

Abstract
In this article we give our perspective on the successes and promise of various molecular and coarse-grained simulation approaches to probing the effect of mechanical forces in the actin cytoskeleton.
1 |. BRIEF OVERVIEW OF FORCES IN THE ACTIN CYTOSKELETON
The actin cytoskeleton is a network of crosslinked polymers in cells that is a major locus of force generation and transmission. 1 Its primary component is the filamentous actin (F-actin) polymer (Fig. 1A), working in concert with numerous actin binding proteins (ABPs) 2;3 that play roles in crosslinking, polymerizing, severing/depolymerizing, or nucleating filaments. 3 Forces on the cytoskeleton can be generated through the action of myosin molecular motors, which are either anchored in place and pull unidirectionally on a filament (as is the case with myosin I) 4 or are combined into a bundle and pull on multiple filaments simultaneously (as in the case of myosin II) 1;5 (Fig. 1B). Actin itself can serve as the origin of force, as the action of polymerizing globular actin (G-actin) from solution can be used to generate pushing forces 6;7;8; this is particularly relevant at the leading edge of cells, where branched actin networks nucleated by Arp2/3 complex are used to propel the cellular plasma membrane forwards (Fig. 1B). 9;10 Generation of forces either through polymerization or motor activity require the conversion of chemical energy to mechanical energy via the hydrolysis and release of ATP molecules. Actin itself in solution contains an ATP molecule, and catalyzes the conversion of this ATP to ADP after undergoing a transition from a twisted to a flat state upon entering into a filament 11;12.
FIGURE 1.
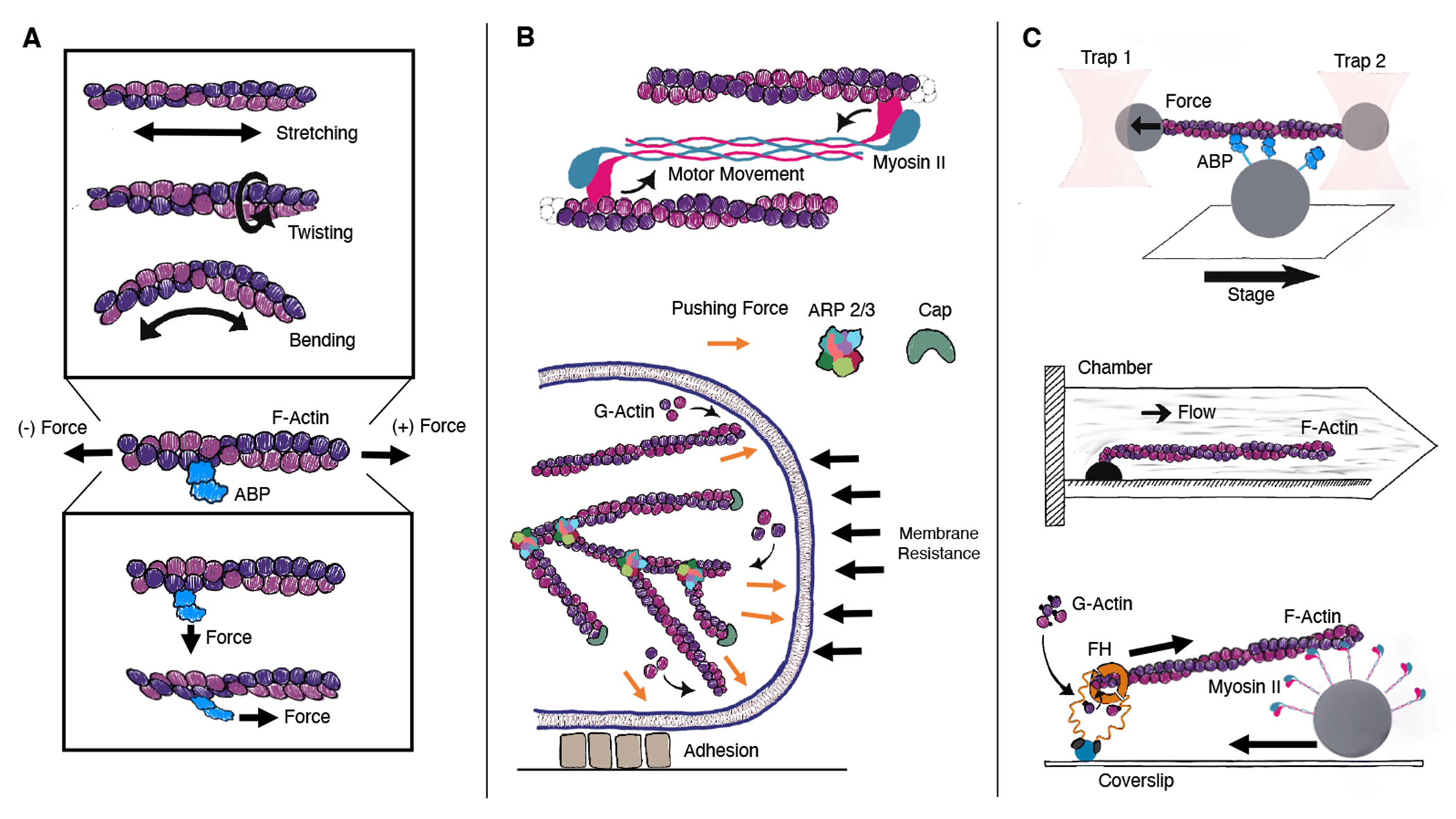
(A) Actin filaments are polar helical non-covalent polymers. Forces applied directly to filaments can cause stretching, twisting, and bending, while forces applied to actin binding proteins (ABPs) perpendicular or parallel can cause stronger or weaker binding. (B) In cells, forces on proteins are caused by motion of motor proteins along filaments, by polymerization of filaments against structures, or through interactions with cellular adhesion proteins. (C) In vitro, the effect of forces on actin and actin binding proteins can be probed by using tweezers, 13 microfluidic devices, 14 or with motors anchored either to substrates or beads. 15
Mechanical forces experienced by actin or actin binding proteins would typically be in the range of zero to several piconewtons (pN). 9 Individual myosin heads are individually capable of producing several piconewtons of force, 16;4;17 meaning that in certain cases, filaments could experience hundreds of piconewtons of tension. 18 Pulling or twisting forces on the pN scale can be translated into down stream signaling effects, meaning it is certainly relevant to be able to predict the molecular affect of these forces on filament structure. 19;20;21;22;23 The effect of forces on the behavior of actin binding proteins in some cases can be probed by in vitro experiments, including those employing molecular tweezers or microfluidic devices to controllably manipulate individual filaments in solution 16;13;24;20 (Fig. 1C). However, it is not possible to observe detailed changes in protein structure while simultaneously applying these forces, and hence here we give our perspective on the use of molecular dynamics simulations to predict the molecular response of small mechanical forces on actin and actin binding proteins.
2 |. INCLUDING FORCE IN MOLECULAR DYNAMICS SIMULATIONS
Molecular dynamics (MD) simulations are a set of techniques where we seek to explicitly model the motion of molecules by treating all the atoms as if they follow the rules of classical mechanics. 25 The intra and intermolecular forces dictating the motion of particles comes from a model for the potential energy of the arrangement of atoms termed a “forcefield.” 26 External mechanical forces can be included through modification of the system’s “Hamiltonian” (the sum of potential and kinetic energy), as
| (1) |
where denotes the positions all atoms in the system, and their momenta.
