

Abstract
Objectives
Purine-rich element-binding protein alpha (PURA) regulates gene expression and is ubiquitously expressed with an enrichment in neural tissues. Pathogenic variants in PURA cause the neurodevelopmental disorder PURA syndrome that has a variable phenotype but typically comprises moderate-to-severe global developmental delay, intellectual disability, early-onset hypotonia and hypothermia, epilepsy, feeding difficulties, movement disorders, and subtle facial dysmorphism. Speech is reportedly absent in most, but the specific linguistic phenotype is not well described.
Methods
We used genome sequencing to identify a pathogenic gene variant as part of a study of children ascertained for severe primary speech disorder in the absence of moderate or severe ID.
Results
The novel PURA c.296G>T (p.Arg99Leu) pathogenic missense variant segregated in the female proband and her affected mother. The proband had dysarthria; phonological disorder; and severe receptive and expressive language impairment, borderline intellect, attention difficulties, oropharyngeal dysmotility, and dysmorphic facial features. Her mother had dysarthria, moderate receptive language impairment, and borderline intellect. Both the proband and her mother completed mainstream schooling with classroom support.
Discussion
This is the first inherited PURA pathogenic germline variant in over 600 unrelated families documented on ClinVar or reported in the literature. PURA testing should be considered in families with primary speech disorder and borderline intellectual disability, given the specific genetic counseling implications.
Introduction
Pathogenic variants in PURA (encoding purine-rich element-binding protein A; MIM#600473) have been associated with an autosomal dominant form of neurodevelopmental disorder often presenting with neonatal onset hypotonia, respiratory insufficiency, and feeding difficulties (NEDRIHF; MIM#616158).1,2 Over 600 individuals with PURA syndrome have been reported on ClinVar3 or in the literature, but the detailed clinical phenotype and genotype-phenotype correlations have been delineated for a relatively small number of cases (n = 54).2 Evaluation of clinical phenotypes from 32 newly identified individuals and 22 previously published individuals with PURA pathogenic variants revealed that all had moderate-to-severe intellectual disability and neonatal-onset symptoms including hypotonia (96%), respiratory problems (57%), feeding difficulties (77%), exaggerated startle response (44%), hypersomnolence (66%), and hypothermia (35%).2 Other commonly observed features included epilepsy, which is often drug resistant (54%), and gastrointestinal (69%), ophthalmologic (51%), and endocrine problems (42%).2 No significant genotype-phenotype correlations between variant classes and disease severity were identified.2 Notably, speech and language disorder phenotypes were not formally assessed, although “absence of speech” was documented for individuals with 4 recurrent PURA pathogenic variants (p.Leu54Cysfs*24, p.Lys97Glu, p.Phe233del, p.Arg245Pro) in this2 and other studies.1,4,5 Many of these findings were more recently replicated in other cohorts.6-9 PURA pathogenic variants documented on ClinVar3 or reported in the literature,1,2,4-9 from over 600 individuals arose de novo, except one transmitted from an unaffected father with low-level mosaicism.10
We describe an Australian family with a mother-child duo with primary speech disorder, borderline intellect, oropharyngeal dysmotility, and facial dysmorphism without other common or severe features of PURA syndrome.10 This study is the first to describe a primary speech disorder and borderline intellect as part of the milder phenotypic spectrum associated with PURA-related disease due to a transmitted pathogenic germline variant.
Methods
The child of this mother-child duo was a proband recruited to a speech gene discovery study of 70 trios ascertained for primary severe speech disorder (childhood apraxia of speech or dysarthria).11
Standard Protocol Approvals, Registrations, and Patient Consents
The study was approved by the Human Research Ethics Committee of the Royal Children's Hospital, Melbourne, Australia (#37353).
Written informed consent was obtained from the parents for themselves and their children.
Sample Collection and DNA Extraction
Venous blood or saliva was obtained from 4 family members and genomic DNA extracted using a Qiagen (Valencia, CA) QIAamp DNA Maxi kit (for blood) or a DNA Genotek Inc (Ontario, Canada) prepIT L2P kit (for saliva).
