

Abstract

COX-2-selective drugs were withdrawn from the market just a few years after their development due to cardiovascular side effects. As a result, developing a selective COX-2 inhibitor as an anti-inflammatory agent with cardioprotective characteristics has become a prominent objective in medicinal chemistry. New 15 diaryl-1,2,4-triazolo[3,4-a]pyrimidine hybrids 8a–o were synthesized and investigated in vitro as dual COX-2/sEH inhibitors. Compounds 8b, 8m, and 8o have the highest potency and selectivity as COX-2 inhibitors (IC50 = 15.20, 11.60, and 10.50 μM, respectively; selectivity index (COX-1/COX-2) = 13, 20, and 25, respectively), compared to celecoxib (COX-2; IC50 = 42 μM; SI = 8). The 5-LOX inhibitory activity of compounds 8b, 8m, and 8o was further examined in vitro. Compounds 8m and 8o, the most effective COX-2 selective inhibitors, demonstrated stronger 5-LOX inhibitory action than the reference quercetin, with IC50 values of 2.90 and 3.05 μM, respectively. Additionally, compounds 8b, 8m, and 8o were the most potent dual COX-2/sEH inhibitors, with IC50 values against sEH of 3.20, 2.95, and 2.20 nM, respectively, and were equivalent to AUDA (IC50 = 1.2 nM). In vivo investigations also demonstrated that these compounds were the most efficacious as analgesic/anti-inflammatory derivatives with a high cardioprotective profile against cardiac biomarkers and inflammatory cytokines. The docking data analysis inquiry helped better understand the binding mechanisms of the most active hybrids within the COX-2 active site and supported their COX-2 selectivity. Compounds 8b, 8m, and 8o exhibited a similar orientation to rofecoxib and celecoxib, with a larger proclivity to enter the selectivity side pocket than the reference compounds.
Introduction
Inflammation is a natural defense mechanism in higher species that evolved to defend them from foreign and harmful stimuli.1,2 An inflammatory response can be acute or chronic. The first is a beneficial process that protects the host organism from infections, damaged cells, and toxic substances. Prolonged and uncontrolled response to inflammatory stimuli, on the other hand, has been linked to the development and progression of various multifactorial disorders.3
Leukotrienes (LTs) from the lipoxygenase (LOX) pathway and prostaglandins (PGs) from the cyclooxygenase (COX) pathway are two pro-inflammatory mediators generated by the arachidonic acid (AA) cascade. The pathogenesis of chronic inflammatory disorders has been connected to these mediators.4,5 Traditional nonsteroidal anti-inflammatory medicines, or tNSAIDs, are the most widely recommended for fever, pain, and inflammation. Their principal mode of action is nonselective cyclooxygenase-1 (COX-1) and cyclooxygenase-2 (COX-2) (COX-1 and COX-2, respectively) inhibition.6,7 These drugs have been linked to several side effects, including gastrointestinal toxicity from COX-1 inhibition8 and cardiovascular disorders.9 Coxibs, a class of highly selective COX-2 inhibitors, were developed to improve the gastrointestinal safety of anti-inflammatory medications.
Nonetheless, these medications raise the risk of cardiovascular events,10,11 while long-term usage increases LOX activity, resulting in raised levels of leukotrienes, which cause bronchoconstriction and increased airway irritation.12,13 Research suggests that blocking the COX-2 or LOX pathways may cause AA metabolism to switch to the other biosynthetic pathway, which could have unfavorable side effects.4,14,15 Thus, there is a critical need for novel anti-inflammatory drugs that prevent the release of prostaglandins and leukotrienes.16 As a result, discovering new, effective, and safe anti-inflammatory drugs could be game changers in modern pharmacy and medicine. A particularly intriguing trend in medicinal chemistry is the development of molecules with dual COX/LOX inhibitory action, which can serve as strong anti-inflammatory therapeutic candidates with much-enhanced safety.16−19
Soluble epoxide hydrolase (sEH) is a ubiquitous enzyme found in all body tissues, with the largest concentrations found in the liver, kidneys, lungs, and vascular tissues.20 This enzyme is specialized for aliphatic epoxides of fatty acids, such as epoxyeicosatrienoic acids (EETs), a metabolic product of AA.21,22 EETs have been found to have analgesic and anti-inflammatory activities and cardiovascular protective benefits.23 Furthermore, EETs displayed pro-angiogenic capabilities associated with a cardioprotective impact in chronic phases. The enzyme mediates the addition of water to EETs, producing dihydroxyeicosatrienoic acids (DHETs) with low biological activity.24 As a result of blocking the enzyme sEH, EET concentration increases, which has anti-inflammatory, pain-relieving, and cardiovascular risk-lowering characteristics.25
On the other hand, triazole-based derivatives are of significant interest as anti-inflammatory agents with low toxicity.26−30 Tozkoparan et al. investigated a series of 5-aryl-3-alkylthio-1,2,4-triazoles and their corresponding sulfones to find improved analgesic-anti-inflammatory drugs with low ulcerogenic risk.29 A number of these drugs had significant activity. Alkyl sulfone compounds (Compound I, Figure 1) were discovered to be far more effective analgesic-anti-inflammatory drugs than their alkylthio counterparts. In contrast to the reference acetylsalicylic acid, these compounds did not cause gastric ulcers in the stomachs of experimental animals at analgesic/anti-inflammatory levels.
Figure 1.

Structures of some 1,2,4-triazole-based anti-inflammatory agents.
In another study, the ibuprofen analogue 1,2,4-triazole II (Figure 1) exhibited a higher rate of edema inhibition (59.8%) than the normal ibuprofen (76.6%) and a lower ulcer index (13.37) than the standard ibuprofen (ulcer index = 21.26).26 Several triazole/heterocyclic hybrids have recently been found to be potent and selective COX-2 inhibitors, such as hybrid III (Figure 1), with IC50 values of 9.81 and 0.91 μM against COX-1 and COX-2 isoenzymes, respectively.31
Pyrimidine-5-carbonitriles have received attention for their anti-inflammatory and analgesic properties.32−34 Compound IV (Figure 2) has been shown to reduce inflammation by 83% at 50 mg/kg.32 Surprisingly, the dual anti-inflammatory and analgesic effects of pyrimidine derivative V, Figure 2, were nearly identical to those of indomethacin, with greater GI safety.34 Moreover, several biological compounds comprising pyrimidine carbonitrile scaffolds have been found to contain a new class of potent and selective COX-2 inhibitors. The potent and selective COX-2 inhibitors quinazolinone-pyrimidine hybrid (VI) and 1,3,4-oxadiazole-pyrimidine hybrid (VII)35,36 are two examples (Figure 2).
Figure 2.

Pyrimidine-5-carbonitriles IV–VII as anti-inflammatory and selective COX-2 inhibitors.
Given this, molecular hybridization is now the most promising treatment method. This technique combines the pharmacophoric units of various bioactive drugs to develop a new single chemical entity with synergistic effects and an improved safety profile. In continuation of our effort to develop safer anti-inflammatory agents,15−18,37−40 we provide here the design, synthesis, and in vitro evaluation of a new series of diaryl-1,2,4-triazolo[3,4-a]pyrimidine hybrids (8a–o) as selective COX-2 inhibitors with potential cardioprotective effect, Figure 3 and Table 1.
Figure 3.

Structures of New Hybrids 8a–o.
Table 1. New Hybrids 8a–o.
| compd. no. | Ar | X | compd. no. | Ar | X | compd. no. | Ar | X |
|---|---|---|---|---|---|---|---|---|
| 8a | Ph | H | 8f | H | 8k | H | ||
| 8b | Ph | Br | 8g | Br | 8l | Br | ||
| 8c | Ph | Cl | 8h | Cl | 8m | Cl | ||
| 8d | Ph | CH3 | 8i | CH3 | 8n | CH3 | ||
| 8e | Ph | CH3O | 8j | CH3O | 8o | CH3O |
The newly synthesized compounds 8a–o were tested for in vitro COX-1/COX-2 inhibition assays where the half-maximal inhibitor concentrations (IC50) were calculated as the mean of three determinations, and the selectivity index (SI) values were calculated as IC50 (COX-1)/IC50 (COX-2). The most potent hybrids were further tested for their 5-LOX and sEH inhibitory actions. Moreover, Compounds with high inhibitory actions were chosen for in vivo testing for analgesic, anti-inflammatory, ulcerogenicity, and cardiovascular effects. Finally, molecular docking analysis were performed to investigate their binding mode of interactions within the COX-2 enzyme isoform.
2. Results and Discussion
2.1. Chemistry
Scheme 1 outlines the synthesis of target compounds 8a–o. The key intermediates 3-amino-5-aryl-1,2,4-triazoles 5a–c were synthesized through two different pathways. Aromatic acids 1a–c were first converted into their ethyl esters 2a–c by the action of ethanol and sulfuric acid. These ethyl esters were then refluxed with hydrazine hydrate to produce aromatic acid hydrazides 3a–c. Acid hydrazides 3a–c reacted with aqueous S-methylisourea sulfate in the presence of sodium hydroxide to give aroylaminoguanidine 4a–c, which were cyclized by fusion at 250 °C to give 3-amino-5-aryl-1,2,4-triazoles 5a-c.41,42 Thermal condensation of aromatic acids 1a–c with aminoguanidine sulfate at 210 °C under solvent-free conditions was used as an alternative method for synthesizing 3-amino-5-aryl-1,2,4-triazoles 5a-c.42
Scheme 1. Synthesis of Compounds 8a–o.

Reagent and reaction conditions: (i) EtOH, H2SO4, reflux 3 h, yield 78–90%; (ii) EtOH, NH2NH2.H2O, reflux 3 h, 70–90%; (iii) S-methylisothioureaa sulfate, NaOH, H2O, stirring, room temp (iv) fusion, 250 °C; (v) fusion, 210 °C; (vi) EtOH, NaOH, reflux.
The synthesis of 5-amino-7,8-dihydro-1,2,4-triazolo[3,4-a]pyrimidines from 3-amino-1,2,4-triazole has been reported in the literature.43−46 This synthesis was accomplished using a multicomponent reaction of 3-amino-1,2,4-triazole, malononitrile, and aldehydes or ketones under various reaction conditions. For example, neat reaction or ethanolic solution in a microwave43,44 or ethanolic solution including triethylamine or sodium hydroxide as a catalyst with reflux or ultrasonic irradiation45 or, ultimately, DABCO-based ionic liquids as a catalyst with heating in a solvent-free situation.46
In the present work, we synthesize compounds 8a–o in high yields by refluxing the multicomponent 3-amino-5-aryl-1,2,4-triazoles 5a–c, aromatic aldehydes 6a–e, and malononitrile 7 in ethanol containing sodium hydroxide as a catalyst (Scheme 1).