The contribution of mechanical forces to the term correspond to doing mechanical work, such that for a static pulling force, , where is the coordinate to which pulling is applied, e.g. the distance between two atoms or the distance between the centers of mass of two residues in a protein. 27;28 Experimentally, single-molecule pulling forces can be applied by attaching the molecule of interest to an AFM or an optical or magnetic trap 29; rather than a constant force, this is typically modeled in simulation by introducing a harmonic restraint centered at a distance . 30 In ‘steered molecular dynamics’ (SMD), is often moved linearly in time to mimic constant velocity experiments. 30;31 While in this article, we are concerned with modeling the effect of true mechanical forces, we also note that these same techniques are often applied to abstract coordinates in order to produce a first guess of a transition trajectory, e.g. to study the activation pathway of Arp2/3 complex or the phosphate release pathway from within filamentous actin. 32;33;34;35;36;37 For now, we and others have treated forces in one of these two modalities, but one lingering question is how best we should apply forces to filaments or binding proteins to mimic how the forces are applied in experiment or in vivo; i.e. we feel it is important to determine whether our current treatments are sufficient to capture the effects produced by shear forces or stochastic motor generated-forces. In other words, is it sufficient to model myosin generated forces, which are produced by sporadic structural transitions, as a constant force, or do we need to take into account their discrete nature? And do in vitro techniques such as micromanipulation by tweezers or the use of microfluidic shear forces distribute forces along the filament/ABPs in the same way, or would we need to take into account how exactly experiments were done if we want to extract underlying molecular details of force sensing?
Typically, the goal of MD simulations is not to study the true dynamics of the system but rather to ‘sample’ configurations from the true equilibrium ensemble; to do so, the equations of motion of the system are modified to include extra terms that keep the system at either constant temperature or at constant temperature and pressure. 25 When this is done, then the configurations seen in the MD simulation should arise with probability proportional to the proper statistical distribution, e.g. in the case of constant temperature, , where is the Boltzmann constant, and kBT ≈ 0.593 kcal/mol ≈ 2.479kJ/mol ≈ 4.114 pN nm at room temperature (298 K). When constant forces are applied, MD simulations performed at constant temperature or at constant temperature and pressure will still sample from this distribution. After doing so, we often want to compute a ‘free energy surface’ (FES) also known as a ‘potential of mean force’ (PMF) which allows us to visualize the relative free energy of configurations along a small number of ‘collective variables’ (CVs) represented by . This FES corresponds to the negative log of the frequency with which configurations have a particular value ,
| (2) |
where is the Dirac delta function. 25
A significant challenge associated with MD simulations is that a small integration time step must be used to propagate the equations of motion, typically 2 femtoseconds for atomistic MD of proteins. 25;38 This limits the amount of sampling to times corresponding to microseconds at most, which is not enough time to sample all relevant configurations from , since relevant biological conformational transitions in which we are interested can have characteristic time scales corresponding to milliseconds or longer. Enhanced sampling methods can help bridge this gap and allow us to compute PMFs and even the rates of slow events within available computational time, as described next. 25;38
3 |. ENHANCED SAMPLING FOR DETERMINING THE EFFECT OF FORCES
In most cases, the use of enhanced sampling techniques will be indispensable for assessing the affect of small forces on actin and actin binding protein structure. We cannot simply rely on Moore’s law scaling of processing power to access realistic time scales. MD simulations of systems consisting of hundreds of thousands of atoms, which is required for modeling even a few actin subunits in solution, are currently limited to microseconds, whereas many cytoskeletal processes take place on the millisecond to seconds scale (e.g. actin polymerization takes place at a rate of 1-100 subunits/second 1, unbinding of ABPs such as vinculin with or without applied force takes place on the seconds time scale, see Sec. 5). To give an more precise idea of current computational power, a moderately large simulation system (170,000 atoms) as might be needed to study an actin trimer in solution would run at ~ 120ns/day using 1 NVIDIA A100 GPU and 16 CPUs using GPU-capable software like GROMACS 39. The special purpose supercomputer Anton3 can reach speeds of 167μs/day for a 300K atom system 40, but this is not generally available, and although it is 100-1000 times faster than what can be achieved on general purpose hardware, it still would require using the entire system for a decade to observe a single unbinding event for the vinculin system discussed below, with an unbinding rate of ~ 1s.
To avert this problem, a vast array of enhanced sampling techniques exist based on different approaches for quickly crossing free energy barriers, allowing us to observe large structural transitions within MD simulations in only nanoseconds rather than miliseconds or longer. 38 We develop and employ these approaches using the PLUMED open-source plugin to many popular MD codes, which allows us to simultaneously apply constant or time dependent forces to our system.41 For large protein complexes, we favor CV-based approaches in which coordinates for biasing are carefully chosen to characterize the states of interest, and ideally the transition state between those states. The original such approach is termed umbrella sampling, wherein the system is constrained (typically by a harmonic potential) to be close to a certain value of a CV; by combining information from many simulations scanned across the physically relevant range of CV values, a PMF can be reconstructed. 25;38
In our work, we typically employ Metadynamics (MetaD) and similar approaches, which have emerged as a very popular method for simultaneously exploring and computing an unknown FES. 44;45 We favor MetaD type approaches because they promote exploration of the system, as they push the system to explore new space rather than restraining the system to be in a specific region (as e.g. done in the method known as umbrella sampling) 44;46. MetaD and other CV-biasing approaches are also preferred over techniques like temperature replica exchange, since these kinds of approaches scale very poorly with system size, and are generally not practical to use with the large systems considered here 47. Furthermore, pN scale forces produce biases that are not large enough on their own to drive a rare event, and thus enhanced sampling approaches are required to observe significant changes in response.
In MetaD, a history-dependent bias potential is added to the system’s Hamiltonian. This bias potential is formed from a sum of Gaussian ‘hills’ that are periodically deposited at the system’s current position in CV space. As a result, the system is driven away from previously explored regions; additionally, the amount of bias applied at each position is then used to estimate the underlying free energy surface, as the amount of bias used is proportional to the negative of the underlying FES. 44 While this allows one to compute free energies much more readily than unbiased sampling, the quality of the result and speed of convergence still depends strongly on the choice of CVs. 44;38 Other variants of MetaD exist that provide distinct advantages; for example the MetaD flavor of OPES (on the fly probability enhanced sampling) progressively updates an estimate of the whole FES rather than building it from a sum of Gaussians, which can give more robust convergence. 45 OPES also permits use of an energy cutoff above which bias is not applied, which can help prevent exploration of unphysical regions of phase space as was shown to be important in the sampling of actin flattening with metabasin MetaD. 46 For constant forces, MetaD can be directly applied to determine how the conformational ensemble of a system changes in response to force. 28;42
MetaD and related methods can also be used to compute the rates of certain very slow processes. 48;49 If bias is added only in the starting basin and not on the transition state, then it can be shown that the effect is to accelerate time by an amount related to the exponential of the bias applied, averaged over the starting basin. 48;49 Many such simulations can be performed and the rate can be computed from the mean of the rescaled times 48;49 or more recently through a maximum likelihood approach. 50 The exponential factor means that rates for processes even on the hours time scale can be obtained from simulations that are only tens of nanoseconds in length, if good CVs are chosen for biasing. We demonstrated that this approach is able to predict the force-dependence of unbinding rates for several different systems ranging from simple potentials to models of protein-ligand complexes (receptor-ligand model) (see Fig. 2); however, difficulty arose when we tested the approach on a larger protein system (Straptavidin-Biotin bond) due to the presence of intermediate states along the unbinding pathway. 42 We have found that the Kramer’s Time Dependent rate approach of Ref. 50 can alleviate some of the error that can arise from choice of CVs by including a scaling factor that accounts for the fact that not all of the energy from biasing goes into promoting the desired transitions, but getting good convergence for large biomolecular complexes such as in the actin cytoskeleton remains a challenge.
FIGURE 2.
