

SUMMARY
Wnt/Wingless (Wg) signaling is critical in development and disease, including cancer. Canonical Wnt signaling is mediated by β-catenin/Armadillo (Arm in Drosophila) transducing signals to the nucleus, with IFT-A/Kinesin 2 complexes promoting nuclear translocation of β-catenin/Arm. Here, we demonstrate that a conserved small N-terminal Arm34–87/β-catenin peptide binds to IFT140, acting as a dominant interference tool to attenuate Wg/Wnt signaling in vivo. Arm34–87 expression antagonizes endogenous Wnt/Wg signaling, resulting in the reduction of its target expression. Arm34–87 inhibits Wg/Wnt signaling by interfering with nuclear translocation of endogenous Arm/β-catenin, and this can be modulated by levels of wild-type β-catenin or IFT140, with the Arm34–87 effect being enhanced or suppressed. Importantly, this mechanism is conserved in mammals with the equivalent β-catenin24–79 peptide blocking nuclear translocation and pathway activation, including in cancer cells. Our work indicates that Wnt signaling can be regulated by a defined N-terminal β-catenin peptide and thus might serve as an entry point for therapeutic applications to attenuate Wnt/β-catenin signaling.
In brief
Vuong and Mlodzik demonstrate that an N-terminal peptide of β-catenin inhibits nuclear translocation of endogenous full-length β-catenin. The inhibitory effect of the peptide is at the level of stable β-catenin, and hence, it can attenuate Wnt signaling in mutant contexts that lead to stable β-catenin or can even stabilize mutations within β-catenin.
Graphical abstract
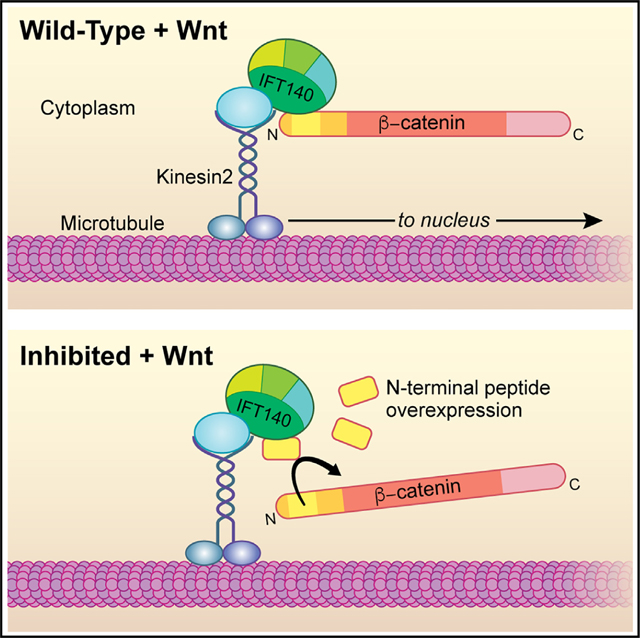
INTRODUCTION
Wnt/Wingless (Wg) pathways are important intercellular signaling pathways in all metazoans. They regulate many processes during embryonic development, including cell growth, migration, fate determination, polarity, stem cell homeostasis, and organogenesis.1–6 Wnt signaling is evolutionarily conserved, and secreted Wnt/Wg proteins activate a variety of signal transduction events across all metazoans.4,6,7 Wnt signaling is also closely related to several diseases, including initiation and progression of various types of cancers.8–13
Canonical Wnt signaling is keyed by nuclear translocation of β-catenin upon pathway activation. β-catenin, Armadillo/Arm in Drosophila, is the “business end” pathway component, with its cytoplasmic stabilization and nuclear translocation setting it up as a transcriptional co-activator, essential for Wnt target activation.4–6,14 Wnt/Wg proteins bind to the Frizzled (Fz) and LRP5/6 (Arrow in Drosophila) co-receptors, with their binding resulting in the disassembly of the “destruction complex” (DC), composed of Axin, APC (adenomatous polyposis coli), GSK3β, and CK1 ∝. In the absence of Wnt ligands, the DC phosphorylates cytoplasmic Arm/β-catenin and targets it for degradation.4–6,14–17 Breakup of the DC is mediated by Disheveled (Dsh/Dvl), causing re-location of Axin to the plasma membrane, where it associates with Dsh/Dvl and the Fz and LRP5/6 co-receptors, leading to Axin-Dsh-Fz-LRP5/6 aggregates, generally referred to as signalosomes.4,6,14,18 Removal of Axin or APC from the DC leads to stabilization of cytoplasmic Arm/β-catenin, allowing its translocation to the nucleus as co-activator of TCF/LEF transcription factors.4,6,14,15,19,20
Arm/β-catenin contains a large region formed by several repeats, the “Arm repeat domain,” which is flanked by distinct N- and C-terminal regions.21–23 The Arm repeats of Arm/β-catenin form a concave groove called “ARM domain,” which binds competitively to cadherins, APC, TCF/LEF1, and Axin.23–25 The ARM domain plays a role in both canonical Wnt/Wg signaling and the formation of adherens junctions (AJs).23–25 The C-terminal region functions specifically in Wnt/Wg signaling, serving as an interaction surface for complexes promoting Arm/β-catenin-mediated transcription, via factors like CBP/p300 and SET-1,21 for example. Unlike the ARM domain, the N-terminal Arm/β-catenin region is less well understood and thus far without known structure.26 It is essential for regulation by the DC, involving phosphorylation by CK1 ∝ and GSK3β.27–29 CK1 family members phosphorylate Arm/β-catenin at serine 45, required as priming phosphorylation for subsequent GSK3β phosphorylation events at residues 33, 37, and 4130 (also Figure 1A). In Drosophila, removal of the N-terminal region or deletion of 53 amino acids (aa 34–87) around the phosphorylation sites (the latter called ArmS10) leads to highly stable cytoplasmic Arm/β-catenin protein, causing constitutive activation of Wnt/β-catenin signaling, independent of ligand activation.29,31
Figure 1. Arm34–87 peptide interferes with Arm/β-catenin signaling in Drosophila wing development.
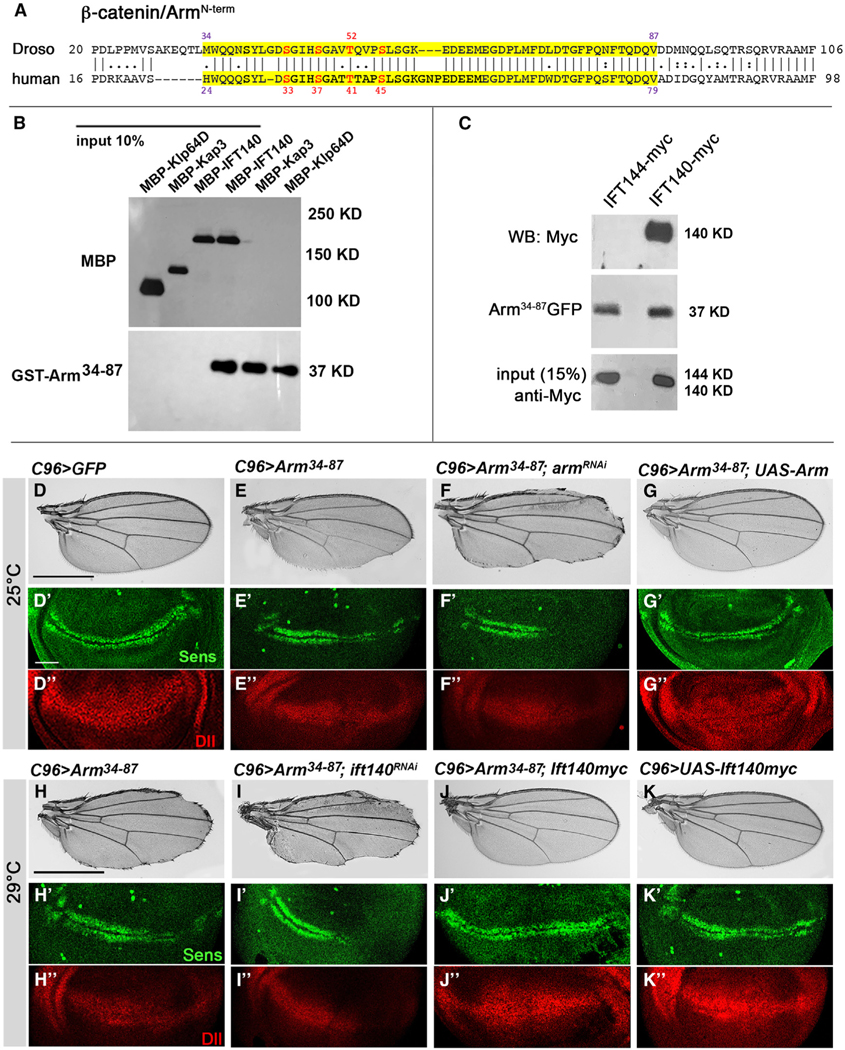
(A) Drosophila Arm and human β-catenin N-terminal sequence with Arm34–87 and β-cat24–79 peptides marked in yellow. Arm34–87 or β-cat24–79 is highly conserved between Drosophila and mammals. Numbered red residues are phosphorylation sites of GSK3β (residues 33, 37, and 45 in human) and CK1 α (residue 41 in human) within this region of β-catenin.
