

Abstract
Reef-building corals depend on an intricate community of microorganisms for functioning and resilience. The infection of coral-associated bacteria by bacteriophages can modify bacterial ecological interactions, yet very little is known about phage functions in the holobiont. This gap stems from methodological limitations that have prevented the recovery of high-quality viral genomes and bacterial host assignment from coral samples. Here, we introduce a size fractionation approach that increased bacterial and viral recovery in coral metagenomes by 9-fold and 2-fold, respectively, and enabled the assembly and binning of bacterial and viral genomes at relatively low sequencing coverage. We combined these viral genomes with those derived from 677 publicly available metagenomes, viromes, and bacterial isolates from stony corals to build a global coral virus database of over 20,000 viral genomic sequences spanning four viral realms. The tailed bacteriophage families Kyanoviridae and Autographiviridae were the most abundant, replacing groups formerly referred to as Myoviridae and Podoviridae, respectively. Prophage and CRISPR spacer linkages between these viruses and 626 bacterial metagenome-assembled genomes and bacterial isolates showed that most viruses infected Alphaproteobacteria, the most abundant class, and less abundant taxa like Halanaerobiia and Bacteroidia. A host–phage–gene network identified keystone viruses with the genomic capacity to modulate bacterial metabolic pathways and direct molecular interactions with eukaryotic cells. This study reveals the genomic basis of nested symbioses between bacteriophage, bacteria, and the coral host and its endosymbiotic algae.
Keywords: viruses, scleractinian corals, bacteria, metabolic genes, symbiosis
Introduction
Microorganisms of the coral holobiont are fundamental to the ecological success of reef-building corals [1]. A complex network of symbiotic interactions connects all coral holobiont entities, including viruses, bacteria, archaea, fungi, and dinoflagellates, broadening the functional capacity of the coral host and influencing the way the coral adapts and interacts with the reef environment [2, 3]. In addition to the well-described coral–Symbiodiniaceae endosymbiosis, prokaryotic communities (bacteria and archaea) play crucial roles in coral health [4]. These assemblages remain flexible in response to a changing environment and can mitigate the effects of environmental stressors [5]. Though we have gained a significant understanding of many coral–bacteria interactions, little is known about the roles of viruses in the holobiont. Currently available metagenomic tools have shown that viruses, particularly double-stranded DNA viruses infecting prokaryotes (herein, phages), are the most abundant and genomically diverse entities within the coral holobiont [6–10]. In other holobionts, phages have been shown to modulate bacterial community composition [11–13] and defend against pathogens by adhering to mucosal surfaces and regulating bacterial colonization through lysis [14]. In corals, the pathogen Vibrio coralliilyticus can weaponize the lytic cycle of prophages in its competitors by triggering induction, gaining a competitive advantage over other symbiotic bacteria [15]. Shifts in phage community composition have also been observed in association with coral disease. For example, bleached and white plague–diseased tissues harbor distinct phage communities [16] and T4-like phages have been associated with black band disease mats [17], yet it remains unclear whether they play a causative role or if their compositional shifts reflect a generalized holobiont response to disease. At the reef scale, phages play a crucial role in shaping coral ecological interactions, including competitive dynamics for benthic space [18] and biogeochemical cycling [19].
Despite these advances, technical limitations in obtaining high-quality viral genomes from corals prevent the determination of phage contributions to holobiont functional diversity [20–22]. Coral host and Symbiodiniaceae genomes overwhelmingly dominate shotgun metagenomes, preventing appropriate coverage of viral sequences necessary for assembly. Moreover, viruses have high sequence diversity, lack hallmark genes, and display high levels of recombination, further complicating the assembly and characterization of viral metagenome–assembled genomes (vMAGs) [10, 23, 24]. Combined with the limited availability of reference viral genomes, most studies to date have relied on the analysis of read data without assembly of viral genomes [10, 22]. These studies also characterized the viral community using a taxonomy framework that uses phage tail morphology (former families Myoviridae, Podoviridae, and Siphoviridae) and the sequence identity of genes in these families to classify unknown viruses. This classification framework was abolished by the International Committee on Taxonomy of Viruses (ICTV) in 2022 in favor of a taxonomy system based on genomic composition, which better captures the evolutionary histories of viral groups [25]. Therefore, an updated perspective on the diversity and functional genomics of coral-associated viruses in light of current approaches and taxonomic framework is due.
Here, we introduce a size fractionation method for enriching viruses and bacteria in coral-associated metagenomes. We combine 33 metagenomes generated with this approach with publicly available datasets in a meta-analysis totaling 710 coral metagenomes, viromes, and bacterial isolates to reveal the genomic repertoire of coral holobiont viruses, focusing on phages. From this robust dataset, we identified 20,397 viral genomic sequences (2,121 vMAGs and 18,098 viral contigs) from 31 coral species distributed across seven oceanographic regions. The phage community was dominated by the class Caudoviricetes (tailed phages, realm Duplodnaviria) but also included Monodnaviria and Varidnaviria phages. The ability to assemble viral and bacterial genomes enabled the matching of phage–host pairs in coral microbiomes via CRISPR spacers. Phages most often infected hosts in the Alphaproteobacteria, Gammaproteobacteria, and Bacteroidia classes and encoded 109 unique metabolic genes and 64 unique bacteria–eukaryote interaction genes with the potential to affect bacterial metabolism and symbiotic interactions. These results transform our knowledge of the coral virome by identifying specific bacteria–phage–gene links involved in quorum sensing, sulfur cycling, DNA methylation, and molecular interactions with eukaryotic cells within the holobiont.
Materials and methods
Development of a viral and bacterial enrichment method
Fragments of the coral Orbicella faveolata (N = 28) were collected via SCUBA in July 2021, along the southwestern coast of Curaçao in the Caribbean (Table S1). Specimens of ~1 cm3 (containing mucus, tissue, and skeleton) were collected using a chisel and hammer and placed in Ziplock polyethylene bags with ambient seawater. The samples were placed on ice for an average of 20 min during transfer to the laboratory at the CARMABI research station, where ambient seawater was removed, and coral samples were flash frozen and stored at −80°C until later processing. The samples were subjected to two DNA extraction protocols for comparison: 19 samples were processed using the viral and bacterial enrichment (VBE) method developed here and 6 underwent bulk extraction and sequencing and were treated as controls. For both extraction methods, coral fragments were thawed, crushed to a fine gravel texture using a sterile mortar and pestle, and suspended in 150 μl of sterile artificial seawater. Control coral homogenates were extracted using a DNeasy PowerSoil Kit (QIAGEN, Germantown, MD), modified with the addition of 20 μl of proteinase K incubated at 56°C for 10 min prior to the kit’s lysis steps. For VBE, the coral homogenate was placed into a tube containing 0.2 g of 425- to 600-μm sterile glass beads (Sigma-Aldrich, St. Louis, MO) and vortexed (VWR Analog Vortex Mixer; VWR, Radnor, PA) at speed 3 (~ 600 rpm) for 5 min to disrupt the larger coral and algal symbiont cells. The supernatant was transferred to a clean tube, brought up to 1 ml with sterile ASW, and treated with DNAse I (20 U/ml final concentration) in DNAse I Buffer (Invitrogen, Waltham, MA) at 25°C for 2 h to degrade the coral host and algal DNA released in the previous step. DNAse activity was stopped with ethylenediaminetetraacetic acid (5 mM final concentration; Genesee Scientific, San Diego, CA), and the sample was filtered through an 8.0-μm membrane (Cytiva, Marlborough, MA). This filter size was intended to allow passage of the bacteria and viruses, while further removing the larger coral and Symbiodiniaceae cells. The flowthrough was collected, transferred to a 100 kDa Amicon Centrifugal Unit (Sigma-Aldrich, St. Louis, MO), and centrifuged at 3,200 g for 30 min to concentrate the bacteria and viruses together. After concentration, each side of the Amicon filter was rinsed and incubated at 56°C for 1 h (per side) in 200 μl Buffer T1 and 20 μl proteinase K from the NucleoSpin Tissue Kit (Macherey-Nagel Inc., Allentown, PA) and DNA extraction proceeded from Step 3 of the kit. DNA was eluted in 100 μl of PCR-grade water and quantified with a Qubit 2.0 Fluorometer (Invitrogen, Waltham, MA). A step-by-step description of the VBE protocol is publicly available on protocols.io [26]. Library preparation and whole-genome sequencing were conducted by Azenta Life Sciences (South Plainfield, NJ). Metagenomic libraries were generated with the NEBNext Ultra DNA Library Preparation kit following the manufacturer’s instructions (New England Biolabs, Ipswich, MA) and paired-end sequenced (2 × 150 bp) on a HiSeq 4000 platform (Illumina, San Diego, CA).
Quantification of viral and bacterial enrichment in coral metagenomes
Raw metagenomic reads were adapter-trimmed, quality-filtered (trimq = 30, maq = 30), and entropy-filtered (entropy = 0.90) using BBDuk [27], generating 594,487,714 quality-controlled (QC) reads. To identify coral host and Symbiodiniaceae reads, the QC reads were mapped to the genome of the coral host, O. faveolata (GCA_001896105.1), and a representative Symbiodiniaceae genome (Symbiodinium sp. clade A Y106; GCA_003297005.1) using Bowtie2 (--mp 4 and -X 1000) [28]. Mapped reads were removed using SAMtools v1.18 [29] and quantified with SeqKit [30], yielding 336,288,370 coral and Symbiodiniaceae-filtered reads (Table S1). The QC reads and coral and symbiont-filtered reads from each sample were assembled separately using metaSPAdes v3.15.5 with default parameters [31]. Bacterial reads were identified with Kaiju v1.9.0 in each group using the proGenomes v3 database [32] and normalized by either the number of QC reads or coral and symbiont-filtered reads in each sample to obtain the percentage of bacterial reads in each metagenome. Bacterial metagenome-assembled genomes (bMAGs) were generated by integrating the single-sample coverage binning outputs from MaxBin2 v2.2.7 [33], MetaBat2 v2.15 [34], and CONCOCT v1.1.0 [35]. The resulting bins were consolidated and improved with the metaWRAP v1.2.1 [36] bin refinement module. The quality of refined bins was assessed with CheckM2 v1.0.2 [37] to select bins with ≥50% completion and ≤10% contamination. Viral identification and quantification were carried out using VIBRANT v1.2.1, which compared genes from all assembled contigs to the Kyoto Encyclopedia of Genes and Genomes (KEGG) KoFam, Pfam (v32), and Virus Orthologous Groups (VOG) [38]. Viral contigs were pooled by DNA extraction method to create two databases, “VBE” and “Control.” The number of reads mapped to their respective databases at 80% identity with SMALT v0.7.6 [39] was normalized by the total number of QC and filtered reads [40]. A summary of the bioinformatic pipeline is shown in Fig. S1. Comparisons between VBE and controls were based on Student’s t-test between the two groups, apart from bMAG assembly, which was compared based on a simple count of bMAGs generated from each group. Taxonomic assignment of phages was performed with the Phage Taxonomy Tool (PTT) [41], with the reference taxonomy edited to reflect updated ICTV classifications [42].
