkers predictive of benefit from ICI therapy.
Numerous biomarkers that predict the response of melanoma to ICIs are under investigation, including those based on clinical characteristics, genomics, transcriptomics, and epigenomics. For genomic data, these predictive biomarkers include tumor mutational burden (TMB; ref. 2), neoantigen load (3), genotypes of HLA-I (3, 4), T-cell repertoire (5), aneuploidy (also known as somatic copy number alterations; ref. 6), and germline variations (7). On the other hand, predictive biomarkers derived from transcriptomic data include tumor oncogene expression signatures, such as genes related to MYC (8), WNT/β-catenin (9, 10), or RAS (11) signaling; gene expression profiles within the tumor immune microenvironment (TIME), such as IFNγ-responsive genes (12), chemokines (13, 14), MHC classes I and II (15); and cytotoxic T-cell and effector T-cell (16, 17) gene expression markers that have been reported to be predictive of ICI response in metastatic melanoma. Unfortunately, the predictive power remains low. For example, in terms of prediction of ICI response, TMB, IFNγ-responsive gene signatures, or the combination of TMB and IFNγ gene signatures produce an area under the receiver operating characteristic (AUROC) curve of 0.60 to 0.84 in melanoma cohorts (18).
Recently, high-throughput transcriptome-, genome-, or amplicon-based sequencing data demonstrated an abundance and variety of microbes’ nucleic acids inside tumors (8). In some cases, 100 of negative controls and paraffin-only blocks were sequenced to ensure a thorough understanding of the background signal and reagent contamination. Furthermore, the presence of microbes has been validated using FISH and IHC (19). The microbes showed cancer specificity (9, 12, 13), and blood-based measurements could predict early-stage disease. These findings suggest that microbes observed in high-throughput sequencing data may also correlate with treatment outcomes. Recent efforts to use these microbes as biomarkers showed that although generally less predictive of prognosis than gene expression, when combined with gene expression, they increase the predictive power (20). Furthermore, the tumor microbiome was predictive of chemotherapy response.
Here, we describe the use of tumor RNA sequencing (RNA-seq) to predict response to ICIs in patients with melanoma (Fig. 1). We demonstrate the presence of microbes within tumors and show different microbial communities in patients whose tumors responded to treatment. We predict treatment response using human gene expression patterns that perform similarly to other ICI response prediction efforts. Finally, we show how the presence of microbes correlates with these signatures, suggesting an interaction with the immune system, and how including tumor microbes in these models improves their predictive accuracy.
Figure 1.

Graphical summary. RNA-seq data from tumor specimens are processed to microbe abundances and human gene expression. Each is associated with ICI response, and then integrative analyses combine them into a model to predict outcomes.
Materials and Methods
Study design
Established in 2014, the Oncology Research Information Exchange Network (ORIEN) is an alliance of 18 US cancer centers. All ORIEN alliance members utilize a standard institutional review board (IRB)-–approved protocol: Total Cancer Care (TCC). As part of the TCC protocol, participants provide written informed consent to have their clinical data followed over time, undergo germline and tumor sequencing, and be contacted in the future by their provider if an appropriate clinical trial or other study becomes available (21). TCC is a prospective cohort study in which a subset of patients elect to be enrolled in the ORIEN Avatar program, which provides research use only–grade whole-exome tumor sequencing, RNA-seq, germline sequencing, and collection of deep longitudinal clinical data with lifetime follow-up. Nationally, more than 325,000 participants have enrolled in TCC. M2GEN, the commercial and operational partner of ORIEN, harmonizes all abstracted clinical data elements and molecular sequencing files into a standardized, structured format to enable the aggregation of de-identified data for sharing across the network. Data access was approved by the IRB in an Honest Broker protocol (2015H0185) and TCC protocol (2013H0199) in coordination with M2GEN and participating ORIEN members. In this study, we assembled RNA-seq data from the tumor samples of 71 patients with metastatic melanoma treated with ICIs.
Primary clinical endpoint
In this real-world dataset, no tumor size measurements were available to assess treatment response according to immune response evaluation criteria in solid tumors (iRECIST) criteria or other standardized scores. Instead, in concordance with other studies with similar cohorts (22–24), we defined durable clinical benefit as OS ≥24 months after the start of ICI treatment (hereafter referred to as responders). Therefore, nonresponders survived for <24 months after the initiation of ICI treatment.
Sequencing methods
ORIEN Avatar specimens underwent nucleic acid extraction and sequencing at HudsonAlpha (Huntsville, AL) or Fulgent Genetics (Temple City, CA). For frozen and optimal cutting temperature (OCT) tissue DNA extraction, Qiagen QIASymphony DNA purification was performed, generating a 213 bp average insert size. For frozen and OCT tissue RNA extraction, the Qiagen RNeasy Plus Mini Kit was used, generating a 216 bp average insert size. For formalin-fixed, paraffin-embedded tissue, a Covaris Ultrasonication FFPE DNA/RNA kit was utilized to extract DNA and RNA, generating a 165 bp average insert size. RNA-seq was performed using the Illumina TruSeq RNA Exome with single library hybridization, cDNA synthesis, library preparation, and sequencing (100 bp paired reads at HudsonAlpha; 150 bp paired reads at Fulgent Genetics) to a coverage of 100 M total reads/50 M paired reads.
Data processing and gene expression analyses
RNA-seq tumor pipeline analysis was processed according to the workflow outlined below using GRCh38/hg38 human genome reference sequencing and GenCode build version 32. Adapter sequences were trimmed from the raw tumor sequencing FASTQ file. Adapter trimming via k-mer matching was performed along with quality trimming and filtering, contaminant filtering, sequence masking, guanosine-cytosine (GC) filtering, length filtering, and entropy filtering. The trimmed FASTQ file was used as input to the read alignment process. The tumor adapter–trimmed FASTQ file was aligned to the human genome reference (GRCh38/hg38) and the GenCode genome annotation v32 using the STAR aligner. The STAR aligner generates multiple output files for gene fusion prediction and gene expression analysis. RNA expression values were calculated and reported using estimated mapped reads, fragments per kilobase of transcript per million mapped reads, and transcripts per million (TPM) mapped reads at both the transcript and gene levels based on transcriptome alignment generated using STAR. RSEM pipeline and gene expressions were quantified as TPM. Gene expressions were log2(TPM + 1)-transformed, and downstream analyses were performed using GE Matrix. To determine differentially expressed genes (DEG) of responders versus nonresponders, we used the limma (v. 3.54.0) and edgeR (v. 3.40.0) packages, in which genes that have log2 fold change greater or less than 1 and adjusted P value ≤0.1 were considered significant DEGs. For gene set enrichment analysis (GSEA) of responders vs. nonresponders, we used the Java version of gsea (v. 4.3.2) using the gene set permutation of 1000 using Hallmark gene sets or TIMEx cell types. Gene sets or cell types that have adjusted P value <0.1 were considered significant. The normalized enrichment score (NES) and adjusted P value were provided in the plot.
Microbe abundance and diversity
RNA-seq reads were used to calculate microbe abundances using the {exotic} pipeline v1.2, which included a 16S validation cohort and described a series of decontamination and filtering steps in detail (25). Briefly, reads were aligned first to the human reference genome, and then unaligned reads were mapped to a database of bacteria, fungi, archaea, viruses, and eukaryotic parasites. The observed microbes were then proceeded through a series of filtering steps to carefully and conservatively remove contaminants, including segmenting the CHM13 human transcriptome into 100 base pairs, with 50 base pair overlaps, running through the exotic pipeline, and filtering out any microbe falsely identified in this process. We used raw counts to calculate diversity measures and for differential abundance analyses using {DESeq2} (26). The stacked bar plot and correlations used a relative abundance value, generated by dividing raw counts by the number of human reads. Diversity measures were estimated by calculating the Shannon and Simpson indices, as well as Chao1, ACE, and inverse Simpson using the R package {vegan} (27).
Signature and pathway analyses
Gene signature scores were calculated using the IOSig and tmesig R packages. In brief, for each published gene signature, we collected and harmonized gene names using the NCBI Entrez gene number. To quantify the published gene expression score, we first transformed the gene expressions across samples within a cohort into a Z-score. Next, we averaged the standardized Z-score across the number of genes in the signature, as previously described (15, 28, 29). This score is used to compare responders and nonresponders of immunotherapies within individual cohorts based on the AUROC, as previously described (28). We performed clustering of gene signatures based on the correlation of AUROC across multiple cohorts. Within a cohort of patients, we stratified the patients into “high” or “low” groups based on the mean of the Z-score. A Mann–Whitney U test was performed by comparing the two groups to determine the difference, and a FDR of <0.05 was deemed to be significant. The list of published gene signatures is available in Supplementary Table S1.