The structures of compounds 8a–o were validated by 1H NMR and 13C NMR, as well as high-resolution mass spectroscopy. The 1H NMR spectra of the newly synthesized hybrids revealed a distinct signal pattern for 5-amino-7,8-dihydro[1,2,4]-triazolo[3,4-a]pyrimidine. The NH triazolopyrimidine proton signals appeared at 9.20–8.80 ppm, NH2 protons signals appeared around 7.25–7.45 ppm, which may be incorporated into the aromatic proton signals, and a distinctive singlet signal appeared at 5.45–5.35 ppm assigned to NHCH2 protons. Moreover, compounds 8e, 8j, and 8o showed singlet signals at 3.74 ppm due to methoxy group protons, whereas compounds 8d, 8i, and 8n showed singlet signals at 2.28 ppm of the methyl group protons. There are also characteristic signal patterns in 13C NMR spectra, such as 53.35, 55.70, and 118.99 ppm signals assigned to CHNH, CC≡N, and C≡N carbons, respectively. In addition, distinctive signals at 160 and 155 ppm were assigned to C5-NH2 and C3–N carbons, respectively. Furthermore, there are characteristic signals at 55 and 20 ppm for CH3 and OCH3 carbons (if present). Finally, the compound’s purity was confirmed through HRMS, and the results were consistent with the product’s molecular formula.
2.2. Biology
2.2.1. In Vitro Inhibition Assays
2.2.1.1. Inhibition Assays for COX-1 and COX-2
Using the COX-1/COX-2 (human) Inhibitor Screening Assay Kit, all newly synthesized compounds 8a–o were tested for in vitro COX-1/COX-2 inhibition assays.16,17 The half-maximal inhibitor concentrations (IC50) were calculated as the mean of three determinations, and the selectivity index (SI) values were calculated as IC50 (COX-1)/IC50 (COX-2), as shown in Table 2. The IC50 values of the screened compounds were determined and compared to the reference drug Celecoxib.
Table 2. IC50 Values of 8a–o against COX-1/COX-2, 5-LOX, and sEHa.

| compd. no. | Ar | X | COX-1 (IC50, μM) | COX-2 (IC50, μM) | SI | 5-LOX (IC50, μM) | sEH (IC50, nM) |
|---|---|---|---|---|---|---|---|
| 8a | Ph | H | 223.80 | 21.70 | 10 | ND | ND |
| 8b | Ph | Br | 198.70 | 15.20 | 13 | 3.50 ± 0.04 | 3.20 ± 0.0.09 |
| 8c | Ph | Cl | 310.50 | 34.50 | 9 | ND | ND |
| 8d | Ph | CH3 | 293.10 | 46.90 | 6 | ND | ND |
| 8e | Ph | CH3O | 237.50 | 29.70 | 8 | ND | ND |
| 8f | 4-pyridyl | H | 303.00 | 51.50 | 6 | ND | ND |
| 8g | 4-pyridyl | Br | 287.50 | 59.60 | 5 | ND | ND |
| 8h | 4-pyridyl | Cl | 267.20 | 23.60 | 11 | ND | ND |
| 8i | 4-pyridyl | CH3 | 293.50 | 55.70 | 5 | ND | ND |
| 8j | 4-pyridyl | CH3O | 308.20 | 42.70 | 7 | ND | ND |
| 8k | 3-pyridyl | H | 283.70 | 37.50 | 8 | ND | ND |
| 8l | 3-pyridyl | Br | 210.50 | 53.70 | 4 | ND | ND |
| 8m | 3-pyridyl | Cl | 235.50 | 11.60 | 20 | 3.05 ± 0.03 | 2.95 ± 0.08 |
| 8n | 3-pyridyl | CH3 | 207.50 | 21.90 | 10 | ND | ND |
| 8o | 3-pyridyl | CH3O | 267.50 | 10.50 | 25 | 2.90 ± 0.03 | 2.10 ± 0.07 |
| Celecoxib | 343 | 42 | 8 | ND | 260 ± 2.00 | ||
| Quercetin | ND | ND | ND | 3.35 ± 0.05 | ND | ||
| AUDA | ND | ND | ND | ND | 1.2 |
ND: Not determined.
Many synthesized compounds demonstrated significant efficacy and selectivity against the COX-2 isoform in vitro. Compounds 8a, 8b, 8h, and 8m–o are potent COX-2 inhibitors with IC50 values in the micromolar range. They showed a definite preference for COX-2 inhibition over COX-1, with respective SIs of 10, 13, 11, 20, 10, and 25. Compounds 8b, 8m, and 8o were particularly attractive because they exhibited the most significant COX-2 inhibitory activity, with IC50 values of 15.20, 11.60, and 10.50 μM, respectively. They had SIs of 13, 20, and 25, being 1.5–3 times higher than celecoxib (SI = 8, Table 2). Except for compounds 8d, 8f, 8g, 8i, 8j, and 8i, all other tested compounds outperform the reference drug celecoxib as COX-2 inhibitors, with IC50 values ranging from 10.50 to 37.50 μM, compared to Celecoxib’s IC50 value of 42 μM.
The structural activity relationship analysis of the new 3,7-diaryl pyrimidine-6-carbonitriles 8a–o revealed that the substitution pattern of the aryl moiety at position 3 was a key component of COX-2 inhibition and hence selectivity. The 7-(4-methoxyphenyl)-3-(pyridine-3-yl) derivative 8o (Ar = pyridin-3-yl, X = OMe) was the most potent of the synthesized derivatives, with an IC50 value of 10.50 μM and SI of 25, and it was more effective than the reference Celecoxib (IC50 = 42 μM, SI = 8). The replacement of the pyridine-3-yl group at the 3-position with other aryl moieties such as pyridine-4-yl as in compound 8j (Ar = pyridin-4-yl, X = OMe) or phenyl as in compound 8e (Ar = phenyl, X = OMe) resulted in a significant decrease in both potency and selectivity against COX-2 isoform. Compounds 8e and 8j had IC50 values of 29.70 and 42.70 μM, respectively, making them 2.8- and 4-fold less potent than compound 8o. Additionally, 8e and 8j had a selectivity index of 8, making them three times less selective than 8o (SI = 25). These findings demonstrated the importance of 3-position substitutions, with activity increasing in the order pyridine-3-yl > phenyl > pyridine-4-yl.
On the other hand, the phenyl group substitution pattern at position 7 was significant for both potency and selectivity. The unsubstituted derivative 8k (Ar = pyridin-3-yl, X = H) was 3.5-fold less potent than 8o, with an IC50 of 37.50 μM against the COX-2 isoform and a SI of 8, whereas the 4-methylphenyl derivative 8n (Ar = pyridin-3-yl, X = Me) had an IC50 value of 21.90 μM, and a SI of 10. The presence of a 4-chlorophenyl moiety of 3,7-diaryl pyrimidine-6-carbonitrile 8m significantly increased COX-2 potency and selectivity over the reference drug celecoxib. Compound 8m (Ar = pyridin-3-yl, X = Cl) ranked second in activity after 8o with an IC50 value of 11.60 μM and a SI of 20. The COX-2 selectivity was reduced by at least 6-fold when the methoxy group in compound 8o was replaced by the bromine atom in compound 8l. Compound 8l (Ar = pyridin-3-yl, X = Br) was the least effective derivative among all synthesized compounds, with an IC50 value of 53.70 μM and a SI of 4. These findings suggested that the nature of the substitution on the phenyl group at position 7 significantly impacts the inhibitory effect of the newly synthesized compounds against COX-2 isoform and that activity increased in the following order: OMe > Cl > CH3 > H > Br.
2.2.1.2. In Vitro 5-LOX Inhibition Assay
Compounds 8b, 8m, and 8o were tested for their ability to inhibit lipoxygenase enzyme using the Abnova lipoxygenase inhibitor assay kit (Catalog No. 760700).47Table 2 shows the results using Quercetin as the reference drug.
The results of this in vitro assay test are consistent with those of the in vitro COX inhibition assay. Compounds 8m and 8o, the most effective COX-2 selective inhibitors, demonstrated stronger 5-LOX inhibitory action than the reference Quercetin, with IC50 values of 2.90 and 3.05 μM, respectively, compared to the reference Quercetin (IC50 = 3.35 μM). Compound 8b inhibited 5-LOX nearly as well as the reference compound Quercetin, with an IC50 value of 3.50 μM. As a result, compounds 8m and 8o had greater dual inhibitory effects against COX-2 and 5-LOX than the reference compounds Celecoxib and Quercetin.
2.2.1.3. sEH Assay
Compounds 8b, 8m, and 8o, the most potent dual inhibitors of COX-2/5-LOX, were examined further for their activity as sEH inhibitors using AUDA as the reference medication.48Table 2 shows the IC50 results.
The results showed that the compounds tested showed good inhibitory activity against sEH, with IC50 values ranging from 2.10 to 3.20 nM. The compounds tested were equipotent to the reference drug, AUDA (IC50 = 1.2 nM) but significantly more effective than Celecoxib (IC50 = 260 nM). The results of the in vitro sEH assay supplemented the results of the COX-2 inhibitory activity experiment, which showed that compounds 8m and 8o with the best COX-2 inhibition and selectivity were the most effective sEH inhibitors, with IC50 values of 2.10 and 2.95 nM, respectively. These findings highlighted the significance of the pyridyl-3-yl moiety at position 3 in inhibiting sEH. The 3-phenyl-substituted derivative, 8b, was the least potent derivative with an IC50 value of 3.20 nM, 2.7-fold less effective than AUDA but 81-fold more potent than celecoxib as sEH inhibitor.
2.2.2. In Vivo Assays
2.2.2.1. Assay for Analgesic Activity
Based on the findings of prior in vitro studies, compounds 8b, 8m, and 8o were chosen to be tested for in vivo analgesic efficacy using the acetic acid-induced writhing method.49 The efficacy and potency of the compounds examined were determined by the reduction in acetic acid-induced writhing episodes. The results are summarized in Table 3. All of the compounds tested showed good analgesic efficacy, with percent inhibition in the number of writhing ranging from 36 to 47%, compared to the reference drug, Celecoxib, which had 13.5% inhibition. Compounds 8m and 8o, the most effective dual COX-2/sEH inhibitors in vitro, also elicited the greatest analgesic effects, with % inhibition of 41 and 47 and potencies of 3 and 3.5, respectively.