Virus identification in global coral datasets
We assembled the global coral virus database (GCVDB) by combining viruses identified in the 28 Curaçao samples described above, 5 additional O. faveolata samples from Miami, FL (processed with VBE), and publicly available metagenomes (N = 355), viromes (N = 12), and genomes of bacteria (N = 310) isolated from reef-building corals (Tables S2 and S3 list the metadata and accession numbers for each dataset). Search terms and combinations of search terms including but not limited to “coral,” “metagenome,” “virome,” and “bacterial isolates” were used to identify publications and their associated datasets through Google Scholar, the National Center for Biotechnology Information (NCBI), the European Nucleotide Archive, and the JGI IMG/M Database [43]. The inclusion of samples was limited to coral metagenomes or bacterial genomes of isolates from corals of the order Scleractinia collected in situ (excluding aquaria and surrounding seawater) and to sequences with associated metadata describing the geographic location of sampling, the taxonomy of the coral host, and the methods used to collect and process samples. Metagenomic samples were restricted to Illumina sequences and excluded early 454 and Sanger sequencing studies.
The quality of the 310 bacterial genomes was assessed using CheckM v.2.0.12 [44]. VIBRANT identified 637 putative viral genomes in these bacterial genomes. After dereplication with Virathon [45], 178 representative viruses were added to the GCVDB. Metagenomes and viromes contained 36,006,392,282 raw metagenomic reads that were QC and assembled with the same parameters described above. No coral or symbiont read filtering was performed on this larger dataset, as not all coral species had available reference genomes. Contigs longer than 1,000 bp were screened by VIBRANT, and the identified viral genomic sequences were grouped by project, location, and host coral species, and co-binned into vMAGs with vRhyme v1.1.0 using the “longest” method, which dereplicates scaffolds, keeping the longest representative sequence [46]. Contigs within bins were subjected to further dereplication using Virathon and then N-linked with 1,000 Ns per link. N-linkages between contigs of a vMAG only indicate that contigs belong to the same vMAG, a step which is necessary for downstream dereplication of viral bins and quality assessment and do not indicate length [46]. N-linked vMAGs were dereplicated with Virathon, and their quality was assessed with CheckV v1.0.1, which estimates completeness and contamination based on the presence of hallmark genes by comparison with VOG, IMG/VR, RVDB, KEGG Orthology, PfamA, PfamB, and TIGRFAM [47]. This process generated 2,121 vMAGs and 18,098 viral contigs that were added to the GCVDB. Combined with the 178 viral contigs derived from bacterial isolates, a total of 20,397 unique putative viral genomes from coral holobionts compose the GCVDB (Table S4).
Diversity of coral-associated viruses
Viral contigs annotated as “complete circular,” “high-quality draft,” and “medium-quality draft” by VIBRANT (N = 317) were combined with vMAGs annotated as “high-quality” and “medium-quality” by CheckV (N = 528) for further analyses (Tables S5 and S6). These genomes were grouped with the NCBI Viral RefSeq v1.1 database (accessed: 13 July 2023), which was first dereplicated using MIUViG-recommended parameters (95% average nucleotide identity, 85% alignment fraction) adopted by the Genomic Standards Consortium [48]. A comparison of genome relatedness between the GCVDB viruses and the RefSeq viruses was performed using GL-UVAB v0.6.pl, which calculates an all-versus-all Dice distance matrix based on the number of shared protein-encoding genes and their identity level [49]. The distance matrix was used to build a neighbor-joining tree containing 845 GCVDB viruses and 257 NCBI viruses. The tree was visualized and annotated using Interactive Tree of Life [50]. Taxonomic assignment of GCVDB viruses was performed by the PTT, which exclusively identifies prokaryotic viruses through protein similarity [41]. The “PTT_virus_taxonomy.tsv” reference data sheet was manually edited to reflect updated ICTV classifications [42]. Adonis and beta-dispersion analysis were performed with the R package Vegan [51].
Bacteriophage host identification
Bacterial MAGs were generated from all metagenomic samples using the methods described above. Briefly, single-sample coverage binning outputs from MaxBin2 v2.2.7 [33], MetaBat2 v2.15 [34], and CONCOCT v1.1.0 [35] were consolidated and improved with metaWRAP v1.2.1 [36], resulting in 910 bins. CheckM2 v1.0.2 [37] was used to select bins with ≥50% completion and ≤10% contamination. Bins with low confidence predictions were removed (N = 6), resulting in 316 bMAGs taxonomically classified with GTDB-Tk v2.3.2 [52] (Table S7). To predict virus–host pairs, we employed a combination of provirus detection and CRISPR spacer matching (Table S8). A database of CRISPR spacers from the 316 bMAGs and 310 bacterial isolates was generated with minCED v0.4.3 [53], a tool derived from CRISPR Recognition Tools (CRT) v1.2 [54]. The resulting CRISPR spacer database was subsequently used to identify sequence homology matches with GCVDB using nucleotide BLAST (BLASTn). Sixty-three high-confidence pairs were generated using thresholds of ≤2 mismatches/gaps, 100% coverage to the spacer, and a sequence length of ≥20 nucleotides. Provirus detection was accomplished by mapping the GCVDB viruses to bMAGs with Minimap2 v2.24-r1122 [55]. Only the bMAG contigs that contained matches of 100% identity (no gaps) along the entire mapping length (N = 448) were selected to be assessed with CheckV, which identified 59 proviruses with host flanking regions. Viral contigs identified in bacterial isolate genomes by VIBRANT were categorized as proviruses (N = 177).
Phage genetic repertoire
The genomes of the GCVDB viruses from metagenomic samples were compared to viruses identified in coral reef seawater metagenomes from the island of Curaçao [56]. These seawater metagenomes are paired with the Curaçao coral metagenomes described above (Table S1). Metabolic genes from viruses identified in seawater are shown in Table S9. We also searched for viral genes involved in bacteria–eukaryote interactions by a protein BLAST (BLASTp) comparison with a curated database of genes that have been experimentally shown to mediate host interactions in bacterial pathogens [57]. Because many of these genes have also been shown to be involved in commensal or mutualistic interactions in other bacterial species [58], here we refer to these genes as bacteria–host interaction genes or symbiosis genes. We used an e-value cut-off of ≤0.00001 and ≥40% identity across ≥20 amino acids to identify conserved domains in the amino acid sequences [59] (Table S10). For multiple quality hits on overlapping regions, the best hit was selected based on the e-value. Only metabolic genes identified in genomes with taxonomic annotations by PTT were included in the analysis of phage–bacteria interactions. Simpson’s Index of Diversity and Pielou’s evenness were used to compare the diversity and evenness of the gene distributions in seawater and corals. GCVDB phage genomes with host linkages and genes of interest (metabolism or symbiosis) were plotted with the R package genoPlotR [60].
Tripartite network
A tripartite network of bacteria–phage–gene linkages was constructed using Cytoscape v3.9.1 [61]. To identify keystone viruses in this network, we treated shared metabolic and symbiosis genes as linkages between viruses. Keystones were identified using values of node degree (k; number of interactions), closeness centrality (cc; distance to all other nodes), and clustering coefficient (clust; connectivity of nearest neighbors) calculated by Cytoscape’s NetworkAnalyzer feature. We ranked each virus by the values of each of these three properties and calculated an average rank across the three to identify keystone viruses [62]. A Sankey diagram depicting the frequency of linkages between bacterial classes and viral families, as well as the frequency of metabolic and symbiosis genes encoded by the viral families, was generated using Google Charts.
Results
Virus and bacteria enrichment increases the recovery of microbial DNA in metagenomes
The VBE (Fig. 1A) metagenomes had 36.39 ± 0.10% (mean, SE) coral or symbiont reads, compared to 69.83 ± 0.13% (mean, SE) in the controls, a 52% reduction in the VBE metagenomes (t-test, t(23) = −5.58, P = 1.124e-05; Fig. 1B). Among quality-controlled reads, 2.72 ± 0.33% (mean, SE) of VBE reads were bacterial, compared to only 0.30 ± 0.03% (mean, SE; here and hereafter) in controls, a 9.01× enrichment in VBE (t-test, t(23) = 4.12, P = 4.15e-04; Fig. 1C). For coral and symbiont-filtered reads, VBE increased bacterial recovery by 5.02× (4.06 ± 0.09% and 0.81 ± 0.06% for VBE and control, respectively; t-test, t(23) = 4.29, P = 2.73e-04; Fig. 1D). High-quality bMAGs could only be generated from VBE metagenomes, with 13 total bMAGs with ≥50% completion and ≤10% contamination identified (Fig. 1E). VBE generated 543 putative viral contigs, compared to 76 from controls, which represented 0.74 ± 0.05% and 0.40 ± 0.05% of QC reads, respectively, a 1.87× increase for VBE (t-test, t(23) = 3.45, P = 2.17e-03; Fig. 1F). Among coral and symbiont-filtered reads, 0.46 ± 0.06% were viral in VBE and 0.16 ± 0.02% in controls, a 2.83× increase in viral recovery by VBE (t-test, t(23) = 2.6878, P = 1.31e-02; Fig. 1G).
Figure 1.

Enrichment of viruses and bacteria from coral samples. (A) Simplified workflow depicting the VBE method for enrichment of viruses and bacteria in coral metagenomes. (B–G) Recovery of coral and symbiont, bacteria, and viruses in VBE and control metagenomes. (B) Percent of coral and symbiont reads, (C) bacterial abundance within QC reads, (D) bacterial abundance within coral and symbiont-filtered (CSF) reads, (E) abundance of bMAGs from QC reads, (F) viral fractional abundance in QC reads and (G) in CSF reads. Stats represent P values from statistical tests: Student’s t-test in panels B, C, D, F, and G (*P < .05, **P < .01, ***P < .001). Workflow created with BioRender.com.