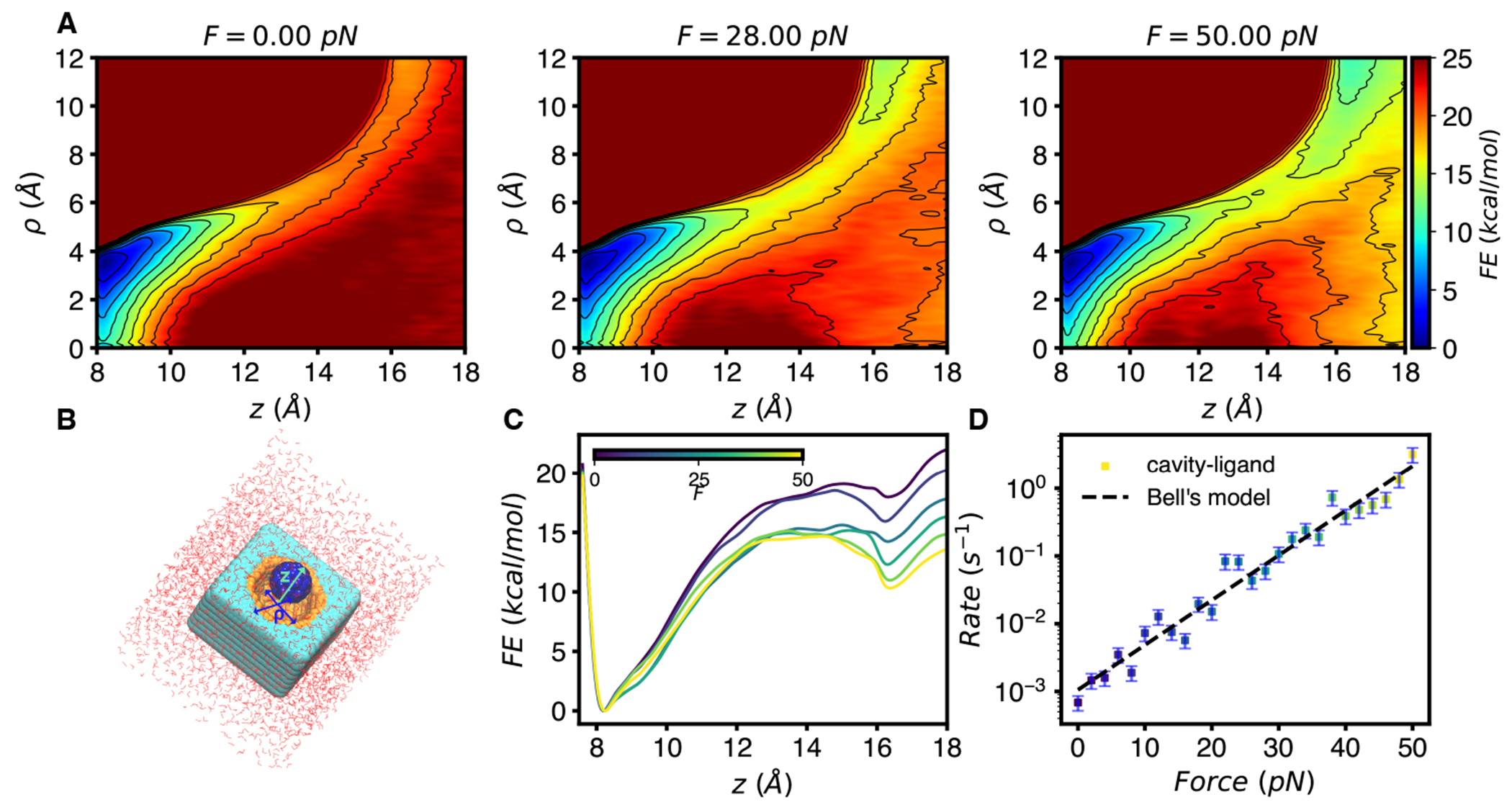
Example of enhanced sampling for a system with applied force, adapted from Ref. 42. (A) Two-dimensional FESs computed from MetaD for a model receptor-ligand system in B. Here, the FES was computed by biasing perpendicular (z) and parallel motion to the cavity surface. Both CVs are distances in Angstroms, FESs in A and C were obtained from 100 ns trajectories at each pulling force. Application of force to z shifts the equilibrium towards unbound. (B) Illustration of the model receptor-ligand system 43, which consists of a lattice of carbon-like atoms bearing a cavity in which surface atoms have an attractive potential with the ligand modeled by a C60 molecule. (C) One-dimensional PMF after integrating out shows how application of force ‘tilts’ the free energy landscape. (D) Rates computed by infrequent MetaD and fit to Bell’s law. Each point is the reciprocal of the mean of rescaled unbinding times at each force. The rate at each force was estimated by 50 simulations of duration less than 20 ns, while predicted rescaled unbinding times for this model range from 100s of miliseconds to 1600 seconds.
In addition to MetaD, it is also possible to develop methods more directly tailored to the effect of force on biomolecular configurations. We previously developed an approach termed Infinite Switch Simulated Tempering in Force, 51 which allows us to assess the effect of a range of forces from a single simulation; we recently demonstrated that convergence of this method can be improved by combination with methods that accelerate sampling through running at multiple temperatures in parallel. 52
For larger biomolecular complexes, these kinds of CV-based approaches may be insufficient, the number of degrees of freedom increase significantly with larger systems, and so there may be many important slow degrees of freedom that cannot be captured by only a small set of biasing coordinates, meaning that sampling is not converged since not all relevant states are sufficiently populated within a trajectory. For probing the pathway between two known states of a system, path-based methods such as the string method with swarm of trajectories 53;54 have been successfully used to make reasonable predictions of minimum free-energy pathways; we believe these approaches could also be employed with and without constant force to determine how force changes the free energy barrier for transition in a much higher dimensional space. For larger systems, coarse-grained (CG) approaches that reduce the dimensionality of the system may be required. 55;56;26;57 Coarse-graining involves mapping multiple atoms to a single interaction site; the aim is to capture the most essential features of a system (such as the protein’s shape and charge density) while removing some possibly irrelevant degrees of freedom (such as specific side chain rotational states). These models sacrifice some chemical detail and perhaps some of the physics contained in atomisticly detailed forcefields, it is the only reasonable approach when tackling biophysical problems at scales beyond thousands of amino-acids (or nucleotides). We believe such models can be leveraged to find useful trends even if they are not quantitative, and that approach can and should be validated by comparison with experiments.
4 |. COMPUTATIONAL INSIGHTS INTO ACTIN MECHANOSENSITIVITY
Here we give a few highlights of how MD simulations coupled to forces have already been used to give insight into actin and actin binding proteins. As previously mentioned, forces can be introduced artificially into MD simulations in order to drive rare transitions, for example promoting unbinding events, as in the release of inorganic phosphate from actin filaments. 35;36;37 Forces have also been used to promote transitions of assemblies of actin and actin binding proteins, such as converting the actin branching Arp2/3 complex from from an inactive to an active state (Fig. 3A), 32;33;34 or transitions between bound states of tropomyosin on actin. 58
FIGURE 3.