For pathway analysis, single-sample GSEA (ssGSEA), via the ssGSEA method in the GSVA R package, was utilized to investigate the enriched gene sets in each sample. GSVA was run using the log2(TPM + 1) gene expression values with the Gaussian kernel. The Hallmark gene sets, TIMEx cell types, and the collected previously published gene expression signatures were used as the gene sets. The Hallmark gene sets are a curated list of gene sets that signify well-understood pathways that display reliable gene expression (30). The TIMEx cell types are formed from pan-cancer single-cell RNA-seq signatures and focus on illuminating immune cell infiltration from bulk RNA-seq data (31). A Spearman correlation analysis was conducted using the differentially expressed microbe data and the three ssGSEA results. The gene sets were clustered according to the Euclidean distance with complete linkage, whereas the microbes were ordered from the highest to the lowest effect size.
Prediction of response to treatment outcomes
To assess the predictive ability of the RNA-seq and microbe data for tumor response to ICIs, random forest classifiers were created using the randomForest R package. Models were based on five sets of input data: (i) microbe data, (ii) 31–gene signature Z-score, (iii) immune-activated gene signature Z-score, (iv) microbe and 31–gene signature Z-score combined, and (v) microbe- and immune-activated gene signature Z-score combined. Models were constructed with 500 trees and fivefold cross-validation. Additionally, 5 seeds were used for each model, resulting in 25 trained models based on each set of input features. The AUROC curve was used to assess the overall performance of the trained models. This metric assesses the model classification accuracy, in which 1 is a perfect classifier and 0.5 is a random classifier. The overall performance for each input feature–based model was taken as the average of the 25 trained models.
Independent validation datasets
We downloaded two independent melanoma RNA-seq datasets from public repositories: Gide and colleagues (BioProject: PRJEB23709, n = 91; ref. 32) and Riaz and colleagues (BioProject: PRJNA356761, n = 105; ref. 33). Both datasets contained patients with melanoma receiving various immunotherapies, similar to the ORIEN Avatar training cohort. Raw RNA-seq files were downloaded and aligned as described in RNA-seq data processing and microbe abundance analyses. We used the random forest models developed in the ORIEN Avatar training set and tested the balanced accuracy of these models. The balanced accuracy is the arithmetic mean of sensitivity and specificity which account for imbalanced data.
Data availability
The Ohio State University IRB approved data access in an Honest Broker protocol (2015H0185) and TCC protocol (2013H0199) in coordination with Aster Insights. The processed data generated in this study are publicly available in Gene Expression Omnibus through the BioProject PRJNA856973. Analysis scripts to regenerate all figures and tables are available at https://github.com/spakowiczlab/exorien-melio.
Results
Patient characteristics
From the ORIEN networks, we included 71 patients with metastatic melanoma in this study (IO_NOVA_Mel). The age of the patients in this cohort ranged from 24 to 83 years; 59% were male; and 55% survived >24 months following the initiation of ICI treatment (Table 1). ICI treatments included anti-CTLA4 (18.8% of nonresponders and 41.0% of responders), anti-PD1/PDL1 (62.5% of nonresponders and 56.4% of responders), and anti-CTLA + anti-PD1/PDL1 (18.8% of nonresponders and 2.6% of responders). The mean OS of responders (49.58 months) and nonresponders (10.82 months) was significantly different (P value <0.001).
Table 1.
Patient demographics stratified by response to ICIs
| Nonresponder (n = 32) | Responder (n = 39) | P value | |||
|---|---|---|---|---|---|
| Age [mean (SD)] | 57.48 (15.85) | 58.62 (13.93) | 0.748 | ||
| Sex = male (%) | 18 (56.2) | 24 (61.5) | 0.835 | ||
| ICI [n (%)] | Anti-CTLA4 | 6 (18.8) | Anti-CTLA4 | 16 (41.0) | 0.022 |
| Anti-PD1/PDL1 | 20 (62.5) | Anti-PD1/PDL1 | 22 (56.4) | ||
| Anti-CTLA4 + anti-PD1/PDL1 | 6 (18.8) | Anti-CTLA4 + anti-PD1/PDL1 | 1 (2.6) | ||
| Sample collected within 1 year of ICI start = Yes (%) | 24 (75.0) | 29 (74.4) | 1.000 | ||
| OS [mean (SD)] months | 10.82 (6.23) | 49.58 (19.24) | <0.001 | ||
Gene expression analysis and its association with response to ICIs
The gene expression profiles for the 71 patients with metastatic melanoma treated with ICIs were obtained from ORIEN. We performed DEG analysis and identified five genes (CLEC12A, GBP1P1, CD96, CCL4, and IDO1) that were overexpressed in the responders compared with the nonresponders with log2 fold change >1 and adjusted P value <0.1 (Fig. 2A; Supplementary Table S2). Interestingly, these five genes were involved in immune modulation and have been previously identified in other studies as predictive biomarkers associated with responders to ICIs. For example, CCL4 has been previously identified as a biomarker in the 12-chemokine signature (13, 14), as well as other gene signatures predictive of neoadjuvant ipilimumab response (34). IDO1 has been identified as a key marker in the IFNγ signature (12), as well as other gene signatures predictive of response to ICIs in lung cancer (35). CD96 is a marker that estimates CD8+ T-cell infiltration (36, 37). CD96 and TIGIT along with the costimulatory receptor CD226 form a pathway that affects the immune response in an analogous way to the CD28/CTLA4 pathway (38). CLEC12A (39, 40) and GBP1P1 (41, 42) were identified in immune-related gene expression signatures predictive of ICI responses.
Figure 2.

Immune-related gene expression associates with the response to ICIs. A, Gene expression differences between the tumors responsive (right) and nonresponsive (left) to ICI treatment. Significantly different genes after FDR correction are colored and labeled. B and C, GSEA comparing responders vs. the nonresponders using the Hallmark gene set and TIMEx cell types. FDR < 0.1 was used as the cutoff. D, Mann–Whitney comparison of responders and nonresponders for signatures reaching the 0.05 FDR threshold.
Next, we investigated which gene sets and pathways were enriched or depleted in responders to ICIs. We performed GSEA using the MSigDB Hallmark gene sets on the RNA-seq and found that several immune-related gene sets were significantly enriched in responders (Fig. 2B), for example, IFNα response (NES = 1.98; FDR < 0.001), IFNγ response (NES = 1.79; FDR < 0.001), and allograft rejection (NES = 1.65; FDR = 0.002). The other two gene sets enriched in responders were spermatogenesis (NES = 1.56; FDR = 0.005) and the pancreas β cell gene sets (NES = 1.40; FDR = 0.036). In contrast, many cell-intrinsic gene sets were enriched in ICI nonresponders, as shown in Fig. 2B. The GSEA results identified in this cohort are similar to previously published studies (28).
We next hypothesized that tumor-infiltrating immune cells could associate with responses to ICIs. To test this hypothesis, we performed cell-type deconvolution of the bulk RNA-seq using CIBERSORT. From CIBERSORT results, we observed that responders had significantly (P value <0.05) higher abundances of CD8+ T cells, activated CD4+ memory T cells, activated NK cells, and M1 macrophages relative to nonresponders, who were shown to have a significantly higher amount of resting mast cells (Supplementary Fig. S1). Similarly, when we performed GSEA using TIMEx gene sets, we observed that 13 CD4+-, CD8+-, and NK-related cell types were enriched in responders (FDR < 0.1), whereas the stromal cell type was enriched in nonresponders (Fig. 2C). This suggests that the tumor microenvironment (TME) of responders had an “immune-inflamed” phenotype, whereas nonresponders had either “immune-excluded” or “immune-desert” TME phenotypes.
To further delineate the immune phenotypes of responders versus nonresponders of ICIs, we used previously published gene signatures. We collected 30 gene expression signatures from the literature that have been implicated to be predictive of ICIs (28). By performing a Z-score for each signature and associating them with responders versus nonresponders, we identified 16 gene signatures (Supplementary Fig. S2) in which high Z-scores are associated with ICI responsiveness in this cohort (FDR <0.05), and the top four gene signatures are illustrated in Fig. 2D. These 16 gene signatures were related to immune activation and inflammation signatures (Supplementary Fig. S2; ref. 28).