Updated taxonomic profile of coral holobiont phages
The 710 publicly available metagenomes, viromes, and bacterial isolates from 31 coral species and 7 ocean regions (Fig. 2A) analyzed here yielded 20,397 viral genomic sequences (18,098 vContigs and 2,121 vMAGs). These viral genomes were combined into the GCVDB, and hereafter, we refer to these genomic sequences as viruses for simplicity. A total of 846 viruses were classified as medium-quality, high-quality, or complete circular genomes (hereafter, high- and medium-quality viruses). A proteomic tree displaying the relationships between high- and medium-quality viruses in the GCVDB (N = 846) and viruses in the ICTV database (N = 99) shows GCVDB viruses spanning four viral realms according to the current ICTV classification (Fig. 2B). These include Duplodnaviria and Varidnaviria double-stranded DNA (dsDNA) viruses, Monodnaviria single-stranded DNA (ssDNA) viruses, and Riboviria RNA viruses similar to retroviruses with a DNA phase. Several branches of ICTV viral families that infect eukaryotes, including Adintoviridae, Polydnaviriformidae, Caulimoviridae, Metaviridae, and Retroviridae, included representatives from GCVDB. Bacteriophages, specifically, belonged to 3 realms and 25 families spanning both dsDNA and ssDNA viruses (Table S11).
Figure 2.

Coral virome diversity. (A) Distribution of sample locations and data types across ocean regions. Samples were grouped by oceanographic region for each pie chart. The radius of each pie chart represents the number of samples with log10(X + 4) transformation. The figure key indicates the type of dataset: metagenome, metavirome, or complete/draft bacterial genomes. (B) Proteomic tree of genomes and genome fragments in the GCVDB and their closest relatives from the NCBI viral RefSeq. The innermost ring (1) describes the data source (metagenome, virome, bacterial isolate), the second ring (2) depicts the family-level taxonomy of the coral host where the virus was identified, and the outermost ring (3) describes the family-level taxonomy of the viral genome (PTT taxonomy for phages and ICTV classification for RefSeq viruses). Viral realms were defined in the tree based on the position of RefSeq viruses. The branch labeled with an asterisk (*) contains Riboviria viruses. Branch lengths were omitted to better display the tree topology. Branches with three or more RefSeq members were collapsed (circles proportionally sized to the number of collapsed nodes). Stars indicate 14 viruses obtained by the VBE method. (C, D) Taxonomic classification of phages in the complete GCVDB (ALL) and in the subset of this database containing viral genomes of high or medium quality (HQMQ) at the realm (C) and family (D) taxonomic levels.
The majority (68.95%) of viruses across the GCVDB were not taxonomically classified due to the low similarity with reference viruses. Among the high- and medium-quality viruses, most were identified as phages (58.85% and 97.96% in metagenomes and bacterial isolate genomes, respectively). Of the phages with taxonomic annotations, Duplodnaviria was the dominant realm regardless of quality (Fig. 2C). In metagenomes, this was followed by Monodnaviria, which represented 2.67% of the classified phages in the full GCVDB and 16.20% of the high- and medium-quality phages. In the genomes of bacterial isolates, Monodnaviria were absent among high and medium quality but comprised 1.24% when including low quality. 46.55% of Duplodnaviria viruses in the high- and medium-quality metagenomes did not have family classification under current taxonomy, resulting in an overrepresentation of ssDNA Monodnaviria families in Fig. 2D. Although these Monodnaviria families, Microviridae and Inoviridae, were the most abundant families identified among the high- and medium-quality metagenomes, dsDNA tailed phages belonging to the class Caudoviricetes were dominant in all other groups. Some dsDNA families annotated as Duplodnaviria were more closely related to viruses from other realms in the proteomic tree. For example, viruses classified as Straboviridae and Ackermannviridae fell within the Varidnaviria, and some Peduoviridae and Ackermannviridae were grouped within the realm Monodnaviria, which may result from the presence of shared genes between these groups.
We estimated the effects of coral host and ocean region in shaping virome composition by using the Jaccard beta diversity index. Viromes were significantly different across coral host families (adonis, R2 = 0.43, P = .001) and ocean regions (adonis, R2 = 0.36, P = .001). However, the project and subproject that generated the data explained the most variation in Jaccard’s beta diversity (60.78% and 71.99%, respectively). As any given project was from a single ocean region, and each subproject includes both a single ocean region and a single coral species, these variables are not independent, and the effects of biology and geography cannot be disentangled. The data displayed different levels of dispersion by ocean region (betadisper, F = 4.4, P = 2.86e-04), coral host family (betadisper, F = 15.7, P = <2.2e-16), project (betadisper, F = 18.3, P = < 2.2e-16), and subproject (betadisper, F = 7.2, P = <2.2e-16), indicating that the Adonis PERMANOVA results are affected by nonhomogeneous dispersion of the data.
Coral phages preferentially infect Alphaproteobacteria, Bacteroidia, and Halanaerobiia
To link viruses with their hosts, we binned and taxonomically classified 316 bMAGs from the metagenomes (belonging to 41 bacterial classes) and combined them with the 310 publicly available bacterial isolate genomes from coral (5 classes) for a total of 626 putative bacterial hosts. We identified 299 putative links between 144 hosts and 275 GCVDB viruses based on the presence of proviruses (N = 236) and CRISPR spacer matches (N = 63) (Table S8). 24.2% of bacterial isolates (N = 75) and 15.5% of bMAGs (N = 49) contained at least one provirus. Alphaproteobacteria, the most abundant bMAG class (28.80%), was overrepresented among phage-linked bMAGs (53.33% of CRISPR spacer links and 54.24% of provirus links; Fig. 3A). Gammaproteobacteria were the second most abundant class of bMAGs representing 15.19% but were underrepresented in their CRISPR spacer (6.67%) and provirus (8.47%) links. The classes Halanaerobiia and Bacteroidia, both comprising obligate anaerobes, represented <13% of the bMAGs combined, yet had over twice as many CRISPR spacer linkages as Gammaproteobacteria bMAGs. Among bacterial isolates, however, Alphaproteobacteria links were underrepresented, accounting for only 5.56% of CRISPR spacer links and 41.24% of provirus links, and Gammaproteobacteria isolates were overrepresented in their linkages, accounting for 94.44% of CRISPR spacer linkages and 55.37% of provirus linkages (Fig. 3B). We also identified viruses predicted to infect the putative coral mutualist Endozoicomonas, which harbored more than one prophage per genome (Table S8).
Figure 3.
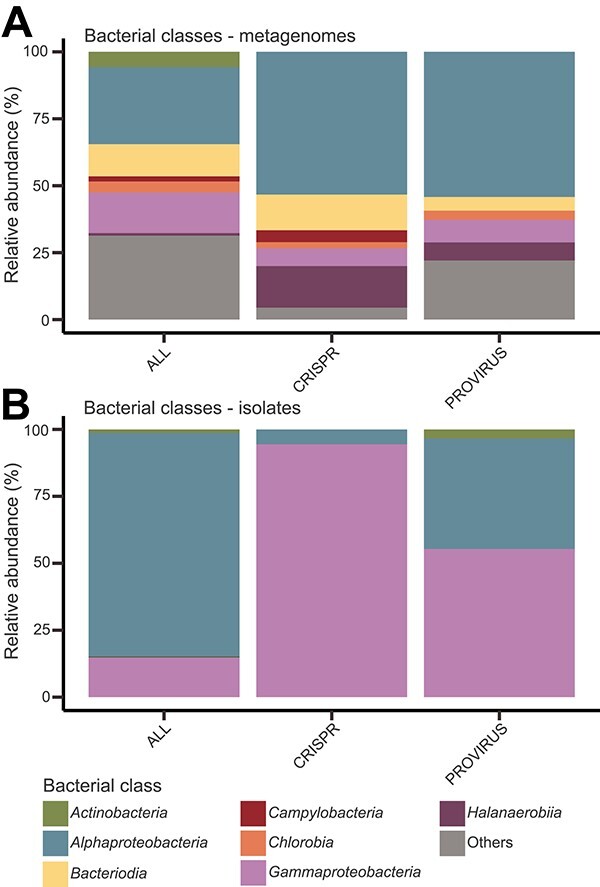
Bacteriophage–bacteria interactions. Frequency of (A) bMAGs and (B) isolates in the metagenomic dataset (ALL) and within those with CRISPR spacer or prophage linkages, at the taxonomic level of class. Classes with abundance <2% (except for Halanaerobiia, which represented <1% of bMAGs, but 7%–16% of linkages) were grouped as “others”.
Metabolic and eukaryote interaction genes differ between coral and seawater viruses
The 3,912 seawater and 6,173 coral viruses classified here as phages encoded 181 unique genes with KEGG ortholog annotations (11.3% of coral and 27.3% of seawater phages). Seawater viruses encoded more unique metabolic genes (N = 72) than corals (N = 51), despite a >22-fold increase in sampling effort (18 seawater metagenomes were used here for comparison versus 400 coral metagenomes; Fig. 4A). Among the 58 metabolic genes shared between coral and seawater viruses, the most common were involved in amino acid metabolism, energy metabolism (photosynthesis), and sulfur relay (Fig. 4B). Only 5 of the 10 most common metabolic genes in coral phages were also among the 10 most common in seawater phages. Metabolic gene α-diversity, as expressed by Simpson’s index of diversity, was higher in seawater (D = 0.95) than in corals (D = 0.61). Additionally, metabolic genes were more evenly distributed in seawater than in coral phages (EPielou = 0.72 in seawater and EPielou = 0.41 in corals), where a single gene, DNA cytosine-5 methyltransferase 1 (DNMT1, dcm), accounted for 60.35% of all metabolic genes identified. DNMT1 was also the most abundant gene in seawater, yet only accounted for 13.25% of the metabolic genes. Together, the four most abundant metabolic genes in coral viruses, DNMT1, dcm; DNA cytosine-5 methyltransferase 3A (DNMT3a; 14.32%); S-(hydroxymethyl)glutathione dehydrogenase/alcohol dehydrogenase (frmA, ADH5, adhC; 3.29%), and nicotinamide phosphoribosyltransferase (NAMPT; 1.57%), accounted for 80% of the metabolic genes encoded by viruses in corals, whereas in seawater, 21 genes constitute that same 80% threshold. These common metabolic genes in coral phages are involved in DNA methylation (DNMT1, dcm; DNMT3a), the oxidation of long-chain alcohols and formaldehyde in several metabolic pathways (frmA, ADH5, adhC), and the metabolism of cofactors and vitamins (nicotinate and nicotinamide metabolism; NAMPT).