Chromosomal Microarray
The proband underwent chromosomal microarray testing on an Illumina (SanDiego, CA) Infinium Global Screening Array platform with a reportable resolution for copy number variants down to 200 Kb. Results were analyzed with KaryoStudio software version 1.3 or 1.4 (Illumina), using genome reference sequence NCBI36/hg18 (v1.3, pre-2013) or GRCh37/hg19 (v1.4, 2013 onward).
Genome Sequencing
The proband and her mother were genome sequenced using Illumina NovaSeq PE150 PCR-free library preparation on the Illumina NovaSeq 6000 to average 30-fold depth with approximately100-Gb data generated per sample. 150-bp sequence pair-end reads were mapped to the hg19 reference genome using the Burrow-Wheeler Aligner (BWA-MEM, bwa v0.7.17) and called and analyzed genome-wide as described previously.11
PCR and Sanger Sequencing
PURA was PCR amplified using specific primers designed to the reference human gene transcript (NCBI Gene). Primer sequences are available on request. Amplification reactions were cycled using a standard protocol on a Veriti Thermal Cycler (Applied Biosystems, Carlsbad, CA). Bidirectional sequencing of all exons and flanking intronic regions including splice sites was completed with a BigDyeTM v3.1 Terminator Cycle Sequencing Kit (Applied Biosystems), according to the manufacturer's instructions. Sequencing products were resolved using a 3730xl DNA Analyzer (Applied Biosystems).
Structural Analysis and Prediction
For the in silico analysis of Arg99Leu mutation, we used a recently published X-ray structure of human PURA I-II fragment (PDB ID: 8CHT)12 and the Drosophila PURA-DNA co-complex (PDB ID: 5FGP).13 The analysis was conducted with the program Coot v 0.9.8.7.14 The figures were prepared with the program Pymol v1.3.15
Data Availability
The PURA variant has been submitted to the LOVD database. Sequencing data are available from the corresponding authors on reasonable request.
Results
Genetic Analysis
No pathogenic copy number variants were detected in the proband by chromosomal microarray. We detected a novel pathogenic missense variant—PURA c.296G>T (p.Arg99Leu)—heterozygous in the proband (III:1) and her mother (II:2) (Figure, A) by genome sequencing analysis. The p.Arg99Leu variant was not present in large public databases including gnomAD, was predicted damaging by PolyPhen (score 0.984) and deleterious by SIFT (score 0), and had a high CADD score (score 28.7). It was classified as Class 4 Likely Pathogenic (PM2, PP1, PP2, PP3) according to ACMG guidelines. There were no other variants of interest on genome-wide analysis. Segregation analysis by Sanger sequencing revealed that the PURA p.Arg99Leu variant was not present in the proband's unaffected grandfather (I:1) or grandmother (I:2) (Figure, B), suggesting it arose de novo in the proband's mother (II:2).
Figure. Pedigree of Australian Family and Segregation of the PURA Variant.