We next used our recently developed IOSig portal (28) to evaluate the predictive values of these 16 gene signatures in our ORIEN cohort (IO_NOVA_Mel), as well as 22 other melanoma cohorts treated with ICIs. We used AUROC to assess the predictive value of these signatures. For the 16 gene signatures, the AUROC values ranged from 0.78 to 0.66 in the IO_NOVA_Mel cohort (Supplementary Fig. S3; Supplementary Table S3). On average, the AUROC values for these 16 gene signatures ranged from 0.61 to 0.68 in the separate 22 melanoma cohorts (Supplementary Fig. S3; Supplementary Table S3).
The melanoma tumor microbiome and its association with response to ICIs
Exogenous taxa were identified in the tumor RNA-seq, including bacteria, fungi, and viruses. A total of 54 phyla were observed, with Firmicutes being the most abundant phylum, followed by Uroviricota (Fig. 3A). Within the tumors responsive to immunotherapy, we found a significant enrichment of the Uroviricota phylum. Comparatively, the cohort of nonresponsive tumors was found to have significant intratumoral enrichment of bacteria and viruses, including Campylobacter jejuni, Acinetobacter calcoaceticus, and the Baculoviridae family (Fig. 3B; Supplementary Table S4). In order to provide further validation, we conducted an analysis checking The Cancer Genome Atlas data for the microbes most significantly enriched in our response groups. All of the microbes that best discriminate between SKCM responders and nonresponders to ICIs were observed in The Cancer Genome Atlas dataset. The prevalences were consistent with the exception of A. calcoaceticus, which was observed but not as prevalent as in the ORIEN dataset (Supplementary Fig. S4). However, the genus Acinetobacter was consistently prevalent between the two datasets. We observed no significant differences between α-diversity metrics of responders and nonresponders (Welch two-sample t tests P value >0.4; Fig. 3C). We found that the random forest classifiers based on microbe diversity measures with five rounds of fivefold cross-validation performed poorly relative to our other microbe-based classifiers (Fig. 3D).
Figure 3.

Melanoma tumors that respond to ICIs have a distinct tumor microbiome. A, The relative abundances of the tumor microbiome at the phylum level showed wide intersample variation in the abundances of Proteobacteria (blue) and viruses [Uroviricota (maroon)], without gross differences between nonresponders (NR) and responders (R). B, Differential abundance analysis of taxa found within tumor RNA-seq data. Colored points represent significantly (P value <0.05) enriched taxa with a high (>1.00) fold difference in abundance between responders and nonresponders to ICIs. C, The diversity of the tumor microbiome between responders and nonresponders shows no significant differences. D, The diversity of the microbiome is a poor predictor of outcomes.
Correlation of tumor RNA-seq (GSEA) with microbes
We next asked whether microbe abundance in the tumor could be associated with tumor-intrinsic pathways or the composition of cell types in the TIME. We focused on the nine microbes identified to be differentially abundant in relation to immunotherapy response in melanoma. For the top nine microbes, one microbe associated with responders to immunotherapy and eight with nonresponders (Fig. 4). To investigate the intrinsic pathways that correlated with the microbes, we performed ssGSEA on patients with melanoma using MSigDB Hallmark gene sets. Uroviricota, which was found to be highly abundant in responders, was correlated with inflammation and immune-related gene sets and pathways (Fig. 4). Conversely, we observed that the microbes that were highly abundant in nonresponders, bacteria in the Moraxellaceae family, correlated most strongly with notch signaling–related, myogenesis-related, and p53 pathway–related genes (Fig. 4). The full list of adjusted P values is available in Supplementary Table S5. These results are consistent with our previous findings, in which we observed similar Hallmark gene sets and pathways enriched in responders versus nonresponders across five melanoma cohorts of immunotherapy-treated patients with pre- and on-treatment tumor biopsies (28).
Figure 4.

Association of microbes and gene signatures. Effect size plot showing the top nine most significantly enriched species. Spearman correlation coefficients between the significantly enriched species, the most significantly correlated signatures, and other gene sets shown in a heatmap.
To further dissect the association of microbe abundance and the composition of cell types in the context of immunotherapy responses in melanoma, we performed cell-type deconvolution using the bulk RNA-seq with TIMEx. We found that Uroviricota was highly correlated with the enrichment of tumor-infiltrated immune cell types, including CD8+ T cells, which are known predictors of immunotherapy response (Fig. 4). In contrast, the lack of tumor-infiltrated immune cell types was correlated with microbes associated with nonresponders. In particular, we observed that stromal cell types, such as fibroblasts and endothelial cells, were enriched in association with the microbes noted in nonresponders, the Moraxellaceae and Microbacteriaceae families (Fig. 4). The tumor immune cell composition corroborated our previous findings (28).
Next, we asked whether the microbe abundance was associated with any gene signatures predictive of immunotherapy responses. To investigate this question, we utilized 31 previously published gene signatures that have been indicated to be associated with immunotherapy responses (28). We correlated microbe abundance with these signatures and found that gene signatures associated with inflammation or immune activation were highly associated with microbes abundant in responders (Fig. 4). On the other hand, gene signatures associated with immune-suppressive or -intrinsic signaling were highly associated with microbes abundant in nonresponders (Fig. 4). These results suggest that microbe abundance could provide a different dimension in understanding the TIME in predicting immunotherapy responsiveness in melanoma.
Prediction of response using tumor gene expression and microbe abundance
We further hypothesized that combining microbe abundance features with gene expression signatures could improve response prediction of melanoma to immunotherapy. To test this hypothesis, we developed an ensemble learning random forest classifier using microbe abundance and gene signatures identified to be associated with immunotherapy responses in melanoma (see Supplementary Fig. S5 for testing of other machine learning approaches). We first developed the random forest classifier based on microbe abundance with 15 input features (microbe) and performed 5 rounds of fivefold cross-validation on the melanoma cohort (Fig. 5). The average AUROC for the microbe classifier was 0.651. We also constructed a random forest classifier based on 31 gene signatures (GeneSig_Z_score) or the 16 immune-activated gene signatures (Imm_Act_Z_score), and the AUROC values for GeneSig or Imm_Act classifier were 0.72 and 0.744, respectively (Fig. 5). Notably, when we combined the microbe abundance and gene signatures to develop the random forest classifier, the ensemble learning random forest classifier for gene signatures plus microbe (GS_Z_microbe) and immune-activated gene signatures plus microbe (Imm_Act_Z_microbe) achieved 0.772 and 0.805, respectively (Fig. 5). Finally, to validate the generalizability of this combined microbe and gene expression signature random forest classifier, we tested this signature in two independent previously published datasets of Gide and colleagues (32) and Riaz and colleagues (33). Raw RNA-seq data were downloaded from BioProject, processed, aligned, and quantified for the gene expression and microbe abundance according to Fig. 1. As illustrated in Fig. 6, we found that the combined microbe and gene signature random forest classifier achieved 69.9% and 69.11% balanced accuracy in Gide and Riaz datasets, respectively. This suggests that microbe abundance features provide a distinct layer of information in predicting response to immunotherapy and, when combined with gene expression signatures, can improve the prediction of response to immunotherapy in melanoma.
Figure 5.

Prediction of response using gene expression and microbes. Mann–Whitney comparisons of the mean AUROC values from random forest model comparisons.
Figure 6.

Prediction of random forest model combining microbe abundance and gene signatures. Balanced accuracies achieved by the microbe abundance, gene expression, and combined models for each of the three datasets.
Discussion
We utilized tumor RNA-seq from patients with melanoma to explore the tumor microbiome’s influence on clinical outcomes, specifically in response to ICIs. We observed microbes in all samples and showed that tumors that responded to ICIs had significantly different taxa present from those that did not respond to treatment. Consistent with previous findings, gene expression seems to be predictive of response to ICIs. In addition, we showed that microbes are also predictive of response to ICIs, particularly when combined with gene expression, suggesting that the inclusion of microbes in these models enhances predictive ability.
A correlation between the gut microbiome and response to ICIs has been consistently indicated in previous research (43–45). Altering the gut microbiome via responder-derived fecal microbiota transplantation has been shown to induce a clinical response to anti-PD1 treatment in patients with melanoma (46, 47). However, many of the efforts in this area have focused solely on the gut microbiome. Therefore, we assessed the tumor microbiome to further explore the impact of microbes on clinical outcomes in body sites beyond the gut.
We observed the presence of microbiota in all 71 tumor samples, as is consistent with previous findings with regard to the tumor microbiome (48, 49). Our study explicitly exhibits the microbial characteristics of tumors in patients with metastatic melanoma. Previous research has shown that the tumor microbiome in this specific subset of cancer is predictive of response to treatment, but these findings have been limited in scope because of samples having been collected before the use of modern ICIs as a standard treatment regimen for metastatic melanoma (20). We showed distinct, significantly enriched taxa, including fungi, at baseline for patients treated with contemporary ICI-based treatment plans.