Figure 4.

Genomic repertoire of coral-associated bacteriophages. (A) Count of unique metabolic genes (defined based on KEGG Orthologs) identified in coral and seawater bacteriophages. An overlap indicates those identified in both sample types. (B) Frequencies of the most common metabolic genes, calculated as the sum of viruses encoding each gene within each sample type and sorted by abundance. The colors indicate the KEGG pathways to which those genes belong. (C) Count of unique symbiosis genes identified in coral and seawater bacteriophages. (D) Frequencies of the 10 most common symbiosis genes, calculated as the sum of the phages containing each gene within each sample type and sorted by the frequency in corals.
Coral and seawater phages also encoded 71 unique genes classified here as eukaryote interaction genes. These genes were previously classified as virulence factors based on their role in known pathogens [57,58], but many have roles in bacteria–eukaryotic interactions in commensal or mutualistic relationships [58] and were referred to here as eukaryote interaction or symbiosis genes. Fifty-one of these genes were unique to coral phages, 13 were shared between corals and seawater, and only 7 were unique to the seawater phages (Fig. 4C). The majority (70.00%) of the genes in coral phages encoded proteins with ankyrin domains involved in bacteria–eukaryote commensal and mutualistic interactions. This includes the most abundant gene, ankY/legA9, encoding an ankyrin motif-containing protein and accounting for 22.86% of the symbiosis gene group in coral phages (Fig. 4D). They were followed by genes involved in stress survival (10.29%) and immune modulation (9.14%). Coral phages carried a variety of other genes, such as pipB2 (T2SS effector; 11.42%) and icmE (DotG T4SS central channel protein; 11.14%), which are components of effector delivery systems that may be involved in the direct molecular interaction between bacteria and eukaryotes. Seawater phages not only primarily encoded genes involved in adherence (35.92%, with csgG, involved in curli production, as the most abundant [32.04%]) and immune modulation (19.42%) but also encoded genes related to stress survival at a similar frequency as in coral viruses (11.65%).
We selected seven high-quality viral genomes encoding genes with putative roles in host metabolism and symbioses to explore in detail (Table S12), six of which have been linked to bacterial hosts (Fig. 5). Among the Duplodnaviria viruses, PRJNA576217_D_bin_78 (genome 1) was linked via CRISPR spacer to a bMAG classified as Amphritea sp. (SRR15960039_bin.6), family Oceanospirillaceae of Gammaproteobacteria. This virus encoded a DNA cytosine-5 methyltransferase 1 (DNMT1, dcm), an acyl-homoserine lactone (AHL) synthase (raiI), and a quorum-sensing system regulator (bjaR1). Another Duplodnaviria, Ga0478965_02 (genome 2), was integrated into the genome of Roseobacter sp. HKCCD5928 (Alphaproteobacteria) isolated from the coral Platygyra acuta. This virus encoded a phosphoadenosine phosphosulfate reductase (cysH) and a CysO sulfur-carrier protein-S-L-cysteine hydrolase (mec). A representative of Ackermannviridae (CUR21_CRL_A_vRhyme_bin_22; genome 3) was not linked to a host but encoded queuosine biosynthesis genes typically involved in the phage–host evolutionary arms race. queD, encoding a 6-pyruvoyltetrahydropterin/6-carboxytetrahydropterin synthase; queE, a 7-carboxy-7-deazaguanine synthase; and GCH1, folE, a GTP cyclohydrolase, all play roles in deazaguanine DNA modifications in phages. SRR8664773_Node_243 (genome 4) was CRISPR spacer linked to a Rhizobiales bMAG (SRR18532180_bin.10) from a Goniastrea minuta metagenome and encoded the metabolic gene (cysH) as well as bacterial cell cycle regulator (gcrA). SRR18532172_Node_13_fragment_1 (genome 5), a Duplodnaviria representative derived from Porites lutea skeleton, was integrated into a bMAG (SRR18532172_bin.9) from the same sample. This virus encoded a sulfotransferase domain (ST domain), cysteine and methionine metabolism PLP-dependent enzyme (Cys/Met Meta PP) as well as virulence factors, including a type IV secretion system effector (lpg2359) and a RhuM family virulence protein (RhuM family VP). Duplodnaviria virus LIQF01000020.1 (Genome 6) was identified in the genome of Marinomonas fungiae JCM18476 isolated from the coral host Fungia echinata and encoded the virulence factor cylR2, a cytolysin regulator. The isolated coral pathogen V. coralliilyticus strain P1 carried an integrated Monodnaviria virus in the Inoviridae family of chronic filamentous viruses (AEQS01000044.1_contig00050; Genome 7) encoding the gene for the zonula occludens toxin (zot).
Figure 5.

Genome plots of GCVDB viruses. Genome annotations of seven viral genomes of interest due to their host link or the presence of metabolic or symbiosis genes. The colors indicate gene classifications, and angled parallel lines indicate separations between contigs within viral MAGs.
Tripartite network
Links between viruses and their putative hosts (N = 299) and the metabolic and symbiosis genes encoded by viruses in the GCVDB (N = 5,219) were used to construct a network where shared genes were the edges connecting phages in addition to phage–bacteria connections via CRISPR and prophage links in a bipartite network (Fig. 6A displays a version of this network where genes are shown as nodes for visualization purposes). Among bacteria, the average clustering coefficient, which describes the connections between neighbors of a node, was zero. Thus, in the case where multiple viruses were linked to the same host, these viruses did not share metabolic or virulence genes. The top six ranks of keystone viruses based on shared metabolic and symbiosis genes included 17 viruses (Table S13), all of which were classified as Duplodnaviria without family-level classification or host linkages, highlighting our lack of information about these important viruses. These viruses primarily encoded the most common genes within the GCVDB, such as DNMT1, dcm, DNMT3A, involved in DNA methylation, and ankY/legA9, involved in bacteria–eukaryote interactions. To better visualize the distribution of the host–phage–gene connections, we displayed these links in an alluvial plot with phages grouped at the family level and hosts at the class level (Fig. 6B). This plot shows the high representation of genes involved in amino acid and energy metabolism (important for phage particle production during infection) and other gene functions related to cofactors and vitamins and carbohydrate metabolisms, encoded by viruses belonging to several families of tailed bacteriophages (Autographiviridae, Herelleviridae, Kyanoviridae, and Straboviridae) infecting Alphaproteobacteria, Gammaproteobacteria, and Halanaerobia, among others.
Figure 6.

Bacteria–phage–gene network. (A) GCVDB viruses were connected to bacterial hosts (isolate and bMAG) according to the host prediction described in the methods and to metabolic and symbiosis genes according to their functional annotations. The ellipse indicates the position of viruses identified as keystones in the network analyses. (B) Alluvial plot displays abundant bacterial classes (left) linked to viral families (center) found to interact with them though CRISPR spacer and provirus linkages identified in this study. On the right, viral families are linked to metabolic genes encoded in their genomes.
Discussion
Here, we introduce a size fractionation method to increase the recovery of viral and bacterial DNA in coral metagenomes. By combining these metagenomes with data generated across coral metagenomic and culture-based studies worldwide, including 710 coral metagenomes, viromes, and bacterial isolates, we identified 531 high- and medium-quality metagenome-assembled viral genomes from corals. These viruses infect diverse hosts and encode genes that contribute to several holobiont functions interactions, specifically in phage defense mechanisms and bacteria–coral symbioses.
Enrichment of viruses and bacteria in coral metagenomes
VBE captured high- and medium-quality viral genomes across four of the six viral realms recognized by the ICTV, except for Adnaviria and Ribozyviria (Fig. 2B). By using size fractionation and sequencing viruses and bacteria together, the VBE method incurred fewer compositional biases compared to previous methods [63]. Chloroform treatment used in previous studies efficiently removes bacteria but selects against enveloped viruses, some nonenveloped viruses, and a third of tailed phages [63–65]. Cesium chloride (CsCl) gradients enhance viral recovery [63] but severely bias viral diversity due to differences in viral capsid density [66] and often require the use of amplification methods that incur further biases [67]. In viral studies that avoided these biases, viral genomes were either not assembled, limiting the analyses to single reads [10] or focused primarily on viruses of eukaryotes [68]. Here, bacterial and viral genome recovery through VBE was possible even at a relatively low-sequencing coverage (37 million reads and 5.5 billion bases per sample on average). This recovery represents an improvement from previous studies that required coverages from 58 to 146 million reads [69–72] to assemble bMAGs and were most successful with samples devoid of coral tissue, such as in studies focused on the skeleton communities [69]. The differences observed in the outputs between VBE and control samples could be partially attributed to differences in sequencing depth, as VBE sample coverage was, by chance, 40% higher than the control samples. However, this difference was not proportional to the 9 times increase in bacterial recovery (800% increase) and 2 times increase in viral recovery (100% increase). VBE uses a coral fragment not much larger than a parrotfish bite, which is minimally invasive to corals. Therefore, this method could be easily adapted for the recovery of bacterial and viral RNA [73], which would presumably require larger amounts of input material. This method also pools all holobiont compartments (skeleton, tissue, and mucus), and adaptations are required for application to specific compartments [70, 74]. Increasing the size of the coral input sample and sequencing depth and incorporating long-read data may lead to the resolution of more complete genomes covering a larger diversity.
A potential caveat of the symbiont filtering approach used here to calculate the efficacy of the VBE method after filtering eukaryotic reads bioinformatically is that reads were mapped to a single Symbiodiniaceae genome at a low-sequence identity threshold with the goal of removing sequences from Symbiodiniaceae or related genera. To test the effect of this approach, we mapped our reads to the genomes of four Symbiodiniaceae genera common in O. faveolata in the Caribbean: Symbiodinium, Breviolum, Cladocopium, and Durusdinium [75]. This increased the number of reads mapped to Symbiodiniaceae from 36% to 44% in the VBE and from 70% to 75% in the control. However, the difference between VBE and control changed only marginally, from a 52% reduction to 58%, confirming that the initial approach removed most Symbiodiniaceae reads. As a result, the enrichment of bacterial reads using VBE was 5.0 times in the older symbiont filtering versus 4.5 times in the filtering with four genera, and for viruses, it changed from 2.8 to 2.5 times. These differences were small, and the statistical difference between VBE and control was maintained.