(B and C) Arm34–87 and IFT140 are physically associated. (B) Direct binding of Arm34–87 and IFT140. IFT140 (MBP fusion, lane 3: input 10%) was pulled down by GST-Arm34–87 (lane 4), whereas full-length MBP-Kap3 and MBP-Klp64D (lanes 1 and 2 from left: input 10%) were not pulled down (lanes 5 and 6). (C) In vivo co-immunoprecipitation assay of IFT144 and IFT140 with Arm34–87 from nub>IFT144myc; Arm34–87GFP (negative control, lane 1) and nub>IFT140myc; Arm34–87GFP (lane 2) wing imaginal discs. Protein extracts from wing discs were co-immunoprecipitated with anti-GFP. Co-immunoprecipitations and input (15% of wing disc lysates used in co-immunoprecipitation) were analyzed by blotting with anti-myc to IFT144 or IFT140.
(D–K″) All wings and wing discs shown use the C96-Gal4 driver (Figure S1C shows expression pattern: C96 is expressed at a slightly higher level in the posterior region of wing discs, right half in picture). Adult wings: anterior is up and distal right; wing discs: dorsal is up and anterior left.
(D–G″) Interaction between Arm34–87 and endogenous Arm. All genotypes were reared at 25°C. (D–D″) UAS-GFP (C96>GFP) control wing and control wing disc with wild-type Sens (green) (D′) and Dll (red) (D″) expression near D/V boundary. (E–E″) C96>Arm34–87 wing and wing disc. Note partial loss of margin (E) and partial loss of Sens (green, E′) and reduction of Dll (red, E″), consistent with adult wing defects. (F–F″) C96>Arm34–87; UAS-armRNAi. Margin loss caused by Arm34–87 was enhanced, and Sens (F′) and Dll (F″) expression was further reduced (cf. to E–E″). (G–G″) C96>Arm34–87 >Armwt: note that phenotype caused by Arm34–87 is suppressed by co-expression of wild-type Arm, and Sens (green, G′) and Dll expression (red, G″) were restored.
(H–K″) Interaction between IFT140 and Arm34–87 (all genotypes reared at 29°C). (H–H″) C96>Arm34–87: note increased margin loss at 29°C (cf. to E) and reduced Sens (H′) and Dll expression (H″). (I–I″) C96>Arm34–87; >Ift140RNAi: wing margin defects, and expression of Sens (I′) and Dll (I″) were reduced, compared to C96>Arm34–87 (cf. to H–H″). (J–J″) C96>Arm34–87; >IFT140myc: co-expression of IFT140myc in C96>Arm34–87 wings suppressed phenotype and restored Sens (J′) and Dll (J″) expression. (K–K″) C96>IFT140myc control: no effect of increased IFT140 levels by itself (wild-type margin and normal Sens and Dll expression in imaginal discs). Scale bar represents 100 μm in adult wings (D–K) and 50 μm in imaginal discs (D′–K″). Figure S1 shows quantification of Sens staining, as well as Figures S1 and S2.
Recently, we have shown that IFT-A forms a complex with Arm/β-catenin via its N-terminal region, facilitating nuclear translocation.29,32 IFT-A (intraflagellar transport A complex, known for its role in ciliogenesis) associates with Kinesin 2/Kif3a and promotes nuclear translocation of Arm/β-catenin upon Wnt/Wg pathway activation.29,32 Loss of function of either IFT-A complex components, and IFT140 in particular, or Kinesin 2 results in impaired Wnt/Wg signaling and developmental defects in Drosophila.29,32 Kinesin 2 interacts with IFT140 through Kap3 and acts as the motor to transport IFT-A along cytoplasmic microtubules. Both single and double mutant clones for kinesin 2 and ift140 fail to activate Wg/Wnt signaling targets in Drosophila.29 Moreover, double mutant clones for IFT140 and axin display high levels of stabilized cytoplasmic Arm/β-catenin, in both wing imaginal disc cells and salivary gland cells, but target gene activation and its nuclear translocation are markedly reduced or lost.29 Here, IFT140 directly binds to Arm/β-catenin through the N-terminal Arm34–87 region in Drosophila or the equivalent β-cat24–79 peptide in mammals. It is thus an intriguing question to determine whether this N-terminal region of Arm/β-catenin, Arm34–87 (or human β-cat24–79), plays a critical role in its nuclear translocation and whether it affects canonical Wnt signaling.
Here, we analyzed the mechanism of how this N-terminal peptide, Arm34–87/β-cat24–79 and IFT140, functions in canonical Wnt signaling. We show that Arm34–87/β-cat24–79 interacts with IFT140, both physically and genetically. Expression of this peptide is sufficient to antagonize endogenous Wnt/Wg signaling activation. We demonstrate that this antagonism is mediated by competitive binding to IFT140, thus inhibiting nuclear translocation of endogenous Arm/β-catenin. This mechanism is conserved in mammalian cells. Our study defines an important role of the N-terminal Arm/β-catenin region, particularly the Arm34–87/β-cat24–79 peptide, in Wnt/Wg signal transduction, in addition to its known function in the DC-mediated process. Our results indicate a mechanism and provide insight into potential therapeutic approaches to attenuate canonical Wnt signaling through (dominant) inhibition of its nuclear translocation.
RESULTS
Arm/β-catenin peptide, Arm34–87, is sufficient to bind to IFT140
IFT-A and Arm/β-catenin are associated in a protein complex with Kinesin 2, in which IFT140 directly interacted with Arm/β-catenin.29 To confirm and further refine the interaction between IFT140 and Arm/β-catenin, we established that the binding of a small protein fragment/peptide within the N-terminal Arm region, residues 34–87 (Figure 1A, shaded in yellow), is specific to and sufficient for IFT140 binding (Figure 1B). Other components of the Kinesin 2 protein complex, Klp64D (Drosophila Kif3a) or Kap3 (kinesin-associated protein 3) did not bind the Arm34–87 fragment, serving as control (Figure 1B). To confirm this in vivo, we performed co-immunoprecipitation assays with Drosophila wing disc extracts expressing Arm34–87-GFP and IFT140 or IFT144 (under nubbin-Gal4 throughout the wing pouch of wing discs33). Extracts from nubbin>IFT140-myc; Arm34–87-GFP and nubbin>IFT144-myc; Arm34–87-GFP wing discs were immunoprecipitated using anti-GFP and probed with anti-myc (Figure 1C), revealing that IFT140 co-immunoprecipitated with Arm34–87, but IFT144 did not (Figure 1C). These data indicate that IFT140 directly binds to Arm/β-catenin within the small N-terminal region encompassing residues 34–87, hence called Arm34–87, and that this Arm peptide is sufficient for interaction with IFT140 in vitro and in vivo.
Arm34–87 affects arm/β-catenin function in Wg signaling
Next, we determined whether expression of the Arm34–87 peptide could impact Wg signaling in vivo. Ubiquitous expression of UAS-Arm34–87 or UAS-Arm34–87-GFP transgenes (both inserted at the same attB-chromosome site34) caused larval lethality, suggesting that the Arm peptide can dominantly interfere with normal development. To define how Arm34–87 affects development, we used Drosophila wing margin patterning as a model system: expression of UAS-Arm34–87 along the dorsal-ventral boundary of wing discs, the future wing margin (under C96-Gal4 control35,36; Figures S1D and S1D′ show expression domain), resulted in partial loss of margin tissue (Figures 1D, 1E, and S1A–S11C). Quantification of this phenotype, measuring the length of margin loss, revealed that Arm34–87 expression resulted in a ~50% loss in margin fate. To ascertain whether Arm34–87 acted as a dominant-negative, interfering with endogenous Arm/β-catenin, we asked whether the Arm34–87 phenotype can be modulated by reducing or increasing Arm/β-catenin levels. Indeed, the effects of Arm34–87 were enhanced by a mild arm knockdown (Figure 1F) or suppressed by co-expression of wild-type Arm/β-catenin (Figure 1G). Distal-less (Dll) and senseless (sens) are transcription targets of Wnt/Wg signaling during wing development, with sens being a high-threshold target, defining the future margin cells, and Dll a general wing pouch target.37–40 Consistent with Arm34–87 interfering with canonical Wnt/Wg signaling, C96>Arm34–87 expression caused either loss of expression in a subset of cells along the wing margin (Sens protein) or markedly reduced Dll expression in distal wing tissue (Figures 1E′, 1E″, S1D–S1K, and S4B for outline of quantification) compared to control discs (Figures 1D′, 1D″, S1D, and S1E). Consistent with Arm34–87 acting like a dominant interfering tool, defects in Sens and Dll protein expression induced by Arm34–87 expression were enhanced or suppressed by arm RNAi knockdown or full-length Arm/β-catenin co-expression (Figures 1F′, 1F″, 1G′, and 1G″). The wing margin loss phenotype induced by Arm34–87 expression was not caused by unspecific cell death, as the Arm34–87 phenotype was not noticeably affected by co-expression of the cell death inhibitor p35,41 while wing margin defects caused by expression of the proapoptotic gene hid (C96>UAS-hid) were rescued by p35 co-expression (Figure S2). Accordingly, Arm34–87 expression did not induce cleaved caspase 3 staining as a marker for cell death (Figure S2).