The mechanisms by which tumor microbes affect response to ICIs may relate to interactions with the immune system or several other established mechanisms (50).The World Health Organization has officially recognized a causal association between 11 microbes and cancer (51). However, in recent years, the number of likely carcinogenic microbes and more loosely related “complicit” microbes has increased dramatically. These have been shown to interact with the host via diverse mechanisms. For example, in colon cancer, Bacteroides fragilis biofilms on colon polyps have been found to secrete a toxin that directly damages DNA (52, 53), as have some Escherichia coli (54). In another mechanism, Helicobacter pylori secrete a series of molecules eliciting an inflammatory cascade shown to drive tumorigenesis in gastric adenocarcinoma and mucosa-associated lymphoma (55, 56). The fungal genus Malassezia caused pancreatic ductal adenocarcinoma growth through activation of the C3 complement pathway (57). The microbe enriched in responders has a precedence for interacting with the human immune system in the context of cancer. Uroviricota, which contains diverse bacteriophages, has been shown to contain antigens with cross-reactivity to melanoma tumors (58). On the other hand, C. jejuni has not been associated with the tumor microbiome or response to ICIs, although it is an established pathogen that has been linked to food-borne illness (59). In our study, it is associated with the distinct immune expression pathways as Uroviricota, suggesting that it acts through a very different mechanism. Furthermore, recent studies have identified bacteria-derived HLA-bound peptides in melanoma presented by tumor cells could elicit immune reactivity. This intratumoral bacteria peptide repertoire could be further explored to understand the mechanism by which microbes modulate the immune system and responses to therapy (60). The demonstration of the utility of high-throughput sequencing to explore these correlations warrants a broader search.
Efforts have been made to identify predictors of response and resistance to ICIs. As previously discussed, expression signatures have been established as predictors of ICI response in metastatic melanoma (9, 12, 14, 15, 28, 61). One such study assessing the model combining IFNγ and TMB found that it was predictive of response but not resistance (61). Another such study developed a multiomics-based classifier that successfully predicted response but was unable to predict resistance (20). We showed significantly enriched taxa in both response groups. We also showed that microbes alone are predictive of response/resistance to immunotherapy and when combined with gene expression, enhance the model’s predictive ability. These accuracies are consistent with other studies. For example, Mihály and colleagues reported that the best performing genes for predicting OS had AUC values of 0.64, 0.62, and 0.62 (62). Liu and colleagues reported that the AUC values for “1-year, 3-year, and 5-year OS were 0.832, 0.850, and 0.768; 0.712, 0.591, and 0.602; and 0.773, 0.702, and 0.673 for the training set, testing set, and whole set, respectively,” for their 6-gene signature (15). Long and colleagues reported that the AUC values of their 4‐gene biomarker prognostic model were 0.7561, 0.7674, 0.7366, 0.7040, and 0.6919 (63). Although these values are too low for clinical relevance, they provide a proof of concept that the inclusion of tumor microbes in predictive models can improve accuracy. Although a more rigorous approach to these predictive models would include a subgroup analysis on the ICI treatment types, we still see predictive power on the broader groups, providing evidence that we could harness yet more predictive power with the inclusion of treatment-type information. Further studies are warranted to combine tumor microbiome abundance with other clinical and “omics” (e.g., genomics and pathomics) for developing an accurate classifier for predicting immunotherapy responses in melanoma. Our findings also warrant further research to evaluate whether these correlations are causally associated with outcomes and their effect on the TIME and immune cell infiltration.
Although there is much utility in assessing a real-world clinical dataset, it also presents significant limitations. Notably, critical information such as stage, progression-free survival, Eastern Cooperative Oncology Group score, and prior treatment were unavailable. Additionally, although the majority of patients were treated with anti-PD1/PDL1 inhibitors, a small subset differed in treatment regimen. This introduces a major limitation as a stratified analysis could provide a meaningful insight, but the study is not sufficiently powered for this because of its small sample size. Previous studies have observed different microbes affecting anti-CTLA4 versus anti-PD1/PDL1 responses owing to distinct mechanisms of action. For instance, Bacteroides may induce an antitumor immune response in anti–CTLA4-treated tumors by activating dendritic cells in the gut to produce IL12, thereby stimulating T helper cells (64). It has been suggested that in anti–PDL1-treated tumors, microbes found in the gut may modulate the immune response through mechanisms such as secretion of small molecules such as inosine or indole-3-acetic acid (65, 66). However, tumors with evidence of immune activation (as measured by T-cell infiltration, PDL1 expression, IFNγ-related gene expression, or high TMB) have been shown to be susceptible to ICIs whether anti-PD1 treatment was used as monotherapy or in combination with anti-CTLA4 antibodies (1, 67). Therefore, intratumoral microbiome features that enhance the immunogenicity of the TME are expected to make these tumors more susceptible to the ICI regimens included in our cohort. Furthermore, we have no means by which microbes are assessed as intra- or extracellular. We note that both have been observed by microscopy (19), though the intra–cancer cell observations have recently been called into question (not, however, the intra–immune cell observations; ref. 68). Further studies are needed to confirm the location of tumoral microbes. Our findings also raise the continued question of whether additional filtering steps to control for contamination are required. Recent work suggests that more stringent human filtering, including the removal or masking of contaminating human reads in draft microbial genomes, is necessary. Many of the associations are conducted on a large number of microbes and genes or signatures, leading to many independent tests. Multiple hypothesis correction is likely insufficient to remove all false discoveries, driving further need for experimental validation. Finally, we validated the combined microbe and gene expression random forest classifier in two independent datasets and showed that the balanced accuracy is about 69%. We acknowledge that there is room for improvement for the machine learning classifier, especially testing the model, on more independent datasets to confirm the value of adding microbes when overfitting can be more accurately assessed.
In conclusion, we found that the tumor microbiome in patients with metastatic melanoma was significantly different in those that responded (>24 months survival) to treatment with ICIs from those who did not respond. Furthermore, the microbial communities had the ability to predict response when incorporated into machine learning models. The tumor microbiome further enhanced models to predict response when combined with gene expression data. Future research has the potential to support therapeutic strategies to modify the tumor microbiome to improve ICI treatment outcomes.