Updated taxonomy of coral-associated viruses
The discontinuation of morphology-based viral taxonomic assignments by the ICTV in 2022 has implications for studying bacterial viruses in corals. This taxonomic reorganization included the creation or relocation of 1 order, 22 families, 30 subfamilies, 321 genera, and 862 species while simultaneously eliminating families such as Podoviridae, Siphoviridae, and Myoviridae, along with the order Caudovirales [42]. In previous studies, these families were consistently the most abundant members of the coral holobiont [9, 10, 20]. Within the GCVDB, we identified viruses in the realms Duplodnaviria, Monodnaviria, Varidnaviria, and Riboviria. Among the phages, the vast majority belonged to the class Caudoviricetes, which includes all tailed bacterial and archaeal viruses with icosahedral capsids and a dsDNA genome [42]. The most abundant families under the new taxonomy were Kyanoviridae and Autographiviridae, which represented 65.0% of Caudoviricetes. The family Kyanoviridae likely represents many of the since-abolished Myoviridae phages previously identified in corals. These “T4-like” phages share the myovirus morphotype and current representatives in this family exclusively infect Cyanobacteria [42]. The Kyanoviridae phages in our dataset encoded many metabolic and symbiosis genes but did not have host linkages, preventing a better understanding of their roles in corals. This lack of linkages could be due to a lack of integration capability, infection of hosts that do not employ CRISPR-Cas defense systems, or low-abundance and genetically diverse hosts for which metagenome-assembled genomes (MAGs) were not assembled [48, 56]. Autographiviridae viruses include cultured representatives that typically exhibit a lytic lifestyle and podophage morphology and primarily infect Gammaproteobacteria hosts [76]. This contrasts with Autographiviridae in our dataset, as two were linked with Alphaproteobacteria hosts.
Coral viruses encode a limited but distinct repertoire of metabolic genes
Through the expression of metabolic genes during infection, phages can impact the function and ecological relationships of their hosts [77, 78]. Despite a massively higher sampling effort of corals (N = 400) in comparison with seawater (N = 18), coral phages had a lower frequency and a less diverse repertoire of metabolic genes, primarily encoding genes involved in amino acid metabolism. These results suggest a strong selection of few metabolic genes in phage genomes associated with corals. Differences in sequencing coverage were likely not an explanation for this pattern, as 6,173 viral genomes were recovered from corals versus 3912 from seawater. Yet, coral–viral genomes were less complete (16.03% average completeness) compared to seawater (51.72%), which could contribute to this difference in metabolic gene recovery. Nevertheless, this completeness estimate relies on hallmark genes and might be biased against novel viruses, particularly those from understudied systems such as coral [47]. The genes encoding DNA (cytosine-5)-methyltransferase 1 (DNMT1, dcm) and DNA (cytosine-5)-methyltransferase 3A (DNMT3A), comprising 60.40% of the metabolic genes in coral viruses, are involved in DNA methylation for evasion of restriction-mediated resistance against phage infection [79]. They were found in viruses infecting eight different bacterial families, indicating a widespread evolutionary arms race between coral viruses and their bacterial hosts [80]. Another possible function of DNMT1, dcm is in the colonization of mucosal surfaces. In Group B Streptococcus specifically, knockout of dcm was found to reduce binding to immobilized mucin [81]. We predict that in corals, these genes are involved in the evasion of bacterial defense during lytic infections and possibly in enhancing bacterial host mucus colonization.
S-(hydroxymethyl) glutathione dehydrogenase (frmA, ADH5, adhC), the next most frequent metabolic gene in coral viruses, plays a role in formaldehyde metabolism, which could be important for biomass and energy production of methylotrophs and other fermenting bacteria [82]. Corals display significant diurnal oxygen fluctuations, with daytime photosynthetic oxygen production followed by rapid consumption by heterotrophic metabolism at night and resultant shifts to anaerobic metabolism [83], which may explain the presence of formaldehyde metabolism genes in coral-associated phages. Viruses carrying frmA, ADH5, adhC genes, if infecting anaerobes that proliferate during nocturnal respiration, may bolster downstream production of formate, a key electron donor in anaerobic respiration [84]. The contribution of fermentation to coral holobiont metabolism during nighttime oxygen depletion and the phage contribution to this shift should be the focus of future studies.
Genes for bacteria–eukaryote interactions are rare in coral–phage genomes
Previous studies indicated that ~40% of viral sequences from corals were related to pathogen–host interactions, annotated as “Virulence, Disease, and Defense” [10]. Although these genes are involved in the virulence of known pathogens, many of the same genes can be involved in commensal or mutualistic bacteria–eukaryote interactions in nonpathogens, showing that they mediate bacteria–host interactions across the full spectrum of symbiosis [58]. Therefore, we refer to this group of genes as symbiosis genes or eukaryote interaction genes. Here, 3.3% of phage genomes in the GCVDB and 2.2% of phage genomes in the seawater samples encoded at least one symbiosis gene. This frequency is close to that observed in coral–seawater boundary layer viruses [57], where 2%–4% of the viral community encoded these genes, an order of magnitude lower than the frequency observed in other coral-associated viruses [10]. This difference may be due to the use of a read-based bioinformatic approach that may recruit bacterial reads and inflate the estimates in the study that found higher frequencies [10], compared to the assembly and MAG-binning approach used here. The distributions and functions of these symbiosis genes also differed between studies. Here, seawater phages often encoded genes related to adherence and invasion, consistent with coral boundary layer samples [57]. In contrast, most symbiosis genes encoded by coral viruses were ankyrin motif-containing proteins (ankY/legA9) that mediate protein–protein interactions between hosts and symbionts [85, 86]. Ankyrin motif-containing proteins transferred by phages can enhance bacteria–host symbioses through the suppression of host immune cells [85, 87]. In other bacterial hosts, these proteins represent a family of Type IV secretion system effectors [88]. The similarly high frequency of Icm/Dot type IV secretion system central channel proteins (icmE/dotG) among coral viruses indicates an important role of effector delivery systems in phage–bacteria–coral interactions, enabling bacteria to directly translocate effectors into prokaryotic competitor cells and eukaryotic host cells through an injection apparatus [89, 90]. Greater microbe–microbe competition in the dense coral microbiome [91] and frequent bacteria–animal interactions may drive the selection of these secretion systems and ankyrin motif-containing proteins in the phages identified here.
Host links and network
Associating uncultivated viruses with their microbial hosts remains a challenge not only for coral microbiomes but also for any phage–host studies [48, 56]. Here, our ability to identify bMAGs enabled predictions of virus–host pairs through CRISPR matches and integrated proviruses. The most abundant bMAG hosts, Alphaproteobacteria and Gammaproteobacteria, displayed an interesting trend where Alphaproteobacteria hosts were overrepresented among bMAGs with linkages and Gammaproteobacteria hosts were underrepresented, suggesting higher frequency of viral infection in the more abundant Alphaproteobacteria, consistent with observations of Alphaproteobacteria in coral reef seawater [56]. Lower completeness of gammaproteobacterial genomes (~7% less complete than Alphaproteobacteria bMAGs, t-test, t(137) = 2.51, P = 1.34e-02) could partially but not fully explain the underrepresentation of Gammaproteobacteria linkages. This may simply indicate a lower frequency of viral infections of Gammaproteobacteria in corals for unknown reasons. Halanaerobiia and Bacteroidia constituted a minor fraction of bMAGs, yet they represented a substantial percentage of the phage linkages. This observation aligns with a previous investigation in coastal seawater environments, wherein an abundance of free rRNA, indicative of recently lysed cells, was noted for copiotrophic and low-abundance bacteria [92]. Here, bMAGs of these bacterial groups had multiple phage linkages, suggesting that these taxa are susceptible to infection by multiple viruses.
A closer inspection of viral genomes with connections with hosts and genes of interest revealed the genomic underpinnings of phage–bacteria–coral interactions. As quantifying viral abundances across datasets produced with different methods was not viable, these genomes were selected based on their high estimated genome quality, in addition to the presence of genes of interest and a host link, to identify possible mechanisms of virus–host interactions based on their high-confidence predictions. Duplodnaviria phage PRJNA576217_D_bin_78 encodes an AHL synthase (raiI) and a LuxR family transcriptional regulator (bjaR1), which are both involved in quorum sensing. RaiI produces AHL, which, upon reaching a critical threshold within a bacterial cell, triggers AHL-signaling and the initiation of the expression of virulence genes [93]. BjaR1 acts as a quorum-sensing transcriptional regulator involved in the response to AHL [94]. The presence of both genes in the same phage suggests that this phage coopts the AHL signaling of its bacterial host (bMAG SRR15960039_Bin.6) to modulate gene expression and/or direct lysis–lysogeny decisions. The host, Amphritea sp., is an Oceanospirillaceae in the Gammaproteobacteria class [95]. A bMAG of the same genus was found across metagenomes of a stony coral tissue loss diseased Stephanocoenia intersepta [70] and has been found in association with black band disease [96]. We speculate that the virus identified here may be involved in the pathogenicity of Amphritea in corals through modulation of quorum-sensing signaling (Fig. 7A).
Figure 7.

Conceptual hypotheses for mechanisms underpinning phage roles on coral-associated microbiomes. (A) Viral infection and gene transfer impact bacterial quorum sensing and defense where phage-encoded methyltransferases (1) protect phage nucleic acids from host-encoded restriction endonucleases and (2) regulate adhesins and metabolic factors with putative roles in carbohydrate metabolism and mucin adherence. Genes involved in the synthesis of queuosine precursors may be used to (3) modify phage DNA as protection against host restriction systems or (4) increase queuosine levels in host tRNAs, improving the host’s translational efficiency. Quorum-sensing genes encoded by phages may influence (5) lysis–lysogeny decisions and (6) the expression of genes related to eukaryotic–host interactions (virulence genes). (B) Phages may contribute to sulfur cycling through (1) impacting production of sulfur-containing metabolites that are effective against oxidative stress, (2) the diversion of sulfur toward cysteine production and assimilation into phage proteins, and (3) interference in the synthesis of DMSP due to the deviation of the DMSP precursor cysteine toward phage proteins. (C) Phages may impact virulence and competition within the coral holobiont by (1) regulating the expression of bacterial cytolysins, (2) expression of exotoxins that increase the permeability of coral host tissues, and (3) encoding type IV secretions system central channel proteins and effectors that can mediate bacteria–bacteria or bacteria–eukaryote interactions. Figure created with BioRender.com.
