

Abstract
Elevated levels of inflammatory cytokines in the vitreous of patients with chronic intraocular inflammatory (uveitis) have long been suspected to contribute to the pathogenesis of retinal degenerative diseases. However, direct connection between chronic inflammation and development of retinal degenerative diseases has been difficult to establish because we lack an appropriate animal model of co-existing chronic intraocular inflammation and neurodegeneration. This report discusses new developments in immunological and diabetic research that suggest that persistent secretion of pro-inflammatory cytokines during uveitis might induce insulin resistance and retinal degenerative changes that contribute to the pathogenesis of Diabetic Retinopathy (DR), a retinal dystrophy of significant public health importance.
Keywords: Uveitis, Retinal degeneration, Insulin resistance, Interferon-γ, Cytokines, Suppressor of cytokine signaling, Experimental autoimmune uveitis, EAU
53.1. Introduction
Retinopathy results from acute or persistent damage to the retina and is a major cause of blindness or severe vision loss, especially if the pathologic changes extend to the macular. Chronic inflammation and inflammatory cytokines or growth factors that promote neovascularization and angiogenesis are suspected to play important role in retinopathies that lead to retinal degeneration and blindness [1]. Although retinal dystrophy such as retinitis pigmentosa can be inherited, most retinopathies are caused by other underlying pathologic conditions such as, diabetes mellitus, sickle cell disease, prematurity of the newborn, retinal vein or artery occlusion or arterial hypertension. Diabetic retinopathy (DR) is of particular public health importance because it is a major cause of vision loss in the developing world and the leading cause of blindness among adults under the age of 65 in the USA [2]. It is projected to affect more than 300 million people worldwide over the next decade. Although the pathogenesis of DR is not known, elevated levels of inflammatory cytokines and chemotactic molecules that recruit leukocytes have been detected in the vitreous of patients with DR in several studies [2], suggesting inflammation mediated etiology.
However, DR is generally considered a glucose-mediated microvascular disease that derives in part from inability of the retina to adapt to metabolic stress and insulin-resistance as a risk factor for developing DR [1, 2]. In animal studies, insulin resistance precedes appearance of early features of DR and neuronal cell death. However, early features of DR such as vascular permeability, edema and increase in inflammatory proteins in the vitreous are also hallmarks of ocular inflammation [1]. Despite the apparent involvement of chronic inflammation and insulin resistance in DR, establishing a direct connection between inflammation, diabetes and the development of retinal degenerative diseases has been difficult because we lack an appropriate animal model of co-existing chronic intraocular inflammation and neurodegeneration.
Recent studies in a mouse model of chronic uveitis have revealed that prolonged inflammation can cause insulin resistance and retinal degeneration [3]. Proof-of-principle studies have also shown that targeted expression of a pro-inflammatory cytokine in the eye induces retinal degenerative changes [4]. Here, I discuss additional data that might provide mechanistic explanation for the link between inflammation, insulin resistance and retinal degeneration.