Supplementary Material
Association of CIBERSORT cell types with the response to ICIs
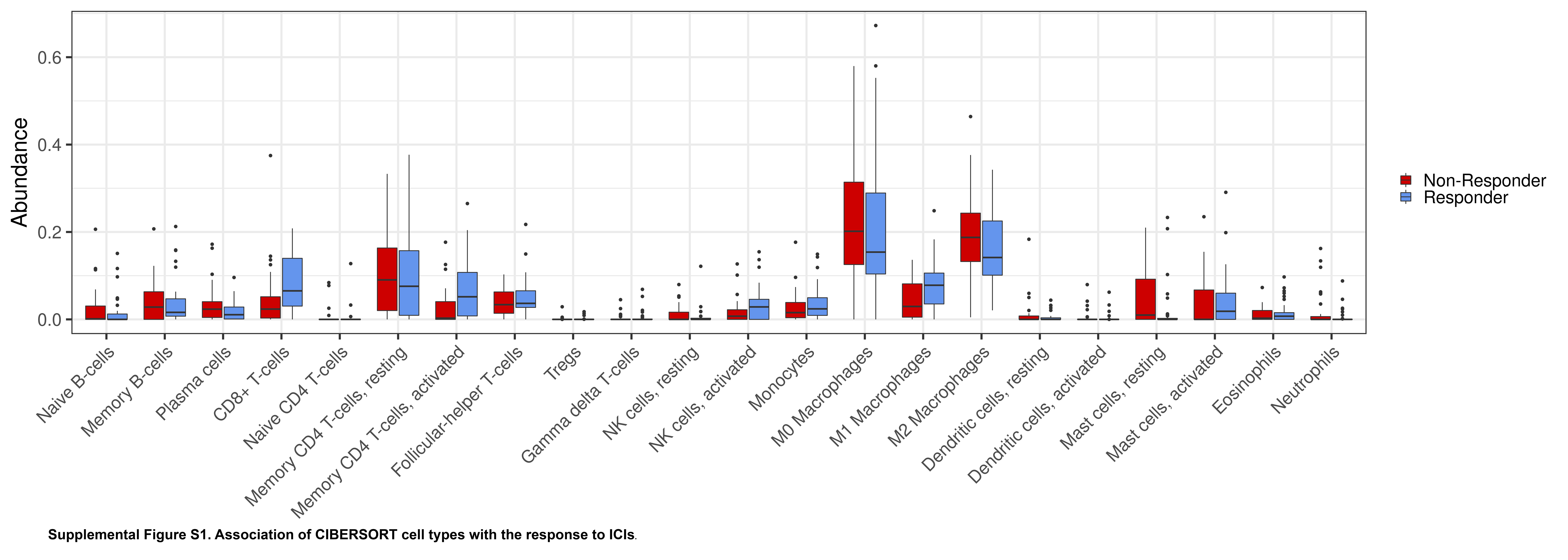{kind=link}
16 gene signatures where high Z-scores are associated with ICI responsiveness in this cohort (FDR < 0.05)
{kind=link}
Predictive values (AUROC) of the 16 gene signatures in the ORIEN cohort (IO_NOVA_Mel) and 22 other melanoma cohorts
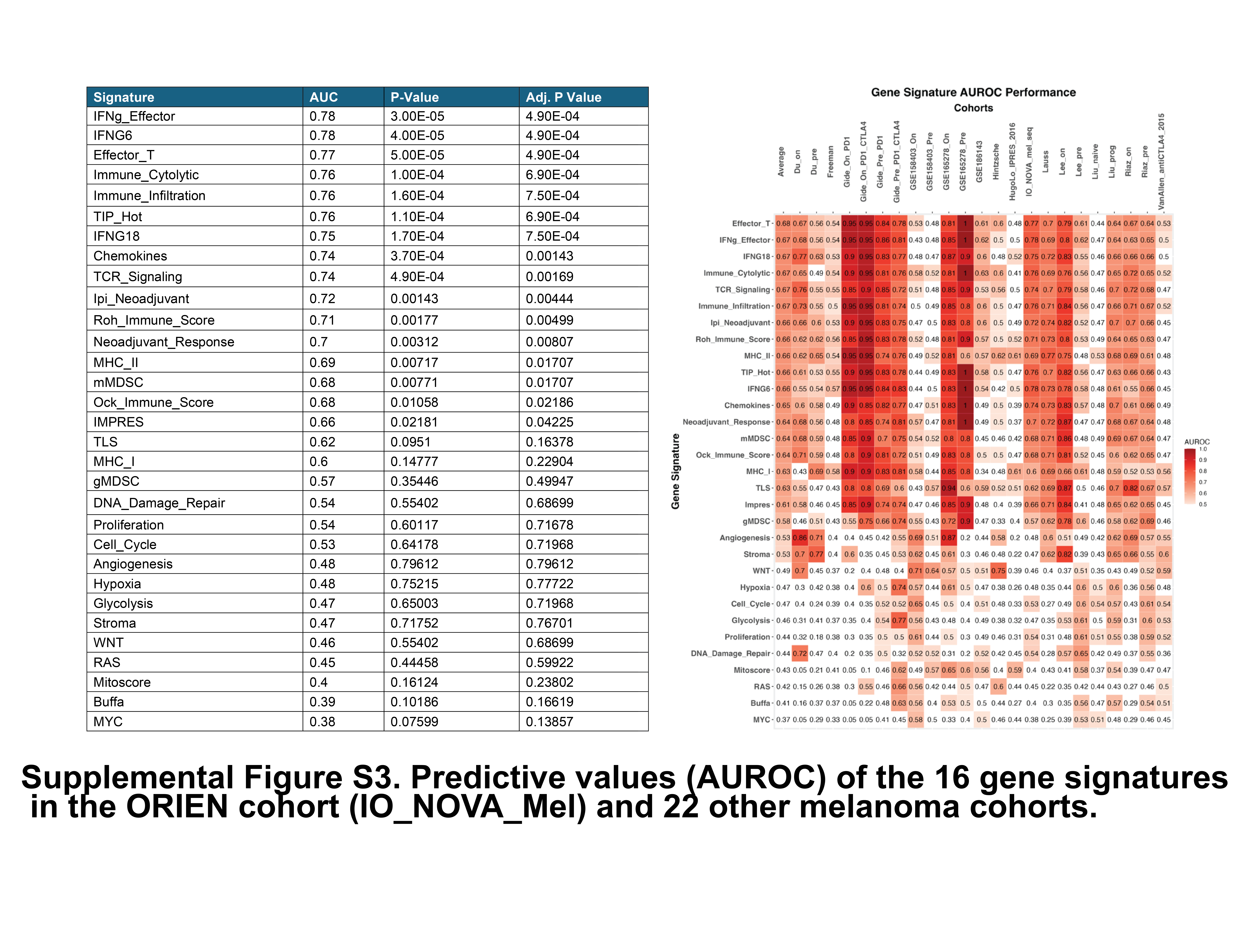{kind=link}
Comparison with TCGA for microbes most significantly enriched in response groups
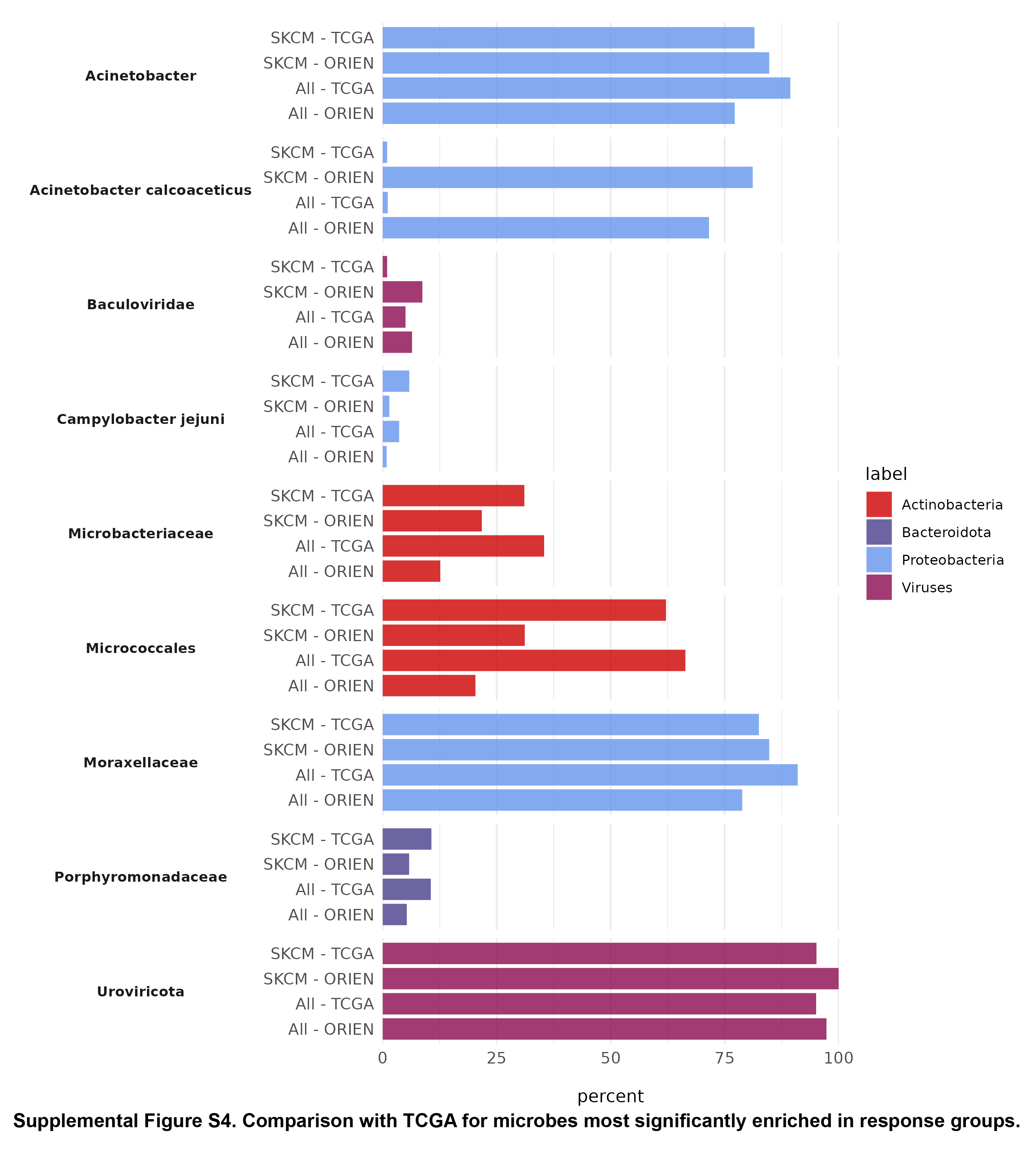{kind=link}
Comparison of alternate machine learning methods
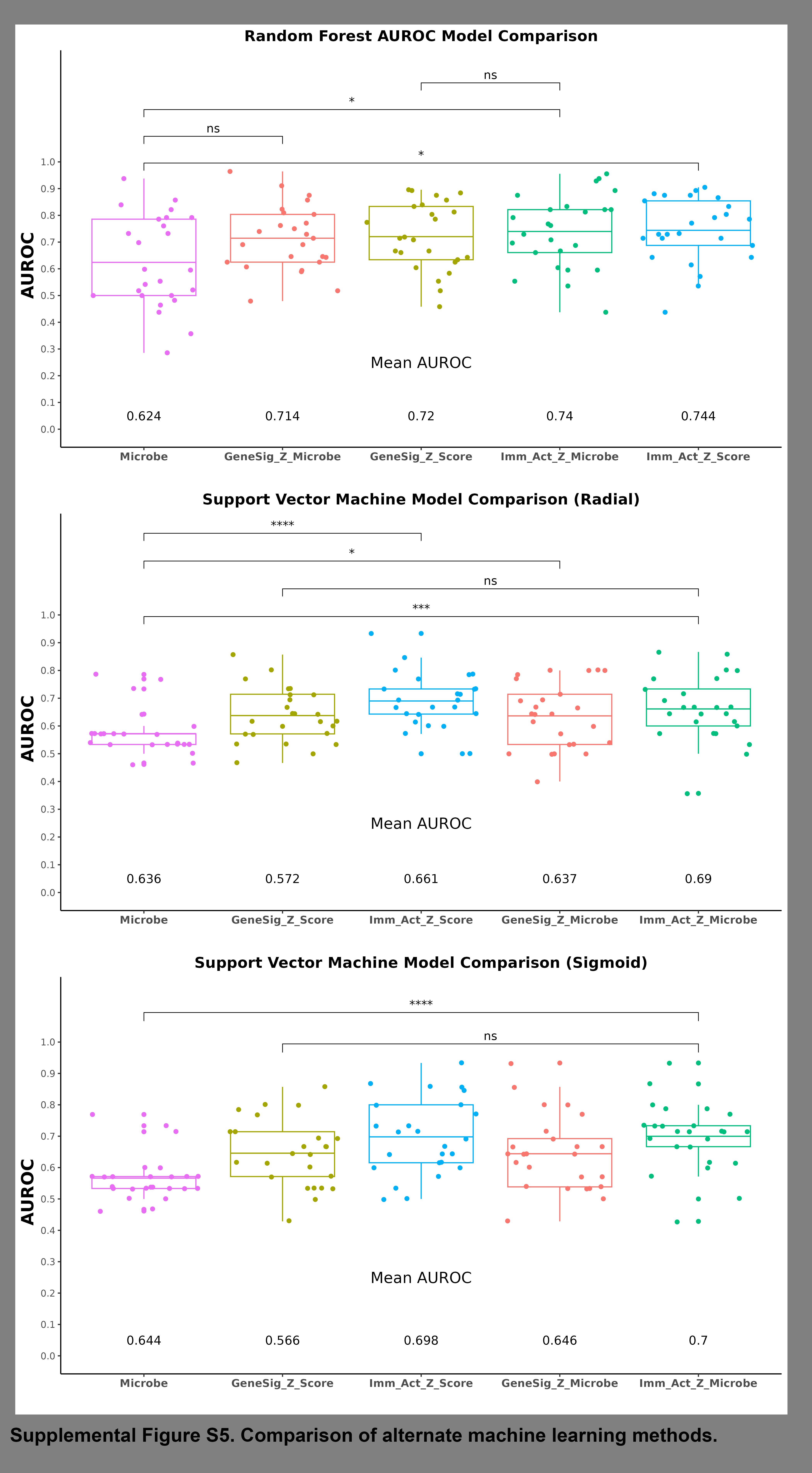{kind=link}
The list of published gene signatures compared between treatment responders and non-responders and to tumor microbe abundances.
Modeling results for gene expression differences between responders and non-responders
Model performance of 31 published gene expression signatures for predicting immunotherapy treatment response
Modeling results for the microbes enriched in the tumors responsive or not-responsive to immunotherapy
Correlations between microbe abundances and MSigDB Hallmark gene sets average Z scores
Acknowledgments
The authors acknowledge the support and resources of the Ohio Supercomputer Center (PAS1695). We would like to thank Angela Dahlberg, Editor, Division of Medical Oncology at The Ohio State University Comprehensive Cancer Center, for editing and proofreading the manuscript. Research reported in this publication was partly supported by The Ohio State University Comprehensive Cancer Center and NIH under grant number P30 CA016058, Huntsman Cancer Institute, and Comprehensive Cancer Center at the University of Utah P30CA042014. This project was partly supported by The Ohio State University Center for Clinical and Translational Science grant, National Center for Advancing Translational Sciences (Grant 8UL1TR000090-05), and the ORIEN NOVA grant 21PRJNOVA009MCC (PI: A.A. Tarhini).
Footnotes
Note: Supplementary data for this article are available at Cancer Research Communications Online (https://aacrjournals.org/cancerrescommun/).
Authors’ Disclosures
M.D. McCarter reports grants from Merck and Taiho outside the submitted work. E.A. Singer reports other support from Aura Biosciences outside the submitted work. G. Tinoco reports personal fees from Deciphera Pharmaceuticals, Daiichi Sankyo Company, and Servier Pharmaceuticals outside the submitted work. C.M. Ulrich reports nonfinancial support from Aster Insights and grants from NIH during the conduct of the study, as well as grants from NIH outside the submitted work. Y. Zakharia reports advisory board: Bristol Myers Squibb, Amgen, Roche Diagnostics, Novartis, Janssen, Eisai, Exelixis, Castle Biosciences, Genzyme