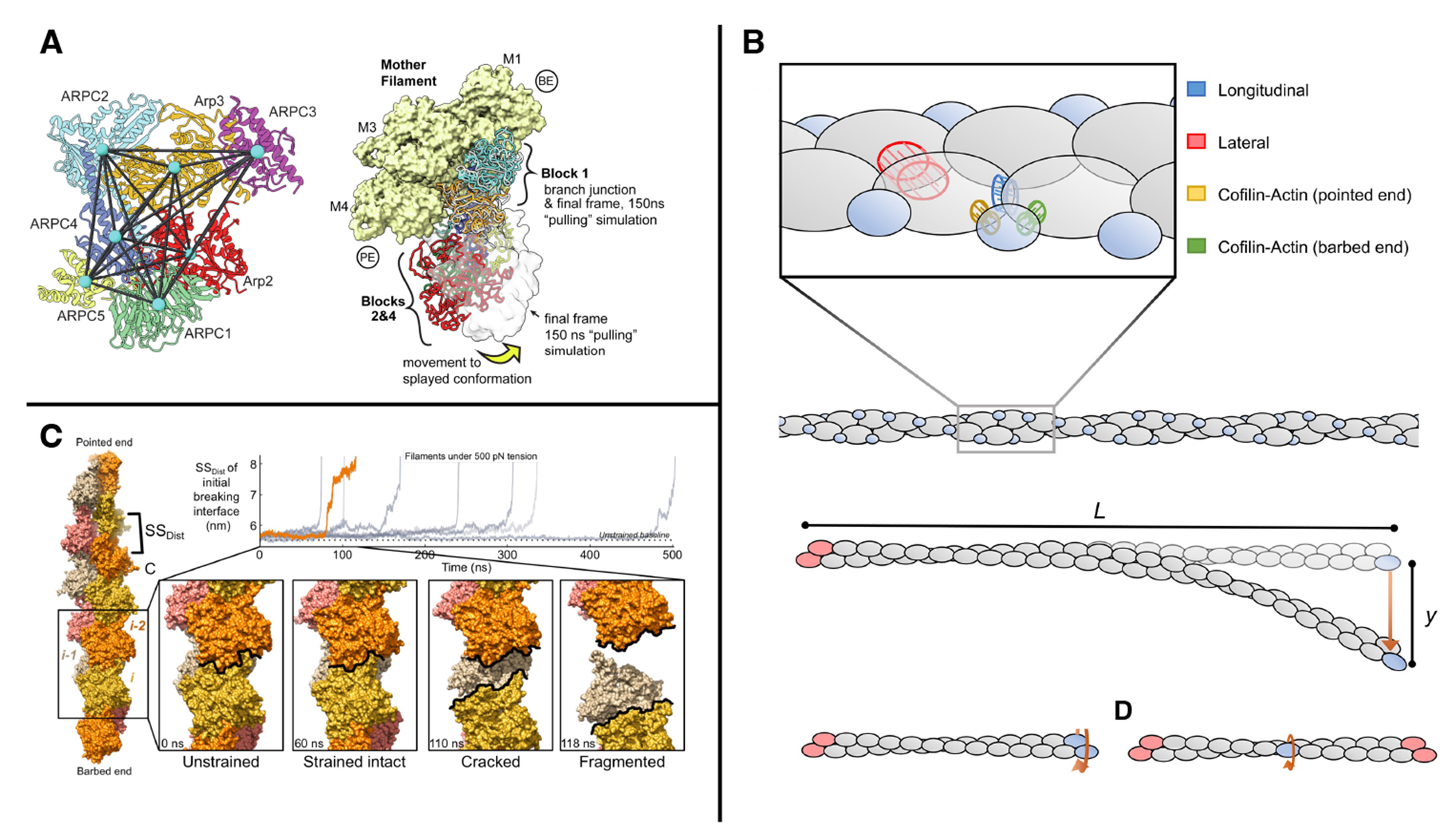
(A) Steered MD applied to pairwise distances between domains of Arp2/3 complex promotes transitions between active and inactive states, adapted from Ref. 33. (B) A CG model for probing the effect of bending and twisting on cofilin-bound actin filaments, adapted from Ref. 65. (C) Forces applied to filaments produced cracks that could be loci for proteins targeted to regions of force, adapted from Ref. 66.
Application of (perhaps unrealistically large) forces have been used as a proxy for predicting the mechanical properties of actin monomers, 59 filaments, 60 or filaments bound to binding proteins such as cofilin. 61 As an alternative to atomistic simulations, coarse grained simulations 61;62 and mechanical models 63;64;65 have also been employed to study the bending, stretching and twisting of F-actin (e.g. Fig. 3B). Only recently have computational approaches been powerful enough to predict the response of atomistic models of actin to larger forces. Studies have shown all-atom MD simulations under 500 pN of extensional strain showed metastable cracked conformations that could relate to the origin of force-activated binding of regulatory factors (e.g. Fig. 3C). 66 Although these forces are large, they may in fact be present when many myosin and filaments are bundled together in a stress fiber 18. We expect many more such studies in the future, and we point to possible areas of opportunity below in Sec. 6.
5 |. CASE STUDY: F-ACTIN/VINCULIN COMPLEX
We argue that we are now poised to leverage enhanced sampling techniques in the presence of small forces to probe the molecular mechanisms underlying mechanosensing behavior in the actin cytoskeleton. Here we give an example of how we are combining several techniques as well as large scale computational resources to tackle a problem of particular interest, namely the origin of catch bonding behavior in the focal adhesion protein vinculin. Vinculin participates in both, focal adhesion and adherens junction assemblies, with particular relevance in cell migration and embryonic development 67. Vinculin has a head and tail domain joined by a linker region, and it performs its functions through its ability to interact with a number of binding partners in each domain, including interactions of the tail with actin and PIP2 lipids, and the head with cytoskeletal proteins such as talin, –actinin and –catenin 67.
When force is applied across a protein-protein interaction, the naive expectation is that it will accelerate the rate of unbinding, or equivalently, decrease the lifetime of the ‘bond’. Simple theoretical arguments show that small forces should accelerate processes exponentially with force, something known as Bell’s law. 28:68 In contrast to this ‘slip bond’ behavior, some biological assemblies show ‘catch’ bond behavior, where the lifetime actually gets longer for small forces 28;69. In focal adhesion and adherens junction assemblies, which help anchor cells to substrates, it is proposed that catch bond behavior engaged by forces applied between the cytoskeleton and the extracellular environment play a role in stabilizing attachment. 13 Single molecule experiments have shown that proteins vinculin and catenin form a catch bond when bound to F-Actin. For vinculin, this behavior has been localized to the tail domain and its interaction with actin 13;70. This catch bonding behavior was found to exist whether the force was applied towards the pointed or barbed end of F-actin, however, the lifetimes of the interactions are larger when pulling towards the pointed end (see Fig. 3A). 13 Furthermore, it was shown that the vinculin tail (Vt, 5-helix bundle) is a key structure for the catch bond behavior, as the directional catch bond was still observed in optical trap experiments where only Vt was bound to F-Actin.
For both catenin and vinculin, a two-state catch bond model 69 was used to describe the force dependence of the unbinding, consisting of an unbound state, a weakly bound state, and a strongly bound state. 13 Within this model, each individual transition can be treated as following Bell’s law, , where is the transition rate from state to state in the absence of force and is a parameter for the distance to the transition barrier between states and . Thus the apparent catch bond behavior arises as a consequence of the rapid force-dependent transition to the strongly bound state. Molecular models proposed for -catenin point to a transition between a four-helix bundle and a five-helix bundle as the key structural motifs underlying this multistate model (see Fig. 4B). 71:70 So far, little has been done from a modeling standpoint to probe the molecular underpinnings of this process or to test the hypothesis proposed through structural biological techniques. Very recently, MD simulations with large forces on Vt bound to two actin subunits were used to predict residues in Vt participating in key interactions (H-bonding with Actin) involved in directional bond strengthening; when Vt was mutated to exclude these key residues, the Vt variants were still able to bind to actin but showed evidence of loss of catch-bonding ability. 72
FIGURE 4.