53.2. Mouse Model of Chronic Intraocular Inflammation (Uveitis)
Uveitis is a group of potentially sight-threatening inflammatory diseases that includes, Behçet’s disease, VKH, ocular sarcoidosis and about 10% of severe visual handicap in the USA are attributed to this group of disorders. It is characterized by progressive photoreceptor cell loss, neovascularization and retinal degeneration. However, investigation of mechanisms underlying the progression of uveitis to retinal degenerative changes in the retina is impeded because of the lack of a suitable animal model. Experimental autoimmune uveitis (EAU) shares essential features with human uveitis and current understanding of the pathophysiology of uveitis derives largely from study of EAU [5]. However, EAU in the mouse is generally considered a self-limiting disease and does not present with clinically manifestations of human chronic uveitis, including neovascularization and retinal degeneration. This limitation is attributable in part to the apparent absence of inflammatory cells in the vitreous of EAU mice 35 days after inception of EAU and this has led to the prevailing view that EAU is mainly an acute disease with complete recovery after 28–35 days. Development and adaptation of non-invasive techniques used in Opthalmology to mouse studies have made it possible to study mouse uveitis over extended period and these include fundoscopy, Optical Coherence Tomography (OCT) and electroretinography (ERG). These techniques have recently been used to monitor EAU over a 7 months period and results from these studies have revealed that the rapid decline of the severe acute retinal inflammation gives rise to a chronic phase of retinal inflammation that persists and did not completely resolve during the 225 days of the study [3]. Features of the chronic phase include severe retinal vasculitis with cuffing, sclerotic vessels, multifocal choroiditis and scaring and/or atrophy of the retina (Fig. 53.1). These pathological changes were observed by fundoscopic examination and histology beginning around day-50 after inception of EAU and persisted. OCT provided unprecedented