Evidence of the phage–host evolutionary arms race was present in several genomes, as many viruses encoded DNA methyltransferases likely for evasion of restriction-mediated resistance. Another mechanism of this restriction evasion was demonstrated in the genome CUR21_CRL_A_vRhyme_bin_22 (Genome 3 in Fig. 5). The queuosine biosynthesis genes encoded by this phage, queD, queE, and folE, are widespread in phages, especially in the genomes of those with pathogenic hosts [97]. These genes are suggested to modify phage DNA as protection against host restriction systems and contribute to the level of queuosine in host tRNAs, improving host translational efficiency [97].
Roseobacters constitute a significant portion of the coral mucous microbiome, and although the precise nature of their interactions with coral remains ambiguous, there is evidence suggesting their potential roles as probiotics in coral reproduction and defense [98]. The degradation and production of dimethylsulfoniopropionate/dimethylsulfide (DMSP/DMS) by Roseobacters is considered a nutrient source for corals [99, 100]. Here, a phage genome (Ga0478965_02) within a Roseobacter isolate (sp. HKCCD5928) encoded metabolic genes related to sulfur metabolism, specifically assimilatory sulfate reduction (phosphoadenosine phosphosulfate reductase; cysH) and the transfer of sulfur during cysteine production (CysO sulfur-carrier protein; mec). Viruses commonly utilize sulfur metabolism genes to increase the rates of cysteine production for viral particle assembly [41]. Yet, these sulfur genes were encoded in a prophage, Ga0478965_02. It is possible that these genes are transcribed only when the prophage enters the lytic cycle [101]. Alternatively, its expression during lysogeny may contribute to the degradation of DMSP and production of sulfur-containing metabolites by the Roseobacter host (Fig. 7B). Other bacterial hosts associated with the skeletal components of coral hosts exhibited connections to viruses that carried multiple genes associated with the metabolism of sulfur-containing amino acids and metabolites, like SRR8664773_Node_243 and SRR18532172_Node_13_fragment_1. Although these phage-encoded genes could potentially play a role in DMSP cycling and phage production, the production of sulfur-containing metabolites has also been linked to resistance against oxidative and nitrosative stress in various bacterial hosts [102].
Genes related to effector delivery systems, with potential roles in bacterial competition and interactions with the coral host, were common symbiosis genes in the GCVDB. Often, these phage-encoded effector proteins are multifunctional, but often, they convert bacterial hosts from nonpathogenic to virulent [103] (Fig. 7C). One of the viral genomes identified here, Duplodnaviria virus LIQF01000020.1, encoded genes involved in the regulation of cytolysin, an exotoxin that lyses prokaryotic and eukaryotic cells [104]. A prophage-encoding zonula occludens toxin (Zot) previously detected in the coral pathogen V. coralliilyticus strain P1 was also observed here. These observations support the idea that certain coral diseases may result from lysogenic conversion [105, 106]. However, some of these virulence genes can also be involved in nonpathogenic functions in the molecular interactions between bacteria and eukaryotic cells when they are called “fitness factors” [58]. Here, these cells may include the coral animal cells, the endosymbiotic algae, or other eukaryotes in the holobiont.
The network analysis revealed several keystone Duplodnaviria viruses based on their shared metabolic genes, which makes them more connected within the network and more able to diversify the functional capacity of bacteria within the holobiont. Low clustering across bacterial members of the network shows that the viruses infecting the same bacterial host do not share metabolic genes. This indicates that different viruses create distinct virocell metabolism upon infection of the same host [107] and supports the idea of viruses as a reservoir of genetic information that can be horizontally acquired by bacteria under changing conditions.
The results described here reveal the basis of mechanisms by which bacteriophages interact with bacteria within the coral holobiont, with potential downstream effects on bacteria–bacteria and bacteria–coral interactions. The large dataset and high resolution gained from the assembly and binning of genomes enabled the use of stringent quality thresholds, increasing confidence in the annotations and predictions made. The gene-resolved phage–host network highlighted phages’ selective repertoire of metabolic genes that can impact bacterial communication, competition, and molecular interactions with the coral host, illuminating the genomic underpinnings of phage–bacteria–coral tripartite symbioses.
Supplementary Material
Acknowledgements
We thank Dr. Mark Vermeij, the director of the CARMABI research station, for field support in Curaçao and for insightful discussions about the corals sampled here and their environment. We thank Captain Rick Armas for field support in Miami. We also thank all researchers who have shared their genomic datasets in public databases; their data were fundamental for the development of this project.
Contributor Information
Bailey A Wallace, Department of Biology, University of Miami, Coral Gables, FL 33146, United States.
Natascha S Varona, Department of Biology, University of Miami, Coral Gables, FL 33146, United States.
Poppy J Hesketh-Best, Department of Biology, University of Miami, Coral Gables, FL 33146, United States; Department Veterinary Population Medicine, University of Minnesota, St. Paul, MN 55108, United States.
Alexandra K Stiffler, Department of Biology, University of Miami, Coral Gables, FL 33146, United States.
Cynthia B Silveira, Department of Biology, University of Miami, Coral Gables, FL 33146, United States; Department of Marine Biology and Ecology, Rosenstiel School of Marine, Atmospheric, and Earth Science, University of Miami, Miami, FL 33149, United States.
Author contributions
Cynthia B. Silveira conceptualized and led the development of the VBE method. Cynthia B. Silveira and Bailey A. Wallace conceptualized the study. Bailey A. Wallace and Natascha S. Varona conducted sampling, method development, and sample processing. Bailey A. Wallace conducted data retrieval, pipeline development, and analyses. Poppy J. Hesketh-Best and Alexandra K. Stiffler conducted analyses. Bailey A. Wallace wrote the first draft, and all authors edited the manuscript.
Conflicts of interest
The authors declare no conflict of interest.
Funding
This work was funded by the University of Miami Provost Research Award (UM PRA 2022-2547 to C.B.S.). B.A.W. was funded by the National Science Foundation Graduate Research Fellowship Program (2023353157), a University of Miami Holmes Fellowship, and the Kushlan Graduate Research Support Award. A.K.S. was funded by the NSF Graduate Research Fellowship Program (2023349872) and the University of Miami Dean’s Fellowship. N.S.V. was funded by the Maytag Fellowship (PG015171). B.A.W., N.S.V., and C.B.S. were partially funded by the National Aeronautics and Space Administration Exobiology Program (80NSSC23K0676).
Data availability
Coral metagenomic sequence data generated for this project are available on NCBI (PRJNA1061506 and PRJNA975592). The code used to conduct this analysis is publicly available on GitHub (https://github.com/Silveira-Lab/Wallace_Coral_Holobiont_Viruses). Assembled viral genomes are available on Figshare: https://doi.org/10.6084/m9.figshare.24968805.v1. Viral genomes with multiple contigs are N-linked. Bacterial MAGs are available on Figshare: https://doi.org/10.6084/m9.figshare.25000160.v1.
References
- 1. Knowlton N, Rohwer F. Multispecies microbial mutualisms on coral reefs: the host as a habitat. Am Nat 2003;162:S51–62. 10.1086/378684 [DOI] [PubMed] [Google Scholar]
- 2. Rohwer F, Seguritan V, Azam Fet al. Diversity and distribution of coral-associated bacteria. Mar Ecol Prog 2002;243:1–10. 10.3354/meps243001 [DOI] [Google Scholar]
- 3. Voolstra CR, Suggett DJ, Peixoto RSet al. Extending the natural adaptive capacity of coral holobionts. Nat Rev Earth Environ 2021;2:747–62. 10.1038/s43017-021-00214-3 [DOI] [Google Scholar]
- 4. Bourne DG, Morrow KM, Webster NS. Insights into the coral microbiome: underpinning the health and resilience of reef ecosystems. Ann Rev Microbiol 2016;70:317–40. 10.1146/annurev-micro-102215-095440 [DOI] [PubMed] [Google Scholar]
- 5. Santoro EP, Borges RM, Espinoza JLet al. Coral microbiome manipulation elicits metabolic and genetic restructuring to mitigate heat stress and evade mortality. Sci Adv 2021;7:eabg3088. 10.1126/sciadv.abg3088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wegley L, Edwards R, Rodriguez-Brito Bet al. Metagenomic analysis of the microbial community associated with the coral Porites astreoides. Environ Microbiol 2007;9:2707–19. 10.1111/j.1462-2920.2007.01383.x [DOI] [PubMed] [Google Scholar]
- 7. Marhaver KL, Edwards RA, Rohwer F. Viral communities associated with healthy and bleaching corals. Environ Microbiol 2008;10:2277–86. 10.1111/j.1462-2920.2008.01652.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Vega Thurber RL, Barott KL, Hall Det al. Metagenomic analysis indicates that stressors induce production of herpes-like viruses in the coral Porites compressa. PNAS 2008;105:18413–8. 10.1073/pnas.0808985105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Weynberg KD, Laffy PW, Wood-Charlson EMet al. Coral-associated viral communities show high levels of diversity and host auxiliary functions. PeerJ 2017;5:e4054. 10.7717/peerj.4054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Cárdenas A, Ye J, Ziegler Met al. Coral-associated viral assemblages from the central Red Sea align with host species and contribute to holobiont genetic diversity. Front Microbiol 2020;11:572534. 10.3389/fmicb.2020.572534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Duerkop BA, Vaishnava S, Hooper LV. Immune responses to the microbiota at the intestinal mucosal surface. Immunity 2009;31:368–76. 10.1016/j.immuni.2009.08.009 [DOI] [PubMed] [Google Scholar]