These data suggest that the dominant-negative in vivo effects of Arm34–87 during development were caused by inhibition of canonical Wnt/Wg signaling at the level of Arm/β-catenin and Wg signaling target gene expression.
Arm34–87 interferes with IFT140 function
To determine whether the effects of Arm34–87 on canonical Wg signaling were caused by interference with IFT140/IFT-A function in canonical Wnt/Wg signaling, we tested whether Arm34–87 was sensitive to IFT140 levels. IFT140 knockdown (via RNAi or in mutant clones) displayed similar margin loss as Arm34–87 expression.29,32 Strikingly, reducing IFT140 levels enhanced the Arm34–87 phenotype, causing an increase of lost margin tissue (Figure 1I), while co-expression of IFT140 with Arm34–87 (increasing IFT140 levels) suppressed Arm34–87-induced margin defects (Figure 1J, cf; Figure 1H; note that C96-driven expression of IFT140 alone [in a wild-type background] had no effect; Figure 1K). Consistent with adult wing margin defects, Arm34–87 effects analyzed in wing discs, revealed reduction or loss of Sens expression (along D/V-boundary) and reduction in Dll protein levels (Figures 1H′ and 1H″, compare to wild-type (WT) control; Figures 1D′ and 1D″ quantified for Sens intensity in Figures S1D and S4A). Arm34–87-induced defects in Sens and Dll expression were consistently enhanced or rescued by IFT140 knockdown (Figures 1I′, 1I′, and S1D) or IFT140 protein co-expression (Figures 1J′ and 1J″), respectively (C96>IFT140 control alone caused no defects in either Sens or Dll expression; Figures 1K′ and 1K″). These data suggest that Arm34–87 is acting as a dominant-negative in canonical Wg signaling by interfering with the interaction between Arm/β-catenin and IFT140.
IFT140 and full-length Arm/β-catenin co-localize in cytoplasmic puncta in Drosophila wing imaginal disc and salivary gland cells upon Wnt/Wg signaling activation.29 As Arm34–87 expression causes a dominant-negative effect on Wg signaling and physically interacts with IFT140, we examined whether Arm34–87 co-localizes with IFT140 and affects the overlapping localization of IFT140 and endogenous Arm/β-catenin. There are no Arm34–87-specific antibodies, so we used tagged Arm34–87-GFP and full-length Arm to distinguish between peptide and endogenous protein, together with IFT140-myc. To allow subcellular localization analyses with sufficient resolution, we used the in vivo Wg signaling assay in the large salivary gland cells.29 In absence of Wg, full-length Arm was detected only at AJs (Figure 2A, magenta), while Arm34–87-GFP (Figure 2A, green) displayed intracellular puncta. Interestingly, these puncta co-stained for IFT140-myc (Figure 2A, red; Figures S3A and S3E), indicating that (1) Arm34–87 is not tethered to AJs, and (2) it interacts with IFT140 in the cytoplasm in absence of Wg signaling, indicating that Arm34–87 forms complexes with IFT-A independent of Wg/Wnt signaling activation, explaining its dominant-negative behavior besides competitive binding with endogenous Arm upon Wnt signaling activation. Upon Wg expression (C805-Gal4>UAS-Wg), full-length Arm was detected both at AJs and in cytoplasmic puncta (Figure 2B; also Vuong et al.29), with Arm, Arm34–87-GFP, and IFT140 triple-positive puncta detected (Figures 2B, S3A–S3C, and S3E). Co-localization of Arm34–87-GFP and endogenous Arm upon Wg signaling induction (Figures S3B and S3C) was dependent on IFT140 presence (it was lost in ift140cx2 mutants; Figure S3D) and thus IFT-A dependent. This observation suggests the formation of large complexes of several IFT-As.
Figure 2. IFT140 and Arm34–87 co-localize in salivary glands and Arm34–87 blocks nuclear translocation of endogenous Arm/β-catenin.
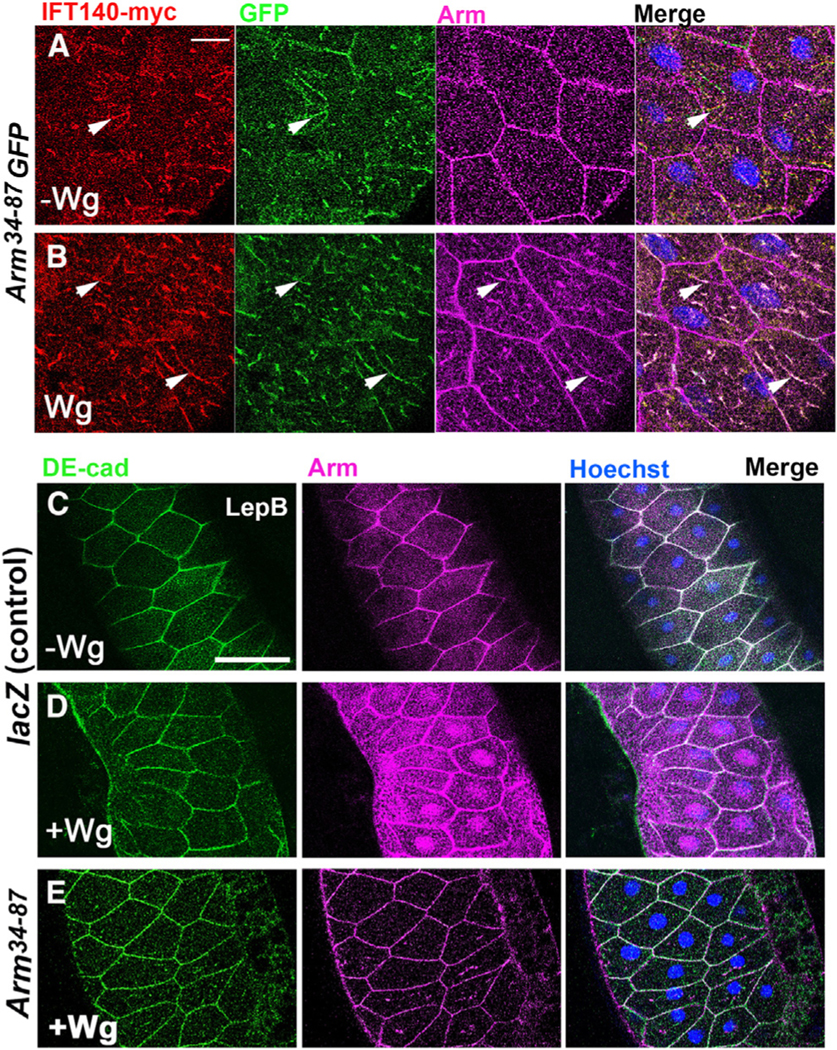
(A and B) Salivary glands (SGs) stained for IFT140-myc (red), Arm34–87-GFP (green, GFP), and endogenous Arm (magenta). (A) Without Wg, Arm/β-catenin localizes to adherens junctions (AJs), and Arm34–87-GFP does not localize to AJs, displaying punctate cytoplasmic staining, partially overlapping with IFT140-myc (arrowheads: examples of co-stained puncta). (B) Upon Wg expression, Arm/β-catenin is found in the cytoplasm, and triple-positive puncta staining for Arm34–87-GFP, IFT140-myc, and endogenous Arm are detected (arrowheads: examples of triple co-stained puncta). Scale bar: 25 μm.
(C–E) Wg-signaling-induced Arm nuclear translocation assay. SGs were exposed to Wg (via C805-Gal4; note mosaic expression, common to all SG drivers) and treated with LepB (inhibiting nuclear export) to enhance nuclear Arm/β-catenin retention. SGs were stained for DE-cad (green, membrane marker), endogenous Arm (magenta), and Hoechst (nuclei, blue). Scale bar represents 100 μm. (C) UAS-lacZ (without Wg, control); Arm/β-catenin mainly localizes to AJs at membrane. (D) Wg expression (>Wg, >LacZ; positive control): note increased cytoplasmic Arm/β-catenin and nuclear localization (uneven Arm/β-catenin levels due to mosaic expression of Gal4-driver). (E) Co-expression of Arm34–87 with Wg (>Wg, >Arm34–87) largely eliminates nuclear translocation of endogenous Arm/β-catenin. Also Figure S3.
Together, these results suggest that (1) Arm34–87-GFP is cytoplasmic and does not associate with AJs, (2) the majority of Arm34–87-GFP co-localized with IFT140 in the presence or absence of Wg signaling activation, and (3) Arm34–87GFP and Arm can display overlapping cytoplasmic localization, dependent on IFT140, which suggests that large complexes of unknown stoichiometry can form.