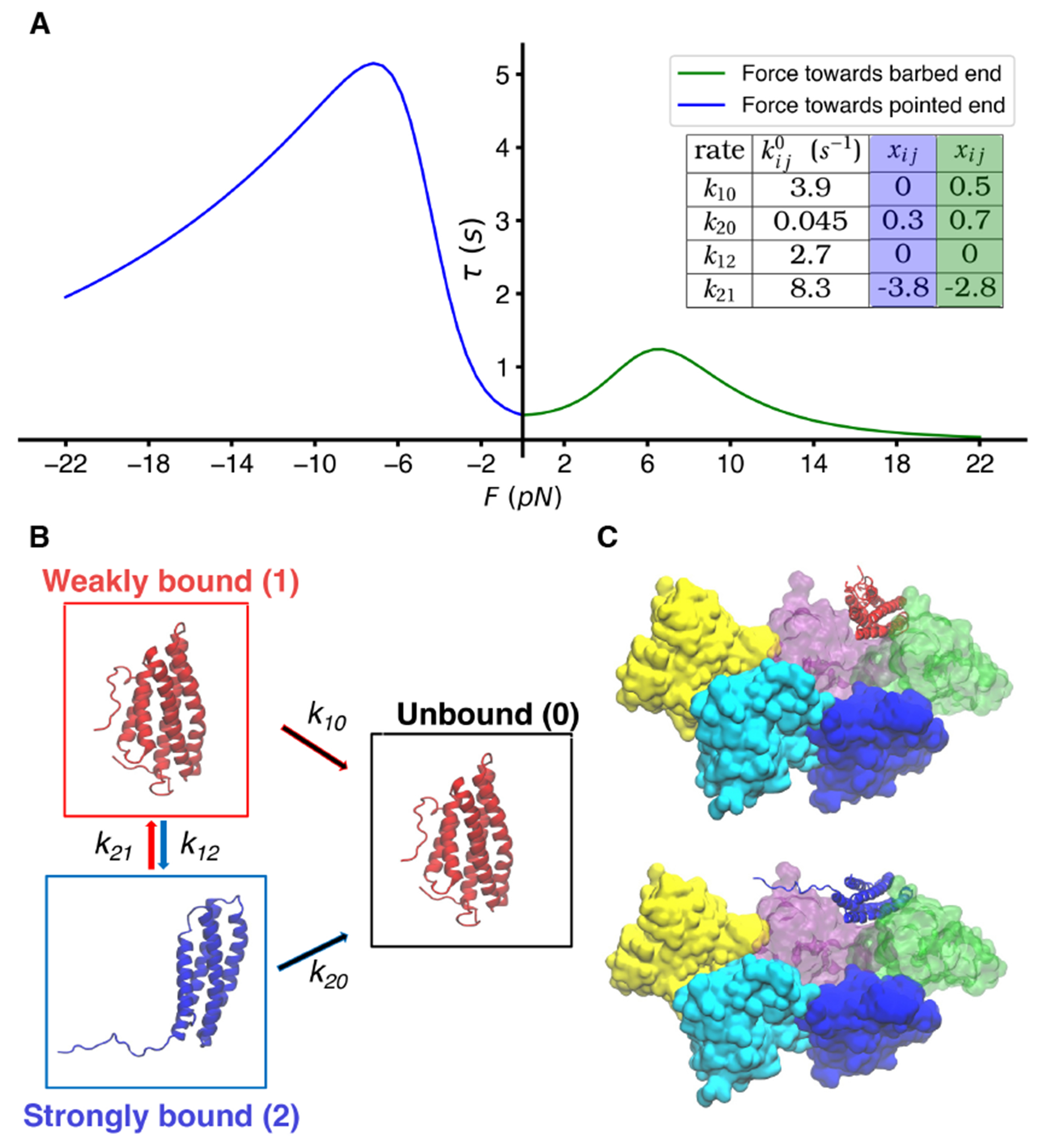
(A) Best-fit curves for the lifetime of Vt bound to actin as assessed in Ref. 13 give rise to a parameterized model (see table) with two bound states and one unbound state. (B) Vinculin four-or five helical bundles corresponding to the putative strong and weak bound states of catenin from Refs. 70 and 71. (C) Some of our models of five actin subunits bound to either four and five-helical bundle states of Vt, for probing the force sensitivity of Vt unbinding.
We see this vinculin/actin interplay as an extremely challenging, yet ideal platform to test the enhanced sampling MD simulation approaches above. These simulations allows us to look at an atomic level what interactions between residues at the binding interface are relevant and how these evolve in time as the unbinding process occurs at different pulling forces. In order to properly tackle all aspects of the problem, we should be able to find the force-dependent rates of unbinding, as well as the free energy barriers dictating changes between states of the protein in its bound pose with actin. Are simulation approaches robust enough to actually predict free energies and rates for this very slow (seconds-time scale) process? Achieving this result requires high quality and large simulations of cytoskeletal assemblies in each of the different putative states, some of which are shown in Fig. 4C. Our group and collaborators are leveraging free energy and rates approaches as described above 45;50, as well as methods for characterizing relevant states of the system and designing high quality CVs requiring minimal bias for producing transitions 73;74 aiming to develop a workflow that is able to provide robust predictions. We are optimistic that such approaches will give detailed insight into how such catch bonding behavior arises in an atomic model of a cytoskeletal system for the first time.
6 |. FUTURE OPPORTUNITIES
Advancing in vitro and structural biological techniques are constantly improving our understanding of the actin cytoskeleton, especially in how actin and several ABPs coordinate in a dynamic fashion to self-organize. 75 For example, single-particle cryo-electron microscopy approaches combined with machine learning approaches are giving new insight into how coupled bending and twisting alter the structure of individual filaments 22, and MD simulations may be able to leverage this information to predict how these changes alter binding affinity for ABPs. We previously showed that the cost of bending actin filaments can lead to sorting of actin crosslinkers in bundles, 76 but these arguments were based on the persistence length of actin filaments, derived from equilibrium bending fluctuations; the ability of filaments to permit bends over shorter length scales is something that could be further detailed through computational modeling.
As another example, EM studies and MD simulations together have given complementary insights into the interface between cofilin-bound and bare actin filaments, 77;78 and ability to accurately model application of force could give a detailed picture of how this leads to filament severing and depolymerization. Tackling larger scale force-induced processes such as the debranching of Arp2/3 complex 79 likely will remain out of reach by atomistic approaches for some time, but could perhaps be investigated through CG modeling.
As described, in vitro experiments show that mechanical forces can produce changes of binding affinity for ABPs. 20 Recent MD studies point to a mechanism for force to expose residues within filaments, which could be a mechanism used by LIM domain proteins to sense stressed filaments, which can also play an important feedback role in maintenance of mechanical properties. 66;80;81;82;83 A major challenge remaining is to demonstrate that a combination of accelerated MD simulations with small forces can have predictive accuracy for such subtle yet important structural changes.
Acknowledgments
GMH would like to thank all past and present collaborators on these topics for many fruitful conversations, in particular Guillaume Stirnemann with whom we are collaborating on the vinculin/actin project described above. FM and GMH were supported by the National Institutes of Health (NIH) via Award No. R35GM138312, and W.J.P.C. by R35GM138312-S1.
references
- [1].Pollard Thomas D. and Cooper John A.. Actin, a Central Player in Cell Shape and Movement. Science, 326(5957):1208–1212, November 2009. doi: 10.1126/science.1175862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Dominguez Roberto. Actin-binding proteins – a unifying hypothesis. Trends in Biochemical Sciences, 29(11):572–578, November 2004. ISSN 0968-0004. doi: 10.1016/j.tibs.2004.09.004. [DOI] [PubMed] [Google Scholar]
- [3].Pollard Thomas D.. Actin and Actin-Binding Proteins. Cold Spring Harb Perspect Biol, 8(8):a018226, August 2016. ISSN , 1943-0264. doi: 10.1101/cshperspect.a018226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Laakso Joseph M., Lewis John H., Shuman Henry, and Ostap E. Michael. Myosin I Can Act As a Molecular Force Sensor. Science, 321(5885):133–136, July 2008. doi: 10.1126/science.1159419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Svitkina Tatyana. The Actin Cytoskeleton and Actin-Based Motility. Cold Spring Harb Perspect Biol, 10(1):a018267, January 2018. ISSN , 1943-0264. doi: 10.1101/cshperspect.a018267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Kovar David R. and Pollard Thomas D.. Insertional assembly of actin filament barbed ends in association with formins produces piconewton forces. Proc. Natl. Acad. Sci, 101(41):14725–14730, October 2004. doi: 10.1073/pnas.0405902101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Marcy Yann, Prost Jacques, Carlier Marie-France, and Sykes Cécile. Forces generated during actin-based propulsion: A direct measurement by micromanipulation. Proc. Natl. Acad. Sci, 101(16):5992–5997, April 2004. doi: 10.1073/pnas.0307704101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Footer Matthew J, Kerssemakers Jacob WJ, Theriot Julie A, and Dogterom Marileen. Direct measurement of force generation by actin filament polymerization using an optical trap. Proc. Natl. Acad. Sci, 104(7):2181–2186, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Blanchoin Laurent, Boujemaa-Paterski Rajaa, Sykes Cécile, and Plastino Julie. Actin Dynamics, Architecture, and Mechanics in Cell Motility. Physiol. Rev, 94(1):235–263, January 2014. ISSN 0031-9333. doi: 10.1152/physrev.00018.2013. [DOI] [PubMed] [Google Scholar]