- 12. Hsu BB, Gibson TE, Yeliseyev Vet al. Dynamic modulation of the gut microbiota and metabolome by bacteriophages in a mouse model. Cell Host Microbe 2019;25:803–814.e5. 10.1016/j.chom.2019.05.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Almeida GMF, Laanto E, Ashrafi Ret al. Bacteriophage adherence to mucus mediates preventive protection against pathogenic bacteria. MBio 2019;10:e01984–19. 10.1128/mBio.01984-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Barr JJ, Auro R, Furlan Met al. Bacteriophage adhering to mucus provide a non–host-derived immunity. Proc Natl Acad Sci USA 2013;110:10771–6. 10.1073/pnas.1305923110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wang W, Tang K, Wang Pet al. The coral pathogen vibrio coralliilyticus kills non-pathogenic holobiont competitors by triggering prophage induction. Nat Ecol Evol 2022;6:1132–44. 10.1038/s41559-022-01795-y [DOI] [PubMed] [Google Scholar]
- 16. Soffer N, Brandt ME, Correa AMet al. Potential role of viruses in white plague coral disease. ISME J. 2014;8:271–83. 10.1038/ismej.2013.137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Buerger P, Weynberg KD, Wood-Charlson EMet al. Novel T4 bacteriophages associated with black band disease in corals. Environ Microbiol 2019;21:1969–79. 10.1111/1462-2920.14432 [DOI] [PubMed] [Google Scholar]
- 18. Roach T, Little M, Arts Met al. A multiomic analysis of in situ coral-turf algal interactions. Proc Natl Acad Sci USA 2020;117:13588–95. 10.1073/pnas.1915455117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Thurber RV, Payet JP, Thurber ARet al. Virus-host interactions and their roles in coral reef health and disease. Nat Rev Microbiol 2017;15:205–16. 10.1038/nrmicro.2016.176 [DOI] [PubMed] [Google Scholar]
- 20. Wood-Charlson EM, Weynberg KD, Suttle CAet al. Metagenomic characterization of viral communities in corals: mining biological signal from methodological noise. Environ Microbiol 2015;17:3440–9. 10.1111/1462-2920.12803 [DOI] [PubMed] [Google Scholar]
- 21. Thurber RV, Haynes M, Breitbart Met al. Laboratory procedures to generate viral metagenomes. Nat Protoc 2009;4:470–83. 10.1038/nprot.2009.10 [DOI] [PubMed] [Google Scholar]
- 22. Thurber RV, Willner-Hall D, Rodriguez-Mueller Bet al. Metagenomic analysis of stressed coral holobionts. Environ Microbiol 2009;11:2148–63. 10.1111/j.1462-2920.2009.01935.x [DOI] [PubMed] [Google Scholar]
- 23. Silveira CB, Gregoracci GB, Coutinho FHet al. Bacterial community associated with the reef coral Mussismilia braziliensis’s momentum boundary layer over a diel cycle. Front Microbiol 2017;8:784. 10.3389/fmicb.2017.00784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Nayfach S, Roux S, Seshadri Ret al. A genomic catalog of Earth’s microbiomes. Nat Biotechnol 2021;39:499–509. 10.1038/s41587-020-0718-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zerbini FM, Siddell SG, Lefkowitz EJet al. Changes to virus taxonomy and the ICTV statutes ratified by the international committee on taxonomy of viruses (2023). Arch Virol 2023;168:175. 10.1007/s00705-023-05797-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Varona N, Wallace B, Silveira C. Virus and prokaryote enrichment in coral DNA metagenomes. protocols.io2023. 10.17504/protocols.io.q26g7p1y3gwz/v1 [DOI]
- 27.Bushnell B. BB Tools software package. DOE Joint Genome Institute; 2014. Available from: https://sourceforge.net/projects/bbmap/
- 28. Langmead B, Salzberg SL. Fast gapped-read alignment with bowtie 2. Nat Methods 2012;9:357–9. 10.1038/nmeth.1923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Li H, Handsaker B, Wysoker Aet al. The sequence alignment/map format and SAMtools. Bioinformatics 2009;25:2078–9. 10.1093/bioinformatics/btp352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Shen W, Le S, Li Yet al. SeqKit: a cross-platform and ultrafast toolkit for FASTA/Q file manipulation. PLoS One 2016;11:e0163962. 10.1371/journal.pone.0163962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Nurk S, Meleshko D, Korobeynikov Aet al. metaSPAdes: a new versatile metagenomic assembler. Genome Res 2017;27:824–34. 10.1101/gr.213959.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Menzel P, Ng KL, Krogh A. Fast and sensitive taxonomic classification for metagenomics with kaiju. Nat Commun 2016;7:11257. 10.1038/ncomms11257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wu YW, Simmons BA, Singer SW. MaxBin 2.0: an automated binning algorithm to recover genomes from multiple metagenomic datasets. Bioinformatics 2016;32:605–7. 10.1093/bioinformatics/btv638 [DOI] [PubMed] [Google Scholar]
- 34. Kang DD, Li F, Kirton Eet al. MetaBAT 2: an adaptive binning algorithm for robust and efficient genome reconstruction from metagenome assemblies. PeerJ 2019;7:e7359. 10.7717/peerj.7359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Alneberg J, Bjarnason BS, de Bruijn Iet al. Binning metagenomic contigs by coverage and composition. Nat Methods 2014;11:1144–6. 10.1038/nmeth.3103 [DOI] [PubMed] [Google Scholar]
- 36. Uritskiy GV, DiRuggiero J, Taylor J. MetaWRAP—a flexible pipeline for genome-resolved metagenomic data analysis. Microbiome 2018;6:158. 10.1186/s40168-018-0541-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Chklovski A, Parks DH, Woodcroft BJet al. CheckM2: a rapid, scalable and accurate tool for assessing microbial genome quality using machine learning. Nat Methods 2023;20:1203–12. 10.1038/s41592-023-01940-w [DOI] [PubMed] [Google Scholar]
- 38. Kieft K, Zhou Z, Anantharaman K. VIBRANT: automated recovery, annotation and curation of microbial viruses, and evaluation of viral community function from genomic sequences. Microbiome 2020;8:90. 10.1186/s40168-020-00867-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wellcome Sanger Institute . Smalt: A Mapper for DNA Sequencing Reads. 2014. Available from: https://sourceforge.net/projects/smalt/
- 40. Cobián Güemes AG, Youle M, Cantú VAet al. Viruses as winners in the game of life. Annu Rev Virol 2016;3:197–214. 10.1146/annurev-virology-100114-054952 [DOI] [PubMed] [Google Scholar]
- 41. Kieft K, Zhou Z, Anderson REet al. Ecology of inorganic sulfur auxiliary metabolism in widespread bacteriophages. Nat Commun 2021;12:3503. 10.1038/s41467-021-23698-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Turner D, Shkoporov AN, Lood Cet al. Abolishment of morphology-based taxa and change to binomial species names: 2022 taxonomy update of the ICTV bacterial viruses subcommittee. Arch Virol 2023;168:74. 10.1007/s00705-022-05694-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Chen IMA, Chu K, Palaniappan Ket al. The IMG/M data management and analysis system v.7: content updates and new features. Nucleic Acids Res 2022;51:D723–32. 10.1093/nar/gkac976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Parks DH, Imelfort M, Skennerton CTet al. CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res 2015;25:1043–55. 10.1101/gr.186072.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Coutinho, FH. Felipehcoutinho/virathon. 2023. Available from: https://github.com/felipehcoutinho/virathon
- 46. Kieft K, Adams A, Salamzade Ret al. vRhyme enables binning of viral genomes from metagenomes. Nucleic Acids Res 2022;50:e83. 10.1093/nar/gkac341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Nayfach S, Camargo AP, Schulz Fet al. CheckV assesses the quality and completeness of metagenome-assembled viral genomes. Nat Biotechnol 2021;39:578–85. 10.1038/s41587-020-00774-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Roux S, Adriaenssens EM, Dutilh BEet al. Minimum information about an uncultivated virus genome (MIUViG). Nat Biotechnol 2019;37:29–37. 10.1038/nbt.4306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Coutinho FH, Edwards RA, Rodríguez-Valera F. Charting the diversity of uncultured viruses of archaea and bacteria. BMC Biol 2019;17:109. 10.1186/s12915-019-0723-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Letunic I, Bork P. Interactive tree of life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucleic Acids Res 2021;49:W293–6. 10.1093/nar/gkab301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Oksanen J, Simpson G, Blanchet F, et al. Vegan: Community Ecology Package. R package Version 2.6-4, 2022. https://CRAN.R-project.org/package=vegan.
- 52. Chaumeil PA, Mussig AJ, Hugenholtz Pet al. GTDB-Tk: a toolkit to classify genomes with the genome taxonomy database. Bioinformatics 2020;36:1925–7. 10.1093/bioinformatics/btz848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Skennerton CT. MinCED—Mining CRISPRs in Environmental Datasets. 2023. https://github.com/ctSkennerton/minced(cited 8 November 2023).
- 54. Bland C, Ramsey TL, Sabree Fet al. CRISPR recognition tool (CRT): a tool for automatic detection of clustered regularly interspaced palindromic repeats. BMC Bioinformatics 2007;8:209. 10.1186/1471-2105-8-209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Li H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 2018;34:3094–100. 10.1093/bioinformatics/bty191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Varona N, Hesketh-Best P, Stiffler A, et al. Productive viral infections in oligotrophic marine waters. Res Sq [Preprint]. 2023;1–56. Available from: https://www.researchsquare.com/article/rs-3040647/v1, cited 16 January 2024.