Table 3. Analgesic Activity of Compounds 8b, 8m, and 8o.
| compound No. | no. of writhesa (mean ± SE) | % inhibition | potencyb |
|---|---|---|---|
| 8b | 26.00 ± 0.70 | 36.00 | 2.70 |
| 8m | 19.00 ± 0.60 | 41.00 | 3.00 |
| 8o | 15.00 ± 0.50 | 47.50 | 3.50 |
| celecoxib | 29.00 ± 0.70 | 13.50 | 1.00 |
| control | 33.50 ± 0.80 |
Values are given as mean ± SE.
Potency are calculated according to the equation of relative potency % = % of inhibition of tested compound/% of inhibition of reference × 100.
2.2.2.2. Assay for Anti-Inflammatory Action
Compounds 8b, 8m, and 8o were chosen to be tested for in vivo anti-inflammatory action utilizing the carrageen-induced paw edema bioassay method developed by Winter et al.50 The efficacy of the compounds was determined as edema inhibition percentage (EI%) after 1, 3, and 5 h of carrageenan injection vs the standard medication Celecoxib. The results showed that the investigated drugs had significant anti-inflammatory effects, with EI% ranging from 27 to 73%. Compounds 8b, 8m, and 8o exceeded Celecoxib in anti-inflammatory activity after 5 h of treatment. They demonstrated a rapid onset of action and a long-lasting impact until the fifth hour after the compounds were administered. Compound 8o was comparable to celecoxib after the first hour but exhibited more anti-inflammatory efficacy than Celecoxib after the third and fifth hours (Table 4). According to our findings, the 1,2,4-triazolopyrimidine scaffold is a viable lead for developing highly efficient COX-2/sEH inhibitors as potent analgesic and anti-inflammatory agents.
Table 4. Anti-Inflammatory Results of Compounds 8b, 8m, and 8o.
| baseline | % of edema inhibition |
|||
|---|---|---|---|---|
| compound no. | paw diameter (mm) ± SE | 1 h | 3 h | 5 h |
| control | 2.80 ± 0.09 | |||
| 8b | 2.20 ± 0.06 | 27 | 49 | 62 |
| 8m | 2.08 ± 0.09 | 32 | 59 | 70 |
| 8o | 2.03 ± 0.09 | 37 | 61 | 73 |
| celecoxib | 2.10 ± 0.07 | 40 | 54 | 22 |
2.2.3. Gastric Ulcerogenic Activity
The two most prevalent unwanted effects of long-term NSAID use are gastrointestinal erosion and ulcers. As a result, we were interested in the ulcerogenic potential of the most effective compounds, 8m and 8o, when given orally. The ulcerogenic effects of compounds 8m and 8o were evaluated by macroscopic examination of rat intestinal mucosa after oral administration of 10 mg/kg of these compounds and indomethacin and celecoxib.39,51
Compound 8o caused no ulceration in the isolated rat stomach, whereas compound 8b caused mild hyperemia but no widespread ulceration (Table 5). Compounds 8m and 8o were discovered to have a strong COX-2/5-LOX/sEH inhibitory profile with no gastrointestinal effects.
Table 5. Ulcerogenic Effects of Compounds 8b and 8o.
| score |
||
|---|---|---|
| groups | no. of gastric ulcers | severity lesions |
| control | 0 | 0 |
| 8b | 0.30 ± 0.01 | 0.50 ± 0.01 |
| 8o | 0 | 0 |
| celecoxib | 2.5 ± 0.10 | 5.80 ± 0.20 |
| indomethacin | 8.5 ± 0.40 | 12.50 ± 0.70 |
2.2.4. Evaluation of Cardiovascular Parameters
Celecoxib-induced cardiotoxicity in rats52,53 was utilized to evaluate the potential cardiovascular risks of the two most active compounds, 8m and 8o. The reaction of the heart to the tested compounds was expressed as a change in serum levels of lactate dehydrogenase (LDH), troponin-I (Tn-I), tumor necrosis factor (TNF-α), and creatine kinase-MB (CK-MB) at a dose of 100 mg/kg of tested compounds plus celecoxib. The results are shown in Table 6.
Table 6. Serum Level of Cardiovascular Parameters of Compounds 8m and 8o.
| groups | troponin-I (pg/mL) | LDH (IU/L) | CK-MB (IU/L) | TNF-α (% inhibition) |
|---|---|---|---|---|
| normal control | 75 ± 05 | 1535 ± 80 | 16 ± 02 | ND |
| celecoxib | 340 ± 10 | 2100 ± 90 | 96 ± 04 | 64 |
| 8m | 128 ± 06 | 1485 ± 60 | 20 ± 02 | 75 |
| 8o | 107 ± 05 | 1390 ± 50 | 17 ± 01 | 79 |
Compared to a control group, treatment with Celecoxib significantly elevated cardiomyopathy diagnostic biomarkers (Tn-I, LDH, and CK-MB).54,55 Contrarily, compared to the control, compounds 8b and 8o had no discernible effects on the levels of two of these biomarkers, CK-MB and LDH, suggesting that they had fewer cardiotoxic side effects. Additionally, compounds 8b and 8o, as demonstrated in Table 6, considerably decreased the serum concentration of TNF-α, a crucial component in the inflammatory response and cardiac depression,56 with % inhibition of 75 and 79%, respectively, in comparison to celecoxib (% inhibition = 64%). Based on these results, the suggested scaffold may be a promising starting point for developing selective COX-2/sEH inhibitors as strong analgesic/anti-inflammatory agents with lower cardiotoxicity.
2.3. Docking Study into COX-2 Active Site
Docking analysis was performed to investigate the plausible 11binding interaction of the newly synthesized compounds within the COX-2 active site. Docking of compounds 8b, 8m, 8o, and the reference drug celecoxib into the crystal structure11 of COX-2 enzyme catalytic domain in complex with ligand inhibitor rofecoxib [PDB ID code 5KIR]57 was performed using Molecular Operating Environment MOE program version 2008.10.58 The most stable docking model was selected according to the best-scored conformation predicted by the MOE scoring function. The docking results are offered in Table 7 and Figures 4–11.
Figure 11.

3D diagram representation of compound 8o docked into COX-2 active site.
Table 7. Binding Scores, Amino Acid Interactions, and Bond Lengths of the Selected Compounds within the COX-2 Active Site.
| compound | binding scores (kcal/mol–1) | ligand atom | residue | interaction | bond length (A°) |
|---|---|---|---|---|---|
| 8b | –14.07 | N of CN | Arg513 | H-bond | 2.82 |
| N of CN | Tyr355 | H-bond | 2.84 | ||
| H of NH2 | Tyr355 | H-bond | 1.69 | ||
| 3-phenyl | Arg120 | arene-cation | |||
| 8m | –14.63 | N of pyridine | Arg513 | H-bond | 3.31 |
| N of CN | Arg120 | H-bond | 2.37 | ||
| N of CN | Tyr355 | H-bond | 2.72 | ||
| 7-(4-Cl·phenyl) | Arg120 | arene-cation | |||
| 8o | –14.95 | N of pyridine | Arg513 | H-bond | 3.30 |
| N of CN | Arg120 | H-bond | 2.37 | ||
| N of CN | Tyr355 | H-bond | 2.74 | ||
| 7-(4-CH3O·phenyl) | Arg120 | arene-cation | |||
| celecoxib | –13.40 | NH of SO2NH2 | His90 | H-bond | 3.18 |
| O of SO2NH2 | Arg513 | H-bond | 2.89 | ||
| rofecoxib | –11.41 | O of SO2NH2 | Arg513 | H-bond | 3.17 |
Figure 4.

2D diagrams represent the overlay of rofecoxib (red) and celecoxib (green) docked into the COX-2 enzyme active site.
Figure 7.

3D diagram representation of compound 8b docked into COX-2 active site.
Figure 8.
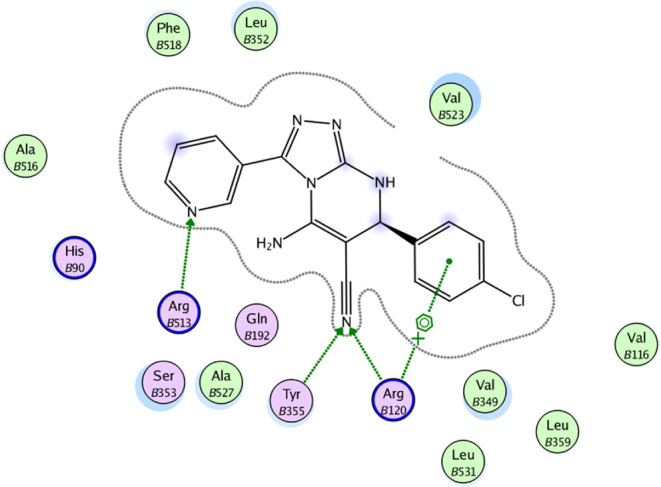
2D diagram representation of compound 8m docked into COX-2 active site.
Figure 9.

3D diagram representation of compound 8m docked into COX-2 active site.
Figure 10.
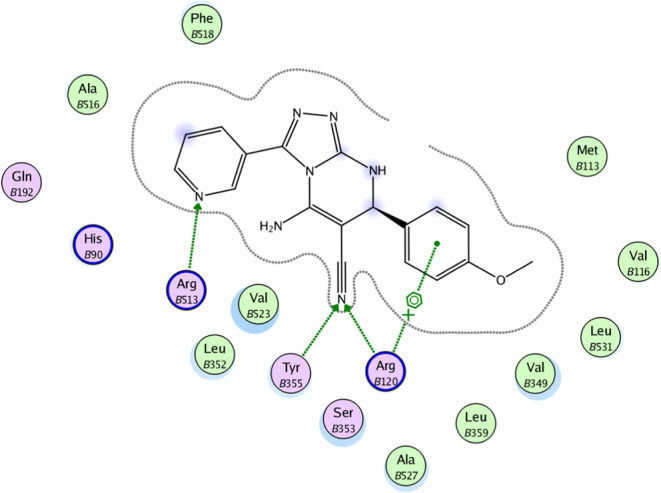
2D diagram representation of compound 8o docked into COX-2 active site.
According to the literature, the COX active binding site is divided into three distinct regions: the carboxylate binding site, the primary pocket region, and the side pocket. The carboxylate binding site is at the entrance and comprises three hydrophilic residues, Arg120, Tyr355, and Glu524, forming a hydrogen bond network. The primary pocket follows the entrance and penetrates deeper into the catalytic domain, forming a lengthy hydrophobic channel. The NSAID binding site is identified by four amino acids: Trp87, Tyr385, Ser530, and Phe518. Val523 expands the side pocket above Arg120 and contains His90 and Arg513 residues, the primary residues responsible for COX-2 selectivity.59
Docking validation was accomplished by self-redocking the cocrystallized conformer rofecoxib into the COX-2 active site and comparing the docking pose to the initial pose using root-mean-square deviation (RMSD). With a docking energy score S = −11.41 kcal/mol, rofecoxib was docked almost at the same position (RMSD = 0.833) and displayed orientations comparable to those previously reported.57 The methyl sulfonyl moiety is inserted into the side pocket, where the sulfonyl oxygen atom is H-bonded to Arg513 (3.17 A°), and its phenyl ring is directed toward the major hydrophobic pocket. Similarly, celecoxib docked into the COX-2 active site, revealing a comparable orientation and binding behavior to rofecoxib with an additional interaction with His90 through H-bonding (3.18 Å); S = −13.40 kcal/mol (Table 7, Figures 4 and 5).