clarity showing evolution of the acute disease to its long-term sequelae characterized by retina edema and neuro retinal degeneration (Fig. 53.2). Serial ERG tracings revealed that prolonged inflammation of the neuroretina adversely affected visual acuity in mouse. These studies established for the first time that, as is the case in human, mice development progressive retina degeneration, severe vision loss and blindness as a result of chronic intraocular inflammation.
Fig. 53.1.
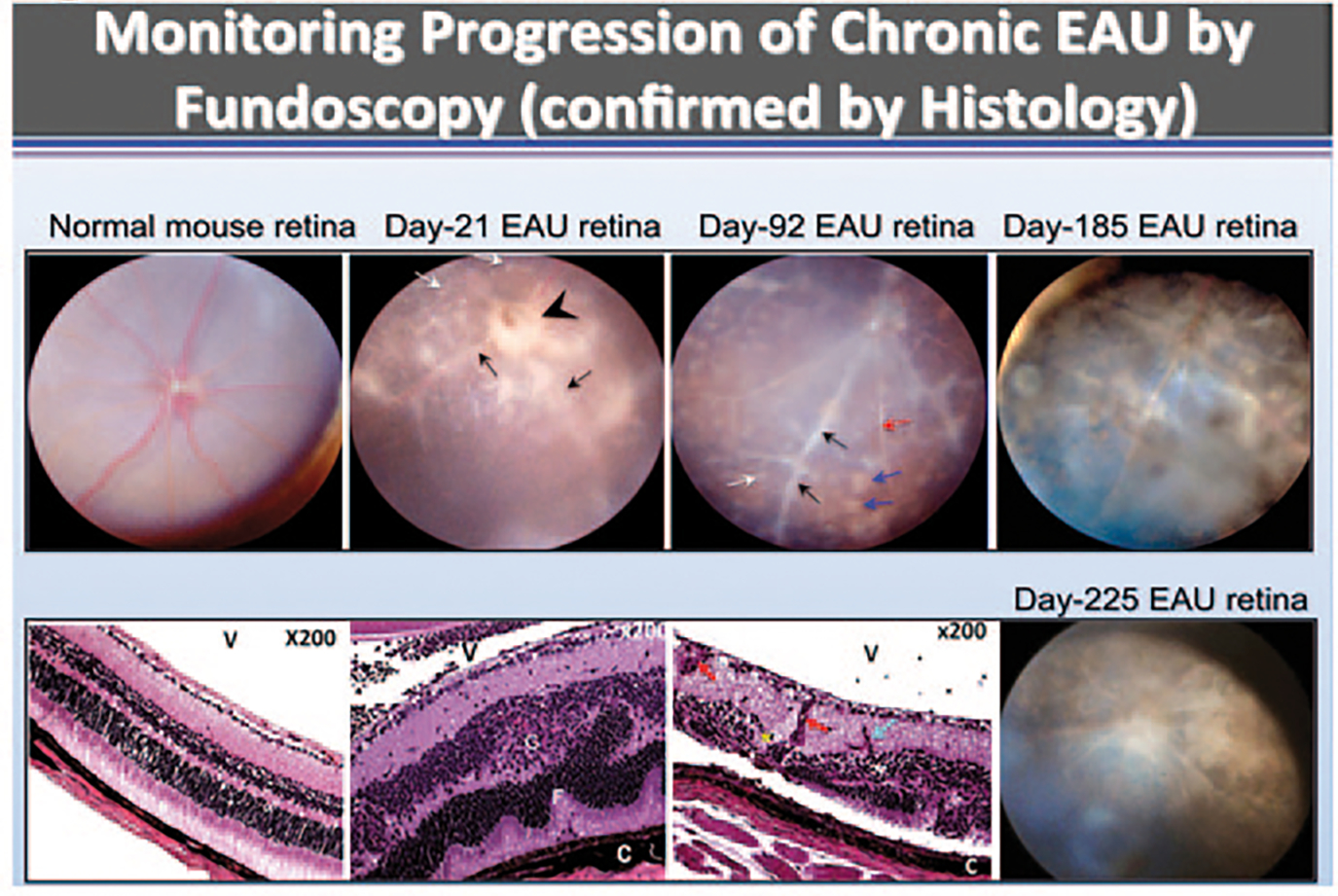
Chronic intraocular inflammation induces retinal degeneration and neovascularization. Fundus images were taken from normal or EAU mice over a 225 days period (top panels). Fundus images reveal blurred optic disc margins and enlarged juxtapapillary area (black arrowhead), retinal vasculitis with moderate cuffing (black arrows) and yellow-whitish retinal and choroidal infiltrates (white arrows). Histological analysis (Lower panels) reveals retinal structural damage, including evidence of atrophic retina (thinning) and sclerotic vessel (red arrow) with multiple whitish infiltrates (white arrow) and brownish chorioretinal scars (blue arrows), photoreceptor cell loss (red asterisk), retinal vasculitis (black arrows), retinal sclerotic vessel (white arrow), choroiditis (black arrowhead), and retinal degeneration (bottom panels). OpN optic nerve, V vitreous, R retina, GCL ganglion cell layer, INL inner nuclear layer, ONL outer nuclear layer, RPE retinal pigment epithelial layer, CH choroid
Fig. 53.2.
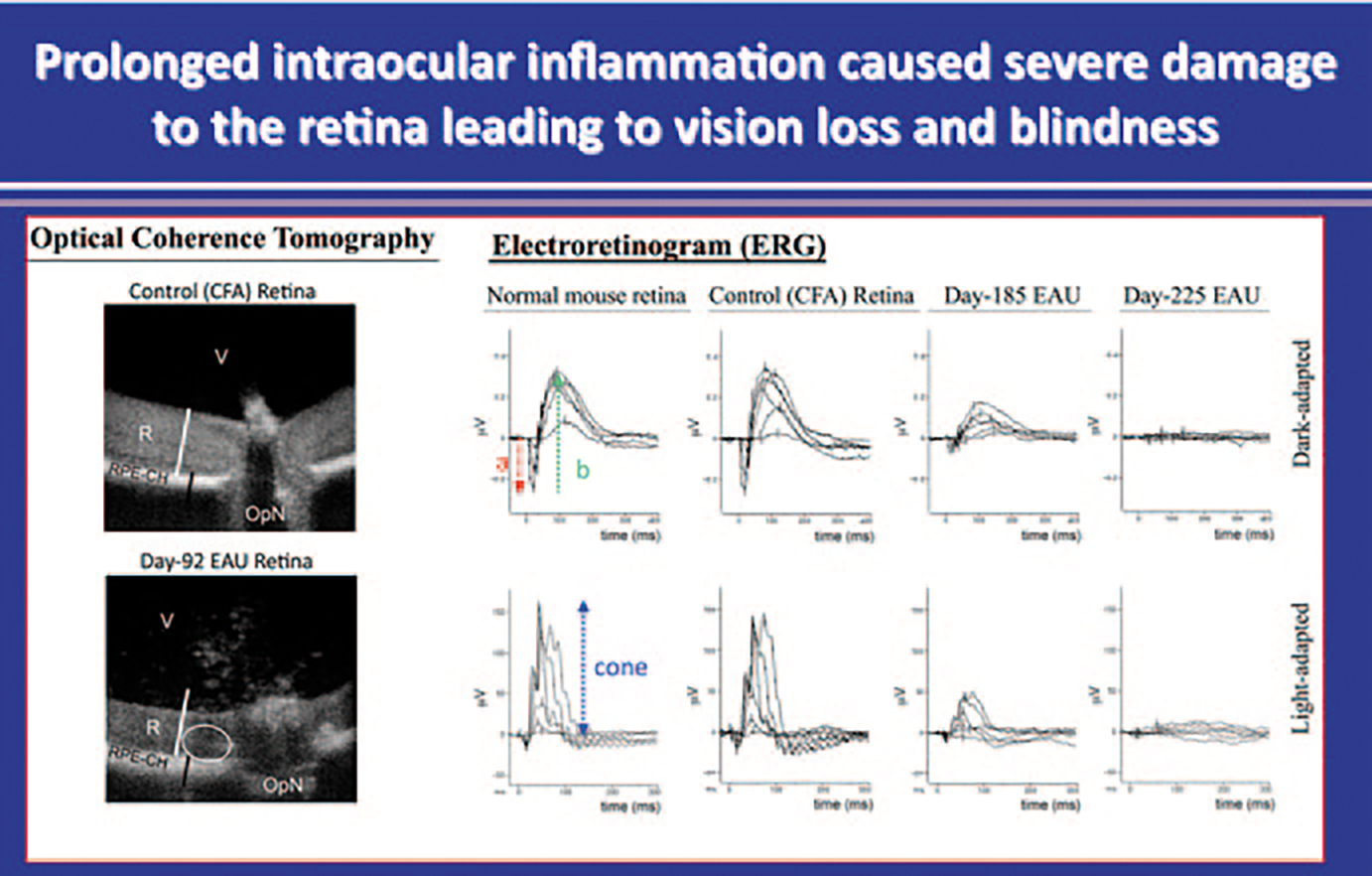
Mice were immunized with CFA alone or IRBP/CFA emulsion and electroretinogram (ERG) recordings were obtained and analyzed 185 or 225 days post-immunization. ERGs of control unimmunized mice or control mice immunized with CFA alone, show the characteristic normal a-, b- and cone waves. In contrast to control mice, light-induced response was barely elicited from retina of day-225 IRBP-induced EAU mice
53.3. Up-Regulation of SOCS Proteins During EAU Induces Insulin Resistance and Inhibits Insulin Signaling in the Retina