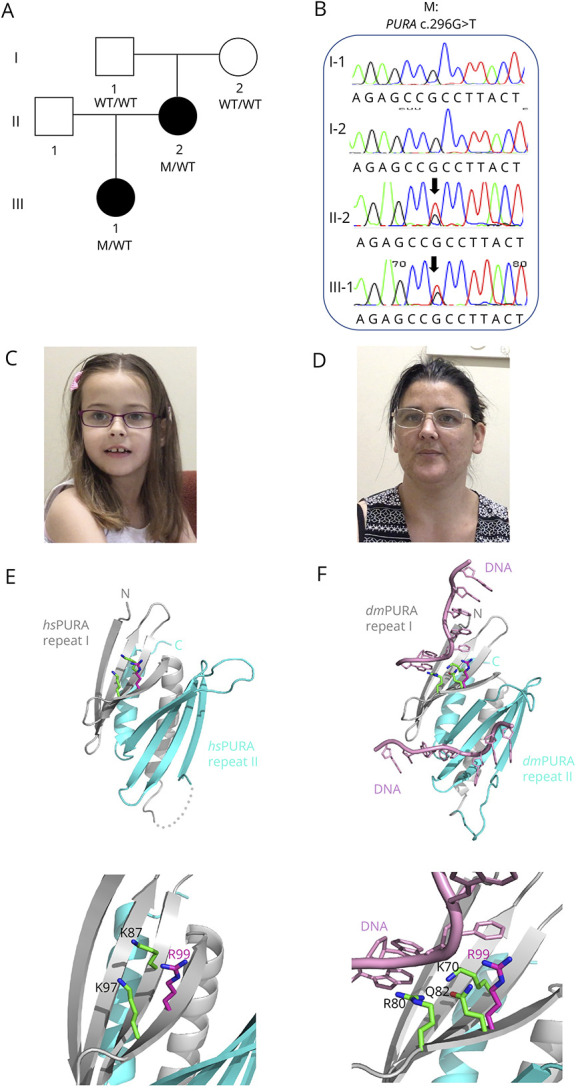
(A) The pedigree of the Australian family showing genotypes based on sequencing analyses. (B) Segregation of PURA c.296G>T (p.Arg99Leu) missense variant by Sanger sequencing. (C and D) Photographs of proband and her mother showing pointed chin and long face. (E) Ribbon model of the nucleic acid–binding domain of human PURA (PDB ID: 8CHT12; amino acids 57–212; repeats I and II shown in grey and cyan, respectively). Side chains of R99 (magenta) and K97 (green) and K87 (green) are shown as sticks. The lower panel in (C) shows magnification of the region of interest. (F) The potential influence of R99 on the interaction with the nucleic acids. The figure shows the X-ray structure of Drosophila PURA (PDB ID: 5FGP13; amino acids 40–183; repeats I and II shown in grey and cyan, respectively) with bound DNA (shown in pink). The amino acids involved in the direct interaction with DNA (K70 and R80) are shown as green sticks. The residue Q82 in Drosophila (green) and its human equivalent R99 (magenta) are shown as well. The position of R99 has been calculated from the superposition of the X-ray structure of human PURA on the Drosophila experimental model. The lower panel in (D) shows magnification of the region of interest.
Phenotypic Analysis
The proband is a 13-year-old adolescent girl born to nonconsanguineous parents (Figure, A). She was born at 38 weeks after an uncomplicated pregnancy. No neonatal concerns were reported except for occasional sleep apnea. The proband was ascertained on the basis of severe speech sound disorder at 7 years of age. She received a diagnosis of dysarthria and phonological disorder (Table 1). It was reported that her first words were delayed (15–18 months of age). She first formed short sentences between 2 and 3 years of age. She had a severe receptive and expressive language impairment as assessed on the Clinical Evaluation of Language Fundamentals 4 (Table 2).16 Comprehensive cognitive assessment could not be completed; however, her perceptual reasoning index was borderline (standard score = 78), and she received a highly supported educational program at mainstream primary and secondary school (Table 2). The proband was reported to have abnormal gait and hypotonia. First steps were at age older than 16 months. She did not have any issues with excessive drooling. Fussy eating was reported by her family, and oropharyngeal dysmotility affected her swallowing function as confirmed by videofluoroscopy, although she did not require any dietary modifications (Table 1). Brain MRI revealed no clinical findings. She had strabismus and a slightly triangular face and pointed chin (Figure, C).
Table 1.
Medical and Neurodevelopmental Features of Family Members
| Family member | Age (y; m) | Sex | Core speech phenotype | Gross motor delay | Fine motor delay | Vision | Hearing loss | MRI findings | Seizures | Other NDDs | Dysmorphic features | Other medical |
| 1:1 | M | N | N | N | N | N | N | N | N | N | N | |
| I:2 | F | N | N | N | N | N | N | N | N | N | N | |
| II:2 | F | Dysarthria | Y | Y | Glasses | N | N | N | N | Pointed china, long facea | N | |
| III:1 | F | Dysarthria phonological disorder | Y | Y | Glasses | N | N | N | Attention difficulties | Pointed china, long facea | Oropharyngeal dysmotility |
F = female; M = male; N = feature not present; NDD = neurodevelopmental disorder; Y = feature present.