Arm34–87 inhibits nuclear localization of endogenous arm/β-catenin
Kinesin 2/IFT-A complexes are required for nuclear localization of Arm/β-catenin.29 Since Arm34–87 is necessary and sufficient for IFT-A binding (via IFT140), we examined whether Arm34–87 could affect nuclear translocation of endogenous Arm upon Wg signaling activation, using the established in vivo salivary gland (SG) assay.29 Wg signaling was activated via the SG-specific C805-Gal4 driving UAS-Wg (note that all known SG drivers display mosaic expression42), inducing nuclear translocation of Arm/β-catenin (detected with anti-Arm; to facilitate nuclear Arm detection, SGs were treated with LepB, a CRM1 inhibitor, blocking nuclear export43; DE-cad and Hoechst were cell membrane and nuclear markers, respectively). Wg expression activated the pathway causing increased Arm levels, both in nuclei and cytoplasm, demonstrating Wg-induced Arm/β-catenin stabilization and nuclear translocation (Figure 2D, note cytoplasmic Arm stabilization and nuclear levels being variable from cell to cell due to mosaic expression of the Gal4-driver). In contrast, Arm/β-catenin was only detected at AJs when Wg was not expressed (Figure 2C). Importantly, co-expression of Arm34–87 with Wg in SGs revealed a loss of nuclear Arm/β-catenin (Figure 2E), indicating that Arm34–87 was interfering with nuclear translocation of endogenous Arm/β-catenin.
Together with the genetic and biochemical results above, these data indicate that Arm34–87 interferes with the Kinesin 2/IFT-A-complex-mediated nuclear translocation of Arm/β-catenin by competing for binding to that complex, thus attenuating Wnt signaling by blocking endogenous Arm/β-catenin nuclear transport.
Function of Arm34–87/β-catenin24–79 is evolutionarily conserved
Sequences across the Arm34–87 region are highly conserved in β-catenin of all higher animals29 (Figure 1A). To determine whether its function in nuclear β-catenin localization is conserved in mammals, we tested the respective mouse peptide, β-cat24–79-GFP, in MEFs (mouse embryonic fibroblasts). WT MEFs transfected with full-length β-catenin-GFP revealed detectable levels of nuclear β-catenin upon Wnt3a stimulation with both the GFP tag (Figure 3A) and β-catenin antibody (Figure 3E). In contrast, under the same Wnt3a conditions, β-cat24–79-GFP-transfected WT MEFs displayed a loss of endogenous nuclear β-catenin (Figures 3G–3J; note β-cat24–79-GFP alone is in the cytoplasm; Figure 3C, like its Drosophila counterpart), indicating a conserved behavior of β-cat24–79 in blocking nuclear translocation of endogenous β-catenin. Consistently with the Drosophila experiments, this function was dependent on Wnt signaling induction, as without Wnt3A treatment (Figures 3B–3D, 3H, and 3J), no effects on β-catenin localization were observed in transfected cells (Figures 3F–3J).
Figure 3. β-catenin34–87 blocks nuclear accumulation of β-catenin in mouse and human cells and inhibits Wnt signaling target expression.
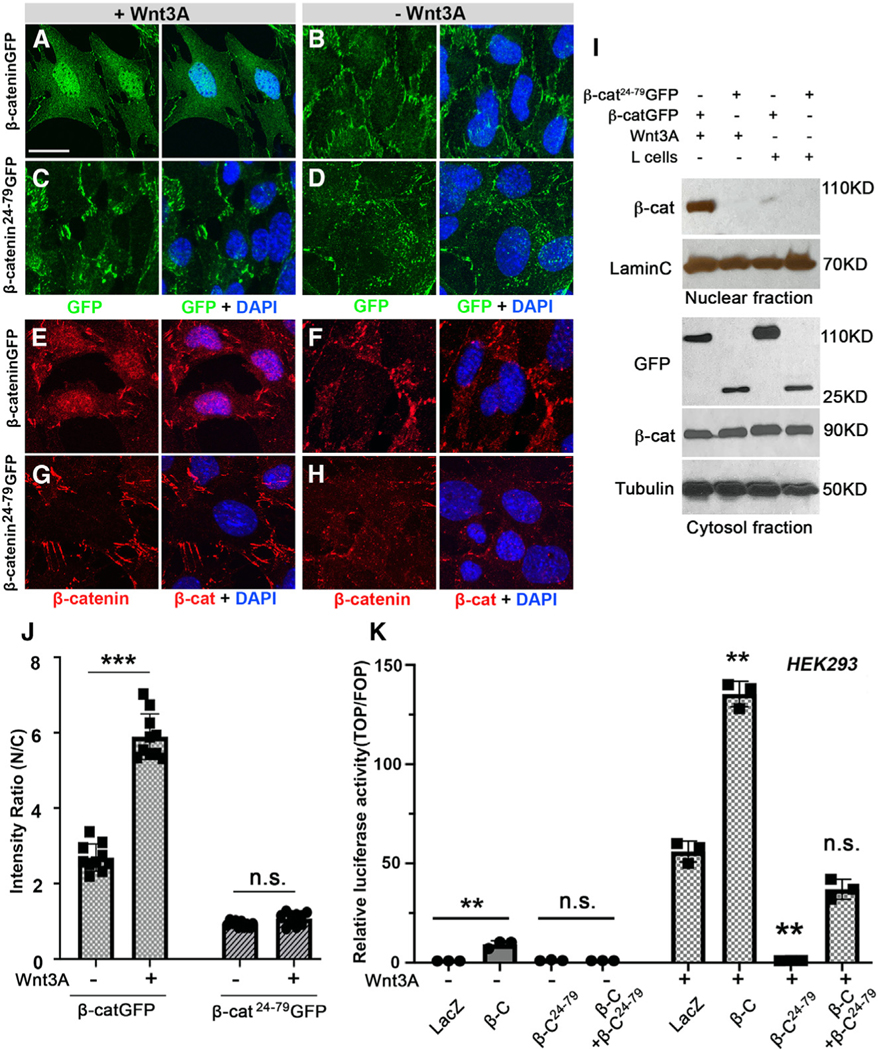
(A–H) Confocal images of β-catenin-GFP or the β-cat24–79-GFP peptide (green) or endogenous β-catenin (red), single channel on left or overlayed with DAPI staining (blue, on right), in MEFs treated with Wnt3A-conditioned media (A, C, E, and G) or without Wnt3A control (L-cell control medium) (B, D, F, and H). Upon 24 h of Wnt3A stimulation, nuclear β-catenin was detectable in wild-type MEFs either by GFP (A; green) or endogenous β-catenin antibody (red; E). In MEFs expressing β-cat24–79, it was undetectable in nuclei, either by GFP (green; C) or β-catenin antibody (red; G). Nuclear β-catenin was not detected in MEFs cultured in control media without Wnt3A (B, D, F, and H). Membrane-associated β-catenin signal was undistinguishable in all MEFs, with Wnt3A-conditioned or control L-cell media (A–H). Scale bar: 50 μm.
(I) Western blots of nuclear and total cytosolic fraction with Wnt3A-conditioned or control L-cell media. γ-Tubulin and LaminC serve as loading controls for cytoplasm and nuclei, respectively. Note nuclear full-length β-catenin upon Wnt treatment and the lack thereof in the presence of β-cat24–79-GFP.
(J) Quantification of nuclear β-catenin and β-cat24–79-GFP signal in MEFs as ratio of nuclear (N) vs. cytoplasmic (C) signal. Y axis denominates ratio of green or red intensity values of selected regions (area of 4.705 μm × 4.843 μm) within nuclei and cytoplasm of individual cells (membrane-associated β-catenin was purposely excluded; see Figure S4C for quantification details). Mean ± SD values, obtained in randomly selected cells (n), are shown from five independent experiments; Student’s t test: ***p < 0.001; ns: not significant.
(K) β-cat24–79 inhibits Wnt signaling target activation in human HEK293 cells. Wnt signaling activity was assayed by comparing Wnt3A-stimulated cells and cells in control media (L-cells without Wnt3A) transfected with either LacZ (control), β-catenin full-length, β-cat24–79, or β-catenin together with β-cat24–79, respectively (indicated on X axis in graph). Relative luciferase activity (Y axis) shows ratio of firefly TOP/FOP reporter and Renilla luciferase control. Wnt3A-conditioned media were added 1 day prior to cell lysis. Overexpression of β-catenin increases luciferase activity, whereas transfection with β-cat24–79 causes a strong reduction of Wnt signaling activity (**p < 0.001 compared to LacZ control, 3 independent assays). Also Figure S4.
To confirm this, we analyzed nuclear β-catenin levels by western blotting. While control WT MEFs displayed a strong accumulation of nuclear β-catenin upon Wnt3A treatment (Figure 3I), MEFs transfected with β-cat24–79-GFP displayed hardly any detectable nuclear β-catenin under the same conditions (Figure 3I; note endogenous cytoplasmic β-catenin levels not being affected; Figure S4A). In summary, these data indicated that N-terminal β-cat24–79 displayed the same behavior as Drosophila Arm34–87, both inhibiting nuclear localization of Arm/β-catenin upon Wnt/Wg signaling activation.