Although direct connection between inflammation and insulin-resistance in etiology of DR has not been established, studies on obesity or metabolic syndrome provide a framework for understanding how these disparate processes can converge in the etiology of DR [6]. Suppressors of cytokine signaling (SOCS) are an eightmember family of intracellular proteins that regulates the intensity and duration of cytokine/growth-factor signals [7, 8]. They are rapidly induced in many cell types in response to cytokines (IFN-γ and IL-6) or growth factors (CNTF, LIF, insulin) and their inhibitory effects derive from direct interaction with cytokine/growth-factor receptors or signaling proteins, leading to proteosomal degradation of the receptor complex and termination of the signal [7, 8]. Because of the relatively short half-life of SOCS proteins, their negative regulatory effects are generally transient. However, constitutive SOCS expression occurs in some tissues due to unabated stimulation by chronic inflammation or cellular stress, leading to silencing of critical cellular pathways. Chronic inflammation and cellular stress induces persistent elevation of SOCS1 and SOCS3 and these SOCS members have been implicated in etiology of diabetes, obesity and metabolic-syndrome [9]. We have shown that inflammatory cytokines produced by inflammatory lymphocytes that mediate EAU induce expression of SOCS1 and SOCS3 (Fig. 53.3) [10]. The increase in SOCS3 protein is accompanied by decrease in pAKT, p85 and IRS proteins, suggesting that elevation of SOCS proteins can inhibit PI3K/AKT-mediated insulin signaling in the retina during EAU (Fig. 53.4) [10]. We have also shown that light damage, hypoxia and metabolic stress caused by type1 diabetes increases SOCS3 expression in the retina [10]. In addition, over-expression of SOCS1 and SOCS3 in human or rodent retinal cells or targeted expression of SOCS1 expression in the retina of transgenic rats inhibited PI3K/AKT pathway [10]. These observations suggest that SOCS3-mediated inhibition during EAU can induce insulin-resistance, an important risk factor of DR and may thus provide a mechanistic link between inflammation, insulin resistance and DR.