Dysmorphic features previously reported in the literature in association with the relevant gene.
Table 2.
Linguistic Phenotypes and Educational Setting of Family Members
| Family member | Oral motor impairment | History of feeding issues | Language: receptivea | Language: expressivea | Reading deficits | Spelling deficits | Speech pathology | IQ | Education setting |
| 1:1 | N | N | N | N | N | N | N | ||
| I:2 | N | N | N | N | N | N | N | Normal intellect | Mainstream |
| II:2 | Y | Y | Moderate | NA | Y | Y | Y | Borderline (PVT4 72) | Mainstream (with classroom support) |
| III:1 | Y | Y | Severe | Severe | Y | Y | Y | Borderline (PRI 78) | Mainstream (with classroom support) |
NA = not assessed; PRI = perceptual reasoning index; Y = feature present.
Language severity rated according to CELF-4 as follows: 86–114 average, 78–85 mild, 71–77 moderate, and <70 severe.
The proband's mother was assessed at 38 years of age (Figure, A). She received a diagnosis of dysarthria and had a moderate receptive language impairment (PPVT = 74) (Table 1 and 2). She was suspected to have borderline intellect and underwent a highly supported educational program at mainstream secondary school (Table 2). Brain MRI revealed no clinical findings. She had some mild gait issues but did not have hip dysplasia or scoliosis. She also had a slightly triangular face and pointed chin (Figure, D).
In Silico Protein Structure Analysis
The substitution of a positively charged arginine side chain for a hydrophobic leucine side chain (p.R99L) occurs in the PURA nucleic acid–binding domain (amino acids 57–212) close to other reported mutations (e.g., p.K97E).1,2 In the experimental model of human PURA,12 R99 is surface-exposed (magenta amino acid with side chain, Figure, E). This surface mutation located on the β-stand changes the local charge of the protein surface but, due to smaller size, should not cause steric clashes in the neighboring side chains. However, R99 is located very close to K97 (green), which on mutation to K97E, results in the full spectrum of PURA syndrome.1,2
To find out whether the R99L mutation could potentially affect PURA's nucleic acid binding, we analyzed the corresponding residues in the previously published costructure of Drosophila PURA with DNA.13 In this experimental X-ray structure, residue R80 (equivalent to K97 in human PURA) is directly involved in the interaction with the base of DNA (Figure, F). Equivalent to R99 in the Drosophila PURA is Q82, which is close to the base of DNA but does not interact directly (distance 6.2 Å). Given the fact that arginine is a larger side chain than glutamine, it seems reasonable that, in human PURA, R99 supports nucleic acid binding, albeit to a minor extent. Therefore, we speculate that a replacement by leucine would have only a minor effect, which is compatible with the mild symptoms of the corresponding proband and her mother (Tables 1 and 2).
Discussion
The findings in our family are not consistent with previous studies (reviewed in Ref. 2) reporting de novo dominant PURA mutations in individuals with classical PURA syndrome symptoms including moderate-to-severe intellectual disability. It was suggested in one study that identification of one individual by targeted Sanger sequencing of PURA pointed toward the clinical recognizability of PURA syndrome.2 However, based on the clinical spectrum defined in this study,2 other cohort studies,6-9 and a recent GeneReviews,10 targeted sequencing of PURA in our proband and her mother would not have been warranted. Although pathogenic PURA missense variants have been reported in numerous studies,1,2,6-9 this is the first report of a transmitted germline pathogenic missense variant in a PURA-related disorder. While, in previous cohort studies,2,6-9 genotype-phenotype analysis showed no significant correlation between variant class and disease severity, our data suggest that inherited missense variants are associated with an atypically mild form of PURA syndrome (Table 1 and 2, Figure, C and D), albeit based on only one family and variant.