Figure 5.

3D representation of the overlay of rofecoxib (red carbon color) and celecoxib (green carbon color) docked into the COX-2 enzyme active site.
The molecular docking results for compounds 8b, 8m, and 8o indicated that these compounds interact with the COX-2 active site in a similar orientation to Rofecoxib and Celecoxib, with docking energy scores of −14.07, −14.63, and −14.95 kcal/mol, respectively. The binding mode of interactions of the investigated compounds revealed that the amino group at position 5, the nitrile group at position 6, and the aryl moiety are inserted in the side pocket, with the amino acids responsible for COX-2 selectivity. In compound 8b, for example, Arg513 is H-bonded to the nitrile group, whereas Tyr355 is H-bonded to both the amino and nitrile groups. Moreover, the phenyl moiety at position 3 interacts with Arg120 through arene-cation interaction. In compounds 8m and 8o, the nitrogen atom of the pyridin-3-yl moiety at position 3 is H-bonded with Arg513, whereas the nitrogen atom of the nitrile group is H-bonded with both Arg120 and Tyr355. Additionally, the phenyl group at position 7 in compounds 8m and 8o interacts with Arg120 via arene-cation interaction (Figures 6–11 and Table 7).
Figure 6.

2D diagram representation of compound 8b docked into COX-2 active site.
In conclusion, the docking data analysis investigation contributed to a better understanding of the binding modes of the most active hybrids within the COX-2 active site and justified their COX-2 selectivity. Compounds 8b, 8m, and 8o exhibited a parallel to the orientation of rofecoxib and celecoxib, with a greater proclivity to enter the selectivity side pocket than the reference compounds.
2.4. Pharmacokinetic Properties
Lipinski’s and Veber’s rules parameters (drug-likeness or ADME properties)60,61 of the tested compounds 8a–o were calculated as possible drug candidates with enzyme inhibitory potential for anti-inflammatory activities. The calculated parameters included MW, HBA, HBD, Nrotb, C log P, and TPSA (Table 8). The prediction of such ADME properties could be of value in expecting the transport properties of the studied molecules through various membranes, such as the gastrointestinal tract and the blood-brain barrier, where the failure of a tested compound to obey two or more of the aforementioned parameters would reflect the possibility of its poor bioavailability. As presented in Table 8, the predicted pharmacokinetic data for all of the tested compounds 8a–o suggested good oral bioavailability by obeying the Lipinski’s rule of five and Veber’s rule for gastrointestinal absorption. In this regard, according to the Lipinski’s rule, the MW of each of these compounds did not exceed 500 Da and no more than 5 HBD or 10 HBA was observed, with their C log P values also less than 5. Moreover, according to the Veber’s rule, none of the compounds displayed more than 10 rotb, more than 12 HBD and HBA, or TPSA of >140 Å2 (Table 8).
Table 8. Lipinski’s Rule of Five and Veber’s Rule for the Tested Compounds 8a–8oa.
| compd. no. | MW (g/mol) | HBA | HBD | C log P | Nrotb | TPSA (Å2) |
|---|---|---|---|---|---|---|
| 8a | 314.352 | 6 | 2 | 3.253 | 3 | 92.55 |
| 8b | 393.248 | 6 | 2 | 4.051 | 3 | 92.55 |
| 8c | 348.797 | 6 | 2 | 3.845 | 3 | 92.55 |
| 8d | 328.379 | 6 | 2 | 3.551 | 3 | 92.55 |
| 8e | 344.378 | 7 | 2 | 3.209 | 4 | 101.78 |
| 8f | 315.34 | 7 | 2 | 2.021 | 3 | 105.44 |
| 8g | 394.236 | 7 | 2 | 2.819 | 3 | 105.44 |
| 8h | 349.785 | 7 | 2 | 2.613 | 3 | 105.44 |
| 8i | 329.367 | 7 | 2 | 2.319 | 3 | 105.44 |
| 8j | 345.366 | 8 | 2 | 1.977 | 4 | 114.67 |
| 8k | 315.34 | 7 | 2 | 2.020 | 3 | 105.44 |
| 8l | 394.236 | 7 | 2 | 2.818 | 3 | 105.44 |
| 8m | 349.785 | 7 | 2 | 2.612 | 3 | 105.44 |
| 8n | 329.367 | 7 | 2 | 2.318 | 3 | 105.44 |
| 8o | 345.366 | 8 | 2 | 1.976 | 4 | 114.67 |
MW: molecular weight; HBA: H-bond acceptor; HBD: H-bond donor; C Log P, calculated log of the partition coefficient; Nrotb: number of rotatable bonds; TPSA, topological polar surface area.
2.5. Molecular Dynamic Simulation
To validate the docking pose of the most potent derivative, 8o inside the active site of COX-2 (PDB ID code 5KIR), it was subjected to 100 ns long molecular dynamic (MD) simulation. As shown in Figure 12A, 8o exhibited good stability inside the active site over the course of the simulation with an average RMSD of 2.2 Å from the starting docking pose.
Figure 12.

(A) RMSDs of 8o inside the active site of COX2 (PDB ID: 5KIR), respectively, over 100 ns long MD simulation. (B) Interaction energy (i.e., electrostatic and van der Waals interaction energies) of 8o inside the active site of COX-2 over 100 ns long MD simulation. (C) Number of H-bonds detected for 8o inside the active sites of COX2 over 100 ns long MD simulation. Cutoff distance for H-bonds was set to 3.0 Å.
Accordingly, the stable binding 8o was translated into stable and significant interaction energy (electrostatic and van der Waals) inside the enzyme’s active site, where it showed total interaction energy averaged around −55.75 (Figure 12B). In addition, their calculated binding free energy in terms of MM-PBSA was −10.9034 kcal/mol indicating a very good affinity toward the active site of COX2 (Table 9).
Table 9. Binding Free Energy (MM-PBSA) of 8o in Complex with COX-2 (PDB ID: 5KIR).
| energy component | 8o-5KIR |
|---|---|
| ΔGgas | –21.5671 |
| ΔGsolv | 10.6637 |
| ΔGTotal | –10.9034 |
Additionally, 8o established stable multiple H-bonds that were found to be around 3 H-bonds throughout the simulation course (Figure 12C).
In conclusion, 8o showed good binding stability inside the active site of COX-2 over the course of 100 ns long MD simulation indicating a possible inhibitory activity.
2.6. Structure–Activity Relationship (SAR) of Compounds 8a–o
SAR studies could be summarized as follows:
Docking studies on COX-2 active sites revealed that the most potent derivatives could interact with the COX-2 active site in a manner similar to Rofecoxib and Celecoxib, with the amino group at position 5, the nitrile group at position 6, and the aryl moiety inserted in the side pocket, with the amino acids responsible for COX-2 selectivity.
The NH2 group is important for activity as it formed hydrogen bond interactions with Tyr355, whereas the nitrogen atom of the nitrile group is H-bonded with both Arg120 and Tyr355.
The aryl moiety at position 3 was a key component of COX-2 inhibition activity and hence selectivity as it interacts with Arg120 through arene-cation interaction, with activity increasing in the order pyridine-3-yl > phenyl > pyridine-4-yl.
The nitrogen atom of the pyridin-3-yl moiety at position 3 is H-bonded with Arg513.
The phenyl group substitution pattern at position 7 was significant for both potency and selectivity due to interactions with Arg120 in the side pocket via arene-cation interaction, with activity increasing in the order OMe > Cl > CH3 > H > Br.
3. Conclusions
New diaryl-1,2,4-triazolo[3,4-a]pyrimidine hybrids were synthesized, validated, and tested in vitro as dual COX-2/5-LOX inhibitors. Among the compounds tested, 8b, 8m, and 8o were the most potent dual COX-2/5-LOX inhibitors. Compounds 8m and 8o could be good candidates for further development as anti-inflammatory agents. Compound 8o, in particular, is a promising leading compound with significant potential for development as a novel selective COX-2/5-LOX inhibitor. The most active dual inhibitors, 8b, 8m, and 8o, demonstrated significant analgesic and anti-inflammatory biological effects in vivo, all of which are greater than celecoxib while having decreased ulcerogenicity. In terms of the cardiovascular system, the results confirmed that 8m and 8o are less cardiotoxic than the standard celecoxib. This was demonstrated by reduced levels of diagnostic indicators of myocardial injury such as LDH, Tn-I, TNF-, and CK-MB. Molecular docking analysis of compounds 8m and 8o revealed high binding affinities toward COX-2 compared to COX-1, which matched the experimental results. Analysis of the binding interactions of the tested compounds into COX-2 revealed hydrogen bonding interactions with key amino acids in COX-2, such as Arg120, Arg513, and Tyr355. These findings support the concept that compounds 8m and 8o are good candidates for developing anti-inflammatory agents with less gastrointestinal and cardiovascular side effects.
4. Materials and Methods
4.1. Chemistry
General details: See Appendix A.
Compounds 5a–c were prepared according to the reported literature.41,42
4.1.1. General Synthesis of Compounds 8a–o
A mixture of 3-amino-1,2,4-triazole derivatives 5a–c (2 mmol), benzaldehyde derivatives 6a–e (2 mmol), malononitrile 7 (2 mmol), and 2 mL of sodium hydroxide 2N was refluxed for 3 h in 10 mL of ethanol. The reaction mixture was cooled before being poured into ice water and acidified with acetic acid. The formed precipitate was filtered and crystallized from ethanol.
4.1.1.1. 5-Amino-7,8-dihydro-3,7-diphenyl-[1,2,4]triazolo[4,3-a]pyrimidine-6-carbonitrile (8a)
Yield 68%; m.p.: 241–43 °C; 1H NMR (400 MHz, DMSO-d6) δ 8.93 (d, J = 2.7 Hz, 1H, NH), 8.01–7.96 (m, 2H, Ar–H), 7.50–7.44 (m, 3H, Ar–H), 7.41–7.31 (m, 5H, Ar–H), 7.24 (s, 2H, NH2), 5.38 (d, J = 2.5 Hz, 1H, CHNH). 13C NMR (101 MHz, DMSO-d6) δ 160.6, 154.6, 146.9, 143.2, 130.2, 129.8, 128.7, 128.6, 128.0, 126.2, 126.1, 119.1, 56.1, 53.9. HRESI-MS m/z calcd for [M + H]+ C18H15N6: 315.1353, found: 315.1347.