- [10].Liu Allen P., Richmond David L., Maibaum Lutz, Pronk Sander, Geissler Phillip L., and Fletcher Daniel A.. Membrane-induced bundling of actin filaments. Nature Phys, 4(10):789–793, October 2008. ISSN 1745-2481. doi: 10.1038/nphys1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Dominguez Roberto and Holmes Kenneth C.. Actin Structure and Function. Annual Review of Biophysics, 40(1):169–186, 2011. doi: 10.1146/annurev-biophys-042910-155359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].McCullagh Martin, Saunders Marissa G, and Voth Gregory A. Unraveling the mystery of atp hydrolysis in actin filaments. J. Am. Chem. Soc, 136(37):13053–13058, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Huang Derek L., Bax Nicolas A., Buckley Craig D., Weis William I., and Dunn Alexander R.. Vinculin forms a directionally asymmetric catch bond with F-actin. Science, 357(6352):703–706, August 2017. doi: 10.1126/science.aan2556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Wioland Hugo, Suzuki Emiko, Cao Luyan, Romet-Lemonne Guillaume, and Jegou Antoine. The advantages of microfluidics to study actin biochemistry and biomechanics. J Muscle Res Cell Motil, 41(1):175–188, March 2020. ISSN 1573-2657. doi: 10.1007/s10974-019-09564-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Zimmermann Dennis, Homa Kaitlin E., Hocky Glen M., Pollard Luther W., De La Cruz Enrique M, Voth Gregory A., Trybus Kathleen M., and Kovar David R.. Mechanoregulated inhibition of formin facilitates contractile actomyosin ring assembly. Nat Commun, 8(1):703, September 2017. ISSN 2041-1723. doi: 10.1038/s41467-017-00445-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Finer Jeffrey T., Simmons Robert M., and Spudich James A.. Single myosin molecule mechanics: piconewton forces and nanometre steps. Nature, 368(6467):113–119, March 1994. ISSN 1476-4687. doi: 10.1038/368113a0. [DOI] [PubMed] [Google Scholar]
- [17].Stam Samantha, Alberts Jon, Gardel Margaret L., and Munro Edwin. Isoforms Confer Characteristic Force Generation and Mechanosensation by Myosin II Filaments. Biophys J, 108(8):1997–2006, April 2015. ISSN 0006-3495. doi: 10.1016/j.bpj.2015.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Quintanilla Melissa A., Patel Hiral, Wu Huini, Sochacki Kem A., Akamatsu Matthew, Rotty Jeremy D., Korobova Farida, Bear James E., Taraska Justin W., Oakes Patrick W., and Beach Jordan R.. Local monomer levels and established filaments potentiate non-muscle myosin 2 assembly, 2023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Enrique M. De La Cruz. Cofilin Binding to Muscle and Non-muscle Actin Filaments: Isoform-dependent Cooperative Interactions. Journal of Molecular Biology, 346(2):557–564, February 2005. ISSN 0022-2836. doi: 10.1016/j.jmb.2004.11.065. [DOI] [PubMed] [Google Scholar]
- [20].Jégou Antoine and Romet-Lemonne Guillaume. Mechanically tuning actin filaments to modulate the action of actin-binding proteins. Current Opinion in Cell Biology, 68:72–80, February 2021. ISSN 0955-0674. doi: 10.1016/j.ceb.2020.09.002. [DOI] [PubMed] [Google Scholar]
- [21].Cossio Pilar and Hocky Glen M.. Catching actin proteins in action. Nature, 611(7935):241–243, November 2022. doi: 10.1038/d41586-022-03343-x. [DOI] [PubMed] [Google Scholar]
- [22].Reynolds Matthew J., Hachicho Carla, Carl Ayala G., Gong Rui, and Alushin Gregory M.. Bending forces and nucleotide state jointly regulate F-actin structure. Nature, 611(7935):380–386, November 2022. ISSN 1476-4687. doi: 10.1038/s41586-022-05366-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Bibeau Jeffrey P., Pandit Nandan G., Gray Shawn, Nejad Nooshin Shatery, Sindelar Charles V., Cao Wenxiang, and De La Cruz Enrique M.. Twist response of actin filaments. Proc. Nat!. Acad. Sci, 120(4):e2208536120, January 2023. doi: 10.1073/pnas.2208536120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Owen Leanna M., Bax Nicolas A., Weis William I., and Dunn Alexander R.. The C-terminal actin-binding domain of talin forms an asymmetric catch bond with F-actin. Proc. Natl. Acad. Sci, 119(10):e2109329119, March 2022. doi: 10.1073/pnas.2109329119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Tuckerman Mark. Statistical Mechanics: Theory and Molecular Simulation. OUP Oxford, February 2010. ISBN 978-0-19152346-5. [Google Scholar]
- [26].Schlick Tamar, Portillo-Ledesma Stephanie, Myers Christopher G., Beljak Lauren, Chen Justin, Dakhel Sami, Darling Daniel, Ghosh Sayak, Hall Joseph, Jan Mikaeel, Liang Emily, Saju Sera, Vohr Mackenzie, Wu Chris, Xu Yifan, and Xue Eva. Biomolecular Modeling and Simulation: A Prospering Multidisciplinary Field. Annu. Rev. Biophys, 50(1):267–301, May 2021. ISSN 1936-122X, 1936-1238. doi: 10.1146/annurev-biophys-091720-102019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Bustamante Carlos, Chemla Yann R., Forde Nancy R., and Izhaky David. Mechanical Processes in Biochemistry. Annual Review of Biochemistry, 73(1):705–748, 2004. doi: 10.1146/annurev.biochem.72.121801.161542. [DOI] [PubMed] [Google Scholar]
- [28].Gomez David, Peña Ccoa Willmor J., Singh Yuvraj, Rojas Enrique, and Hocky Glen M.. Molecular Paradigms for Biological Mechanosensing. J. Phys. Chem. B, 125(44):12115–12124, November 2021. ISSN 1520-6106. doi: 10.1021/acs.jpcb.1c06330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Bustamante Carlos, Alexander Lisa, Maciuba Kevin, and Kaiser Christian M.. Single-Molecule Studies of Protein Folding with Optical Tweezers. Annual Review of Biochemistry, 89(1):443–470, 2020. doi: 10.1146/annurev-biochem-013118-111442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Lu Hui and Schulten Klaus. Steered molecular dynamics simulations of force-induced protein domain unfolding. Proteins: Structure, Function, and Bioinformatics, 35(4):453–463, 1999. ISSN 1097-0134. doi: . [DOI] [PubMed] [Google Scholar]
- [31].Stirnemann Guillaume. Recent Advances and Emerging Challenges in the Molecular Modeling of Mechanobiological Processes. J. Phys. Chem. B, 126(7):1365–1374, February 2022. ISSN 1520-6106. doi: 10.1021/acs.jpcb.1c10715. [DOI] [PubMed] [Google Scholar]
- [32].Ding Bojian, Narvaez-Ortiz Heidy Y., Singh Yuvraj, Hocky Glen M., Chowdhury Saikat, and Nolen Brad J.. Structure of Arp2/3 complex at a branched actin filament junction resolved by single-particle cryo-electron microscopy. Proc. Natl. Acad. Sci, 119(22):e2202723119, May 2022. doi: 10.1073/pnas.2202723119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Singh Yuvraj, Hocky Glen M., and Nolen Brad J.. Molecular dynamics simulations support a multistep pathway for activation of branched actin filament nucleation by Arp2/3 complex. J. Biol. Chem, 299(9):105169, September 2023. ISSN 0021-9258. doi: 10.1016/j.jbc.2023.105169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Dalhaimer Paul and Pollard Thomas D.. Molecular Dynamics Simulations of Arp2/3 Complex Activation. Biophysical Journal, 99(8):2568–2576, October 2010. ISSN 0006-3495. doi: 10.1016/j.bpj.2010.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Wriggers Willy and Schulten Klaus. Investigating a back door mechanism of actin phosphate release by steered molecular dynamics. Proteins: Structure, Function, and Bioinformatics, 35(2):262–273, 1999. ISSN 1097-0134. doi: . [DOI] [PubMed] [Google Scholar]
- [36].Okazaki Kei-ichi and Hummer Gerhard. Phosphate release coupled to rotary motion of F1-ATPase. Proc. Natl. Acad. Sci, 110(41):16468–16473, October 2013. doi: 10.1073/pnas.1305497110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Wang Yihang, Zsolnay Vilmos, Pollard Thomas D., and Voth Gregory A.. Mechanism of Phosphate Release from Actin Filaments, August 2023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Hénin Jérôme, Lelievre Tony, Shirts Michael R., Valsson Omar, and Delemotte Lucie. Enhanced Sampling Methods for Molecular Dynamics Simulations [Article v1.0]. Living Journal of Computational Molecular Science, 4(1):1583–1583, December 2022. ISSN 2575-6524. doi: 10.33011/livecoms.4.1.1583. [DOI] [Google Scholar]
- [39].Gromacs performance on different GPU types – hpc.fau.de https://hpc.fau.de/2022/02/10/gromacs-performance-on-different-gpu-types/, 2022. [Accessed 20-01-2024].