While severe neonatal and early-onset symptoms are common and well recognized in PURA-related disorder,10 speech and language abilities are less commonly formally assessed, although “absent speech” has been documented in several individuals.1,2,4,5 Unfortunately, the term absent speech does not delineate whether children are truly nonverbal or minimally verbal or whether their communication difficulties are due to severe speech motor or cognitive-linguistic presentations. With this study, we expand the reported cases to include a family with an inherited missense variant and considerably milder neurodevelopmental features including both a motor speech profile of dysarthria and cognitive-linguistic deficits characterized by both phonological disorder and a broader receptive and expressive language impairment (Tables 1 and 2). Given the relatively small cohorts of individuals with PURA syndrome available, we expect the inclusion of such milder symptoms to improve diagnosis and management of PURA syndrome. Because the phenotypes of PURA syndrome have been reported as difficult to recognize in daily clinical practice,2 formal assessment of speech and language disorder and consideration of mild intellectual disability and family history are likely to enhance clinical diagnosis. Strikingly, some features of the milder inherited syndrome—gastrointestinal disorder (oropharyngeal dysmotility) and facial dysmorphism (Table 1, Figure, C and D)—are also commonly observed in individuals with classical PURA syndrome.
Our family shows that a PURA pathogenic variant may be inherited and that variants in PURA should be considered in a broader spectrum of phenotypes including speech disorder and borderline intellect. Recognition of this diagnosis will also inform reproductive counseling because the recurrence risk is 50%.
Acknowledgment
The authors thank the family for their participation in the study and PURA Foundation Australia for their support.
Appendix. Authors
| Name | Location | Contribution |
| Michael S. Hildebrand, PhD | Epilepsy Research Centre, Department of Medicine, The University of Melbourne, Austin Health, Heidelberg; Neuroscience Group, Murdoch Children’s Research Institute, Royal Children’s Hospital, Parkville, Victoria, Australia | Drafting/revision of the manuscript for content, including medical writing for content; major role in the acquisition of data; study concept or design; analysis or interpretation of data |
| Ruth O. Braden, PhD | Speech and Language, Murdoch Children’s Research Institute, Royal Children’s Hospital, Parkville; Department of Audiology and Speech Pathology, The University of Melbourne, Carlton, Victoria, Australia | Major role in the acquisition of data; analysis or interpretation of data |
| Mariana L. Lauretta, MSP | Speech and Language, Murdoch Children’s Research Institute, Royal Children’s Hospital, Parkville; Department of Audiology and Speech Pathology, The University of Melbourne, Carlton, Victoria, Australia | Analysis or interpretation of data |
| Antony Kaspi, PhD | Population Health and Immunity Division, The Walter and Eliza Hall Institute of Medical Research; Department of Medical Biology, The University of Melbourne, Parkville, Victoria, Australia | Analysis or interpretation of data |
| Richard J. Leventer, MBBS, PhD | Neuroscience Group, Murdoch Children’s Research Institute; Department of Paediatrics, The University of Melbourne; Department of Neurology, Royal Children’s Hospital, Parkville, Victoria, Australia | Study concept or design; analysis or interpretation of data |
| Melinda Anderson | PURA Foundation Australia Ltd, Plenty, Victoria, Australia | Analysis or interpretation of data |
| Himanshu Goel, MBBS, MD | Hunter Genetics, John Hunter Hospital, New Lambton Heights, New South Wales, Australia | Analysis or interpretation of data |
| Melanie Bahlo, PhD | Population Health and Immunity Division, The Walter and Eliza Hall Institute of Medical Research; Department of Medical Biology, The University of Melbourne, Parkville, Victoria, Australia | Analysis or interpretation of data |