4.1.1.2. 5-Amino-7-(4-bromophenyl)-7,8-dihydro-3-phenyl-[1,2,4]triazolo[3,4-a]pyrimidine-6-carbonitrile (8b)
Yield 78%; m.p.: 282–84 °C; 1H NMR (400 MHz, DMSO-d6) δ 8.95 (s, 1H, NH), 8.03–7.95 (m, 2H, Ar–H), 7.59 (d, J = 8.1 Hz, 2H, Ar–H), 7.50–7.43 (m, 3H, Ar–H), 7.36–7.25 (m, 4H, NH2, Ar–H), 5.42 (d, J = 2.0 Hz, 1H, CHNH). 13C NMR (101 MHz, DMSO-d6) δ 160.6, 154.5, 147.0, 142.5, 131.7, 130.1, 129.9, 128.6, 128.5, 126.2, 121.2, 119.0, 55.7, 53.3. HRESI-MS m/z calcd for [M + H]+ C18H14BrN6: 393.0458, found: 393.0451.
4.1.1.3. 5-Amino-7-(4-chlorophenyl)-7,8-dihydro-3-phenyl-[1,2,4]triazolo[3,4-a]pyrimidine-6-carbonitrile (8c)
Yield 75%; m.p.: 274–76 °C; 1H NMR (400 MHz, DMSO-d6) δ 8.96 (d, J = 2.6 Hz, 1H, NH), 8.03–7.96 (m, 2H, Ar–H), 7.51–7.35 (m, 7H, Ar–H), 7.30 (s, 2H, NH2), 5.44 (d, J = 2.4 Hz, 1H, CHNH). 13C NMR (101 MHz, DMSO-d6) δ 160.7, 154.5, 147.0, 142.1, 132.7, 130.1, 129.9, 128.7, 128.6, 128.1, 126.2, 119.0, 55.8, 53.3. HRESI-MS m/z calcd for [M + H]+ C18H14ClN6: 349.0963, found: 349.0958.
4.1.1.4. 5-Amino-7,8-dihydro-3-phenyl-7-p-tolyl-[1,2,4]triazolo[4,3-a]pyrimidine-6-carbonitrile (8d)
Yield 66%; m.p.: 252–54 °C; 1H NMR (400 MHz, DMSO-d6) δ 8.87 (d, J = 2.5 Hz, 1H, NH), 8.01–7.95 (m, 2H, Ar–H), 7.51–7.41 (m, 3H, Ar–H), 7.26–7.15 (m, 6H, NH2, Ar–H), 5.32 (d, J = 2.5 Hz, 1H, CHNH), 2.28 (s, 3H, CH3). 13C NMR (101 MHz, DMSO-d6) δ 160.5, 154.5, 146.9, 140.3, 137.3, 130.2, 129.8, 129.2, 128.6, 126.2, 126.1, 119.1, 56.3, 53.7, 20.7. HRESI-MS m/z calcd for [M + H]+ C19H17N6: 329.1509, found: 329.1508.
4.1.1.5. 5-Amino-7,8-dihydro-7-(4-methoxyphenyl)-3-phenyl-[1,2,4]triazolo[4,3-a]pyrimidine-6-carbonitrile (8e)
Yield 70%; m.p.: 120–22 °C; 1H NMR (400 MHz, DMSO-d6) δ 8.84 (d, J = 2.4 Hz, 1H, NH), 8.00–7.95 (s, 2H, Ar–H), 7.51–7.41 (m, 3H, Ar–H), 7.27 (d, J = 8.5 Hz, 2H, Ar–H), 7.20 (s, 2H, NH2), 6.95 (d, J = 8.5 Hz, 2H, Ar–H), 5.32 (d, J = 2.5 Hz, 1H, CHNH), 3.74 (s, 3H, OCH3). 13C NMR (101 MHz, DMSO-d6) δ 160.5, 159.1, 154.5, 146.8, 135.2, 130.2, 129.8, 128.6, 127.5, 126.2, 119.1, 114.0, 56.5, 55.2, 53.4. HRESI-MS m/z calcd for [M + H]+ C19H17N6O: 345.1458, found: 345.1457.
4.1.1.6. 5-Amino-7,8-dihydro-7-phenyl-3-(pyridin-4-yl)-[1,2,4]triazolo[4,3-a]pyrimidine-6-carbonitrile (8g)
Yield 65%; m.p.: 254–56 °C; 1H NMR (400 MHz, DMSO-d6) δ 9.04 (s, 1H, NH), 8.69 (d, J = 5.0 Hz, 2H, Ar–H), 7.87 (d, J = 5.1 Hz, 2H, Ar–H), 7.42–7.28 (m, 7H, NH2, Ar–H), 5.42 (s, 1H, CHNH). 13C NMR (101 MHz, DMSO-d6) δ 158.7, 154.7, 150.3, 146.7, 143.1, 137.3, 128.8, 128.2, 126.2, 120.2, 118.9, 57.0, 54.0. HRESI-MS m/z calcd for [M + H]+ C17H14N7: 316.1305, found: 316.1300.
4.1.1.7. 5-Amino-7-(4-bromophenyl)-7,8-dihydro-3-(pyridin-4-yl)-[1,2,4]triazolo-[4,3-a]pyrimidine-6-carbonitrile (8h)
Yield 78%; m.p.: 260–262 °C; 1H NMR (400 MHz, DMSO-d6) δ 9.06 (s, 1H, NH), 8.69 (d, J = 5.2 Hz, 2H, Ar–H), 7.87 (d, J = 5.2 Hz, 2H, Ar–H), 7.58 (d, J = 8.1 Hz, 2H, Ar–H), 7.45–7.29 (m, 4H, NH2, Ar–H), 5.47 (s, 1H, CHNH). 13C NMR (101 MHz, DMSO-d6) δ 158.8, 154.6, 150.3, 146.8, 142.3, 137.3, 131.7, 128.6, 121.4, 120.3, 118.8, 56.5, 53.6. HRESI-MS m/z calcd for [M + H]+ C17H13BrN7: 394.0410, found: 394.0407.
4.1.1.8. 5-Amino-7-(4-chlorophenyl)-7,8-dihydro-3-(pyridin-4-yl)-[1,2,4]triazolo-[4,3-a]pyrimidine-6-carbonitrile (8i)
Yield 65%; m.p.: 254–257 °C; 1H NMR (400 MHz, DMSO-d6) δ 9.05 (d, J = 2.6 Hz, 1H, NH), 8.69 (d, J = 5.2 Hz, 2H, Ar–H), 7.85 (d, J = 5.2 Hz, 2H, Ar–H), 7.46 (d, J = 8.4 Hz, 2H, Ar–H), 7.42–7.33 (m, 4H, NH2, Ar–H), 5.46 (d, J = 2.3 Hz, 1H, CHNH). 13C NMR (101 MHz, DMSO-d6) δ 158.7, 154.6, 150.3, 146.7, 142.0, 137.2, 132.7, 128.8, 128.2, 120.2, 118.7, 56.5, 53.4. HRESI-MS m/z calcd for [M + H]+ C17H13ClN7: 350.0915, found: 350.0913.
4.1.1.9. 5-Amino-7,8-dihydro-3-(pyridin-4-yl)-7-p-tolyl-[1,2,4]triazolo[4,3-a]pyrimidine-6-carbonitrile (8i)
Yield 68%; m.p.: 244–246 °C; 1H NMR (400 MHz, DMSO-d6) 8.98 (d, J = 2.6 Hz, 1H, NH), 8.69 (d, J = 5.1 Hz, 2H, Ar–H), 7.86 (d, J = 5.0 Hz, 2H, Ar–H), 7.30 (s, 2H, NH2), 7.25–7.16 (m, 4H, Ar–H), 5.36 (d, J = 2.5 Hz, 1H, CHNH), 2.28 (s, 3H, CH3). 13C NMR (101 MHz, DMSO-d6) δ 158.7, 154.7, 150.3, 146.6, 140.1, 137.5, 137.3, 129.3, 126.2, 120.2, 118.9, 57.0, 53.8, 20.7. HRESI-MS m/z calcd for [M + H]+ C18H16N7: 330.1462, found: 330.1456.
4.1.1.10. 5-Amino-7,8-dihydro-7-(4-methoxyphenyl)-3-(pyridin-4-yl)-[1,2,4]-triazolo[4,3-a]pyrimidine-6-carbonitrile (8j)
Yield 70%; m.p.: 244–246 °C; 1H NMR (400 MHz, DMSO-d6) δ 8.96 (s, 1H, NH), 8.69 (d, J = 5.1 Hz, 2H, Ar–H), 7.87 (d, J = 5.1 Hz, 2H, Ar–H), 7.34–7.25 (m, 4H, NH2, Ar–H), 6.94 (d, J = 8.3 Hz, 2H, Ar–H), 5.38 (s, 1H, CHNH), 3.73 (s, 3H, OCH3). 13C NMR (101 MHz, DMSO-d6) δ 159.2, 158.7, 154.7, 150.3, 146.6, 137.4, 135.1, 127.7, 120.3, 119.0, 114.1, 57.3, 55.2, 53.7. HRESI-MS m/z calcd for [M + H]+ C18H16N7O: 346.1411, found: 346.1405.
4.1.1.11. 5-Amino-7,8-dihydro-7-phenyl-3-(pyridin-3-yl)-[1,2,4]triazolo[4,3-a]pyrimidine-6-carbonitrile (8k)
Yield 70%; m.p.: 276–278 °C; 1H NMR (400 MHz, DMSO-d6) δ 9.17 (s, 1H, NH), 9.04 (d, J = 2.6 Hz, 1H, Ar–H), 8.65 (d, J = 4.8 Hz, 1H, Ar–H), 8.27 (d, J = 8.0 Hz, 1H, Ar–H), 7.50 (dd, J = 8.0, 4.9 Hz, 1H, Ar–H), 7.43–7.29 (m, 7H, NH2, Ar–H), 5.42 (d, J = 2.4 Hz, 1H, CHNH). 13C NMR (101 MHz, DMSO-d6) δ 158.6, 154.7, 150.6, 147.3, 146.8, 143.1, 133.5, 128.8, 128.1, 126.2, 126.1, 123.9, 119.1, 56.4, 54.0. HRESI-MS m/z calcd for [M + H]+ C17H14N7: 316.1305, found: 316.1300..
