

Abstract
Aberrant signaling in tumor cells induces nonmetabolic functions of some metabolic enzymes in many cellular activities. As a key glycolytic enzyme, the nonmetabolic function of hexokinase 2 (HK2) plays a role in tumor immune evasion. However, whether HK2, dependent of its nonmetabolic activity, plays a role in human pancreatic ductal adenocarcinoma (PDAC) tumorigenesis remains unclear. Here, we demonstrated that HK2 acts as a protein kinase and phosphorylates IκBα at T291 in PDAC cells, activating NF‐κB, which enters the nucleus and promotes the expression of downstream targets under hypoxia. HK2 nonmetabolic activity‐promoted activation of NF‐κB promotes the proliferation, migration, and invasion of PDAC cells. These findings provide new insights into the multifaceted roles of HK2 in tumor development and underscore the potential of targeting HK2 protein kinase activity for PDAC treatment.
Keywords: HK2, IκBα, nonmetabolic activity, pancreatic ductal adenocarcinoma, tumor progression
HK2‐mediated phosphorylation of IκBα at T291 in PDAC cells leads to IκBα degradation and subsequent activation of NF‐κB for the upregulation of downstream target transcription. HK2 nonmetabolic activity‐promoted activation of NF‐κB stimulates the proliferation, migration, and invasion of PDAC cells.
1. INTRODUCTION
Hexokinases (HKs) are conserved enzymes that catalyze the first step of glycolysis to produce the metabolic intermediate glucose‐6‐phosphate (G6P). Mammals have four HK isozymes: hexokinase 1 (HK1), hexokinase 2 (HK2), hexokinase 3 (HK3), and hexokinase 4 (HK4). 1 , 2 Among these, HK1 and HK2, which bind to the mitochondrial outer membrane through an N‐terminal motif absent in other isoforms, have a high affinity for glucose. 3 HK1 and HK2 have different kinetic properties and structures contributing to their function and regulation. HK1 is highly expressed in all adult tissues, whereas HK2 is expressed only in adult muscle and heart tissues and cells derived from embryos. 4 HK2 is often overexpressed in tumor cells and is considered important for maintaining cancer malignancy. 5 , 6 In particular, the latest research showed that the functional importance of HK2 extends beyond glucose phosphorylation. HK2 is also a protein kinase for IκBα and stimulates the activation of NF‐κB and upregulation of programmed death‐ligand 1 (PD‐L1) expression to regulate tumor immune evasion in glioblastoma (GBM) cells. 7 , 8 In addition, the nonmetabolic function of HK2 promotes small‐cell lung cancer stemness by increasing ubiquitin‐specific protease 11 (USP11)‐mediated CD133 stability. 9 From the published structural information of HK2, the D209/D657A mutant lost its ability to bind glucose and thus to convert glucose to G6P. 10 It was recently reported that HK2 acted as a protein kinase to phosphorylate IkBa, and the HK2 S340/S788A mutant lost the protein kinase activity probably by impairing the transfer of γ‐phosphate of ATP to the protein substrate. 7 However, whether HK2, dependent of its nonmetabolic activity, plays a role in human pancreatic ductal adenocarcinoma (PDAC) tumorigenesis is unclear.
Pancreatic ductal adenocarcinoma, the main type of primary pancreatic cancer, is a common malignant tumor of the digestive system and is predicted to become the second leading cause of cancer‐related deaths by 2030. 11 Hypoxia, a characteristic of the cancer microenvironment, occurs in most malignancies and affects carcinogenesis and tumorigenesis. The rapid proliferation of PDAC, abundant fibrosis, and extracellular matrix deposition can easily cause oxidative stress and gradually form a hypoxic microenvironment. 12 , 13 Enhanced HK2‐mediated glycolysis to support tumor cell growth in a hypoxic microenvironment is found in PDAC cells. 14 Selective pharmacological inhibition of glycolysis by inactivating HK2 does not significantly slow or inhibit PDAC progression. 15 More importantly, studying the HK2 nonmetabolic functions may offer new perspectives on PDAC treatment.
The nuclear factor kappa B (NF‐κB) family of transcription factors directly and indirectly plays essential roles in multiple physiological and pathological processes. 16 There are two general types of NF‐κB signaling pathways: the classical (canonical) and alternative (noncanonical), with different activating mechanisms. 17 , 18 Notably, NF‐κB activation is also prevalent in carcinomas, in which NF‐κB activation is mainly driven by cytokines in the tumor microenvironment. 19 , 20 The activation of NF‐κB is regulated by the canonical pathway in response to cytokines resulting from IκB kinase (IKK)‐mediated phosphorylation of IκBα at S32 and S36, leading to IκBα polyubiquitylation and proteasome‐dependent degradation. We recently showed that HK2‐mediated IκBα phosphorylation at T291 promotes the binding of IκBα to m‐calpain and its subsequent degradation by m‐calpain in response to high glucose. 7 However, the precise mechanism of how PDAC hypoxia microenvironment regulates NF‐κB activation has been a hitherto long‐standing question with no clear answer.
In this study, we demonstrated that HK2 is highly expressed in PDAC specimens and positively correlated with lymph node involvement, advanced stages of PDAC, and low survival rates in patients with PDAC. Exposure of PDAC cells to hypoxia induces HK2 transcription and mitochondrial dissociation. HK2 nonmetabolic functions‐mediated IκBα T291 phosphorylation, IκBα degradation, and NF‐κB activation, thereby promoting PDAC cell proliferation, migration, and invasion.
2. MATERIALS AND METHODS
2.1. Materials
Antibody against Flag (F1804) was obtained from Sigma. Antibodies against HK2 (ab209847), α‐tubulin (ab210797), HIF‐1α (ab51608), β‐actin (ab8224), Ki67 (ab92742), and p65 (ab32536) were obtained from Abcam. Antibodies against Lamin B (12255), c‐Myc (#18583), COX IV (4850 T), IκBα (4814) were purchased from Cell Signaling Technology. Antibody against IκBα pT291 was purchased from Signalway Biotechnology. CCK‐8 kit was purchased from DOJINDO. Anti‐FLAG® M2 Beads, EDTA‐free Protease Inhibitor Cocktail, and PhosSTOP were purchased from Sigma. 3‐Bromopyruvate (3‐BP), JSH‐23, and puromycin were purchased from Selleck. GN44028 was purchased from MCE.
2.2. Cell lines and transfection
Human pancreatic cancer cell lines (PANC‐1, SW1990, and AsPC‐1) were purchased from the Cell Bank of the Chinese Science Academy. HPDE cells were obtained from American Type Culture Collection (ATCC). HPDE and PANC‐1 cells were maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (HyClone). SW1990 and AsPC‐1 cells were maintained in RPMI 1640 medium supplemented with 10% fetal bovine serum (HyClone). All cell lines were routinely monitored for mycoplasmas and tested negative.
Transfection was performed using Lipofectamine 3000 transfection reagent (Invitrogen) according to the manufacturer's instructions. Stable cell lines with HK2 knocked down or HK2 mutation were produced as previously described. 21 In brief, HK2 gene was depleted through HK2 targeting shRNA‐containing pLKO.1 lentiviral transfection in PDAC cells, while RNAi‐resistant HK2 gene was reconstituted within the lentivector carrying HK2 or mutant (D209/D657A or S340/S788A) plasmid in PDAC cells using lentiviral infections.