Corporation, AstraZeneca, Array, Bayer, Pfizer, Clovis, EMD Serono, and Myovant; grant/research support: institution clinical trial support from NewLink Genetics, Pfizer, Exelixis, and Eisai; Data Safety and Monitoring Committee: Janssen Research and Development; and consultant honorarium: Pfizer and Novartis. D. Spakowicz reports grants from Lung Cancer Foundation of America, National Institutes on Aging, American Lung Association, Pelotonia, National Comprehensive Cancer Center Network, Oncology Nursing Foundation, Sarcoma Foundation of America, NCI, and American Cancer Society outside the submitted work. A.A. Tarhini reports grants from Bristol Myers Squibb, Merck, Novartis, Genentech-Roche, Regeneron, Sanofi-Genzyme, Pfizer, Acrotech, InflaRx, Scholar Rock, Simcha, Iovance/Clinigen, Agenus, and OncoSec and personal fees from Bristol Myers Squibb, Merck, Regeneron, Sanofi-Genzyme, ConcertAI, Teva, Instil Bio, and Pfizer outside the submitted work. No disclosures were reported by the other authors.
Authors’ Contributions
C.E. Dravillas: Data curation, software, formal analysis, supervision, visualization, methodology, writing-original draft, writing-review and editing. S.S. Coleman: Data curation, software, formal analysis, visualization, methodology, writing-original draft, writing-review and editing. R. Hoyd: Data curation, software, formal analysis, methodology, writing-review and editing. G. Caryotakis: Formal analysis, methodology. L. Denko: Data curation, software, writing-review and editing. C.H.F. Chan: Respurces, writing-review and editing. M.L. Churchman: Resources, writing-original draft, project administration, writing-review and editing. N. Denko: Writing-review and editing. R.D. Dodd: Resources, writing-review and editing. I. Eljilany: Writing-review and editing. S. Hardikar: Resources, writing-review and editing. M. Husain: Writing-review and editing. A.P. Ikeguchi: Resources, writing-review and editing. N. Jin: Writing-review and editing. Q. Ma: Resources, writing-review and editing. M.D. McCarter: Resources, writing-review and editing. A.E.G. Osman: Resources, writing-review and editing. L.A. Robinson: Resources, writing-review and editing. E.A. Singer: Resources, writing-review and editing. G. Tinoco: Writing-review and editing. C.M. Ulrich: Resources, writing-review and editing. Y. Zakharia: Resources, writing-review and editing. D. Spakowicz: Conceptualization, resources, software, formal analysis, methodology, writing-original draft, project administration, writing-review and editing. A.A. Tarhini: Conceptualization, resources, project administration, writing-review and editing. A.C. Tan: Conceptualization, resources, software, formal analysis, supervision, methodology, writing-original draft, project administration, writing-review and editing.