4.1.1.12. 5-Amino-7-(4-bromophenyl)-7,8-dihydro-3-(pyridin-3-yl)-[1,2,4]triazolo [4,3-a]pyrimidine-6-carbonitrile (8l)
Yield 82%; m.p.: 290–292 °C; 1H NMR (400 MHz, DMSO-d6) δ 9.15 (s, 1H, NH), 9.04 (s, 1H, Ar–H), 8.64 (s, 1H, Ar–H), 8.26 (d, J = 7.9 Hz, 1H, Ar–H), 7.66–7.28 (m, 8H, NH2, Ar–H), 5.45 (s, 1H, CHNH). 13C NMR (101 MHz, DMSO-d6) δ 158.6, 154.6, 150.6, 147.3, 146.9, 142.4, 133.5, 131.7, 128.5, 126.0, 123.9, 121.3, 118.9, 56.0, 53.4. HRESI-MS m/z calcd for [M + H]+ C17H13BrN7: 394.0410, found: 394.0405.
4.1.1.13. 5-Amino-7-(4-chlorophenyl)-7,8-dihydro-3-(pyridin-3-yl)-[1,2,4]triazolo [4,3-a]pyrimidine-6-carbonitrile (8m)
Yield 72%; m.p.: 244–246 °C; 1H NMR (400 MHz, DMSO-d6) δ 9.14 (s, 1H, NH), 9.04 (s, 1H, Ar–H), 8.64 (s, 1H, Ar–H), 8.26 (d, J = 8.0 Hz, 1H, Ar–H), 7.57–7.27 (m, 7H, NH2, Ar–H), 5.45 (s, 1H, CHNH). 13C NMR (101 MHz, DMSO-d6) δ 158.6, 154.6, 150.7, 147.2, 146.9, 142.0, 133.4, 132.7, 128.8, 128.2, 126.0, 123.9, 118.9, 56.0, 53.3. HRESI-MS m/z calcd for [M + H]+ C17H13ClN7: 350.0915, found: 350.0909.
4.1.1.14. 5-Amino-7,8-dihydro-3-(pyridin-3-yl)-7-p-tolyl-[1,2,4]triazolo[4,3-a]pyrimidine-6-carbonitrile (8n)
Yield 60%; m.p.: 246–248 °C; 1H NMR (400 MHz, DMSO-d6) δ 9.14 (s, 1H, NH), 8.97 (s, 1H, Ar–H), 8.64 (d, J = 4.8 Hz, 1H, Ar–H), 8.26 (d, J = 7.9 Hz, 1H, Ar–H), 7.54–7.48 (m, 1H, Ar–H), 7.31–7.17 (m, 6H, NH2, Ar–H), 5.35 (s, 1H, CHNH), 2.28 (s, 3H, CH3). 13C NMR (101 MHz, DMSO-d6) δ 158.5, 154.6, 150.6, 147.2, 146.7, 140.2, 137.4, 133.4, 129.3, 126.1, 126.1, 123.9, 119.0, 56.5, 53.7, 20.7. HRESI-MS m/z calcd for [M + H]+ C18H16N7: 330.1462, found: 330.1457.
4.1.1.15. 5-Amino-7,8-dihydro-7-(4-methoxyphenyl)-3-(pyridin-3-yl)-[1,2,4] triazolo[4,3-a]pyrimidine-6-carbonitrile (8o)
Yield 73%; m.p.: 124–126 °C; 1H NMR (400 MHz, DMSO-d6) δ 9.14 (d, J = 2.1 Hz, 1H, NH), 8.94 (d, J = 2.4 Hz, 1H, Ar–H), 8.67–8.60 (m, 1H, Ar–H), 8.26 (d, J = 8.2 Hz, 1H, Ar–H), 7.51 (dd, J = 7.9, 4.9 Hz, 1H, Ar–H), 7.30–7.23 (m, 4H, NH2, Ar–H), 6.95 (d, J = 8.3 Hz, 2H, Ar–H), 5.35 (d, J = 2.3 Hz, 1H, CHNH), 3.74 (s, 3H, OCH3). 13C NMR (101 MHz, DMSO-d6) δ 159.1, 158.5, 154.6, 150.6, 147.2, 146.7, 135.1, 133.4, 127.6, 126.1, 123.9, 119.0, 114.1, 56.7, 55.2, 53.5. HRESI-MS m/z calcd for [M + H]+ C18H16N7O: 346.1411, found: 346.1407.
4.2. Biology
4.2.1. In Vitro Inhibition Assay of COX-1/COX-2 Isoenzymes
All newly synthesized compounds 8a–o were tested for in vitro COX-1/COX-2 inhibition assays using the COX-1/COX-2 (human) Inhibitor Screening Assay Kit.16,17 The half-maximal inhibitor concentrations (IC50) were calculated as the mean of three determinations, and the selectivity index (SI) values were calculated as IC50 (COX-1)/IC50 (COX-2), as shown in Table 2. The IC50 values of the screened compounds were determined and compared to the reference drug celecoxib.
4.2.2. In Vitro 5-LOX Inhibition Assay
Compounds 8b, 8m, and 8o were tested for their ability to inhibit lipoxygenase enzyme using the Abnova lipoxygenase inhibitor assay kit (Catalog No. 760700).47 For more details, see Appendix A.
4.2.3. In Vitro sEH Inhibition Assay
Compounds 8b, 8m, and 8o, the most active dual COX-2/5-LOX inhibitors, were tested for their activity as sEH inhibitors using AUDA as the reference drug.48 Refer to Appendix A for more details
4.2.4. In Vivo Analgesic Assay
Compounds 8b, 8m, and 8o were chosen to be tested for in vivo analgesic efficacy using the acetic acid-induced writhing method.49 The efficacy and potency of the compounds examined were determined by the reduction in acetic acid-induced writhing episodes. See Appendix A for more details.
4.2.5. In Vivo Anti-Inflammatory Assay
Compounds 8b, 8m, and 8o were chosen to be tested for in vivo anti-inflammatory action utilizing the carrageen-induced paw edema bioassay method developed by Winter et al.50 The efficacy of the compounds was determined as edema inhibition percentage (EI%) after 1, 3, and 5 h of carrageenan injection vs the standard medication celecoxib. Refer to Appendix A for details.
4.2.6. Assessment of Inflammatory Cytokines
PGE2, IL-6, and TNF-α were determined using specific ELISA kits according to the manufacturer’s instructions. All of the parameters are measured using OD 450 nm.50 Refer to Appendix A.
4.2.7. Ulcerogenic Effect Assay
The ulcerogenic effects of compounds 8m and 8o were evaluated by macroscopic examination of rat intestinal mucosa after oral administration of 10 mg/kg of these compounds and indomethacin and celecoxib.39,51 See Appendix A for details.
4.2.8. Cardiovascular Parameters Assays
The reaction of the heart to compounds 8m and 8o was expressed as a change in serum levels of LDH, Tn-I, TNF-α, and CK-MB at a dose of 100 mg/kg of tested compounds plus celecoxib.54,55
4.3. Docking Experiment
Chem. Draw develops a two-dimensional structure of compounds 8b, 8m, and 8o. The tested compounds were then transferred to the MOE software and subjected to energy minimization.15,57 The cocrystallized structures of COX 2 cocrystallized with a rofecoxib inhibitor were downloaded using PDB codes 5KIR.62,63 Refer to Appendix A for more details.
4.4. Pharmacokinetic Properties and Drug-Likeness
Lipinski’s rule of five and Veber rule calculation were carried out using Molecular Operating Environment (MOE version 2008.10).63 The 15 tested compounds in their MOE database were applied for Lipinski’s rule of five and Veber rule through open compute, descriptor, and select the codes needed by Lipinski’s rule of five and Veber rule and calculate.
4.5. Molecular Dynamics Simulation
NAMD 3.0.0. software was used for performing MD simulation.64,65 This software applies the Charmm-36 force field. Protein systems were built using the QwikMD toolkit of the VMD software,65,66 where the protein structure was checked for any missing hydrogens, the protonation states of the amino acid residues were set (pH = 7.4), and the cocrystallized water molecules were removed. For more details, see Appendix A.
4.6. Binding Free Energy Calculations
Molecular Mechanics Poisson–Boltzmann Surface Area (MM-PBSA) embedded in the MMPBSA.py module of AMBER18 was utilized to calculate the binding free energy of the docked complex.67 Refer to Appendix A for more information.
Acknowledgments
The authors acknowledge the support by Princess Nourah bint Abdulrahman University Researchers Supporting Project Number (PNURSP2024R3), Princess Nourah bint Abdulrahman University, Riyadh, Saudi Arabia. They also acknowledge support from the KIT-Publication Fund of the Karlsruhe Institute of Technology.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.4c00870.
1H NMR and 13C NMR spectra of compounds 8a–o (Figures S1–S30); LCMS of compounds 8a–o (Figures S31–S45); and Appendix A (PDF)
This work was funded by Princess Nourah bint Abdulrahman University Researchers Supporting Project Number (PNURSP2024R3), Princess Nourah bint Abdulrahman University, Riyadh, Saudi Arabia.
The authors declare no competing financial interest.