- 57. Silveira CB, Coutinho FH, Cavalcanti GSet al. Genomic and ecological attributes of marine bacteriophages encoding bacterial virulence genes. BMC Genomics 2020;21:126. 10.1186/s12864-020-6523-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Brüssow H, Canchaya C, Hardt WD. Phages and the evolution of bacterial pathogens: from genomic rearrangements to lysogenic conversion. Microbiol Mol Biol Rev 2004;68:560–602. 10.1128/mmbr.68.3.560-602.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Grundy WN. Homology detection via family pairwise search. J Comput Biol 1998;5:479–91. 10.1089/cmb.1998.5.479 [DOI] [PubMed] [Google Scholar]
- 60. Guy L, Kultima JR, Andersson SGE. genoPlotR: comparative gene and genome visualization in R. Bioinformatics 2010;26:2334–5. 10.1093/bioinformatics/btq413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Shannon P, Markiel A, Ozier Oet al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 2003;13:2498–504. 10.1101/gr.1239303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Quinn RA, Whiteson K, Lim YWet al. Ecological networking of cystic fibrosis lung infections. NPJ Biofilms Microbi 2016;2:1–11. 10.1038/s41522-016-0002-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Weynberg KD, Wood-Charlson EM, Suttle CAet al. Generating viral metagenomes from the coral holobiont. Front Microbiol 2014;5:206. 10.3389/fmicb.2014.00206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Calendar R. The Bacteriophages. New York (USA): Oxford University Press, 2006, 761. [Google Scholar]
- 65. Conceição-Neto N, Zeller M, Lefrère Het al. Modular approach to customise sample preparation procedures for viral metagenomics: a reproducible protocol for virome analysis. Sci Rep 2015;5:16532. 10.1038/srep16532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Kleiner M, Hooper LV, Duerkop BA. Evaluation of methods to purify virus-like particles for metagenomic sequencing of intestinal viromes. BMC Genomics 2015;16:7. 10.1186/s12864-014-1207-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Karlsson OE, Belák S, Granberg F. The effect of preprocessing by sequence-independent, single-primer amplification (SISPA) on metagenomic detection of viruses. Biosecur Bioterror 2013;11:S227–34. 10.1089/bsp.2013.0008 [DOI] [PubMed] [Google Scholar]
- 68. Messyasz A, Rosales SM, Mueller RSet al. Coral bleaching phenotypes associated with differential abundances of nucleocytoplasmic large DNA viruses. Front Mar Sci 2020;7:555474. 10.3389/fmars.2020.555474 [DOI] [Google Scholar]
- 69. Cárdenas A, Raina JB, Pogoreutz Cet al. Greater functional diversity and redundancy of coral endolithic microbiomes align with lower coral bleaching susceptibility. ISME J 2022;16:2406–20. 10.1038/s41396-022-01283-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Rosales SM, Huebner LK, Clark ASet al. Bacterial metabolic potential and micro-eukaryotes enriched in stony coral tissue loss disease lesions. Front Mar Sci 2022;8:776859. 10.3389/fmars.2021.776859 [DOI] [Google Scholar]
- 71. Robbins SJ, Singleton CM, Chan CXet al. A genomic view of the reef-building coral Porites lutea and its microbial symbionts. Nat Microbiol 2019;4:2090–100. 10.1038/s41564-019-0532-4 [DOI] [PubMed] [Google Scholar]
- 72. Sun F, Yang H, Wang Get al. Combination analysis of metatranscriptome and metagenome reveal the composition and functional response of coral symbionts to bleaching during an El Niño event. Front Microbiol 2020;11:448. 10.3389/fmicb.2020.00448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Levin RA, Voolstra CR, Weynberg KDet al. Evidence for a role of viruses in the thermal sensitivity of coral photosymbionts. ISME J. 2017;11:808–12. 10.1038/ismej.2016.154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Lima LFO, Weissman M, Reed Met al. Modeling of the coral microbiome: the influence of temperature and microbial network. MBio 2020;11:e02691–19. 10.1128/mBio.02691-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Kemp DW, Thornhill DJ, Rotjan RDet al. Spatially distinct and regionally endemic Symbiodinium assemblages in the threatened Caribbean reef-building coral Orbicella faveolata. Coral Reefs 2015;34:535–47. 10.1007/s00338-015-1277-z [DOI] [Google Scholar]
- 76. Boeckman J, Korn A, Yao Get al. Sheep in wolves’ clothing: temperate T7-like bacteriophages and the origins of the Autographiviridae. Virology 2022;568:86–100. 10.1016/j.virol.2022.01.013 [DOI] [PubMed] [Google Scholar]
- 77. Breitbart M, Thompson L, Suttle Cet al. Exploring the vast diversity of marine viruses. Oceanography 2007;20:135–9. 10.5670/oceanog.2007.58 [DOI] [Google Scholar]
- 78. Ankrah NYD, May AL, Middleton JLet al. Phage infection of an environmentally relevant marine bacterium alters host metabolism and lysate composition. ISME J. 2014;8:1089–100. 10.1038/ismej.2013.216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Murphy J, Mahony J, Ainsworth Set al. Bacteriophage orphan DNA methyltransferases: insights from their bacterial origin, function, and occurrence. Appl Environ Microbiol 2013;79:7547–55. 10.1128/AEM.02229-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Stern A, Sorek R. The phage-host arms-race: shaping the evolution of microbes. BioEssays 2011;33:43–51. 10.1002/bies.201000071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Manzer HS, Brunetti T, Doran KS. Identification of a DNA-cytosine methyltransferase that impacts global transcription to promote group B streptococcal vaginal colonization. MBio 2023;14:e0230623. 10.1128/mbio.02306-23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Oren A. The Order Halanaerobiales, and the Families Halanaerobiaceae and Halobacteroidaceae. The Prokaryotes: Firmicutes and Tenericutes, 2014, 153–77. [Google Scholar]
- 83. Ricci F, Rossetto Marcelino V, Blackall LLet al. Beneath the surface: community assembly and functions of the coral skeleton microbiome. Microbiome 2019;7:159. 10.1186/s40168-019-0762-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Hughes ER, Winter MG, Duerkop BAet al. Microbial respiration and formate oxidation as metabolic signatures of inflammation-associated dysbiosis. Cell Host Microbe 2017;21:208–19. 10.1016/j.chom.2017.01.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Siozios S, Ioannidis P, Klasson Let al. The diversity and evolution of Wolbachia ankyrin repeat domain genes. PLoS One 2013;8:e55390. 10.1371/journal.pone.0055390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Mosavi LK, Cammett TJ, Desrosiers DCet al. The ankyrin repeat as molecular architecture for protein recognition. Prot Sci 2004;13:1435–48. 10.1110/ps.03554604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Jahn MT, Arkhipova K, Markert SMet al. A phage protein aids bacterial symbionts in eukaryote immune evasion. Cell Host Microbe 2019;26:542–550.e5. 10.1016/j.chom.2019.08.019 [DOI] [PubMed] [Google Scholar]
- 88. Habyarimana F, Al-khodor S, Kalia Aet al. Role for the Ankyrin eukaryotic-like genes of legionella pneumophila in parasitism of protozoan hosts and human macrophages. Environ Microbiol 2008;10:1460–74. 10.1111/j.1462-2920.2007.01560.x [DOI] [PubMed] [Google Scholar]
- 89. Sgro GG, Oka GU, Souza DPet al. Bacteria-killing type IV secretion systems. Front Microbiol 2019;10:1078. 10.3389/fmicb.2019.01078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Wallden K, Rivera-Calzada A, Waksman G. Type IV secretion systems: versatility and diversity in function. Cell Microbiol 2010;12:1203–12. 10.1111/j.1462-5822.2010.01499.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Lima LFO, Alker AT, Papudeshi Bet al. Coral and seawater metagenomes reveal key microbial functions to coral health and ecosystem functioning shaped at reef scale. Microb Ecol 2023;86:392–407. 10.1007/s00248-022-02094-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Zhong KX, Wirth JF, Chan AMet al. Mortality by ribosomal sequencing (MoRS) provides a window into taxon-specific cell lysis. ISME J 2023;17:105–16. 10.1038/s41396-022-01327-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Schaefer AL, Val DL, Hanzelka BLet al. Generation of cell-to-cell signals in quorum sensing: acyl homoserine lactone synthase activity of a purified Vibrio fischeri LuxI protein. Proc Natl Acad Sci 1996;93:9505–9. 10.1073/pnas.93.18.9505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Lindemann A, Pessi G, Schaefer ALet al. Isovaleryl-homoserine lactone, an unusual branched-chain quorum-sensing signal from the soybean symbiont Bradyrhizobium japonicum. PNAS 2011;108:16765–70. 10.1073/pnas.1114125108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Gärtner A, Wiese J, Imhoff JF. Amphritea atlantica gen. Nov., sp. nov., a gammaproteobacterium from the Logatchev hydrothermal vent field. Int J Syst Evol Microbiol 2008;58:34–9. 10.1099/ijs.0.65234-0 [DOI] [PubMed] [Google Scholar]
- 96. Séré M, Wilkinson DA, Schleyer MHet al. Characterisation of an atypical manifestation of black band disease on Porites lutea in the western Indian Ocean. PeerJ 2016;4:e2073. 10.7717/peerj.2073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Ma R, Lai J, Chen Xet al. A novel phage infecting Alteromonas represents a distinct group of siphophages infecting diverse aquatic copiotrophs. mSphere 2021;6:10.1128. 10.1128/mSphere.00454-21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Luo H, Moran MA. Evolutionary ecology of the marine Roseobacter clade. Microbiol Mol Biol Rev 2014;78:573–87. 10.1128/MMBR.00020-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Raina JB, Tapiolas D, Willis BLet al. Coral-associated bacteria and their role in the biogeochemical cycling of sulfur. Appl Environ Microbiol 2009;75:3492–501. 10.1128/AEM.02567-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Kuek FWI, Motti CA, Zhang Jet al. DMSP production by coral-associated bacteria. Front Mar Sci 2022;9:869574. 10.3389/fmars.2022.869574 [DOI] [Google Scholar]
- 101. Fornelos N, Bamford JKH, Mahillon J. Phage-borne factors and host LexA regulate the lytic switch in phage GIL01. J Bacteriol 2011;193:6008–19. 10.1128/JB.05618-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Senaratne RH, De Silva AD, Williams SJet al. 5′-Adenosinephosphosulphate reductase (CysH) protects Mycobacterium tuberculosis against free radicals during chronic infection phase in mice. Mol Microbiol 2006;59:1744–53. 10.1111/j.1365-2958.2006.05075.x [DOI] [PubMed] [Google Scholar]
- 103. Boyd EF. Bacteriophage-encoded bacterial virulence factors and phage-pathogenicity island interactions. Adv Virus Res 2012;82:91–118. [DOI] [PubMed] [Google Scholar]
- 104. Haas W, Shepard BD, Gilmore MS. Two-component regulator of enterococcus faecalis cytolysin responds to quorum-sensing autoinduction. Nature 2002;415:84–7. 10.1038/415084a [DOI] [PubMed] [Google Scholar]
- 105. Weynberg KD, Voolstra CR, Neave MJet al. From cholera to corals: viruses as drivers of virulence in a major coral bacterial pathogen. Sci Rep 2015;5:17889. 10.1038/srep17889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Rubio-Portillo E, Martin-Cuadrado AB, Caraballo-Rodríguez AMet al. Virulence as a side effect of interspecies interaction in Vibrio coral pathogens. MBio 2020;11:e00201–20. 10.1128/mBio.00201-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Howard-Varona C, Lindback MM, Bastien GEet al. Phage-specific metabolic reprogramming of virocells. ISME J 2020;14:881–95. 10.1038/s41396-019-0580-z [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Coral metagenomic sequence data generated for this project are available on NCBI (PRJNA1061506 and PRJNA975592). The code used to conduct this analysis is publicly available on GitHub (https://github.com/Silveira-Lab/Wallace_Coral_Holobiont_Viruses). Assembled viral genomes are available on Figshare: https://doi.org/10.6084/m9.figshare.24968805.v1. Viral genomes with multiple contigs are N-linked. Bacterial MAGs are available on Figshare: https://doi.org/10.6084/m9.figshare.25000160.v1.