2.3. DNA constructs and mutagenesis
Polymerase chain reaction (PCR)‐amplified human HK2 was cloned into a p3‐BPti/puro(+)‐3 Flag vector. rHK2 WT, HK2 D209/D657A, and HK2 S340/S788A (kinase‐dead mutant) were generated using the QuikChange site‐directed mutagenesis kit. All sequences are listed in Table S1. An shRNA‐targeting HK2 (5′‐GGATGTGTGTGAACATGGAAT‐3′) was inserted into the pLKO.1 vector.
2.4. Hypoxia exposure
Hypoxia experiments were performed in a sealed hypoxia chamber (Billups‐Rothenburg) filled with 1% O2, 5% CO2, and balanced N2 at 37°C for the indicated periods.
2.5. Transwell assay
For the transwell assay, pancreatic cancer cells were seeded in the upper chamber in a serum‐free medium. The chamber was covered or uncovered with Matrigel (Corning) to investigate cell invasion or migration, respectively. The lower chamber was filled with 500 μL of medium containing 10% FBS. After 24 h (migration assay) or 30 h (invasion assay) of incubation, penetrating cells were stained with 4% methyl alcohol for 20 min and then with 0.1% crystal violet solution. Noninvading cells were wiped off with a cotton swab. Membranes were randomly photographed using an optic microscope.
2.6. Immunoblotting analysis
Proteins were extracted using radioimmunoprecipitation assay (RIPA) buffer containing protease inhibitor and PhosSTOP phosphatase inhibitor cocktails (Roche). Proteins were separated using sodium dodecyl sulfate‐polyacrylamide gel electrophoresis (SDS‐PAGE) and transferred to polyvinylidene fluoride (PVDF) membranes. After blocking, membranes were probed with various primary antibodies overnight at 4°C, followed by incubation with horseradish peroxidase (HRP)‐linked secondary antibodies for 1 h at room temperature. Finally, the signals were detected using the electrochemiluminescence ECL method.
2.7. Reverse‐transcription and quantitative RT‐PCR
Total RNA was extracted using TRIzol reagent (TaKaRa) and reverse transcribed with Maxima Reverse Transcriptase (Thermo Fisher Scientific) following the manufacturer's instructions. cDNAs were quantified by quantitative RT‐PCR using SYBR Green dye (TaKaRa). The results were normalized to GAPDH mRNA levels. The RT‐PCR primer sequences are listed in Table S2.
2.8. Proliferation assays
For the cell counting kit 8 (CCK8) assay, 5000 cells were plated in 96‐well plates. Then 10 μL of CCK8 solution was added to each well, followed by incubation for 30 min. Cell viability was determined by measuring absorbance at 450 nm. Colony formation assays were performed by seeding cells at a 500‐cells/well density and incubating them for 2 weeks. Then, cells were washed in phosphate‐buffered saline (PBS), fixed in 4% paraformaldehyde, and stained with 0.1% crystal violet. Colonies were photographed and counted (each colony contained at least 50 cells).
2.9. Subcellular fractionation
Mitochondrial and cytosolic fractions were isolated using a mitochondria/cytosol fractionation kit (Abcam) according to the manufacturer's protocol. Nuclear and cytosolic fractions were extracted using a nucleoprotein extraction kit (Sangon, China) according to the manufacturer's protocol.
2.10. Immunofluorescence (IF) analysis
Briefly, cells grown on chambered coverslips were washed with PBS, fixed with 4% paraformaldehyde, and permeabilized with 0.25% Triton X‐100 for 15 min. The cells were then blocked with 5% bovine serum albumin (BSA) for 1 h. Cells were probed with indicated antibodies overnight at 4°C. The cells were then washed, and secondary antibodies with specific fluorescent dyes were added. Finally, the nuclei were stained with a DAPI solution. Images were acquired using a microscope (Leica).
2.11. Luciferase reporter assay
NF‐κB activity was measured using a luciferase assay system as previously described. 22 Briefly, cells at a density of 50%–70% confluence in 24‐well plates were cotransfected with NF‐κB activity reporter luciferase plasmids, pRL‐TK vector (a plasmid encoding Renilla luciferase), and other plasmids using Lipofectamine 3000 (Invitrogen). After transfection, the cells were cultured under hypoxic or normoxic conditions for 12 h. Luciferase activity was measured using a Dual‐Luciferase Reporter Assay kit (Promega) with a luminometer, and the ratio of firefly to Renilla luciferase was determined.
2.12. Immunohistochemical (IHC) staining and scoring
All PDAC speciments were paraffin embedded and collected from the Beijing Luhe Hospital. Paraffin‐embedded xenograft tissue sections were stained with an antibody against Ki67 or nonspecific immunoglobulin G (IgG) as a negative control. IHC staining was performed using the VECTASTAIN ABC kit (Vector Laboratories) according to the manufacturer's instructions. We quantitatively scored the tissue sections according to the percentage of positive cells (1, 0%–25%; 2, 26%–50%; 3, 51%–75%; and 4, >75%) and staining intensity (0, no staining; 1, weak staining; 2, moderate staining; and 3, strong staining). These numbers were then multiplied, resulting in a score of 0–12 as described previously. 23
Detailed clinicopathological information was obtained according to the tumor classification of the American Joint Committee on Cancer staging system. Detailed information on the patient characteristics is presented in Table S3.
2.13. Mice
Male nude mice, 6‐week‐old (n = 6 for each group), were injected with 3 × 106 gene‐modified PANC‐1 cells in a volume of 100 μL of PBS. Injections were administered subcutaneously into the left axilla. The tumor volume was calculated using the formula V = ab 2/2, where a represents the base diameter of the tumor and b represents the corresponding perpendicular plate.
The mice were treated in accordance with relevant institutional and national guidelines and regulations. The use of animals was approved by the Institutional Animal Care and Use Committee of the Capital Medical University. No blinding was done. Mice were randomly placed in separate group cages for the experiments.
2.14. Hexokinase 2 activity assay
The activity of HK2 WT and HK2 S340/S788A mutant was tested by employing the method as previously described. 24 In brief, PANC‐1 cells with depleted HK2 and reconstituted expression of rWT or rHK2 S340/S788A cultured in 60‐mm dishes were harvested with 400 uL lysis buffer (50 mM potassium phosphate, 2 mM EDTA, 2 mM dithiothreitol, and 20 mM sodium fluoride) and homogenized at 4°C. The homogenate was centrifuged at 18,000 g at 4°C for 15 min, and 40 uL of supernatant was added to 1 mL of reaction buffer (100 mM Tris–HCl (pH 8.0), 0.5 mM EDTA, 10 mM ATP, 10 mM MgCl2, 2 mM glucose, 0.2 mM NADP+, and 30 ug of G6PD). HK2 activity was determined by following the G6PD dependent conversion of NADP to NADPH spectrophotometrically at 340 nm at 37°C. HK2 activities of different samples in the same experiment were normalized to the protein contents in the lysates.