| Ingrid E. Scheffer, MBBS, PhD | Epilepsy Research Centre, Department of Medicine, The University of Melbourne, Austin Health, Heidelberg; Neuroscience Group, Murdoch Children’s Research Institute; Department of Paediatrics, The University of Melbourne, Royal Children’s Hospital; The Florey Institute, Parkville, Victoria, Australia | Drafting/revision of the manuscript for content, including medical writing for content; analysis or interpretation of data |
| David J. Amor, MD | Department of Paediatrics, The University of Melbourne; Neurodisability and Rehabilitation Group, Murdoch Children’s Research Institute, Royal Children’s Hospital, Parkville, Victoria, Australia | Drafting/revision of the manuscript for content, including medical writing for content; analysis or interpretation of data |
| Robert Janowski, PhD | Institute of Structural Biology, Helmholtz Zentrum Muenchen – German Research Centre for Environmental Health, Neuherberg, Germany | Analysis or interpretation of data |
| Dierk Niessing, PhD | Institute of Structural Biology, Helmholtz Zentrum Muenchen – German Research Centre for Environmental Health, Neuherberg, Germany | Drafting/revision of the manuscript for content, including medical writing for content; major role in the acquisition of data; study concept or design; analysis or interpretation of data |
| Angela T. Morgan, PhD | Speech and Language, Murdoch Children’s Research Institute, Royal Children’s Hospital, Parkville; Department of Audiology and Speech Pathology, The University of Melbourne, Carlton, Victoria, Australia | Drafting/revision of the manuscript for content, including medical writing for content; major role in the acquisition of data; study concept or design; analysis or interpretation of data |
Study Funding
This work was made possible through the Victorian State Government Operational Infrastructure Support and Australian Government National Health and Medical Research Council (NHMRC) independent research Institute Infrastructure Support Scheme (IRIISS). M. Bahlo was supported by an NHMRC Investigator Grant (1195236). I.E. Scheffer was supported by a NHMRC Investigator Grant (1172897). D. Niessing was supported by the Science Award of the Care-for-Rare Foundation. A.T. Morgan was supported by a NHMRC Investigator Grant (1195955). M.S. Hildebrand, I.E. Scheffer, D.J. Amor., and A.T. Morgan were supported by a NHMRC Centre of Research Excellence Translational Centre for Speech Disorders Grant (2015757).
Disclosure
I.E. Scheffer has served on scientific advisory boards for BioMarin, Chiesi, Eisai, Encoded Therapeutics, GlaxoSmithKline, Knopp Biosciences, Nutricia, Rogcon, Takeda Pharmaceuticals, UCB, Xenon Pharmaceuticals, Cerecin; has received speaker honoraria from GlaxoSmithKline, UCB, BioMarin, Biocodex, Chiesi, Liva Nova, Nutricia, Zuellig Pharma, Stoke Therapeutics and Eisai; has received funding for travel from UCB, Biocodex, GlaxoSmithKline, Biomarin, Encoded Therapeutics, Stoke Therapeutics and Eisai; has served as an investigator for Anavex Life Sciences, Cerevel Therapeutics, Eisai, Encoded Therapeutics, EpiMinder Inc, Epygenyx, ES-Therapeutics, GW Pharma, Marinus, Neurocrine BioSciences, Ovid Therapeutics, Takeda Pharmaceuticals, UCB, Ultragenyx, Xenon Pharmaceuticals, Zogenix and Zynerba; and has consulted for Care Beyond Diagnosis, Epilepsy Consortium, Atheneum Partners, Ovid Therapeutics, UCB, Zynerba Pharmaceuticals, BioMarin, Encoded Therapeutics and Biohaven Pharmaceuticals; and is a Non-Executive Director of Bellberry Ltd and a Director of the Australian Academy of Health and Medical Sciences and the Australian Council of Learned Academies Limited. She may accrue future revenue on pending patent WO61/010176 (filed: 2008): Therapeutic Compound; has a patent for SCN1A testing held by Bionomics Inc and licensed to various diagnostic companies; has a patent molecular diagnostic/theranostic target for benign familial infantile epilepsy (BFIE) [PRRT2] 2011904493 and 2012900190 and PCT/AU2012/001321 (TECH ID:2012-009). The remaining authors declare no competing interests. Go to Neurology.org/NG for full disclosures.