Fig. 53.3.
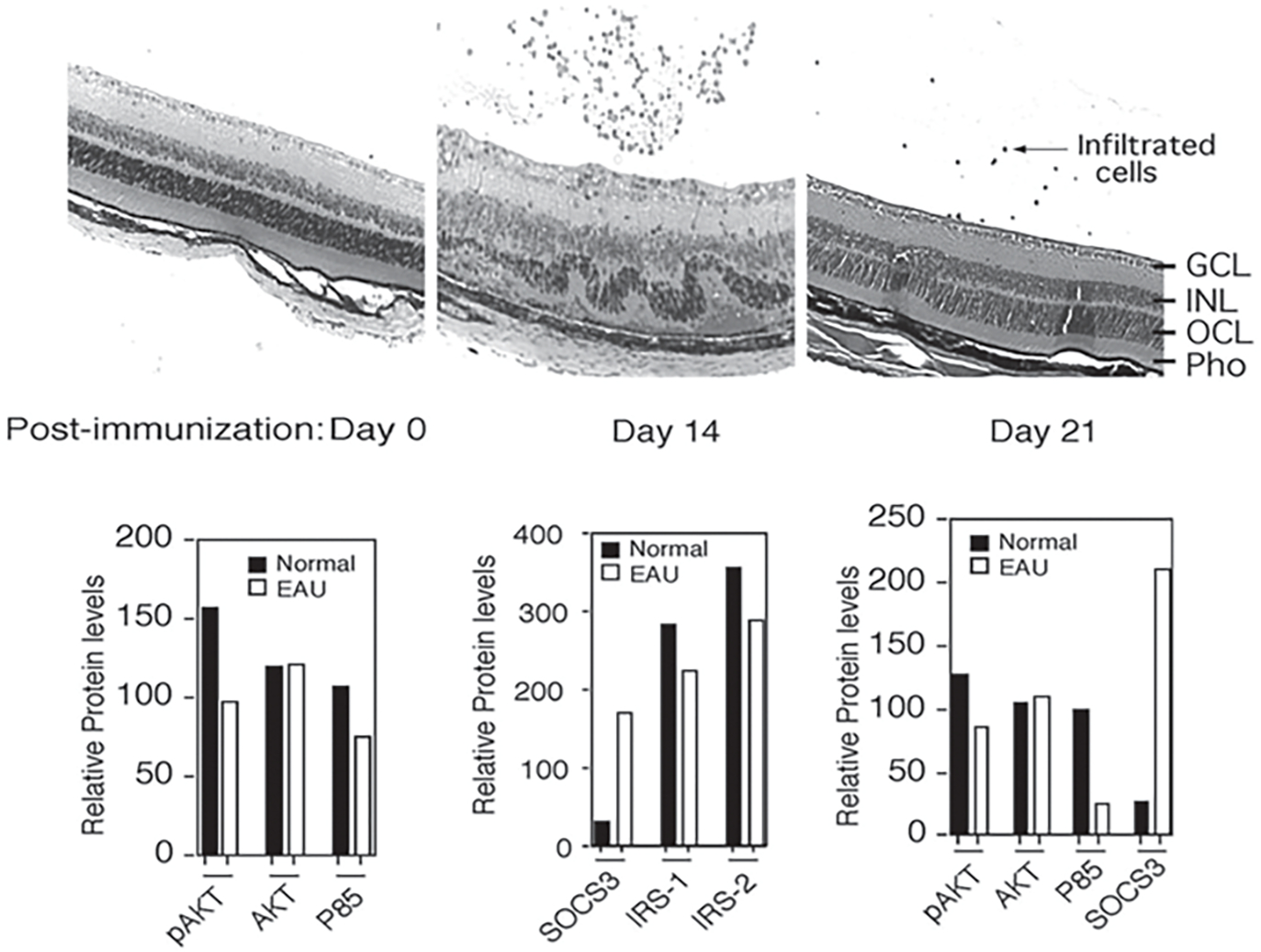
Induction of SOCS1 and SOCS3 during EAU correlates with inhibition of insulin signaling in the retina. a Six-week-old mouse was immunized with IRBP in CFA and eyes were enucleated and analyzed by histology at various times. Four microns thick sections were cut through the retina and stained with H&E. b Corresponding Western Blot analysis was performed. Relative Intensities of the western blot bands were analyzed in a densitometer to quantify the relative expression levels of SOCS3, IRS-1 and IRS-2
Fig. 53.4.
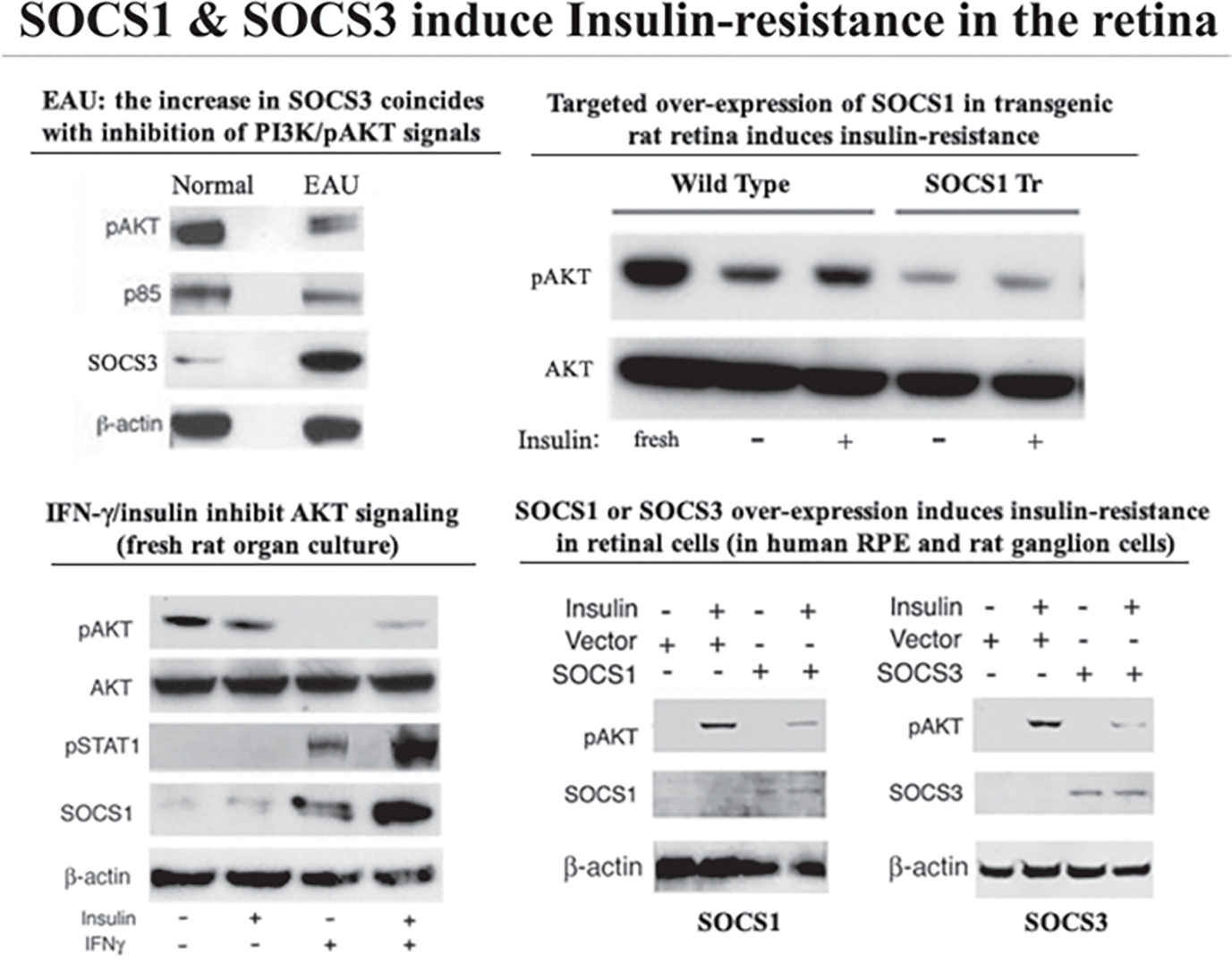
SOCS1 & SOCS3 induce Insulin-resistance in the retina
53.4. Targeted Expression of Interferon-γ (IFN-γ) in the Eye Induced Retinal Degeneration
During uveitis Th1 and Th17 lymphocytes are recruited into the retina and mediate ocular pathology through the production of inflammatory cytokines including IFN-γ, TNF-α, IL-6 and IL-17 [11, 12]. These cytokines are also detected at elevated levels in the vitreous of patients with uveitis. Several of these factors induce production of SOCS1 and SOCS3. In a proof-of-principle experiment we sought to directly test the notion that chronic inflammation leading to persistent production of pro-inflammatory cytokines that up-regulate SOCS1 or SOCS3 