Arm34–87/β-cat24–79 affects nuclear localization and target gene activation of stable β-catenin during development and in cancer cell lines
The dominant-negative effect of Arm34–87 in Drosophila wing development and its effects on nuclear β-catenin translocation in MEFs suggested that it functions as an inhibitor of Wnt signaling, possibly also in aberrant human Wnt signaling contexts. We thus carried out Wnt pathway activation assays using the TOP/FOP reporter (luciferase reporter under TCF/β-catenin transcriptional activation control) to measure target gene activation in human cells. We assayed HEK293 cells (human embryonic kidney cells) transfected with LacZ as control in the absence or presence of Wnt, with Wnt3A-containing medium added 1 day after transfection to induce signaling. Without Wnt3A, reporter activation was near zero. Addition of Wnt3A increased relative luciferase activity significantly (Figure 3K), and this was further increased by overexpressing β-catenin (Figure 3K). Conversely, β-cat24–79 transfection inhibited Wnt signaling to the basal level (compare to no Wnt3A, Figure 3K). When β-catenin and β-cat24–79 were co-expressed, Wnt signaling activity was markedly suppressed (Figure 3K), indicating that β-cat24–79 strongly inhibits Wnt signaling target gene activation in human cells.
The N-terminal β-catenin region is unstructured. Interestingly, the Arm34–87/β-cat24–79 peptide is a small region of exon 3 (amino acid residues 5–80 in mouse and human), which is known as a “mutation hotspot.”44–46 To evaluate whether the peptide has an effect on non-phosphorylatable, stable β-catenin mutations, we used first Drosophila ArmT52A, a stable Arm/β-catenin isoform.27 ArmT52A carries a mutation in the CK1 priming phospho-target site, T52A27,31 (Figure 1A), which is the same mutant residue as in colorectal cancer cell lines (SNU407 and CCK81)47,48 and lung cancer line (LXF289).46,49,50 These mutant isoforms are stable because they cannot be phosphorylated by kinases associated with the “destruction complex.”27,31 ArmT52A expression in wings with C96-Gal4 induced a Wg signaling gain-of-function (GOF) effect with extra margin bristles and associated increase in cells expressing sens (Figures 4A, 4C, 4A′, and 4C′). While C96>Arm34–87 caused a wing margin loss phenotype and loss of sens expression (Figures 4B and 4B′, also Figure 1), the Arm34–87 peptide reversed the ArmT52A GOF phenotype with sens expression back to near WT (Figures 4D and 4D′, quantified in Figures S4B and S4C).
Figure 4. β-catenin24–79 affects stable β-catenin mutations, inhibiting Wnt signaling and cell proliferation in human cancer cell lines.

(A–D′) In vivo inhibition of a stable Arm mutation. Arm34–87 rescues the phenotype caused by ArmT52A point mutation in the destruction complex target site. (A and A′) UAS-GFP (C96>GFP) control wing (A) and wing disc with wild-type Sens expression (magenta, A′) near D/V boundary. (B and B′) C96>Arm34–87 wing and wing disc. Note partial loss of margin (B) and Sens expression (B′). (C and C′) C96>ArmT52A: note ectopic margin bristle phenotype (C) and ectopic Sens expression (C′). (D and D′) C96>ArmT52A > Arm34–87: ectopic margin bristle phenotype is suppressed by Arm34–87 co-expression (D), and Sens level is restored (D′).
(E–H) Stable β-catS33Y is suppressed by β-cat24–79. Confocal images of β-catS33Y (red, single channel on left) and nuclei (blue in overlay) in MEFs (E) Nuclear β-catenin was detectable in β-catS33Y-transfected MEFs by β-catenin antibody (red), but it was undetectable in MEFs with β-cat24–79 transfection (F). No nuclear β-catenin was detected in any MEFs co-transfected with both β-catS33Y and inhibitory β-cat24–79 peptide, without (G) or with Wnt3A treatment (H). Membrane-associated β-catenin was undistinguishable in all conditions. Scale bar: 50 μm. (I–K) Proliferation of two colorectal cancer lines with constitutively active Wnt signaling, HCT116 (I) and DLD1 (J), was suppressed by β-cat24–79. Cells were transfected with LacZ (control, left in I and J) or β-cat24–79 (right in I and J; in absence of Wnt3A-conditioned media). Proliferation was assessed via the “cell proliferation assay” (STAR Methods). Note markedly reduced proliferation in β-cat24–79-transfected cells (quantified in K, left for HCT116 cells, and right panel for DLD1 cells). *p < 0.01; **p < 0.001; ***p < 0.0001, three independent assays), also visible as cell density with photographs (I and J; 72 h after transfection. Scale bar: 500 μm.
(L) Relative luciferase activity assay in HCT116 (left) and DLD1 cells (right panel). β-cat24–79-transfected cells show reduced reporter activity in absence or presence of Wnt3A treatment (**p < 0.001, ***p < 0.0001 three independent assays). Also Figures S4 and S5.
Similarly, β-catS33Y is a non-phosphorylatable mutation in the GSK3β phospho-target site S33 and a stable (“activated”) β-catenin mammalian mutant,51 with the same mutation associated with the SW48 colorectal cancer cell line.46 The WT MEFs transfected with β-catS33Y showed nuclear β-catenin accumulation without Wnt3A treatment (Figure 4E, quantified in Figure S4E; also Figures S4E′–S4F). Strikingly, MEFs co-transfected with β-catS33Y and β-cat24–79 showed a marked reduction of nuclear β-catenin without or upon Wnt3A treatment (Figures 4G, 4H, S4E, and S4F), indicating that β-cat24–79 inhibits nuclear localization of stable β-catenin (note that control MEFs transfected with β-cat24–79 alone also showed no detectable nuclear β-catenin, Figures 4F and S4E). To investigate the inhibitory potential of β-cat24–79 on Wnt/β-catenin signaling in disease contexts, we tested its effect on colorectal cancer (CRC) cell lines (HCT116 and DLD1)52–55 in which the Wnt pathway is locked in the “activated” state. While in HCT116, a mutant β-catenin isoform has Ser45 deleted in one allele,56,57 the DLD1 line carries a truncated APC protein, breaking apart the destruction complex and hence stabilizing β-catenin.56,58 We asked whether in the HCT116 and DLD1 cell lines transfection of β-cat24–79 could (1) reduce Wnt3A-induced gene expression as measured in the TOP/FOP luciferase assay and (2) inhibit cell proliferation. In HCT116 cells, addition of Wnt3A or transfection of full-length β-catenin increased luciferase activity when comparing to controls (Figure 4L), which is explained by HCT116 cells retaining one WT β-catenin allele. Expression of β-cat24–79 inhibited Wnt signaling to almost basal level in both HCT116 scenarios, with or without Wnt3A treatment (Figure 4L). Similar to the TOP/FOP assay, transfection of β-cat24–79 markedly reduced proliferation of HCT116 cells (Figures 4I and 4K). In DLD1 cells, mutant for APC, Wnt pathway activity is not changed when transfecting full-length β-catenin or adding Wnt3A (Figure 4L, right graph). However, Wnt pathway activity was again markedly decreased when transfected with β-cat24–87 (Figure 4L, right graph) with cell proliferation also reduced (Figures 4J and 4K). These results indicated that β-cat24–79 acts as a dominant-negative inhibiting Wnt signaling in cell lines with a constitutively active Wnt pathway.
To corroborate this, we assessed the impact of the peptide in two lung cancer cell lines (H1299 and H2009), breast cancer line (HCC1395), and a brain tumor line (SF295). Western blot analysis was used to determine endogenous Wnt3A levels in these cancer lines (Figure S5D). Wnt3A was expressed at high levels in these cell lines, with the A549 line serving as negative control (Figure S5D). Similar to the CRC lines, luciferase reporter activation by β-cat/TCF was suppressed in the presence of β-cat24–79 in H2009 and H1299 lung cancer lines and the SF295 brain tumor line (Figures S5A–S5C; these cancer lines express Wnt3A endogenously in an autocrine manner; Figure S5D). Similarly, transfection of β-cat24–79 markedly reduced proliferation of the breast cancer line HCC1395 to very similar levels as the LGK-974 drug, which targets Porcupine, thus inhibiting Wnt secretion and autocrine signaling59 (Figures S5F and S5G). Importantly, combining the drug and β-cat24–74 caused similar levels of suppression (Figure S5F), consistent with the notion that β-cat24–79 acts exclusively through Wnt signaling inhibition. β-cat24–79 also eliminated activation of the Wnt signaling reporter (Figure S5E). Co-transfection of full-length β-catenin partially rescued the effect caused by β-cat24–79 on Wnt3A activity (Figure S5E), consistent with the notion that β-cat24–79 competes with endogenous β-catenin (different levels of suppression or rescue of full-length β-catenin co-transfection are cell line dependent and thus likely β-catenin concentration dependent). Together, these results argue that β-cat24–79, equivalent to Arm34–87, is effective at reducing or even blocking Wnt signaling in a multitude of tumors.