2.15. Measurements of glucose consumption and lactate production
For the glucose consumption, 2000 cells were cultured in 96‐well plates, and the medium was changed after 6 h with no‐serum DMEM. Cells were treated with or without 3‐BP (25 μM) for 24 h. The cells were then collected for measurement of glucose concentrations. Glucose levels were determined using a glucose (GO) assay kit (ab136955) according to the manufacturer's protocol. A lactate assay kit (ab65330) was used to determine lactate levels according to the manufacturer's protocol.
3. RESULTS
3.1. HK2 is highly expressed in PDAC and correlated with poor prognosis of patients with PDAC
The GEPIA database was used to determine HK2 expression in PDAC and normal tissues. HK2 expression was higher in PDAC tissues (red box) than in normal tissues (gray box) (Figure S1A). To study the effect of HK2 on the malignant phenotype of patients with PDAC, we investigated the relationship between HK2 expression and overall survival (OS) and disease‐free survival (DFS) of patients with PDAC in the GEPIA database. High HK2 expression was associated with worse OS and DFS in patients with PDAC in the GEPIA database (Figure S1B,C).
Considering the prognostic value of HK2 in the database, we performed IHC analyses of 60 primary PDAC tumor specimens and their adjacent normal tissues using an anti‐HK2 antibody. We observed that HK2 expression levels were substantially higher in PDAC samples than in adjacent normal tissues (Figure 1A,B). We then analyzed the correlation between HK2 and the clinical features of patients with PDAC. HK2 levels were positively correlated with lymph node involvement and advanced TNM stage in PDAC (p < 0.001) (Figure 1C). We also assessed the survival duration of 60 patients with PDAC corresponding to these obtained samples. Kaplan–Meier analysis showed that high HK2 expression was significantly associated with decreased OS durations of the patients (Figure 1D). Consistent results showed that HK2 expression levels in AsPC‐1, PANC‐1, and SW1990 human PDAC cells were higher than those in normal human pancreatic duct epithelial (HPDE) cells (Figure 1E). These results demonstrate that HK2 is highly expressed in PDAC cells and positively correlated with the clinical aggressiveness of human PDAC.
FIGURE 1.
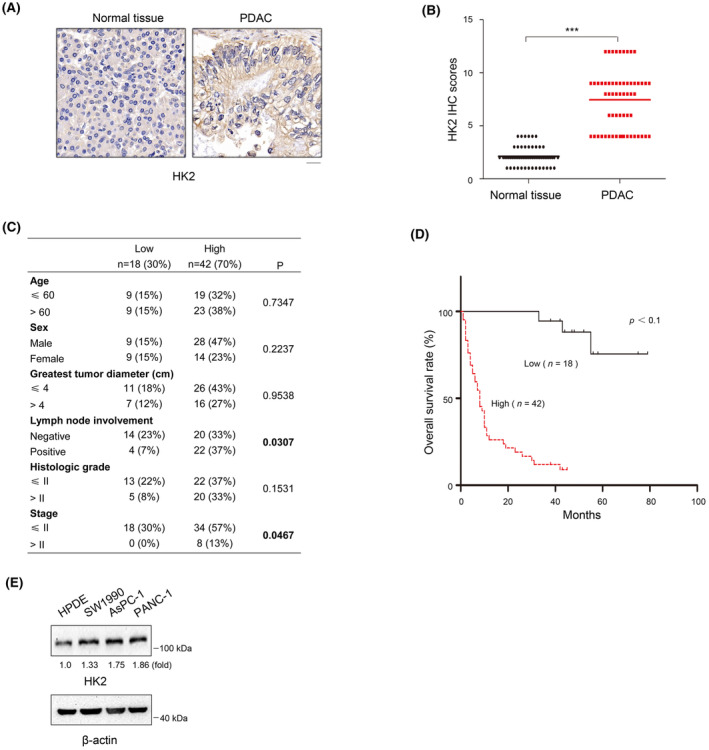
HK2 is highly expressed in PDAC and correlated with poor prognosis of patients with PDAC. (A) Expression of HK2 in 60 samples of human PDAC tissues and matched adjacent normal tissues by immunohistochemical (IHC) staining with an anti‐HK2 antibody. Representative images are shown. Scale bars, 20 μm. (B) Comparative analysis of HK2 expression between PDAC tissues and matched adjacent normal tissues. ***p < 0.001. (C) Correlations between HK2 expression levels and PDAC clinicopathological parameters. (D) Kaplan–Meier plots and p‐values of the log‐rank test for comparing survivals of PDAC patients with high (staining score, 5–12) and low (staining score, 0–4) expression of HK2. (E) Lysates of the indicated cells were prepared. Immunoblot analyses were performed with the indicated antibodies.
3.2. HK2 promotes PDAC cell proliferation, migration, and invasion
Metabolic enzymes possess noncanonical functions critical for many cellular activities, such as cancer stemness and gene expression. 9 To determine the role of HK2 in PDAC cell proliferation, migration, and invasion, we depleted HK2 in PANC‐1 and AsPC‐1 cells by expressing three HK2 shRNAs (Figure 2A). The results showed that HK2 depletion inhibited cell proliferation (Figure 2B) and colony formation (Figure 2C). Transwell assays further verified that HK2 depletion reduced the invasive and migratory abilities of PANC‐1 and AsPC‐1 cells (Figure 2D). In addition, treatment of the cells with 3‐BP, a small‐molecule compound that inhibits glycolysis by inactivating HK, inhibited glucose uptake (Figure S2A), lactate production (Figure S2B), cell proliferation (Figure 2B), and colony formation (Figure 2C) and reduced the invasive and migratory abilities of PANC‐1 and AsPC‐1 cells (Figure 2D). These results indicated that HK2 promoted the proliferation, migration, and invasion of PDAC cells.
FIGURE 2.
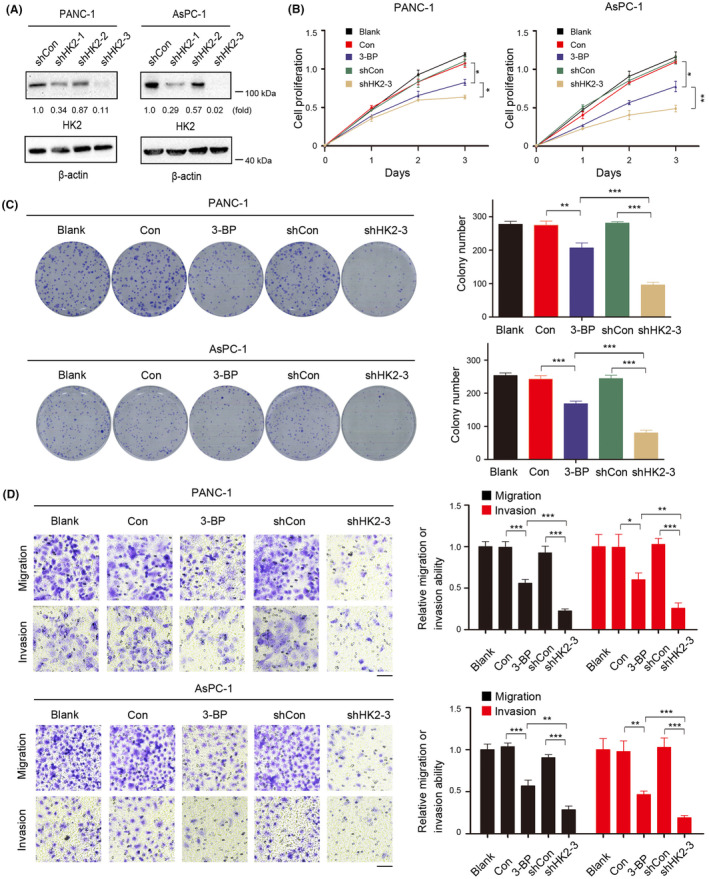
HK2 promotes pancreatic ductal adenocarcinoma (PDAC) cell proliferation, migration, and invasion. (A) HK2 was depleted in the indicated cells by expressing HK2 shRNA. (B) Indicated cells were cultured in the presence or absence of 3‐bromopyruvate (3‐BP) (25 μM) for the indicated time. Cell proliferation was examined using a Cell Counting Kit‐8 (CCK‐8) assay. Data are presented as the means ± SD from three independent experiments (n = 3). *p < 0.05; **p < 0.01. (C) Indicated PANC‐1 and AsPC‐1 cells were plated for 2 weeks in the presence or absence of 3‐BP (25 μM) before counting colony numbers. Data are presented as the means ± SD from three independent experiments (n = 3). **p < 0.01; ***p < 0.001. (D) The migration and invasion of the indicated cells in the presence or absence of 3‐BP (25 μM) were examined by transwell assay. The membrane was photographed using a digital camera mounted onto a microscope. Scale bars, 50 μm. Data are presented as mean ± SD *p < 0.05; **p < 0.01; ***p < 0.001.