References
- 1. Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Cowey CL, Lao CD, et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med 2015;373:23–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Yarchoan M, Hopkins A, Jaffee EM. Tumor mutational burden and response rate to PD-1 inhibition. N Engl J Med 2017;377:2500–1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. McGranahan N, Furness AJS, Rosenthal R, Ramskov S, Lyngaa R, Saini SK, et al. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science 2016;351:1463–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Iafolla MAJ, Yang C, Chandran V, Pintilie M, Li Q, Bedard PL, et al. Predicting toxicity and response to pembrolizumab through germline genomic HLA class 1 analysis. JNCI Cancer Spectr 2020;5:pkaa115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Postow MA, Manuel M, Wong P, Yuan J, Dong Z, Liu C, et al. Peripheral T cell receptor diversity is associated with clinical outcomes following ipilimumab treatment in metastatic melanoma. J Immunother Cancer 2015;3:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Davoli T, Uno H, Wooten EC, Elledge SJ. Tumor aneuploidy correlates with markers of immune evasion and with reduced response to immunotherapy. Science 2017;355:eaaf8399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sayaman RW, Saad M, Thorsson V, Hu D, Hendrickx W, Roelands J, et al. Germline genetic contribution to the immune landscape of cancer. Immunity 2021;54:367–86.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Casey SC, Baylot V, Felsher DW. The MYC oncogene is a global regulator of the immune response. Blood 2018;131:2007–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Spranger S, Bao R, Gajewski TF. Melanoma-intrinsic β-catenin signalling prevents anti-tumour immunity. Nature 2015;523:231–5. [DOI] [PubMed] [Google Scholar]
- 10. Luke JJ, Bao R, Sweis RF, Spranger S, Gajewski TF. WNT/β-catenin pathway activation correlates with immune exclusion across human cancers. Clin Cancer Res 2019;25:3074–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Coelho MA, de Carné Trécesson S, Rana S, Zecchin D, Moore C, Molina-Arcas M, et al. Oncogenic RAS signaling promotes tumor immunoresistance by stabilizing PD-L1 mRNA. Immunity 2017;47:1083–99.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ayers M, Lunceford J, Nebozhyn M, Murphy E, Loboda A, Kaufman DR, et al. IFN-γ-related mRNA profile predicts clinical response to PD-1 blockade. J Clin Invest 2017;127:2930–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Coppola D, Nebozhyn M, Khalil F, Dai H, Yeatman T, Loboda A, et al. Unique ectopic lymph node-like structures present in human primary colorectal carcinoma are identified by immune gene array profiling. Am J Pathol 2011;179:37–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Messina JL, Fenstermacher DA, Eschrich S, Qu X, Berglund AE, Lloyd MC, et al. 12-Chemokine gene signature identifies lymph node-like structures in melanoma: potential for selection for immunotherapy? Sci Rep 2012;2:765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Liu D, Lin J-R, Robitschek EJ, Kasumova GG, Heyde A, Shi A, et al. Evolution of delayed resistance to immunotherapy in a melanoma responder. Nat Med 2021;27:985–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Fehrenbacher L, Spira A, Ballinger M, Kowanetz M, Vansteenkiste J, Mazieres J, et al. Atezolizumab versus docetaxel for patients with previously treated non-small-cell lung cancer (POPLAR): a multicentre, open-label, phase 2 randomised controlled trial. Lancet 2016;387:1837–46. [DOI] [PubMed] [Google Scholar]
- 17. Bolen CR, McCord R, Huet S, Frampton GM, Bourgon R, Jardin F, et al. Mutation load and an effector T-cell gene signature may distinguish immunologically distinct and clinically relevant lymphoma subsets. Blood Adv 2017;1:1884–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Litchfield K, Reading JL, Puttick C, Thakkar K, Abbosh C, Bentham R, et al. Meta-analysis of tumor- and T cell-intrinsic mechanisms of sensitization to checkpoint inhibition. Cell 2021;184:596–614.e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Nejman D, Livyatan I, Fuks G, Gavert N, Zwang Y, Geller LT, et al. The human tumor microbiome is composed of tumor type–specific intracellular bacteria. Science 2020;368:973–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hermida LC, Gertz EM, Ruppin E. Predicting cancer prognosis and drug response from the tumor microbiome. Nat Commun 2022;13:2896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Dalton WS, Sullivan D, Ecsedy J, Caligiuri MA. Patient enrichment for precision-based cancer clinical trials: using prospective cohort surveillance as an approach to improve clinical trials. Clin Pharmacol Ther 2018;104:23–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hodi FS, Chesney J, Pavlick AC, Robert C, Grossmann KF, McDermott DF, et al. Combined nivolumab and ipilimumab versus ipilimumab alone in patients with advanced melanoma: 2-year overall survival outcomes in a multicentre, randomised, controlled, phase 2 trial. Lancet Oncol 2016;17:1558–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sun L, Bleiberg B, Hwang W-T, Marmarelis ME, Langer CJ, Singh A, et al. Association between duration of immunotherapy and overall survival in advanced non–small cell lung cancer. JAMA Oncol 2023;9:1075–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. McDermott D, Haanen J, Chen T-T, Lorigan P, O’Day S; MDX010-20 Investigators . Efficacy and safety of ipilimumab in metastatic melanoma patients surviving more than 2 years following treatment in a phase III trial (MDX010-20). Ann Oncol 2013;24:2694–8. [DOI] [PubMed] [Google Scholar]
- 25. Hoyd R, Wheeler CE, Liu Y, Jagjit Singh MS, Muniak M, Jin N, et al. Exogenous sequences in tumors and immune cells (exotic): a tool for estimating the microbe abundances in tumor RNA-seq data. Cancer Res Commun 2023;3:2375–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 2014;15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Oksanen J, Blanchet FG, Friendly M, Kindt R, Legendre P, McGlinn D, et al. vegan: community ecology package [internet] 2018[cited 2024 May 21]. Available from:https://CRAN.R-project.org/package=vegan.