DISCUSSION
We have identified a peptide in the N-terminal region of Arm/β-catenin, residues 34–87 in Drosophila and 24–79 in humans, referred to as Arm34–87 or β-cat24–79, as sufficient for its physical interaction with IFT140/IFT-A. As previously shown, the Kinesin 2/IFT-A complex is required for nuclear translocation of Arm/β-catenin.29 Expression of Arm34–87 in wings in Drosophila causes Wg signaling loss-of-function defects, manifesting in wing margin loss and loss or reduction of expression of the Wg signaling targets Sens and Dll. Arm34–87 expression inhibits nuclear translocation of endogenous Arm/β-catenin, and the same inhibitory behavior on Wnt signaling is observed in mammalian contexts, with β-cat24–79 transfection blocking Wnt3A-induced nuclear translocation of endogenous β-catenin and Wnt reporter expression in MEFs, HEK293 cells, and several human cancer lines. We conclude that Arm34–87/β-cat24–79 generally exerts its effects by blocking Wnt-induced nuclear translocation of endogenous Arm/β-catenin via binding to IFT140, without affect junctional Arm/β-catenin.
Arm/β-catenin is a multifunctional protein associated with cell adhesion at AJs, linking these to the actin cytoskeleton, and canonical Wnt signaling, here acting as the key nuclear effector. Entry of cytoplasmic Arm/β-catenin into the nucleus is critical in Wnt/β-catenin signaling. Since Arm/β-catenin has no nuclear localization sequence, it has long been speculated on how it gets translocated into the nucleus.60 Recently, the requirement of a Kinesin 2/IFT-A complex was identified for its nuclear translocation,29 with IFT140 and Arm/β-catenin directly interacting through a small N-terminal Arm/β-catenin region (Arm34–87). Deletion of these residues results in a stable isoform, called ArmS10,31 as it also contains all phosphorylation sites targeted by the destruction complex. ArmS10 is commonly used in Drosophila as a “constitutively active” Arm/β-catenin isoform.31 In fact, ArmS10 can enter the nucleus independently of the Kinesin 2/IFT-A function.29 While this is surprising at first, Kinesin 2/IFT140-independent, alternative nuclear translocation mechanisms have been recently proposed,61 and ArmS10 does not require further protection from association with the destruction complex due to the deletion. Our data argue strongly that Kinesin 2/IFT140-dependent nuclear translocation is the primary mechanism, as functional studies with the Arm34–87/β-cat24–79 peptide reduced or eliminated nuclear β-catenin translocation in Drosophila and mammalian cells.
The Arm34–87 peptide is necessary and sufficient for IFT140 binding. Functional in vivo assays suggest that Arm34–87 displays dominant-negative behavior on the IFT-A interaction of endogenous Arm/β-catenin, which is supported by the suppression or enhancement of Arm34–87 phenotypes by increasing or reducing IFT140 levels, respectively. Similarly, increasing Arm/β-catenin levels suppresses the dominant effect of Arm34–87, largely rescuing the phenotype and restoring Sens and Dll expression. Our biochemical data and co-localization studies indicate its dominant effect is mediated by competitive binding to IFT140, competing with endogenous Arm/β-catenin and suggesting that the Arm34–87 fragment is critical for the IFT140-β-catenin interaction and essential for normal Arm/β-catenin nuclear translocation during development and disease.
As β-catenin is the key effector of Wnt signaling responsive for signal transduction to the nucleus, our data suggest its nuclear translocation is explorable as a therapeutic target to attenuate a Wnt response. In Drosophila, Arm34–87 can rescue ectopic Wg signaling caused by a non-phosphorylatable β-catenin mutation (ArmT52A). Furthermore, this peptide also affects the nuclear localization and target gene activation of the stable β-catenin mutation S33Y in MEFs and in cancer lines with either stabilizing β-catenin mutations (HCT116) or mutations in APC, removing the destruction complex protein (DLD1). Similarly, Wnt-addicted (autocrine) cancer lines (H1299, H2009, SF25, and HCC1395) are all inhibited by expression of β-cat24–79. We thus not only define the function of a peptide within the N-terminal region of Arm/β-catenin, but our data should serve as an entry point for potential new diagnostics and therapeutic applications to detect and inhibit overactive Wnt/β-catenin signaling in disease contexts, including cancer. This is of particular significance, as there are currently no approved drugs that inhibit canonical Wnt signaling at the level of β-catenin or any level in the intracellular signaling relay.
Limitations of the study
Our work provides insight into a function of a conserved peptide within the N-terminal region of Arm/β-catenin that inhibits Wnt/Wg signaling. Although all presented data are consistent with the proposed model, the 50-aa peptide is relatively long as a potential therapeutic agent to treat Wnt-signaling-associated diseases, and our data rely on cell-based assays with cancer lines in mammalian contexts rather than real tumors. Confirmation in tumor models (human organoids or mouse models) and expansion of the work to define a minimal peptide will be important to push it further to the potential discovery of a therapeutic agent.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Marek Mlodzik (marek.mlodzik@mssm.edu).
Materials availability
All Drosophila strains generated in this study are available upon request.
Data and code availability
All data reported in this paper will be shared by the lead contact upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND STUDY PARTICIPANT DETAILS
Drosophila melanogaster
The Gal4/UAS system was used for expression of RNAi constructs (sometimes in combination with UAS-Dcr2) and other transgenes. Gal4-driver for wing margin during wing development was C96-Gal4 expressed around the dorsal-ventral compartment boundary of wing imaginal discs, and the Gal4-driver for salivary glands was C805-Gal4. All crosses were set up at 25°C or at 29°C, as indicated in the Figures.
METHOD DETAILS
Immunostaining and histology
Imaginal discs were dissected at 3rd instar larval stage in PBS and fixed in PBS, 4%PFA. Discs were washed 2 times in PBS 0.1% Triton X-100 (PBT), incubated in primary antibodies o/n at 4°C. After washing in PBT, incubation with secondary antibodies was at RT for 2hrs. Samples were mounted in Vectashield. Wing disc images were acquired with a confocal microscope (20X-40X, oil immersion, Leica SP8 or Zeiss LSM880 system). Images were processed with ImageJ (National Institutes of Health) and assembled in Photoshop (Adobe).
Salivary glands were dissected at 3rd instar larval stage in PBS and treated with 0.1% Leptomycin B (Sigma) in 5–10 min before fixation in PBS, 4% PFA. All subsequent steps were as described for wing imaginal discs.
Analyses of adult wings: wings were removed, incubated in PBT, and mounted on a slide in 80% glycerol in PBS, and imaged using Zeiss Axioplan microscope. All adult images were acquired using Zeiss Axiocam color-type 412–312 camera and the Zeiss axiocam Zen software.
Transgene construction
To generate transgenic flies, Arm34−87 was amplified by PCR using DGRC LD23131 cDNA (for Arm) and cloned into pUAS-attB and pUAS-attB-GFP vectors (VK1, second chromosome 2R 59D3) using NotI and XbaI sites. The following primers were used to make Arm34−87 constructs:
AR34−87: 5′- ATGTGGCAGCAGAATTCGTACTTGGGCGAC - 3′ and. 5′- CACTTGGTCTTGTGTGAAATTCTGCGGGAA - 3′
GST pull down
For GST pull-downs, IPTG-inducible E. coli R2 cells (BL21) were transformed with plasmid constructs for fusion proteins MBP-Kap3, MBP-Klp64D, MBP-IFT140 and GST-Arm34–87. Bacterial lysates were prepared and the fusion proteins were purified. An equal amount of glutathione Sepharose 4B beads with GST, GST fusion protein or beads alone were incubated with lysates containing MBP-fusion proteins O/N at 4°C. After several washes with pull-down buffer (20 mM Tris pH 7.5, 150 mM NaCl, 0.5 mM EDTA, 10% glycerol, 0.1% Triton X-100, 1mM DTT, and protease inhibitor cocktail), sample buffer was added, beads were boiled, and protein were resolved by SDS-PAGE. For Western blotting, proteins were transferred onto nitrocellulose, blocked in 5% skim milk and incubated with primary goat anti-GST or rabbit anti-MBP antibody. Protein bands were visualized using Immobilon Forte Western HRP Substrate kit.
Immunoprecipitation
Lysates from 30 wing imaginal discs of nub>IFT140myc, Arm34−87GFP, C96> IFT144myc, Arm34–87GFP were precleared by incubating with protein A-Agarose beads for 1 h at 4°C followed by centrifugation. A-Agarose beads were immune-precipitated with specific antibodies at 4°C for 1hr. Polyclonal anti-GFP antibody was used. Immunoprecipitates were resuspended in SDS sample buffer, boiled for 5 min, separated by SDS-PAGE, and transferred to nitrocellulose for immunoblotting. Protein was detected by Immobilon Forte Western HRP Substrate kit.
MEF immunofluorescence staining and Wnt3a-induced β-catenin localization
For Wnt3A-induced β-catenin nuclear translocation: MEF were grown at 70% confluence in DMEM medium supplemented with 10% fetal bovine serum. β-cat24−79GFP construct was generated from the original plasmid MSCV-β-catenin-IRES-GFP. β-catenin and β-cat24−79 were cloned into pCMV6-GFP vector using HindIII and NotI. These constructs were transfected into MEFs (20μg/μL) by Lipofectamine LTX with PLUS Reagent. Cells were treated with Wnt3A conditioned medium or L-cells medium (as control) a day after transfection. Cells were collected 12–16h after treatment with medium. Cells were then fixed in cold methanol for 15 min at −20°C and labeled with primary antibodies for GFP or β-catenin diluted 1:100 in 2% bovine serum albumin/PBS for 2 h, washed in PBS, incubated with fluorescent secondary antibodies diluted 1:500 in PBS for 1 h, and mounted with Vectashield mounting medium.