3.3. Hypoxia promotes the transcription and the mitochondrial dissociation of HK2
Hypoxia regulates glycolysis; therefore, a connection between hypoxia/HIF‐1α and HK2 in PDAC may exist. Using The GEPIA database, we found that HIF‐1α was positively correlated with HK2 expression in PDAC (Figure S3A). In addition, we examined HK2 expression in PDAC cells following hypoxic stimulation. Western blot analysis of PANC‐1 and AsPC‐1 human PDAC cells demonstrated that hypoxia increased HK2 protein levels in a time‐dependent manner (Figure 3A). RT‐PCR analyses of PDAC cells indicated that enhanced hypoxia increased the HK2 mRNA levels (Figure 3B). Next, treatment of PDAC cells with GN44028, a small‐molecule compound that inhibits HIF‐1α transcriptional activity, decreased HK2 mRNA and protein expression, as demonstrated by RT‐PCR (Figure 3B) and Western blot analyses (Figure 3C).
FIGURE 3.
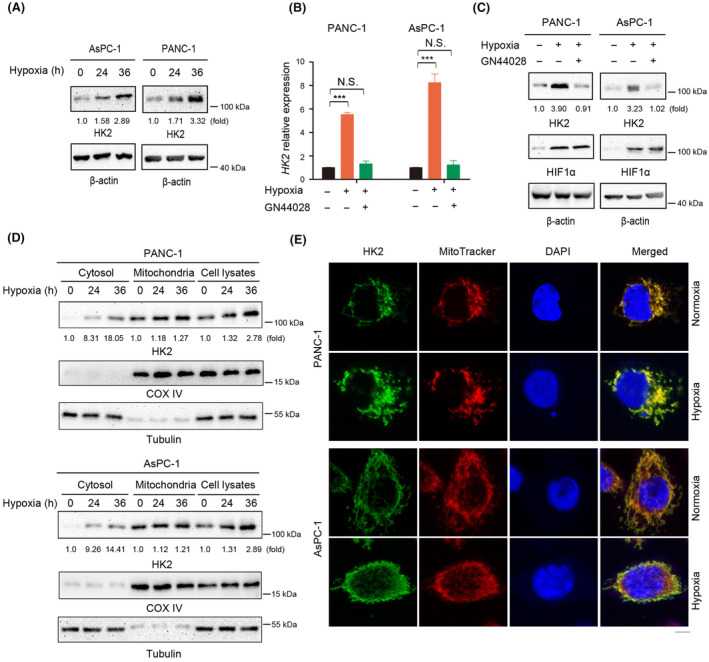
Hypoxia promotes the transcription and the mitochondrial dissociation of HK2. (A, C, D) Immunoblot analyses were performed with the indicated antibodies. (A) PANC‐1 and AsPC‐1 cells were cultured under normoxic or hypoxic condition for the indicated time. (B, C) PANC‐1 and AsPC‐1 cells were cultured under normoxic or hypoxic condition for 24 h in the absence or presence of GN44028 (20 nM). RT‐PCR (B) and immunoblot analyses (C) were performed. N.S., not significant for the indicated comparison; ***p < 0.001. (D) Mitochondrial and cytosolic fractions of PANC‐1 and AsPC‐1 cells cultured under normoxic or hypoxic condition for 24 h were prepared. (E) PANC‐1 and AsPC‐1 cells were stimulated with or without hypoxia for 24 h. Immunofluorescence analyses were performed with an anti‐HK2 antibody. Scale bars, 5 μm.
The dynamic movement of HK2 between the cytosolic compartments and mitochondria is regulated by various factors, including physiological concentrations of ATP, G6P, and intracellular pH. 3 , 25 , 26 To determine whether hypoxia influences the translocation of HK2 from mitochondria to the cytosol, we examined the cellular distribution of HK2 in PANC‐1 and AsPC‐1 cells. Cell fractionation analysis showed that hypoxia induced HK2 translocation from mitochondria to the cytosol (Figure 3D). In line with this finding, IF analyses of PANC‐1 and AsPC‐1 human PDAC cells demonstrated a hypoxia‐induced cytosol accumulation of HK2 (Figure 3E). These results suggest that hypoxia promotes the transcription and dissociation of HK2 from the mitochondria.
3.4. Hypoxia promotes NF‐κB activation dependent on an HK2‐mediated IκBα T291 phosphorylation
Metabolic enzymes exhibit protein kinase activities. We showed that in human GBM cells, high glucose promotes HK2 dissociation from the mitochondria and the subsequent binding and phosphorylation of IκBα at T291, leading to increased IκBα degradation and NF‐κB activation. 7 In addition, hypoxia has recently been reported to trigger NF‐κB activation in GBM cells. 27 Hypoxia induced the nuclear translocation of p65, with a corresponding decrease in cytosolic IκBα expression in PANC‐1 and AsPC‐1 cells, as demonstrated by cell fractionation analyses (Figure 4A). IF analyses of PANC‐1 and AsPC‐1 cells demonstrated a hypoxia‐induced nuclear accumulation of p65 (Figure 4B). Additionally, the luciferase reporter assay results showed that hypoxia induced the transcriptional activity of NF‐κB. The NF‐κB activation was notably inhibited by HK2 depletion but was not affected by the 3‐BP treatment (Figure 4C).
FIGURE 4.