- [40].Shaw David E, Adams Peter J, Azaria Asaph, Bank Joseph A, Batson Brannon, Bell Alistair, Bergdorf Michael, Bhatt Jhanvi, Butts J Adam, Correia Timothy, et al. Anton 3: twenty microseconds of molecular dynamics simulation before lunch. In Proceedings of the International Conference for High Performance Computing, Networking, Storage and Analysis, pages 1–11, 2021. [Google Scholar]
- [41].Bonomi Massimiliano, Bussi Giovanni, Camilloni Carlo, Tribello Gareth A., Banáš Pavel, Barducci Alessandro, Bernetti Mattia, Bolhuis Peter G., Bottaro Sandro, Branduardi Davide, Capelli Riccardo, Carloni Paolo, Ceriotti Michele, Cesari Andrea, Chen Haochuan, Chen Wei, Colizzi Francesco, De Sandip, De La Pierre Marco, Donadio Davide, Drobot Viktor, Ensing Bernd, Ferguson Andrew L., Filizola Marta, Fraser James S., Fu Haohao, Gasparotto Piero, Luigi Gervasio Francesco, Giberti Federico, Gil-Ley Alejandro, Giorgino Toni, Heller Gabriella T., Hocky Glen M., lannuzzi Marcella, Invernizzi Michele, Jelfs Kim E., Jussupow Alexander, Kirilin Evgeny, Laio Alessandro, Limongelli Vittorio, Lindorff-Larsen Kresten, Löhr Thomas, Marinelli Fabrizio, Martin-Samos Layla, Masetti Matteo, Meyer Ralf, Michaelides Angelos, Molteni Carla, Morishita Tetsuya, Nava Marco, Paissoni Cristina, Papaleo Elena, Parrinello Michele, Pfaendtner Jim, Piaggi Pablo, Piccini GiovanniMaria, Pietropaolo Adriana, Pietrucci Fabio, Pipolo Silvio, Provasi Davide, Quigley David, Raiteri Paolo, Raniolo Stefano, Rydzewski Jakub, Salvalaglio Matteo, Sosso Gabriele Cesare, Spiwok Vojtěch, Šponer Jiří, Swenson David W. H., Tiwary Pratyush, Valsson Omar, Vendruscolo Michele, Voth Gregory A., White Andrew, and The PLUMED consortium. Promoting transparency and reproducibility in enhanced molecular simulations. Nat Methods, 16(8):670–673, August 2019. ISSN 1548-7105. doi: 10.1038/s41592-019-0506-8.. [DOI] [PubMed] [Google Scholar]
- [42].Willmor J. Peña Ccoa and Hocky Glen M.. Assessing models of force-dependent unbinding rates via infrequent metadynamics. The Journal of Chemical Physics, 156(12):125102, March 2022. ISSN 0021-9606. doi: 10.1063/5.0081078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Mondal Jagannath, Morrone Joseph A, and Berne BJ. How hydrophobic drying forces impact the kinetics of molecular recognition. Proc. Natl. Acad. Sci, 110(33):13277–13282, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Bussi Giovanni and Laio Alessandro. Using metadynamics to explore complex free-energy landscapes. Nat Rev Phys, 2(4):200–212, April 2020. ISSN 2522-5820. doi: 10.1038/s42254-020-0153-0. [DOI] [Google Scholar]
- [45].Invernizzi Michele, Piaggi Pablo M., and Parrinello Michele. Unified Approach to Enhanced Sampling. Phys. Rev. X, 10(4):041034, November 2020. doi: 10.1103/PhysRevX.10.041034. [DOI] [Google Scholar]
- [46].Dama James F., Hocky Glen M., Sun Rui, and Voth Gregory A.. Exploring Valleys without Climbing Every Peak: More Efficient and Forgiving Metabasin Metadynamics via Robust On-the-Fly Bias Domain Restriction. J. Chem. Theory Comput, 11(12):5638–5650, December 2015. ISSN 1549-9618. doi: 10.1021/acs.jctc.5b00907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Sugita Yuji and Okamoto Yuko. Replica-exchange molecular dynamics method for protein folding. Chem. Phys. Lett, 314(1-2):141–151, 1999. [Google Scholar]
- [48].Tiwary Pratyush and Parrinello Michele. From Metadynamics to Dynamics. Phys. Rev. Lett, 111(23):230602, December 2013. doi: 10.1103/PhysRevLett.111.230602. [DOI] [PubMed] [Google Scholar]
- [49].Ray Dhiman and Parrinello Michele. Kinetics from Metadynamics: Principles, Applications, and Outlook. J. Chem. Theory Comput, 19(17):5649–5670, September 2023. ISSN 1549-9618. doi: 10.1021/acs.jctc.3c00660. [DOI] [PubMed] [Google Scholar]
- [50].Palacio-Rodriguez Karen, Vroylandt Hadrien, Stelzl Lukas S., Pietrucci Fabio, Hummer Gerhard, and Cossio Pilar. Transition Rates and Efficiency of Collective Variables from Time-Dependent Biased Simulations. J. Phys. Chem. Lett, 13(32):7490–7496, August 2022. doi: 10.1021/acs.jpclett.2c01807. [DOI] [PubMed] [Google Scholar]
- [51].Hartmann Michael J., Singh Yuvraj, Vanden-Eijnden Eric, and Hocky Glen M.. Infinite switch simulated tempering in force (FISST). The Journal of Chemical Physics, 152(24):244120, June 2020. ISSN 0021-9606. doi: 10.1063/5.0009280. [DOI] [PubMed] [Google Scholar]
- [52].Singh Yuvraj and Hocky Glen M.. Improved prediction of molecular response to pulling by combining force tempering with replica exchange methods. J. Phys. Chem. B, 2024. doi: 10.1021/acs.jpcb.3c07081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Weinan E, Ren Weiqing, and Vanden-Eijnden Eric. String method for the study of rare events. Phys. Rev. B, 66(5):052301, August 2002. doi: 10.1103/PhysRevB.66.052301. [DOI] [PubMed] [Google Scholar]
- [54].Pan Albert C., Sezer Deniz, and Roux Benoît. Finding Transition Pathways Using the String Method with Swarms of Trajectories. J. Phys. Chem. B, 112(11):3432–3440, March 2008. ISSN 1520-6106. doi: 10.1021/jp0777059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Marrink Siewert J., Risselada H. Jelger, Yefimov Serge, Tieleman D. Peter, and deVries Alex H.. The MARTINI Force Field: Coarse Grained Model for Biomolecular Simulations. J. Phys. Chem. B, 111(27):7812–7824, July 2007. ISSN 1520-6106. doi: 10.1021/jp071097f. [DOI] [PubMed] [Google Scholar]
- [56].Noid WG. Perspective: Coarse-grained models for biomolecular systems. The Journal of Chemical Physics, 139(9):090901, September 2013. ISSN 0021-9606. doi: 10.1063/1.4818908. [DOI] [PubMed] [Google Scholar]
- [57].Jin Jaehyeok, Pak Alexander J., Durumeric Aleksander E. P., Loose Timothy D., and Voth Gregory A.. Bottom-up Coarse-Graining: Principles and Perspectives. J. Chem. Theory Comput, 18(10):5759–5791, October 2022. ISSN 1549-9618. doi: 10.1021/acs.jctc.2c00643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Williams Michael R., Tardiff Jil C., and Schwartz Steven D.. Mechanism of Cardiac Tropomyosin Transitions on Filamentous Actin As Revealed by All-Atom Steered Molecular Dynamics Simulations. J. Phys. Chem. Lett, 9(12):3301–3306, June 2018. doi: 10.1021/acs.jpclett.8b00958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Mehrafrooz Behzad and Shamloo Amir. Mechanical differences between ATP and ADP actin states: A molecular dynamics study. Journal of Theoretical Biology, 448:94–103, July 2018. ISSN 0022-5193. doi: 10.1016/j.jtbi.2018.04.010. [DOI] [PubMed] [Google Scholar]
- [60].Shamloo Amir and Mehrafrooz Behzad. Nanomechanics of actin filament: A molecular dynamics simulation. Cytoskeleton, 75(3):118–130, 2018. ISSN 1949-3592. doi: 10.1002/cm.21429. [DOI] [PubMed] [Google Scholar]