β-catenin nuclear fraction assay
β-cat24−79GFP transfected into MEFs, stimulated with Wnt3A or unstimulated (control supernatant), were gently washed with PBS and cells were minced on ice by sharp scalpel and collected by centrifugation at 5000rpm/4°C. Samples were resuspended in 500μm buffer comprising 250mM sucrose, 50mM Tris-Cl pH7.4, 5mM MgCl2, 1M EDTA, 1% Triton X-100 and protease inhibitor cocktail and gently homogenized for 1 min on ice using homogenizer. Samples were kept for 30 min at 4°C (on ice) in microfuge tubes, and supernatants were cleared by 20 min centrifugation at 4°C and saved as the cytoplasmic fraction. The pellet, or nuclear fraction, was washed by the lysis buffer without protease inhibitor cocktail. Nuclear fraction was incubated for 30 min at 4°C with nuclear extract buffer containing 20mM HEPES ph7.9, 15mM MgCl2, 0.5M NaCl, 1M EDTA, 20% glycerol, 1% Triton X-100 protease inhibitor cocktail and sonicated for 10″ after incubation. The resulting supernatant was collected by 30min centrifugation at 4°C as the nuclear fraction. Protein extracts were boiled for 5 min at 95°C in SDS-sample buffer, separated by 10% SDS-page gel and transferred to nitrocellulose. Protein levels were analyzed by immunoblotting with the corresponding antibodies.
Cell lines and culture conditions
HEK293, Wnt3a-expressing L-cells and control L-cells were grown in Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with 10% fetal bovine serum (FBS), and 1% penicillin/streptomycin. Wnt3A and control L-cells were prepared as described by a protocol provided by ATCC. Human cancer cell lines DLD1, HCT116 were grown in DMEM supplemented with 10%FBS. H1299, H2009, HCC1395 and SF295 cell lines were grown in RPMI 1640 medium supplemented with 10% FBS. All cells were cultured at 37°C in 5% CO2.
Luciferase reporter assay for β-catenin activity (TOP/FOP reporter assay)
Human full-length β-catenin, β-catenin24−79 or lacZ were cloned into pcDNA4/TO plasmids. For HEK293 cell and all cancer cells, cells were seeded at a density of 4×105 cells/12-well plate one day before transfection. The cells were then transfected with plasmids constructs (LacZ, full-length β-catenin or β-catenin24−79:2 μ g each) together with 3 μ g pGL-TOP or its negative control vector pGL-FOP. Renilla reporter plasmid pRL-CMV was used for normalization. All transfections were done using 1 mg/ml Lipofectamine. For HEK293, HCT116 or DLD1 cell lines, after 24h, Wnt3A condition medium was added to the cells and incubated for additional 24h. The treated cells were then washed and lysed using 20 μl luciferase lysis buffer per well, and luciferase activities were performed and measured in 96-well plates using Dual-Luciferase Reporter Assay system according to the manufacturer’s protocol. The measurement was conducted on Synergy MX luminometer. Experiments were carried out in triplicate and repeated at least three times as the mean ± SD of the ratio between the TOP/FOP and renilla reporters.
Cell proliferation assay
The cell proliferation assay was determined by 3-(4, 5-dimethylthiazol-2-yl)-2,5-diphenyl-tetrazolium bromide (MTT) assay (Sigma). All the cancer cell lines were seeded in 24-well plates (2×104 cells/well). After 24hr, cells were transfected with full length β-catenin or β-cat24–79. In HCC1395 cell line, the cells were then incubated with medium containing LGK-974 (10 μM) for 48h and then change back to RPMI 1640 medium for cell proliferation assay. Absorbance at 570nm was read on a microplate reader. All assays were performed in triplicate.
QUANTIFICATION AND STATISTICAL ANALYSIS
Quantitative analysis of wing discs
Wing imaginal discs staining images were processed using ImageJ. To establish an appropriate quantification of signal, Sens intensity was normalized by subtracting the signal in the Sens negative (–) cells from the signal obtained in Sens positive region, “(+) cells”. Mean measurements were plotted for 10 wing discs. For statistical analyses, a two tailed t test was performed on normalized mean intensity measurements to compare genotypes.
Quantification of β-catenin nuclear translocation via microscopy analysis
Confocal imaging was performed using a Zeiss LSM880 microscope. Z stacks of 6 (1.083μm) to 12 optical slices (4.362μm) at 8-bit were captured and analyzed via ImageJ-Fiji. β-catenin translocation was evaluated through optical density assays of mean gray values between the nucleus and cytoplasm of individual cells. To maintain uniformity in all acquisitions, measurements were obtained under an image size of 1024×1024 pixels, FITC (green) channel or TRITC (red) channel, maximum projection (Z-project on ImageJ), and a standardized region of interest at 4.705μm × 4.843μm avoiding cell membrane regions. Intensity ratios (nucleus/cytoplasm), standard deviations, and student’s two-tailed test were performed using Microsoft Excel and Prism.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| Mouse anti-Arm | DSHB | N2 7A1 RRID: AB 528089 |
| Mouse anti-Myc | Santa Cruz | Cat# sc-40 RRID:AB 627268 |
| Mouse anti-GFP | Roche | Cat# 11814460001 RRID:AB 390913 |
| Mouse anti-Axin | R.Nusse | N/A |
| Mouse anti-γ-tubulin | Sigma-Aldrich | Cat# T6557 RRID:AB 477584 |
| Mouse anti-GST | Invitrogen | Cat# MA4–004-HRP RRID:AB 2537634 |
| Rat anti-DE cad | DSHB | DCAD2 RRID: AB 528120 |
| Rat anti-Dll | Jun Wu | N/A |
| Rabbit anti-Myc | Santa Cruz | Cat# d1–717 RRID:AB 647957 |
| Rabbit anti-MBP | Sigma-Aldrich | Cat# SAB2104171 RRID:AB 10668141 |
| Rabbit anti-β-catenin | Invitrogen | Cat# AHO0462 RRID:AB 1500389 |
| Rabbit anti Caspase 3 | Cell signaling Technology | Cat# 9661 RRID: AB 2341188 |
| Guinea pig anti-Sens | H. Bellen | N/A |
| Mouse anti Wg | DSHB | 4D4 RRID: AB 528512 |
| Rabbit anti Wnt3A | Abcam | AB 219412 |
| Rabbit anti LaminC | Abcam | AB 125679 |
| Chicken anti-GFP | Aves Labs | Cat# GFP-1020 RRID:AB 10000240 |
| Anti Rat-FITC | Jackson ImmunoResearch | Cat# 712–095-153 RRID:AB 2340652 |
| Anti Chicken FITC | Jackson ImmunoResearch | Cat# 703–095-155 RRID:AB 2340356 |
| Anti Guinea pig TRITC | Jackson ImmunoResearch | Cat# 706–025-148 RRID:AB 2340445 |
| Anti Mouse FITC | Jackson ImmunoResearch | Cat# 715–095-151 RRID:AB 2335588 |
| Anti Rat TRITC | Jackson ImmunoResearch | Cat# 712–025-153 RRID:AB 2340636 |
| Anti Rabbit Alexa Fluor 647 | Jackson ImmunoResearch | Cat# 711–605-152 RRID:AB 2492288 |
| Anti Rat Alexa Fluor 647 | Jackson ImmunoResearch | Cat# 712–605-150 RRID:AB 2340693 |
| Anti Rabbit Alexa Flour 488 | Life Technologies | Cat# A-11008 RRID:AB 143165 |
| Anti Rabbit HRP | Jackson ImmunoResearch | Cat# 711–165-152 RRID:AB 2307433 |
| Anti Rat HRP | Jackson ImmunoResearch | Cat# 712–035-153 RRID:AB 2340639 |
| Anti Mouse HRP | Jackson ImmunoResearch | Cat# 715–035-150 RRID:AB 2340770 |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| Leptomycine B | Sigma-Aldrich | L2913–5UG |
| Porcn Inhibitor IV (LGK-974) | Sigma-Aldrich | 1243244–14-5 |
| Lipofectamine LTX with PLUS Reagent | Invitrogen | A12621 |
| Paraformaldehyde 32% | EMS | 15714-S |