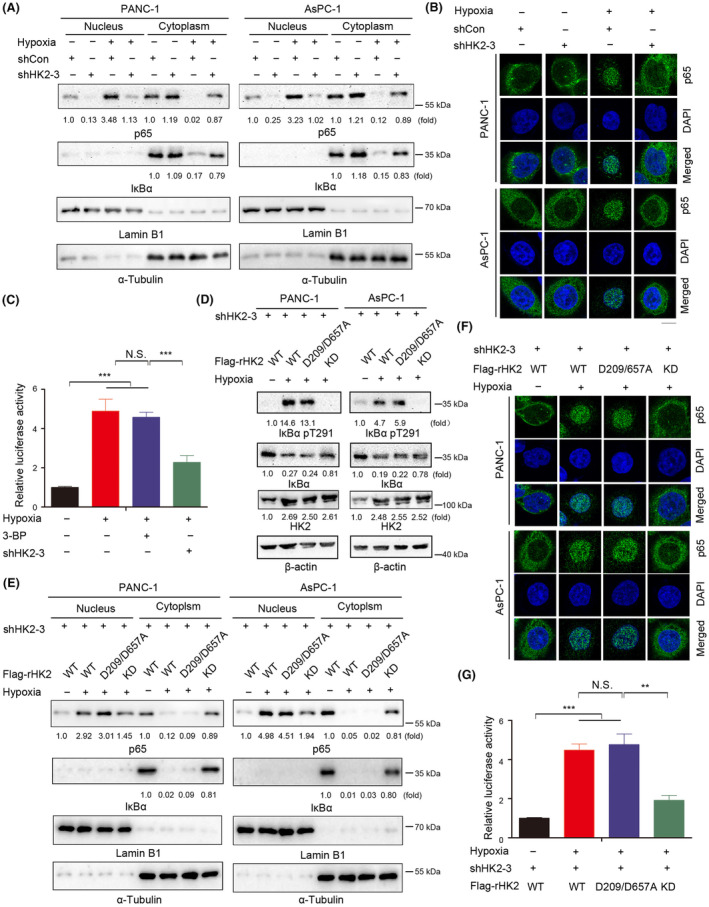
Hypoxia promotes NF‐κB activation dependent on a HK2‐mediated IκBα T291 phosphorylation. (A, D, E) Immunoblot analyses were performed with the indicated antibodies. (A, B) PANC‐1 and AsPC‐1 cells with or without HK2 depletion were cultured for 24 h under normoxia or hypoxia. Cytosolic and nuclear fractions of the indicated cells were prepared (A). PANC‐1 and AsPC‐1 cells with or without HK2 depletion were stimulated with or without hypoxia for 24 h. Immunofluorescence analyses were performed with an anti‐p65 antibody (B). Scale bars, 10 μm. (C) PANC‐1 cells with or without HK2 depletion were cultured in the absence or presence of 3‐bromopyruvate (3‐BP) (25 μM). The indicated cells were transfected with luciferase reporter plasmid (NF‐κB‐Luc) and stimulated with or without hypoxia for 12 h. The data are presented as the mean ± SD of triplicate samples. N.S., not significant for the indicated comparison; ***p < 0.001. (D) PANC‐1 and AsPC‐1 cells with depleted HK2 and reconstituted expression of WT rHK2, rHK2 D209/D657A, or KD were cultured with or without hypoxia for 24 h. Cytosolic and nuclear fractions of the indicated cells were prepared. (E, F) PANC‐1 cells with depleted HK2 and reconstituted expression of WT rHK2, rHK2 D209/D657A, or KD were cultured with or without hypoxia for 24 h. Cytosolic and nuclear fractions of the indicated cells were prepared (E). Immunofluorescence analyses were performed with an anti‐p65 antibody. Scale bars, 10 μm. (G) WT rHK2, rHK2 D209/D657A, or KD was expressed in PANC‐1 cells with the depletion of HK2. The indicated cells were transfected with luciferase reporter plasmid (NF‐κB‐Luc) and stimulated with or without hypoxia for 12 h. The data are presented as the mean ± SD of triplicate samples. N.S., not significant for the indicated comparison; **p < 0.01; ***p < 0.001.
To determine whether hypoxia‐induced transactivation of NF‐κB was mediated by HK2‐mediated phosphorylation of IκBα, we expressed Flag‐WT rHK2, glucose‐catalytic‐dead mutant rHK2 D209/D657A (that lost its ability to bind glucose), or protein kinase‐dead (KD) mutant rHK2 S340/S788A (that lost its ability to phosphorylate IkBα) in PANC‐1 and AsPC‐1 cells with depletion of endogenous HK2 (Figure S4A). To test whether the HK2 KD mutant has some effect on the metabolic activity of HK2, we performed the enzyme activity assay and showed that the HK2 KD mutant also largely reduced the metabolic activity of HK2 to convert glucose to G6P (Figure S4B). WT rHK2 and rHK2 D209/D657A expression induced IκBα T291 phosphorylation in HK2‐depleted PANC‐1 and AsPC‐1 cells under hypoxic conditions. However, the rHK2 KD mutant lost its ability to phosphorylate IκBα T291 (Figure 4D). Additionally, HK2‐mediated IκBα T291 phosphorylation decreased IκBα expression (Figure 4D). Consistent with this finding, WT rHK2 and rHK2 D209/D657A expression induced the nuclear translocation of p65, with a corresponding decrease in cytosolic IκBα expression under hypoxia, as detected by Western blot analysis and IF (Figure 4E,F), and activation of the transcriptional activity of NF‐κB, as detected by a luciferase reporter assay (Figure 4G). The HK2 D209/D657A mutant that lost its ability to bind glucose retained its ability to activate NF‐κB under hypoxic conditions, whereas the rHK2 KD mutant clearly reduced this effect. These results indicated that hypoxia promotes NF‐κB activation via HK2‐mediated IκBα T291 phosphorylation.
3.5. NF‐κB inhibitor abrogates HK2 nonmetabolic function‐induced proliferation, migration, and invasion of PDAC cells in vitro
The NF‐κB signaling pathway activates downstream targets and plays an important role in several solid cancers, including PDAC. 17 , 28 To further verify whether HK2 nonmetabolic functions promote malignant progression in PDAC through the NF‐κB signaling pathway, we treated PANC‐1 and AsPC‐1 cells after HK2 D209/D657A overexpression with JSH‐23, an inhibitor of the NF‐κB signaling pathway. CCK‐8 and colony formation assays demonstrated that HK2 D209/D657A overexpression promoted the proliferation of PANC‐1 and AsPC‐1 cells; however, this effect was abrogated by JSH‐23 treatment (Figure 5A,B). Furthermore, the transwell assay showed that HK2 D209/D657A overexpression promoted tumor invasion and migration, and the promoting effects were abrogated after JSH‐23 administration (Figure 5C). HK2 D209/D657A overexpression increased the expression levels of genes of NF‐κB downstream, such as c‐Myc and cyclin D1, but this increase was completely abrogated by JSH‐23 treatment (Figure 5D). In addition, WT rHK2 and rHK2 D209/D657A induced c‐Myc and cyclin D1 expression in HK2‐depleted PANC‐1 and AsPC‐1 cells under hypoxic conditions. However, the ability of the rHK2 KD mutant to induce NF‐κB downstream genes was notably weakened (Figure 5E).
FIGURE 5.