- 28. Coleman S, Xie M, Tarhini AA, Tan AC. Systematic evaluation of the predictive gene expression signatures of immune checkpoint inhibitors in metastatic melanoma. Mol Carcinog 2023;62:77–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Tarhini AA, Lee SJ, Tan A-C, El Naqa IM, Stephen Hodi F, Butterfield LH, et al. Improved prognosis and evidence of enhanced immunogenicity in tumor and circulation of high-risk melanoma patients with unknown primary. J Immunother Cancer 2022;10:e004310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Liberzon A, Birger C, Thorvaldsdóttir H, Ghandi M, Mesirov JP, Tamayo P. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst 2015;1:417–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Xie M, Lee K, Lockhart JH, Cukras SD, Carvajal R, Beg AA, et al. TIMEx: tumor-immune microenvironment deconvolution web-portal for bulk transcriptomics using pan-cancer scRNA-seq signatures. Bioinformatics 2021;37:3681–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Gide TN, Quek C, Menzies AM, Tasker AT, Shang P, Holst J, et al. Distinct immune cell populations define response to anti-PD-1 monotherapy and anti-PD-1/anti-CTLA-4 combined therapy. Cancer Cell 2019;35:238–55.e6. [DOI] [PubMed] [Google Scholar]
- 33. Riaz N, Havel JJ, Makarov V, Desrichard A, Urba WJ, Sims JS, et al. Tumor and microenvironment evolution during immunotherapy with nivolumab. Cell 2017;171:934–49.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Tarhini AA, Lin Y, Lin H-M, Vallabhaneni P, Sander C, LaFramboise W, et al. Expression profiles of immune-related genes are associated with neoadjuvant ipilimumab clinical benefit. Oncoimmunology 2016;6:e1231291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Thompson JC, Hwang W-T, Davis C, Deshpande C, Jeffries S, Rajpurohit Y, et al. Gene signatures of tumor inflammation and epithelial-to-mesenchymal transition (EMT) predict responses to immune checkpoint blockade in lung cancer with high accuracy. Lung Cancer 2020;139:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJM, Robert L, et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 2014;515:568–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Şenbabaoğlu Y, Gejman RS, Winer AG, Liu M, Van Allen EM, de Velasco G, et al. Tumor immune microenvironment characterization in clear cell renal cell carcinoma identifies prognostic and immunotherapeutically relevant messenger RNA signatures. Genome Biol 2016;17:231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Dougall WC, Kurtulus S, Smyth MJ, Anderson AC. TIGIT and CD96: new checkpoint receptor targets for cancer immunotherapy. Immunol Rev 2017;276:112–20. [DOI] [PubMed] [Google Scholar]
- 39. Cristescu R, Nebozhyn M, Zhang C, Albright A, Kobie J, Huang L, et al. Transcriptomic determinants of response to pembrolizumab monotherapy across solid tumor types. Clin Cancer Res 2022;28:1680–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Huang AC, Orlowski RJ, Xu X, Mick R, George SM, Yan PK, et al. A single dose of neoadjuvant PD-1 blockade predicts clinical outcomes in resectable melanoma. Nat Med 2019;25:454–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ock C-Y, Hwang J-E, Keam B, Kim S-B, Shim J-J, Jang H-J, et al. Genomic landscape associated with potential response to anti-CTLA-4 treatment in cancers. Nat Commun 2017;8:1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Xiong D, Wang Y, You M. A gene expression signature of TREM2hi macrophages and γδ T cells predicts immunotherapy response. Nat Commun 2020;11:5084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Matson V, Fessler J, Bao R, Chongsuwat T, Zha Y, Alegre M-L, et al. The commensal microbiome is associated with anti-PD-1 efficacy in metastatic melanoma patients. Science 2018;359:104–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Gopalakrishnan V, Spencer CN, Nezi L, Reuben A, Andrews MC, Karpinets TV, et al. Gut microbiome modulates response to anti–PD-1 immunotherapy in melanoma patients. Science 2018;359:97–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Routy B, Le Chatelier E, Derosa L, Duong CPM, Alou MT, Daillère R, et al. Gut microbiome influences efficacy of PD-1–based immunotherapy against epithelial tumors. Science 2018;359:91–7. [DOI] [PubMed] [Google Scholar]
- 46. Davar D, Dzutsev AK, McCulloch JA, Rodrigues RR, Chauvin J-M, Morrison RM, et al. Fecal microbiota transplant overcomes resistance to anti–PD-1 therapy in melanoma patients. Science 2021;371:595–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Baruch EN, Youngster I, Ben-Betzalel G, Ortenberg R, Lahat A, Katz L, et al. Fecal microbiota transplant promotes response in immunotherapy-refractory melanoma patients. Science 2021;371:602–9. [DOI] [PubMed] [Google Scholar]
- 48. Poore GD, Kopylova E, Zhu Q, Carpenter C, Fraraccio S, Wandro S, et al. Microbiome analyses of blood and tissues suggest cancer diagnostic approach. Nature 2020;579:567–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Geller LT, Barzily-Rokni M, Danino T, Jonas OH, Shental N, Nejman D, et al. Potential role of intratumor bacteria in mediating tumor resistance to the chemotherapeutic drug gemcitabine. Science 2017;357:1156–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Cullin N, Azevedo Antunes C, Straussman R, Stein-Thoeringer CK, Elinav E. Microbiome and cancer. Cancer Cell 2021;39:1317–41. [DOI] [PubMed] [Google Scholar]
- 51. Centre International de Recherche sur le Cancer . A review of human carcinogens. Lyon: International Agency for Research on Cancer; 2012. [Google Scholar]
- 52. Cheng WT, Kantilal HK, Davamani F. The mechanism of Bacteroides fragilis toxin contributes to colon cancer formation. Malays J Med Sci 2020;27:9–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Chung L, Thiele Orberg E, Geis AL, Chan JL, Fu K, DeStefano Shields CE, et al. Bacteroides fragilis toxin coordinates a pro-carcinogenic inflammatory cascade via targeting of colonic epithelial cells. Cell Host Microbe 2018;23:203–14.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Pleguezuelos-Manzano C, Puschhof J, Rosendahl Huber A, van Hoeck A, Wood HM, Nomburg J, et al. Mutational signature in colorectal cancer caused by genotoxic pks+E. coli. Nature 2020;580:269–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Park JB, Koo JS. Helicobacter pylori infection in gastric mucosa-associated lymphoid tissue lymphoma. World J Gastroenterol 2014;20:2751–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Sierra JC, Piazuelo MB, Luis PB, Barry DP, Allaman MM, Asim M, et al. Spermine oxidase mediates Helicobacter pylori-induced gastric inflammation, DNA damage, and carcinogenic signaling. Oncogene 2020;39:4465–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Aykut B, Pushalkar S, Chen R, Li Q, Abengozar R, Kim JI, et al. The fungal mycobiome promotes pancreatic oncogenesis via activation of MBL. Nature 2019;574:264–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Fluckiger A, Daillère R, Sassi M, Sixt BS, Liu P, Loos F, et al. Cross-reactivity between tumor MHC class I-restricted antigens and an enterococcal bacteriophage. Science 2020;369:936–42. [DOI] [PubMed] [Google Scholar]
- 59. Young KT, Davis LM, DiRita VJ. Campylobacter jejuni: molecular biology and pathogenesis. Nat Rev Microbiol 2007;5:665–79. [DOI] [PubMed] [Google Scholar]
- 60. Kalaora S, Nagler A, Nejman D, Alon M, Barbolin C, Barnea E, et al. Identification of bacteria-derived HLA-bound peptides in melanoma. Nature 2021;592:138–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Newell F, Pires da Silva I, Johansson PA, Menzies AM, Wilmott JS, Addala V, et al. Multiomic profiling of checkpoint inhibitor-treated melanoma: identifying predictors of response and resistance, and markers of biological discordance. Cancer Cell 2022;40:88–102.e7. [DOI] [PubMed] [Google Scholar]
- 62. Mihály Z, Kormos M, Lánczky A, Dank M, Budczies J, Szász MA, et al. A meta-analysis of gene expression-based biomarkers predicting outcome after tamoxifen treatment in breast cancer. Breast Cancer Res Treat 2013;140:219–32. [DOI] [PubMed] [Google Scholar]
- 63. Long J, Zhang L, Wan X, Lin J, Bai Y, Xu W, et al. A four-gene-based prognostic model predicts overall survival in patients with hepatocellular carcinoma. J Cell Mol Med 2018;22:5928–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Vétizou M, Pitt JM, Daillère R, Lepage P, Waldschmitt N, Flament C, et al. Anticancer immunotherapy by CTLA-4 blockade relies on the gut microbiota. Science 2015;350:1079–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Mager LF, Burkhard R, Pett N, Cooke NCA, Brown K, Ramay H, et al. Microbiome-derived inosine modulates response to checkpoint inhibitor immunotherapy. Science 2020;369:1481–9. [DOI] [PubMed] [Google Scholar]
- 66. Bender MJ, McPherson AC, Phelps CM, Pandey SP, Laughlin CR, Shapira JH, et al. Dietary tryptophan metabolite released by intratumoral Lactobacillus reuteri facilitates immune checkpoint inhibitor treatment. Cell 2023;186:1846–62.e26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Wolchok JD, Chiarion-Sileni V, Gonzalez R, Grob J-J, Rutkowski P, Lao CD, et al. Long-term outcomes with nivolumab plus ipilimumab or nivolumab alone versus ipilimumab in patients with advanced melanoma. J Clin Oncol 2022;40:127–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Ghaddar B, Biswas A, Harris C, Omary MB, Carpizo DR, Blaser MJ, et al. Tumor microbiome links cellular programs and immunity in pancreatic cancer. Cancer Cell 2022;40:1240–53.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Association of CIBERSORT cell types with the response to ICIs
16 gene signatures where high Z-scores are associated with ICI responsiveness in this cohort (FDR < 0.05)
Predictive values (AUROC) of the 16 gene signatures in the ORIEN cohort (IO_NOVA_Mel) and 22 other melanoma cohorts
Comparison with TCGA for microbes most significantly enriched in response groups
Comparison of alternate machine learning methods
The list of published gene signatures compared between treatment responders and non-responders and to tumor microbe abundances.
Modeling results for gene expression differences between responders and non-responders
Model performance of 31 published gene expression signatures for predicting immunotherapy treatment response
Modeling results for the microbes enriched in the tumors responsive or not-responsive to immunotherapy
Correlations between microbe abundances and MSigDB Hallmark gene sets average Z scores
Data Availability Statement
The Ohio State University IRB approved data access in an Honest Broker protocol (2015H0185) and TCC protocol (2013H0199) in coordination with Aster Insights. The processed data generated in this study are publicly available in Gene Expression Omnibus through the BioProject PRJNA856973. Analysis scripts to regenerate all figures and tables are available at https://github.com/spakowiczlab/exorien-melio.