- [61].Kim Jae In, Kwon Junpyo, Baek Inchul, and Na Sungsoo. Steered molecular dynamics analysis of the role of cofilin in increasing the flexibility of actin filaments. Biophysical Chemistry, 218:27–35, November 2016. ISSN 0301-4622. doi: 10.1016/j.bpc.2016.08.002. [DOI] [PubMed] [Google Scholar]
- [62].Chu Jhih-Wei and Voth Gregory A.. Coarse-Grained Modeling of the Actin Filament Derived from Atomistic-Scale Simulations. Biophysical Journal, 90(5):1572–1582, March 2006. ISSN 0006-3495. doi: 10.1529/biophysj.105.073924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Li Si, Zhang Jin, Wang Chengyuan, and Nithiarasu Perumal. Atomistic Modeling of F-Actin Mechanical Responses and Determination of Mechanical Properties. ACS Biomater. Sci. Eng, 4(8):2794–2803, August 2018. doi: 10.1021/acsbiomaterials.8b00640. [DOI] [PubMed] [Google Scholar]
- [64].Schramm Anthony C., Hocky Glen M., Voth Gregory A., Blanchoin Laurent, Martiel Jean-Louis, and De La Cruz Enrique M.. Actin Filament Strain Promotes Severing and Cofilin Dissociation. Biophys J, 112(12):2624–2633, June 2017. ISSN 1542-0086. doi: 10.1016/j.bpj.2017.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Schramm Anthony C., Hocky Glen M., Voth Gregory A., Martiel Jean-Louis, and De La Cruz Enrique M.. Plastic Deformation and Fragmentation of Strained Actin Filaments. Biophys J, 117(3):453–463, August 2019. ISSN 0006-3495. doi: 10.1016/j.bpj.2019.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Zsolnay Vilmos, Gardel Margaret L., Kovar David R., and Voth Gregory A.. Cracked actin filaments as mechanosensitive receptors. bioRxiv, page 2023.06.26.546553, June 2023. doi: 10.1101/2023.06.26.546553. [DOI] [PubMed] [Google Scholar]
- [67].Bays Jennifer L and DeMali Kris A. Vinculin in cell–cell and cell–matrix adhesions. Cell. Mol. Life Sci, 74:2999–3009, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Bell George I.. Models for the Specific Adhesion of Cells to Cells. Science, 200(4342):618–627, May 1978. doi: 10.1126/science.347575. [DOI] [PubMed] [Google Scholar]
- [69].Chakrabarti Shaon, Hinczewski Michael, and Thirumalai D. Phenomenological and microscopic theories for catch bonds. Journal of Structural Biology, 197(1):50–56, January 2017. ISSN 1047-8477. doi: 10.1016/j.jsb.2016.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Wang Amy, Dunn Alexander R, and Weis William I. Mechanism of the cadherin–catenin F-actin catch bond interaction. eLife, 11:e80130, August 2022. ISSN 2050-084X. doi: 10.7554/eLife.80130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Mei Lin, de los Reyes Santiago Espinosa, Reynolds Matthew J, Leicher Rachel, Liu Shixin, and Alushin Gregory M. Molecular mechanism for direct actin force-sensing by alpha-catenin. eLife, 9:e62514, September 2020. ISSN 2050-084X. doi: 10.7554/eLife.62514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Chirasani Venkat R., Khan Mohammad Ashhar I., Malavade Juilee N., Dokholyan Nikolay V., Hoffman Brenton D., and Campbell Sharon L.. Elucidation of the Molecular Basis and Cellular Functions of Vinculin-Actin Directional Catch Bonding. Res Sq, pages rs.3.rs–2334490, January 2023. doi: 10.21203/rs.3.rs-2334490/v1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Klem Heidi, Hocky Glen M., and McCullagh Martin. Size-and-Shape Space Gaussian Mixture Models for Structural Clustering of Molecular Dynamics Trajectories. J. Chem. Theory Comput, 18(5):3218–3230, May 2022. ISSN 1549-9618. doi: 10.1021/acs.jctc.1c01290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Sasmal Subarna, McCullagh Martin, and Hocky Glen M.. Reaction Coordinates for Conformational Transitions Using Linear Discriminant Analysis on Positions. J. Chem. Theory Comput, 19(14):4427–4435, July 2023. ISSN 1549-9618. doi: 10.1021/acs.jctc.3c00051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Kadzik Rachel S., Homa Kaitlin E., and Kovar David R.. F-Actin Cytoskeleton Network Self-Organization Through Competition and Cooperation. Annual Review of Cell and Developmental Biology, 36(1):35–60, 2020. doi: 10.1146/annurev-cellbio-032320-094706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Freedman Simon L., Suarez Cristian, Winkelman Jonathan D., Kovar David R., Voth Gregory A., Dinner Aaron R., and Hocky Glen M.. Mechanical and kinetic factors drive sorting of F-actin cross-linkers on bundles. Proc. Natl. Acad. Sci, 116(33):16192–16197, August 2019. doi: 10.1073/pnas.1820814116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Huehn Andrew R., Bibeau Jeffrey P., Schramm Anthony C., Cao Wenxiang, De La Cruz Enrique M., and Sindelar Charles V.. Structures of cofilin-induced structural changes reveal local and asymmetric perturbations of actin filaments. Proc. Natl. Acad. Sci, 117(3):1478–1484, January 2020. doi: 10.1073/pnas.1915987117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Hocky Glen M., Sindelar Charles V., Cao Wenxiang, Voth Gregory A., and De La Cruz Enrique M.. Structural basis of fast- and slow-severing actin–cofilactin boundaries. Journal of Biological Chemistry, 296, January 2021. ISSN 0021-9258, 1083–351X. doi: 10.1016/j.jbc.2021.100337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Pandit Nandan G., Cao Wenxiang, Bibeau Jeffrey, Johnson-Chavarria Eric M., Taylor Edwin W., Pollard Thomas D., and De La Cruz Enrique M.. Force and phosphate release from Arp2/3 complex promote dissociation of actin filament branches. Proc. Natl. Acad. Sci, 117(24):13519–13528, June 2020. doi: 10.1073/pnas.1911183117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Winkelman Jonathan D., Anderson Caitlin A., Suarez Cristian, Kovar David R., and Gardel Margaret L.. Evolutionarily diverse LIM domain-containing proteins bind stressed actin filaments through a conserved mechanism. Proc. Natl. Acad. Sci, 117(41):25532–25542, October 2020. doi: 10.1073/pnas.2004656117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Anderson Caitlin A., Kovar David R., Gardel Margaret L., and Winkelman Jonathan D.. LIM domain proteins in cell mechanobiology. Cytoskeleton, 78(6):303–311, 2021. ISSN 1949-3592. doi: 10.1002/cm.21677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Sun Xiaoyu, Phua Donovan Y. Z., Axiotakis Lucas, Smith Mark A., Blankman Elizabeth, Gong Rui, Cail Robert C., de los Reyes Santiago Espinosa, Beckerle Mary C., Waterman Clare M., and Alushin Gregory M.. Mechanosensing through Direct Binding of Tensed F-Actin by LIM Domains. Developmental Cell, 55(4):468–482.e7, November 2020. ISSN 1534-5807. doi: 10.1016/j.devcel.2020.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Sun Xiaoyu and Alushin Gregory M.. Cellular force-sensing through actin filaments. The FEBS Journal, 290(10):2576–2589, 2023. ISSN 1742-4658. doi: 10.1111/febs.16568. [DOI] [PMC free article] [PubMed] [Google Scholar]