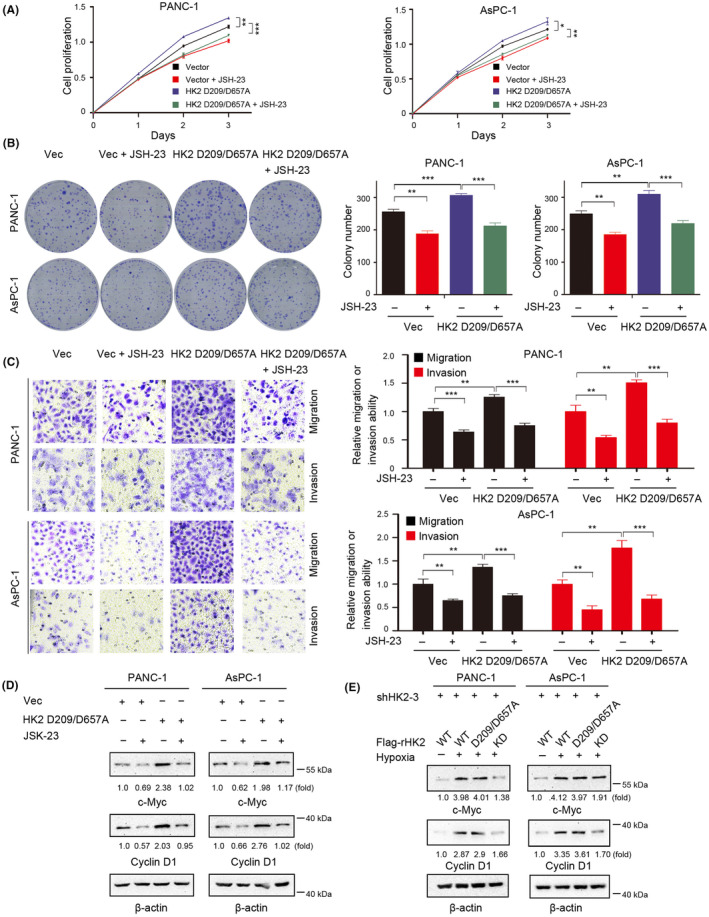
NF‐κB inhibitor abrogates HK2 nonmetabolic function‐induced proliferation, migration, and invasion of pancreatic ductal adenocarcinoma (PDAC) cells in vitro. (D, E) Immunoblot analyses were performed with the indicated antibodies. (A) PANC‐1 and AsPC‐1 with or without HK2 D209/D657A expression were cultured in the presence or absence of JSH‐23 (10 μM) for the indicated time. Cell proliferation was examined using a Cell Counting Kit‐8 (CCK‐8) assay. Data are presented as the means ± SD from three independent experiments (n = 3). *p < 0.05; **p < 0.01; ***p < 0.001. (B) PANC‐1 and AsPC‐1 cells with or without HK2 D209/D657A expression were cultured in the presence or absence of JSH‐23 (10 μM) for 2 weeks before counting colony numbers. Data are presented as the means ± SD from three independent experiments (n = 3). **p < 0.01; ***p < 0.001. (C) The migration and invasion of the indicated cells in the presence or absence of JSH‐23 (10 μM) were examined by transwell assay. The membrane was photographed using a digital camera mounted onto a microscope. Scale bars, 50 μm. Data are presented as mean ± S.D. **p < 0.01; ***p < 0.001. (D) PANC‐1 and AsPC‐1 cells with or without HK2 D209/D657A expression were cultured in the presence or absence of JSH‐23 (10 μM) for 24 h. (E) PANC‐1 and AsPC‐1 cells with depleted HK2 and reconstituted expression of WT rHK2, rHK2 D209/D657A, or KD were cultured with or without hypoxia for 24 h.
3.6. HK2 nonmetabolic functions promote PDAC development in vivo
To better understand the HK2 nonmetabolic functions in PDAC proliferation, PANC‐1 cells with endogenous HK2 depletion and reconstituted expression of WT rHK2, rHK2 D209/D657A or KD were subcutaneously injected into athymic nude mice. We found that tumor growth (Figure 6A), tumor volume (Figure 6B), and tumor weight (Figure 6C) were substantially lower in rHK2 KD than in rHK2 WT or rHK2 D209/D657A. In addition, the Ki67 and IκBα pT291 expressions were substantially less in rHK2 KD than in rHK2 WT or rHK2 D209/D657A (Figure 6D). These results indicated that HK2 nonmetabolic functions promote tumor growth in mice.
FIGURE 6.
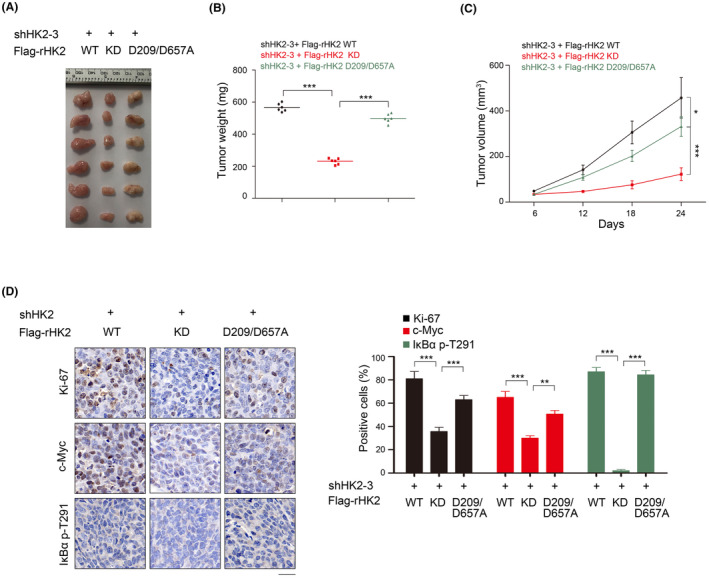
HK2 nonmetabolic functions promote PDAC development in vivo. (A–D) PANC‐1 cells with depleted HK2 and reconstituted expression of WT rHK2, rHK2 D209/D657A, or KD were intracranially injected into athymic nude mice. Tumor growth was examined 21 days after injection. Representative tumor xenografts were shown (n = 6 mice per group) (A). Tumor weights were calculated (B). Tumor growth was measured every other day beginning on day 6, and tumor volumes were calculated (C). Immunohistochemical analyses of tumor sections with the indicated antibodies were performed. Representative images are shown (D). Scale bar, 10 mm. The percentages of Ki67‐, c‐Myc‐, and IκBα pT291‐positive cells were calculated. *p < 0.05; **p < 0.01; ***p < 0.001.
4. DISCUSSION
Metabolism comprises chemical reactions that are ultimately responsible for biological processes; the synthesis of ATP and building blocks for proteins, nucleotides, and lipids; and the elimination of nitrogenous wastes. Metabolic enzymes catalyze these reactions under strict regulation at specific subcellular locations in metabolic cascades. Several metabolic enzymes possess nonmetabolic activities critical for cancer progression. These nonmetabolic activities can be divided into two categories: First, several metabolic enzymes can act as protein kinases and phosphorylate multiple protein substrates, thereby regulating diverse functions. For instance, pyruvate kinase M2 isoform (PKM2), 29 , 30 , 31 phosphoglycerate kinase 1 (PGK1), 32 , 33 phosphoenolpyruvate carboxykinase1 (PCK1), 34 and choline kinase a (CHKa) 35 phosphorylate various protein substrates, promoting oncogenic gene expression and tumor development. Second, several metabolic enzymes can translocate from their original subcellular compartments to different organelles, where metabolite production is directly used to modify or regulate other proteins. For instance, mitochondrial α‐ketoglutarate dehydrogenase (α‐KGDH) translocates to the nucleus and produces succinyl‐coenzyme A, which is used by histone acetyltransferase 2A (KAT2A) to succinylate histone H3 succinylation on lysine 79, with maximum frequency around the transcription‐starting sites of genes. 21 , 36 The nonmetabolic activities of metabolic enzymes make them promising therapeutic targets for cancer treatment.