Supplementary Material
References
- Qu X.; Tang Y.; Hua S. Immunological approaches towards cancer and inflammation: a cross talk. Front. Immunol. 2018, 9, 563. 10.3389/fimmu.2018.00563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashley N. T.; Weil Z. M.; Nelson R. J. Inflammation: mechanisms, costs, and natural variation. Ann. Rev. Ecol., Evol., Syst. 2012, 43, 385–406. 10.1146/annurev-ecolsys-040212-092530. [DOI] [Google Scholar]
- Chen L.; Deng H.; Cui H.; Fang J.; Zuo Z.; Deng J.; Li Y.; Wang X.; Zhao L. Inflammatory responses and inflammation-associated diseases in organs. Oncotarget 2018, 9 (6), 7204. 10.18632/oncotarget.23208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sala A.; Proschak E.; Steinhilber D.; Rovati G. E. Two-pronged approach to anti-inflammatory therapy through the modulation of the arachidonic acid cascade. Biochem. Pharmacol. 2018, 158, 161–173. 10.1016/j.bcp.2018.10.007. [DOI] [PubMed] [Google Scholar]
- Bennett M.; Gilroy D. W. Lipid mediators in inflammation. Myeloid Cells Health Dis.: A Synth. 2017, 343–366. 10.1128/9781555819194.ch19. [DOI] [Google Scholar]
- Jahnavi K.; Reddy P. P.; Vasudha B.; Narender B. Non-steroidal anti-inflammatory drugs: an overview. J. Drug Delivery Ther. 2019, 9 (1-s), 442–448. 10.22270/jddt.v9i1-s.2287. [DOI] [Google Scholar]
- Fokunang C.; Fokunang E. T.; Frederick K.; Ngameni B.; Ngadjui B. Overview of non-steroidal anti-inflammatory drugs (nsaids) in resource limited countries. Moj Toxicol. 2018, 4 (1), 5–13. 10.15406/mojt.2018.04.00081. [DOI] [Google Scholar]
- Wolfe M. M.; Lichtenstein D. R.; Singh G. Gastrointestinal toxicity of nonsteroidal antiinflammatory drugs. N. Engl. J. Med. 1999, 340 (24), 1888–1899. 10.1056/NEJM199906173402407. [DOI] [PubMed] [Google Scholar]
- Varga Z.; rafay ali Sabzwari S.; Vargova V. Cardiovascular risk of nonsteroidal anti-inflammatory drugs: an under-recognized public health issue. Cureus 2017, 9 (4), e1144. 10.7759/cureus.1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cairns J. A. The coxibs and traditional nonsteroidal anti-inflammatory drugs: a current perspective on cardiovascular risks. Can. J. Cardiol. 2007, 23 (2), 125–131. 10.1016/S0828-282X(07)70732-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee D.; Nissen S. E.; Topol E. J. Risk of cardiovascular events associated with selective COX-2 inhibitors. Jama 2001, 286 (8), 954–959. 10.1001/jama.286.8.954. [DOI] [PubMed] [Google Scholar]
- Qandeel N. A.; El-Damasy A. K.; Sharawy M. H.; Bayomi S. M.; El-Gohary N. S. Synthesis, in vivo anti-inflammatory, COX-1/COX-2 and 5-LOX inhibitory activities of new 2, 3, 4-trisubstituted thiophene derivatives. Bioorg. Chem. 2020, 102, 103890 10.1016/j.bioorg.2020.103890. [DOI] [PubMed] [Google Scholar]
- Ben-Noun L. Drug-induced respiratory disorders: incidence, prevention and management. Drug Saf. 2000, 23, 143–164. 10.2165/00002018-200023020-00005. [DOI] [PubMed] [Google Scholar]
- Manju S. L.; Ethiraj K. R.; Elias G. Safer anti-inflammatory therapy through dual COX-2/5-LOX inhibitors: A structure-based approach. Eur. J. Pharm. Sci. 2018, 121, 356–381. 10.1016/j.ejps.2018.06.003. [DOI] [PubMed] [Google Scholar]
- Abdel-Aziz S. A.; Taher E. S.; Lan P.; Asaad G. F.; Gomaa H. A.; El-Koussi N. A.; Youssif B. G. Design, synthesis, and biological evaluation of new pyrimidine-5-carbonitrile derivatives bearing 1, 3-thiazole moiety as novel anti-inflammatory EGFR inhibitors with cardiac safety profile. Bioorg. Chem. 2021, 111, 104890 10.1016/j.bioorg.2021.104890. [DOI] [PubMed] [Google Scholar]
- Abdelrahman M. H.; Youssif B. G.; Abdelazeem A. H.; Ibrahim H. M.; Abd El Ghany A. M.; Treamblu L.; Bukhari S. N. A. Synthesis, biological evaluation, docking study and ulcerogenicity profiling of some novel quinoline-2-carboxamides as dual COXs/LOX inhibitors endowed with anti-inflammatory activity. Eur. J. Med. Chem. 2017, 127, 972–985. 10.1016/j.ejmech.2016.11.006. [DOI] [PubMed] [Google Scholar]
- Mohassab A. M.; Hassan H. A.; Abdelhamid D.; Gouda A. M.; Gomaa H. A.; Youssif B. G.; Radwan M. O.; Fujita M.; Otsuka M.; Abdel-Aziz M. New quinoline/1, 2, 4-triazole hybrids as dual inhibitors of COX-2/5-LOX and inflammatory cytokines: Design, synthesis, and docking study. J. Mol. Struct. 2021, 1244, 130948 10.1016/j.molstruc.2021.130948. [DOI] [Google Scholar]
- Shawky A. M.; Almalki F. A.; Abdalla A. N.; Youssif B. G.; Abdel-Fattah M. M.; Hersi F.; El-Sherief H. A.; Nashwa A. I.; Gouda A. M. Discovery and optimization of 2, 3-diaryl-1, 3-thiazolidin-4-one-based derivatives as potent and selective cytotoxic agents with anti-inflammatory activity. Eur. J. Med. Chem. 2023, 259, 115712 10.1016/j.ejmech.2023.115712. [DOI] [PubMed] [Google Scholar]
- Chen W.; Xu Q.; Ma X.; Mo J.; Lin G.; He G.; Chu Z.; Li J. Synthesis and biological evaluation of N-(benzene sulfonyl) acetamide derivatives as anti-inflammatory and analgesic agents with COX-2/5-LOX/TRPV1 multifunctional inhibitory activity. Bioorg. Med. Chem. Lett. 2023, 80, 129101 10.1016/j.bmcl.2022.129101. [DOI] [PubMed] [Google Scholar]
- Enayetallah A. E.; French R. A.; Thibodeau M. S.; Grant D. F. Distribution of soluble epoxide hydrolase and of cytochrome P450 2C8, 2C9, and 2J2 in human tissues. J. Histochem. Cytochem. 2004, 52 (4), 447–454. 10.1177/002215540405200403. [DOI] [PubMed] [Google Scholar]
- Imig J. D.; Hammock B. D. Soluble epoxide hydrolase as a therapeutic target for cardiovascular diseases. Nat. Rev. Drug Discovery 2009, 8 (10), 794–805. 10.1038/nrd2875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spector A. A.; Fang X.; Snyder G. D.; Weintraub N. L. Epoxyeicosatrienoic acids (EETs): metabolism and biochemical function. Prog. Lipid Res. 2004, 43 (1), 55–90. 10.1016/S0163-7827(03)00049-3. [DOI] [PubMed] [Google Scholar]
- Nithipatikom K.; Gross G. J. Epoxyeicosatrienoic acids: novel mediators of cardioprotection. J. Cardiovasc. Pharmacol. Ther. 2010, 15 (2), 112–119. 10.1177/1074248409358408. [DOI] [PubMed] [Google Scholar]
- Yang T.; Peng R.; Guo Y.; Shen L.; Zhao S.; Xu D. The role of 14, 15-dihydroxyeicosatrienoic acid levels in inflammation and its relationship to lipoproteins. Lipids Health Dis. 2013, 12, 151. 10.1186/1476-511X-12-151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner K. M.; McReynolds C. B.; Schmidt W. K.; Hammock B. D. Soluble epoxide hydrolase as a therapeutic target for pain, inflammatory and neurodegenerative diseases. Pharmacol. Ther. 2017, 180, 62–76. 10.1016/j.pharmthera.2017.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassan G. S.; Hegazy G. H.; Ibrahim N. M.; Fahim S. H. New ibuprofen derivatives as H2S and NO donors as safer anti-inflammatory agents. Future Med. Chem. 2019, 11 (23), 3029–3045. 10.4155/fmc-2018-0467. [DOI] [PubMed] [Google Scholar]
- Kharb R.; Sharma P. C.; Yar M. S. Pharmacological significance of triazole scaffold. J. Enzyme Inhib. Med. Chem. 2011, 26 (1), 1–21. 10.3109/14756360903524304. [DOI] [PubMed] [Google Scholar]
- Paprocka R.; Wiese M.; Eljaszewicz A.; Helmin-Basa A.; Gzella A.; Modzelewska-Banachiewicz B.; Michalkiewicz J. Synthesis and anti-inflammatory activity of new 1, 2, 4-triazole derivatives. Bioorg. Med. Chem. Lett. 2015, 25 (13), 2664–2667. 10.1016/j.bmcl.2015.04.079. [DOI] [PubMed] [Google Scholar]
- Tozkoparan B.; Küpeli E.; Yeşilada E.; Ertan M. Preparation of 5-aryl-3-alkylthio-l, 2, 4-triazoles and corresponding sulfones with antiinflammatory–analgesic activity. Bioorg. Med. Chem. 2007, 15 (4), 1808–1814. 10.1016/j.bmc.2006.11.029. [DOI] [PubMed] [Google Scholar]
- Angajala K. K.; Vianala S.; Macha R.; Raghavender M.; Thupurani M. K.; Pathi P. J. Synthesis, anti-inflammatory, bactericidal activities and docking studies of novel 1, 2, 3-triazoles derived from ibuprofen using click chemistry. SpringerPlus 2016, 5 (1), 423. 10.1186/s40064-016-2052-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fadaly W. A.; Elshaier Y. A.; Hassanein E. H.; Abdellatif K. R. New 1, 2, 4-triazole/pyrazole hybrids linked to oxime moiety as nitric oxide donor celecoxib analogs: Synthesis, cyclooxygenase inhibition anti-inflammatory, ulcerogenicity, anti-proliferative activities, apoptosis, molecular modeling and nitric oxide release studies. Bioorg. Chem. 2020, 98, 103752 10.1016/j.bioorg.2020.103752. [DOI] [PubMed] [Google Scholar]
- Undare S. S.; Valekar N. J.; Patravale A. A.; Jamale D. K.; Vibhute S. S.; Walekar L. S.; Kolekar G. B.; Deshmukh M.; Anbhule P. V. One-pot synthesis and in vivo biological evaluation of new pyrimidine privileged scaffolds as potent anti-inflammatory agents. Res. Chem. Intermed. 2016, 42, 4373–4386. 10.1007/s11164-015-2281-1. [DOI] [Google Scholar]