References
- 1.Lalani SR, Zhang J, Schaaf CP, et al. Mutations in PURA cause profound neonatal hypotonia, seizures, and encephalopathy in 5q31.3 microdeletion syndrome. Am J Hum Genet. 2014;95(5):579-583. doi: 10.1016/j.ajhg.2014.09.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Reijnders MRF, Janowski R, Alvi M, et al. PURA syndrome: clinical delineation and genotype-phenotype study in 32 individuals with review of published literature. J Med Genet. 2018;55(2):104-113. doi: 10.1136/jmedgenet-2017-104946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.NCBI ClinVar. Accessed July 30, 2023. ncbi.nlm.nih.gov/clinvar/
- 4.Tanaka AJ, Bai R, Cho MT, et al. De novo mutations in PURA are associated with hypotonia and developmental delay. Cold Spring Harb Mol Case Stud. 2015;1(1):a000356. doi: 10.1101/mcs.a000356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hunt D, Leventer RJ, Simons C, et al. Whole exome sequencing in family trios reveals de novo mutations in PURA as a cause of severe neurodevelopmental delay and learning disability. J Med Genet. 2014;51(12):806-813. doi: 10.1136/jmedgenet-2014-102798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Johannesen KM, Gardella E, Gjerulfsen CE, et al. PURA-related developmental and epileptic encephalopathy: phenotypic and genotypic spectrum. Neurol Genet. 2021;7(6):e613. doi: 10.1212/NXG.0000000000000613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Choi SA, Lee HS, Park TJ, et al. Expanding the clinical phenotype and genetic spectrum of PURA-related neurodevelopmental disorders. Brain Dev. 2021;43(9):912-918. doi: 10.1016/j.braindev.2021.05.009 [DOI] [PubMed] [Google Scholar]
- 8.Dai W, Sun Y, Fan Y, et al. A 25 Mainland Chinese cohort of patients with PURA-related neurodevelopmental disorders: clinical delineation and genotype-phenotype correlations. Eur J Hum Genet. 2023;31(1):112-121. doi: 10.1038/s41431-022-01217-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lee BH, Reijnders MRF, Abubakare O, et al. Expanding the neurodevelopmental phenotype of PURA syndrome. Am J Med Genet A. 2018;176(1):56-67. doi: 10.1002/ajmg.a.38521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Reijnders MRF, Leventer RJ, Lee BH, et al. PURA-related neurodevelopmental disorders. 2017. In: Adam MP, Mirzaa GM, Pagon RA, et al., eds. GeneReviews® [Internet]. University of Washington, Seattle; 1993-2023. [PubMed] [Google Scholar]
- 11.Kaspi A, Hildebrand MS, Jackson VE, et al. Correction: genetic aetiologies for childhood speech disorder: novel pathways co- expressed during brain development. Mol Psychiatry. 2023;28(4):1664-1666. doi: 10.1038/s41380-022-01879-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Proske M, Janowski R, Bacher S, et al. PURA syndrome-causing mutations impair PUR- domain integrity and affect P-body association. Elife. 2024;13:RP93561. doi: 10.7554/eLife.93561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Weber J, Bao H, Hartlmüller C, et al. Structural basis of nucleic-acid recognition and double-strand unwinding by the essential neuronal protein Pur-alpha. Elife. 2016;5(5):e11297. doi: 10.7554/eLife.11297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60(Pt 12 Pt 1):2126-2132. doi: 10.1107/S0907444904019158 [DOI] [PubMed] [Google Scholar]
- 15.Schrödinger L, DeLano W. PyMOL; 2020. Accessed February 15, 2024. pymol.org/pymol [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The PURA variant has been submitted to the LOVD database. Sequencing data are available from the corresponding authors on reasonable request.