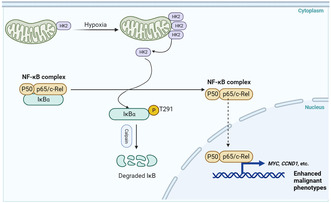
Because the high glucose utilization distinguishes many cancer cells from their normal counterparts, the selective inhibition of cancer‐driven glycolysis has been investigated for clinical cancer therapy, and HK2 has been proposed as a therapeutic target. 15 , 37 Recently, several HK inhibitors have been investigated, including the glucose analogue competitive inhibitor 2‐Deoxy‐D‐glucose (2‐DG) and the catalytic inhibitor 3‐BP, whether they can be used against cancer. 38 2‐DG can sensitize chemoresistant cancer cells to cisplatin in vitro and in vivo; however, its use has been abandoned owing to its severe toxicity during clinical trials. In addition, 3‐BP was ineffective in phase II and III clinical trials. 39 Several possible reasons why they did not observe an increase in cell death after 3‐BP treatment exist. First, the cells express HK1, which is sufficient to protect them against cell death. Second, HK2 regulates critical cellular activities with unidentified noncanonical functions. Here, we showed that HK2, acting as a protein kinase, phosphorylates IκBα at T291 in PDAC cancer cells, leading to IκBα degradation and subsequent activation of NF‐κB for the upregulation of downstream target transcription. HK2 nonmetabolic activity‐promoted activation of NF‐κB stimulates the proliferation, migration, and invasion of PDAC cells (Figure 7). HK2 metabolic enzyme activity and nonmetabolic enzyme activity depend on different sites. Given this, further investigation of the mechanisms underlying the regulation of HK2 function is required so as to better understand how HK2 signaling is kept well tuned in tumor cells.
FIGURE 7.
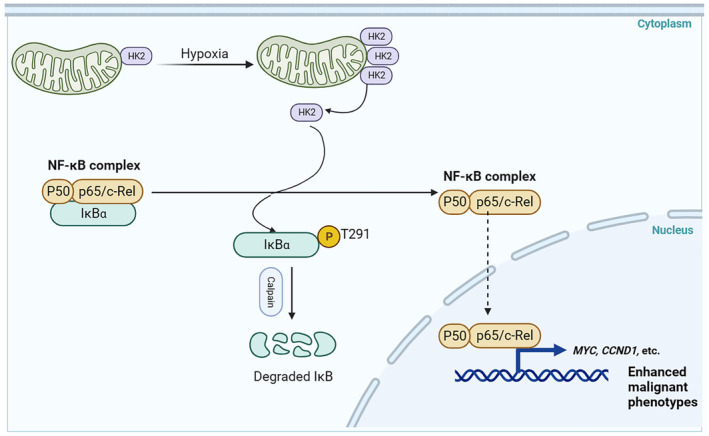
Diagram displaying the regulation of NF‐κB signaling by the nonmetabolic function of HK2. HK2‐mediated phosphorylation of IκBα at T291 in PDAC cells, leading to IκBα degradation and subsequent activation of NF‐κB for the upregulation of downstream target transcription. HK2 nonmetabolic activity‐promoted activation of NF‐κB stimulates the proliferation, migration, and invasion of PDAC cells.
Pancreatic ductal adenocarcinoma is a leading cause of cancer‐related death, and therapeutic interventions are only marginally beneficial. Here, we reveal a novel mechanism underlying the enhanced proliferation, migration, and invasion of PDAC cells, in which NF‐κB activation by HK2‐dependent IκBα phosphorylation plays a critical role. Our study provides a molecular basis for developing a therapeutic strategy against PDAC tumors with aberrant HK2 nonmetabolic activity.
AUTHOR CONTRIBUTIONS
Yingying Tong: Investigation. Xin Liu: Investigation. Lihui Wu: Investigation. Yaoxian Xiang: Investigation. Jing Wang: Investigation. Yurong Cheng: Investigation. Chan Zhang: Investigation. Baojuan Han: Investigation; methodology. Li Wang: Investigation. Dong Yan: Project administration.
FUNDING INFORMATION
This work was supported by grants from the R&D Program of Beijing Municipal Education Commission (no. KM202110025002 to Y.T., KM202010025005 to D.Y.), Beijing Municipal Natural Science Foundation (no. 7222100), and the National Natural Science Foundation of China (no. 82203576).
CONFLICT OF INTEREST STATEMENT
The authors declare no conflict of interest.
ETHICS STATEMENT
Approval of the research protocol by an Institutional Review Board: This study was approved by the Ethics Committee of Beijing Luhe Hospital (Beijing, China).
Informed Consent: All patients involved in this article signed informed consent.
Registry and the Registration No. of the study/trial: N/A.
Animal Studies: All the animal experiments in this study were approved by the Institutional Animal Care and Use Committee (IACUC) of Capital Medical University.
Supporting information
Appendix S1.
ACKNOWLEDGMENTS
We thank Dr. Dong Guo (Zhejiang University School of Medicine, Hangzhou, China) for technical suggestions.
Tong Y, Liu X, Wu L, et al. Hexokinase 2 nonmetabolic function‐mediated phosphorylation of IκBα enhances pancreatic ductal adenocarcinoma progression. Cancer Sci. 2024;115:2673‐2685. doi: 10.1111/cas.16204
Yingying Tong and Xin Liu contributed equally to this work.