- Anjos J. V. d.; Srivastava R. M.; Costa-Silva J. H.; Scotti L.; Scotti M. T.; Wanderley A. G.; Leite E. S.; de Melo S. J.; Mendonca Junior F. J. Comparative computational studies of 3, 4-dihydro-2, 6-diaryl-4-oxo-pyrimidine-5-carbonitrile derivatives as potential antinociceptive agents. Molecules 2012, 17 (1), 809–819. 10.3390/molecules17010809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alam M. M.; Akhter M.; Husain A.; Marella A.; Tanwar O. P.; Ali R.; Hasan S. M.; Kumar H.; Haider R.; Shaquiquzzaman M. Anti-inflammatory and antimicrobial activity of 4,5-dihydropyrimidine-5-carbonitrile derivatives: Their synthesis and spectral elucidation. Acta Pol. Pharm. 2012, 69 (6), 1077–1085. [PubMed] [Google Scholar]
- Abbas S. E.; Awadallah F. M.; Ibrahin N. A.; Said E. G.; Kamel G. M. New quinazolinone–pyrimidine hybrids: Synthesis, anti-inflammatory, and ulcerogenicity studies. Eur. J. Med. Chem. 2012, 53, 141–149. 10.1016/j.ejmech.2012.03.050. [DOI] [PubMed] [Google Scholar]
- Alfayomy A. M.; Abdel-Aziz S. A.; Marzouk A. A.; Shaykoon M. S. A.; Narumi A.; Konno H.; Abou-Seri S. M.; Ragab F. A. Design and synthesis of pyrimidine-5-carbonitrile hybrids as COX-2 inhibitors: anti-inflammatory activity, ulcerogenic liability, histopathological and docking studies. Bioorg. Chem. 2021, 108, 104555 10.1016/j.bioorg.2020.104555. [DOI] [PubMed] [Google Scholar]
- Abdelazeem A. H.; El-Saadi M. T.; Said E. G.; Youssif B. G.; Omar H. A.; El-Moghazy S. M. Novel diphenylthiazole derivatives with multi-target mechanism: Synthesis, docking study, anticancer and anti-inflammatory activities. Bioorg. Chem. 2017, 75, 127–138. 10.1016/j.bioorg.2017.09.009. [DOI] [PubMed] [Google Scholar]
- Abdel-Aziz S. A.; Taher E. S.; Lan P.; El-Koussi N. A.; Salem O. I.; Gomaa H. A.; Youssif B. G. New pyrimidine/thiazole hybrids endowed with analgesic, anti-inflammatory, and lower cardiotoxic activities: Design, synthesis, and COX-2/sEH dual inhibition. Arch. Pharm. 2022, 355 (7), 2200024 10.1002/ardp.202200024. [DOI] [PubMed] [Google Scholar]
- Hendawy O.; Gomaa H. A.; Alzarea S. I.; Alshammari M. S.; Mohamed F. A.; Mostafa Y. A.; Abdelazeem A. H.; Abdelrahman M. H.; Trembleau L.; Youssif B. G. Novel 1, 5-diaryl pyrazole-3-carboxamides as selective COX-2/sEH inhibitors with analgesic, anti-inflammatory, and lower cardiotoxicity effects. Bioorg. Chem. 2021, 116, 105302 10.1016/j.bioorg.2021.105302. [DOI] [PubMed] [Google Scholar]
- Youssif B. G.; Mohamed M. F.; Al-Sanea M. M.; Moustafa A. H.; Abdelhamid A. A.; Gomaa H. A. Novel aryl carboximidamide and 3-aryl-1, 2, 4-oxadiazole analogues of naproxen as dual selective COX-2/15-LOX inhibitors: Design, synthesis and docking studies. Bioorg. Chem. 2019, 85, 577–584. 10.1016/j.bioorg.2019.02.043. [DOI] [PubMed] [Google Scholar]
- Dolzhenko A. V.; Pastorin G.; Dolzhenko A. V.; Chui W. K. An aqueous medium synthesis and tautomerism study of 3 (5)-amino-1, 2, 4-triazoles. Tetrahedron Lett. 2009, 50 (18), 2124–2128. 10.1016/j.tetlet.2009.02.172. [DOI] [Google Scholar]
- El-Koussi N. A.; Omar F. A.; Abdel-Aziz S. A.; Radwan M. F. Synthesis and Antibacterial Screening Of Some 2, 5, 7triaryl-1, 2, 4-Triazolo [1, 5-a] PYRIMIDINES. Bull. Pharm. Sci., Assiut Univ. 2004, 27 (1), 144–154. 10.21608/bfsa.2004.65432. [DOI] [Google Scholar]
- Dandia A.; Sarawgi P.; Arya K.; Khaturia S. Mild and ecofriendly tandem synthesis of 1, 2, 4-triazolo [4, 3-a] pyrimidines in aqueous medium. Arkivoc 2006, 16, 83–92. [Google Scholar]
- Dandia A.; Singh R.; Singh D.; Arya K. Facile regioselective green synthesis of triazolo [4, 3-a] pyrimidines in aqueous medium. Lett. Org. Chem. 2009, 6 (1), 100–105. 10.2174/157017809787003124. [DOI] [Google Scholar]
- Ablajan K.; Kamil W.; Tuoheti A.; Wan-Fu S. An efficient three component one-pot synthesis of 5-amino-7-aryl-7, 8-dihydro-[1, 2, 4] triazolo [4, 3-a]-pyrimidine-6-carbonitriles. Molecules 2012, 17 (2), 1860–1869. 10.3390/molecules17021860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jamasbi N.; Irankhah-Khanghah M.; Shirini F.; Tajik H.; Langarudi M. S. N. DABCO-based ionic liquids: introduction of two metal-free catalysts for one-pot synthesis of 1, 2, 4-triazolo [4, 3-a] pyrimidines and pyrido [2, 3-d] pyrimidines. New J. Chem. 2018, 42 (11), 9016–9027. 10.1039/C8NJ01455H. [DOI] [Google Scholar]
- Chagas-Paula D. A.; Oliveira T. B.; Faleiro D. P. V.; Oliveira R. B.; Da Costa F. B. Outstanding anti-inflammatory potential of selected Asteraceae species through the potent dual inhibition of cyclooxygenase-1 and 5-lipoxygenase. Planta Med. 2015, 81 (14), 1296–1307. 10.1055/s-0035-1546206. [DOI] [PubMed] [Google Scholar]
- Schmelzer K. R.; Inceoglu B.; Kubala L.; Kim I.-H.; Jinks S. L.; Eiserich J. P.; Hammock B. D. Enhancement of antinociception by coadministration of nonsteroidal anti-inflammatory drugs and soluble epoxide hydrolase inhibitors. Proc. Natl. Acad. Sci. U.S.A. 2006, 103 (37), 13646–13651. 10.1073/pnas.0605908103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koster R. In Acetic Acid for Analgesics Screening, Fed Proc 1959, pp 412–417.
- Winter C. A.; Risley E. A.; Nuss G. W. Carrageenin-induced edema in hind paw of the rat as an assay for antiinflammatory drugs. Proc. Soc. Exp. Biol. Med. 1962, 111 (3), 544–547. 10.3181/00379727-111-27849. [DOI] [PubMed] [Google Scholar]
- Manivannan E.; Chaturvedi S. Analogue-based design, synthesis and molecular docking analysis of 2, 3-diaryl quinazolinones as non-ulcerogenic anti-inflammatory agents. Bioorg. Med. Chem. 2011, 19 (15), 4520–4528. 10.1016/j.bmc.2011.06.019. [DOI] [PubMed] [Google Scholar]
- Ahmad S.; Panda B. P.; Kohli K.; Fahim M.; Dubey K. Folic acid ameliorates celecoxib cardiotoxicity in a doxorubicin heart failure rat model. Pharm. Biol. 2017, 55 (1), 1295–1303. 10.1080/13880209.2017.1299768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmad S.; Panda B. P.; Fahim M.; Dhyani N.; Dubey K. Ameliorative effect of beraprost sodium on celecoxib induced cardiotoxicity in rats. Iran J. Pharm. Res.: IJPR 2018, 17 (1), 155. [PMC free article] [PubMed] [Google Scholar]
- Olatidoye A. G.; Wu A. H.; Feng Y.-J.; Waters D. Prognostic role of troponin T versus troponin I in unstable angina pectoris for cardiac events with meta-analysis comparing published studies. Am. J. Cardiol. 1998, 81 (12), 1405–1410. 10.1016/S0002-9149(98)00200-8. [DOI] [PubMed] [Google Scholar]
- Farvin K. S.; Anandan R.; Kumar S. H.; Shiny K. S.; Sankar T. V.; Thankappan T. K. Effect of squalene on tissue defense system in isoproterenol-induced myocardial infarction in rats. Pharmacol. Res. 2004, 50 (3), 231–236. 10.1016/j.phrs.2004.03.004. [DOI] [PubMed] [Google Scholar]
- Zhou Q.; Pan X.; Wang L.; Wang X.; Xiong D. The protective role of neuregulin-1: a potential therapy for sepsis-induced cardiomyopathy. Eur. J. Pharmacol. 2016, 788, 234–240. 10.1016/j.ejphar.2016.06.042. [DOI] [PubMed] [Google Scholar]
- Orlando B. J.; Malkowski M. G. Crystal structure of rofecoxib bound to human cyclooxygenase-2. Acta Crystallogr., Sect. F: Struct. Biol. Commun. 2016, 72 (10), 772–776. 10.1107/S2053230X16014230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molecular Operating Environment (MOE); Chemical Computing Group Inc: Montreal, QC, Canada, 2016. [Google Scholar]
- Kurumbail R. G.; Stevens A. M.; Gierse J. K.; McDonald J. J.; Stegeman R. A.; Pak J. Y.; Gildehaus D.; Iyashiro J. M.; Penning T. D.; Seibert K.; et al. Structural basis for selective inhibition of cyclooxygenase-2 by anti-inflammatory agents. Nature 1996, 384 (6610), 644–648. 10.1038/384644a0. [DOI] [PubMed] [Google Scholar]
- Lipinski C. A.; Lombardo F.; Dominy B. W.; Feeney P. J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Delivery Rev. 1997, 23 (1–3), 3–25. 10.1016/S0169-409X(96)00423-1. [DOI] [PubMed] [Google Scholar]
- Veber D. F.; Johnson S. R.; Cheng H.-Y.; Smith B. R.; Ward K. W.; Kopple K. D. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002, 45 (12), 2615–2623. 10.1021/jm020017n. [DOI] [PubMed] [Google Scholar]
- Molecular Operating Environment (MOE); Chemical Computing Group Inc: 1010 Sherbooke St. West, Suite, 2016; Vol. 910. [Google Scholar]
- Molecular Operating Environment (MOE); Chem Comput Group Inc.: Montreal, QC, Canada, 2016. [Google Scholar]
- Phillips J. C.; Braun R.; Wang W.; Gumbart J.; Tajkhorshid E.; Villa E.; Chipot C.; Skeel R. D.; Kale L.; Schulten K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005, 26 (16), 1781–1802. 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribeiro J. V.; Bernardi R. C.; Rudack T.; Schulten K.; Tajkhorshid E. QwikMD-Gateway for Easy Simulation with VMD and NAMD. Biophys. J. 2018, 114 (3), 673a–674a. 10.1016/j.bpj.2017.11.3632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphrey W.; Dalke A.; Schulten K. VMD: visual molecular dynamics. J. Mol. Graphics 1996, 14 (1), 33–38. 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- Miller B. R. III; McGee T. D. Jr; Swails J. M.; Homeyer N.; Gohlke H.; Roitberg A. E. MMPBSA. py: an efficient program for end-state free energy calculations. J. Chem. Theory Comput. 2012, 8 (9), 3314–3321. 10.1021/ct300418h. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.