can cause uveitis and retinal degeneration [13]. IFN-γ expression was targeted to the lens of TR rats and the lens served as a depot to release IFN-γ to retina. The IFN-γ expression induced degeneration of the lens and as the transgenic lens continued to degenerate it began to release copious amounts of the cytokine into the vitreous (Fig. 53.5). By 3 months of age this led to significant degeneration of the retina characterized by significant apoptotic death of ganglion cells and photoreceptors [13].
Fig. 53.5.

Phenotypes of wild-type (WT) and aAcry/IFN-γ transgenic (TR) Lewis rat eyes. Note the time-dependent disintegration of the lens capsule and inception of retinal degenerative changes. Arrow in middle panel shows areas of retinal in-folding, a hallmark of severe uveitis. Arrows in lower panel show TUNEL-stained apoptotic ganglion cells
53.5. Conclusions
Retinal dystrophies including DR cause severe vision impairment or blindness and understanding of cellular pathways that promote the survival or repair of damaged neurons is essential to efforts aimed at developing effective treatment for retinopathies. Although molecular cues that influence the decision to allow death or survival of partially damaged neurons and photoreceptors are largely unknown, neurotrophic factors are thought to be involved in reparative processes in retina. Retinal neurotrophic factors such as CNTF, OSM or LIF activate and utilize JAK/STAT pathways and are therefore under negative-feedback regulation by SOCS proteins. Data presented here indicate that retinal cells respond to chronic inflammation, light-damage, oxidative-stress or metabolic stress due diabetes by up-regulating expression of SOCS1 and SOCS3 [10]. Although the induction of SOCS proteins undoubtedly confer protection from cytotoxic effects of pro-inflammatory cytokines or deleterious effects of excessive cellular stress, persistently high SOCS levels in retina is potentially pathogenic because it would inhibit survival signals that emanate from neurotrophic factors. In this report I have also presented evidence showing that persistent secretion of pro-inflammatory cytokines during uveitis elevated SOCS1 and SOCS3 levels in the retina, resulting in insulin resistance retinal degeneration. The recent establishment of a chronic EAU model showing that, as is the case with humans, that chronic intraocular inflammation induces retina degeneration, severe vision loss and blindness, now provides a useful system to fully investigate the mechanistic link between chronic inflammation, insulin resistance and pathogenesis of RD.