REFERENCES
- 1. Hay N. Reprogramming glucose metabolism in cancer: can it be exploited for cancer therapy? Nat Rev Cancer. 2016;16:635‐649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Wilson JE. Isozymes of mammalian hexokinase: structure, subcellular localization and metabolic function. J Exp Biol. 2003;206:2049‐2057. [DOI] [PubMed] [Google Scholar]
- 3. Robey RB, Hay N. Mitochondrial hexokinases, novel mediators of the antiapoptotic effects of growth factors and Akt. Oncogene. 2006;25:4683‐4696. [DOI] [PubMed] [Google Scholar]
- 4. Xu S, Catapang A, Doh HM, et al. Hexokinase 2 is targetable for HK1 negative, HK2 positive tumors from a wide variety of tissues of origin. J Nucl Med. 2018;60:212‐217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Mathupala SP, Ko YH, Pedersen PL. Hexokinase II: cancer's double‐edged sword acting as both facilitator and gatekeeper of malignancy when bound to mitochondria. Oncogene. 2006;25:4777‐4786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ros S, Schulze A. Glycolysis back in the limelight: systemic targeting of HK2 blocks tumor growth. Cancer Discov. 2013;3:1105‐1107. [DOI] [PubMed] [Google Scholar]
- 7. Guo D, Tong Y, Jiang X, et al. Aerobic glycolysis promotes tumor immune evasion by hexokinase2‐mediated phosphorylation of IκBα. Cell Metab. 2022;34:1312‐1324.e6. [DOI] [PubMed] [Google Scholar]
- 8. Fang J, Luo S, Lu Z. HK2: gatekeeping microglial activity by tuning glucose metabolism and mitochondrial functions. Mol Cell. 2023;83:829‐831. [DOI] [PubMed] [Google Scholar]
- 9. Wang J, Shao F, Yang Y, et al. A non‐metabolic function of hexokinase 2 in small cell lung cancer: promotes cancer cell stemness by increasing USP11‐mediated CD133 stability. Cancer Commun. 2022;42:1008‐1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Nawaz MH, Ferreira JC, Nedyalkova L, et al. The catalytic inactivation of the N‐half of human hexokinase 2 and structural and biochemical characterization of its mitochondrial conformation. Biosci Rep. 2018;38:BSR20171666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Rahib L, Smith BD, Aizenberg R, Rosenzweig AB, Fleshman JM, Matrisian LM. Projecting cancer incidence and deaths to 2030: the unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 2014;74:2913‐2921. [DOI] [PubMed] [Google Scholar]
- 12. Yamasaki A, Yanai K, Onishi H. Hypoxia and pancreatic ductal adenocarcinoma. Cancer Lett. 2020;484:9‐15. [DOI] [PubMed] [Google Scholar]
- 13. Park W, Chawla A, O'Reilly EM. Pancreatic cancer: a review. JAMA. 2021;326:851‐862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Guillaumond F, Leca J, Olivares O, et al. Strengthened glycolysis under hypoxia supports tumor symbiosis and hexosamine biosynthesis in pancreatic adenocarcinoma. Proc Natl Acad Sci USA. 2013;110:3919‐3924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ciscato F, Ferrone L, Masgras I, Laquatra C, Rasola A. Hexokinase 2 in cancer: a prima Donna playing multiple characters. Int J Mol Sci. 2021;22:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Pramanik KC, Makena MR, Bhowmick K, Pandey MK. Advancement of NF‐κB signaling pathway: a novel target in pancreatic cancer. Int J Mol Sci. 2018;19:3890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Taniguchi K, Karin M. NF‐kappaB, inflammation, immunity and cancer: coming of age. Nat Rev Immunol. 2018;18:309‐324. [DOI] [PubMed] [Google Scholar]
- 18. Hou Y, Liang H, Rao E, et al. Non‐canonical NF‐kappaB antagonizes STING sensor‐mediated DNA sensing in radiotherapy. Immunity. 2018;49:490‐503.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Karin M, Greten FR. NF‐kappaB: linking inflammation and immunity to cancer development and progression. Nat Rev Immunol. 2005;5:749‐759. [DOI] [PubMed] [Google Scholar]
- 20. Rius J, Guma M, Schachtrup C, et al. NF‐kappaB links innate immunity to the hypoxic response through transcriptional regulation of HIF‐1alpha. Nature. 2008;453:807‐811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Tong Y, Guo D, Yan D, et al. KAT2A succinyltransferase activity‐mediated 14‐3‐3ζ upregulation promotes β‐catenin stabilization‐dependent glycolysis and proliferation of pancreatic carcinoma cells. Cancer Lett. 2020;469:1‐10. [DOI] [PubMed] [Google Scholar]
- 22. Li X, Qian X, Wang B, et al. Programmable base editing of mutated TERT promoter inhibits brain tumour growth. Nat Cell Biol. 2020;22:282‐288. [DOI] [PubMed] [Google Scholar]
- 23. Hirsch FR, Varella‐Garcia M, Bunn PA, et al. Epidermal growth factor receptor in non‐small‐cell lung carcinomas: correlation between gene copy number and protein expression and impact on prognosis. J Clin Oncol. 2003;21:3798‐3807. [DOI] [PubMed] [Google Scholar]
- 24. Zhang J, Wang S, Jiang B, et al. c‐Src phosphorylation and activation of hexokinase promotes tumorigenesis and metastasis. Nat Commun. 2017;8:13732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Shangguan X, He J, Ma Z, et al. SUMOylation controls the binding of hexokinase 2 to mitochondria and protects against prostate cancer tumorigenesis. Nat Commun. 2021;12:1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zuurbier CJ, Eerbeek O, Meijer AJ. Ischemic preconditioning, insulin, and morphine all cause hexokinase redistribution. Am J Physiol Heart Circ Physiol. 2005;289:H496‐H499. [DOI] [PubMed] [Google Scholar]
- 27. Gorlach A, Bonello S. The cross‐talk between NF‐kappaB and HIF‐1: further evidence for a significant liaison. Biochem J. 2008;412:e17‐e19. [DOI] [PubMed] [Google Scholar]
- 28. Gao S, Sun Y, Zhang X, et al. IGFBP2 Activates the NF‐kappaB Pathway to Drive Epithelial‐Mesenchymal Transition and Invasive Character in Pancreatic Ductal Adenocarcinoma. 2016. [DOI] [PMC free article] [PubMed]
- 29. Jiang Y, Wang Y, Wang T, et al. PKM2 phosphorylates MLC2 and regulates cytokinesis of tumour cells. Nat Commun. 2014;5:5566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Liang J, Cao R, Zhang Y, et al. PKM2 dephosphorylation by Cdc25A promotes the Warburg effect and tumorigenesis. Nat Commun. 2016;7:12431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Yang W, Xia Y, Hawke D, et al. PKM2 phosphorylates histone H3 and promotes gene transcription and tumorigenesis. Cell. 2012;150:685‐696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Li X, Zheng Y, Lu Z. PGK1 is a new member of the protein kinome. Cell Cycle. 2016;15:1803‐1804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Qian X, Li X, Lu Z. Protein kinase activity of the glycolytic enzyme PGK1 regulates autophagy to promote tumorigenesis. Autophagy. 2017;13:1246‐1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Xu D, Wang Z, Xia Y, et al. The gluconeogenic enzyme PCK1 phosphorylates INSIG1/2 for lipogenesis. Nature. 2020;580:530‐535. [DOI] [PubMed] [Google Scholar]
- 35. Liu R, Lee JH, Li J, et al. Choline kinase alpha 2 acts as a protein kinase to promote lipolysis of lipid droplets. Mol Cell. 2021;81:2722‐2735.e9. [DOI] [PubMed] [Google Scholar]
- 36. Wang Y, Guo YR, Liu K, et al. KAT2A coupled with the alpha‐KGDH complex acts as a histone H3 succinyltransferase. Nature. 2017;552:273‐277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. DeWaal D, Nogueira V, Terry AR, et al. Hexokinase‐2 depletion inhibits glycolysis and induces oxidative phosphorylation in hepatocellular carcinoma and sensitizes to metformin. Nat Commun. 2018;9:446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Heneberg P. Redox regulation of hexokinases. Antioxid Redox Signal. 2019;30:415‐442. [DOI] [PubMed] [Google Scholar]
- 39. Garcia SN, Guedes RC, Marques MM. Unlocking the potential of HK2 in cancer metabolism and therapeutics. Curr Med Chem. 2019;26:7285‐7322. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1.