References
- 1.Antonetti DA, Barber AJ, Bronson SK, Freeman WM, Gardner TW, Jefferson LS et al. (2006) Diabetic retinopathy: seeing beyond glucose-induced microvascular disease. Diabetes 55(9):2401–2411 [DOI] [PubMed] [Google Scholar]
- 2.Meleth AD, Agron E, Chan CC, Reed GF, Arora K, Byrnes G et al. (2005) Serum inflammatory markers in diabetic retinopathy. Invest Ophthalmol Vis Sci 46(11):4295–4301 [DOI] [PubMed] [Google Scholar]
- 3.Oh HM, Yu CR, Lee Y, Chan CC, Maminishkis A, Egwuagu CE (2011) Autoreactive memory CD4+ T lymphocytes that mediate chronic uveitis reside in the bone marrow through STAT3-dependent mechanisms. J Immunol 187(6):3338–3346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Egwuagu CE, Mahdi RM, Chan CC, Sztein J, Li W, Smith JA et al. (1999) Expression of interferon-gamma in the lens exacerbates anterior uveitis and induces retinal degenerative changes in transgenic Lewis rats. Clin Immunol 91(2):196–205 [DOI] [PubMed] [Google Scholar]
- 5.Nussenblatt RB (1991) Proctor lecture. Experimental autoimmune uveitis: mechanisms of disease and clinical therapeutic indications. Invest Ophthalmol Vis Sci 32(13):3131–3141 [PubMed] [Google Scholar]
- 6.Taniguchi CM, Emanuelli B, Kahn CR (2006) Critical nodes in signalling pathways: insights into insulin action. Nat Rev Mol Cell Biol 7(2):85–96 [DOI] [PubMed] [Google Scholar]
- 7.Naka T, Fujimoto M, Kishimoto T (1999) Negative regulation of cytokine signaling: STAT-induced STAT inhibitor. Trends Biochem Sci 24(10):394–398 [DOI] [PubMed] [Google Scholar]
- 8.Hilton DJ (1999) Negative regulators of cytokine signal transduction. Cell Mol Life Sci 55(12):1568–1577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Emanuelli B, Glondu M, Filloux C, Peraldi P, Van Obberghen E (2004) The potential role of SOCS-3 in the interleukin-1beta-induced desensitization of insulin signaling in pancreatic beta-cells. Diabetes 53(Suppl 3):S97–S103 [DOI] [PubMed] [Google Scholar]
- 10.Liu X, Mameza MG, Lee YS, Eseonu CI, Yu CR, Kang Derwent JJ et al. (2008) Suppressors of cytokine-signaling proteins induce insulin resistance in the retina and promote survival of retinal cells. Diabetes 57(6):1651–1658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu X, Lee YS, Yu CR, Egwuagu CE (2008) Loss of STAT3 in CD4+ T cells prevents development of experimental autoimmune diseases. J Immunol 180(9):6070–6076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Amadi-Obi A, Yu CR, Liu X, Mahdi RM, Clarke GL, Nussenblatt RB et al. (2007) T(H)17 cells contribute to uveitis and scleritis and are expanded by IL-2 and inhibited by IL-27/STAT1. Nat Med 13(6):711–718 [DOI] [PubMed] [Google Scholar]
- 13.Egwuagu CE, Mahdi RM, Chan CC, Sztein J, Li W, Smith JA et al. (1999) Expression of interferon-gamma in the lens exacerbates anterior uveitis and induces retinal degenerative changes in transgenic Lewis rats. Clin Immunol 91(2):196–205 [DOI] [PubMed] [Google Scholar]