| Vectashield with DAPI | Vector Lab | H-1200 |
| Glutathione Sepharose 4B | GE Healthcare | 17–0756-01 |
| Immobilon Forte Western HRP Substrate | Millipore | WBLUF0500 |
| Pierce™ Protein A Agarose | Thermo Scientific™ | 20333 |
| Protease Inhibitor Tablets, EDTA-Free | Thermo Scientific™ | A32965 Lot# UL2876847 |
| Dulbecco’s Modified Eagle Medium (DMEM) | Gibco | 11965092 |
| RPMI 1640 | Millipore Sigma | R8758 |
| Fetal Bovine Serum | Thermo Scientific™ | A5256701 |
| G418 | Gibco | 11811–098 |
| Hoechst 33342 | Thermo Scientific™ | H3570 |
| Penicillin/Streptomycin | Gibco | Cat# 15140–122 Lot# 1322643 |
| Dual-Luciferase Reporter Assay system | Promega | E1910 |
| MTT assay | Abcam | Ab211091 |
|
| ||
| Experimental models: Cell lines | ||
|
| ||
| HEK293 | Stuart Aaronson | N/A |
| H2009 | Stuart Aaronson | N/A |
| H1299 | Stuart Aaronson | N/A |
| SF295 | Stuart Aaronson | N/A |
| HCC1395 | Stuart Aaronson | N/A |
| DLD1 | Stuart Aaronson | N/A |
| HCT116 | Stuart Aaronson | N/A |
|
| ||
| Experimental models: Organisms/strains | ||
|
| ||
| D.melanogaster:UAS-IFT140 myc | T.Avidor-Reiss | N/A |
| D.melanogaster:UAS-ArmT52A | K. Cadigan | N/A |
| D.melanogaster:IFT140cx2 (rempAl(2)21Ci1)l | M.Kernan | N/A |
| D.melanogaster:UAS-IFT144 myc | T.Avidor-Reiss | N/A |
| D.melanogaster:UAS-IFT140 RNAi (rempA) | VDRC | VDRC: v31575 Flybase: FBst0459093 |
| D.melanogaster:UAS-ArmRNAi | VDRC | VDRC: v107344 Flybase: FBst0479166 |
| D.melanogaster:UAS-Wg | BDSC | BDSC: BL108487 Flybase: FBst0307132 |
| D.melanogaster:UAS-hid | BDSC | BDSC: BL65403 Flybase: FBst0065403 |
| D.melanogaster:UAS-GFP | BDSC | BDSC: BL32184 Flybase: FBti0131930 |
| D.melanogaster:UAS-lacZ | BDSC | BDSC: BL1776 Flybase: FBtp0000355 |
| D.melanogaster:UAS-p35 | BDSC | BDSC: BL8651 Flybase: FBti0012594 |
| D.melanogaster:C805-Gal4 | BDSC | BDSC: BL6986 Flybase: FBst006986 |
| D.melanogaster:Pc135-Gal4 | BDSC | BDSC: BL6978 Flybase: FBst006978 |
| D.melanogaster:UAS-dcr2; C96-Gal4 | BDSC | BDSC: BL25757 Flybase: FBst0025757 |
| D.melanogaster:C96-Gal4 | BDSC | BDSC: BL43343 Flybase: FBst0043343 |
|
| ||
| Oligonucleotides | ||
|
| ||
| Kap3 forward primer: 5′ ATGCAACGACAACAACAAACA |
This paper Eurofins |
N/A |
| Kap3 reverse primer: 5′ TTAAGCCATCAGCAGCTCCTC |
This paper Eurofins |
N/A |
| IFT140 forward primer: 5′ ATGATTGTGTGGGGTGAGCCC |
This paper Eurofins |
N/A |
| IFT140 reverse primer: 5′ CTAATGCAGCTCCTCGGTGAT |
This paper Eurofins |
N/A |
| Armaa34−87 forward primer: 5′ ATGTGGCAGCAGAATTCGTACTTGGGCGAC |
This paper Eurofins |
N/A |
| Armaa34−87 reverse primer: 5′ CACTTGGTCTTGTGTGAAATTCTGCGGGAA |
This paper Eurofins |
N/A |
| beta-cateninaa24−79 (mouse and human) forward primer: 5′ CACTGGCAGCAGCA GTCTTACTTGATTCTG |
This paper Eurofins |
N/A |
| beta-cateninaa24−79 (mouse and human) reverse primer: 5′ TACTTGCTCTTGCGTGA AGGACTCGGAAAA |
This paper Eurofins |
N/A |
| beta-catenin-FL (mouse and human) forward primer: 5′ ATGGCTACTCAAG CTGACCTGATGGAGTTG |
This paper Eurofins |
N/A |
| beta-catenin-FL (mouse and human) reverse primer: 5′ TAGCAGGTCAGTA TCAAACCAGGCCAGCTG |
This paper Eurofins |
N/A |
|
| ||
| Recombinant DNA | ||
|
| ||
| pcDNA4/TO | Stuart Aaronson | N/A |
| pCMV6-GFP | Stuart Aaronson | N/A |
| PGL-TOP | Stuart Aaronson | N/A |
| pGL-FOP | Stuart Aaronson | N/A |
| pRL-CMV | Stuart Aaronson | N/A |
| LacZ | Stuart Aaronson | N/A |
| cDNA: Kap3 | Drosophila Genomics Resource Center | DGRC: 5090 Flybase: FBcl 0157674 |
| cDNA:Arm | Drosophila Genomics Resource Center | DGRC: LD23131 Flybase: FBcl 0179217 |
| pGEX-4T-1 | Milipore-Sigma | Cat #GE28–9545-49 |
| pMAL-c2X | Addgene | Cat# 75286 RRID: Addgene_75286 |
| pUAST-attb-GFP-V5-His | Addgene | Cat# 85621 RRID: Addgene_85621 |
| MSCV-beta catenin IRES GFP | Addgene | Cat #14717 RRID: Addgene_14717 |
| PMXs-beta-catenin-S33Y | Addgene | Cat #13371 RRID: Addgene_13371 |
| Human beta-catenin pcDNA3 | Addgene | Cat #16828 RRID: Addgene_16828 |
| Human beta-catenin GFP | Addgene | Cat#71367 RRID: Addgene_71367 |
| GST-Arm | K.W.Choi | N/A |
| MBP-Klp64D | K.W.Choi | N/A |
| GST-Klp64D | K.W.Choi | N/A |
Highlights.
N-terminal β-catenin fragment acts as a dominant interference tool to inhibit Wnt signaling
It inhibits Wnt signaling by interfering with nuclear translocation of endogenous β-catenin
It serves as entry point for therapeutic applications to attenuate Wnt signaling
ACKNOWLEDGMENTS
We are grateful to Carlo Iomini, Davide Esposito, and Stuart A. Aaronson for reagents and advice and Bo Chen for letting us use their tissue culture facility. We thank all Mlodzik lab members for helpful suggestions and Prashanth Rangan and Sam Sidi for helpful comments on the manuscript. We thank the Bloomington Drosophila Stock Center for fly strains and Developmental Studies Hybridoma Bank for antibodies. We would like to thank the ISMMS Microscopy CoRE, where confocal microscopy was performed, which was in part supported by the Tisch Cancer Institute P30 CA196521 grant from the NCI. This work was supported by NIH grant R35 GM127103 to M.M. and an NYSTEM science training award (postdoctoral fellowship to L.T.V.).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2024.114362.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- 1.Logan CY, and Nusse R. (2004). The Wnt signaling pathway in development and disease. Annu. Rev. Cell Dev. Biol 20, 781–810. 10.1146/annurev.cellbio.20.010403.113126. [DOI] [PubMed] [Google Scholar]
- 2.Clevers H. (2006). Wnt/beta-catenin signaling in development and disease. Cell 127, 469–480. 10.1016/j.cell.2006.10.018. [DOI] [PubMed] [Google Scholar]
- 3.Clevers H, and Nusse R. (2012). Wnt/β-catenin signaling and disease. Cell 149, 1192–1205. 10.1016/j.cell.2012.05.012. [DOI] [PubMed] [Google Scholar]
- 4.MacDonald BT, Tamai K, and He X. (2009). Wnt/beta-catenin signaling: components, mechanisms, and diseases. Dev. Cell 17, 9–26. 10.1016/j.devcel.2009.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Macdonald BT, Semenov MV, and He X. (2007). SnapShot: Wnt/beta-catenin signaling. Cell 131, 1204. 10.1016/j.cell.2007.11.036. [DOI] [PubMed] [Google Scholar]
- 6.Cadigan KM, and Waterman ML (2012). TCF/LEFs and Wnt signaling in the nucleus. Cold Spring Harb. Perspect. Biol 4, a007906. 10.1101/cshperspect.a007906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Valenta T, Hausmann G, and Basler K. (2012). The many faces and functions of beta-catenin. Embo J 31, 2714–2736. 10.1038/emboj.2012.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Reya T, and Clevers H. (2005). Wnt signalling in stem cells and cancer. Nature 434, 843–850. 10.1038/nature03319. [DOI] [PubMed] [Google Scholar]
- 9.Raslan AA, and Yoon JK (2020). WNT Signaling in Lung Repair and Regeneration. Mol. Cells 43, 774–783. 10.14348/molcells.2020.0059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pecina-Slaus N, and Kafka A. (2015). Wnt signaling and astrocytic brain tumors. CNS Oncol 4, 369–370. 10.2217/cns.15.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhao H, Ming T, Tang S, Ren S, Yang H, Liu M, Tao Q, and Xu H. (2022). Wnt signaling in colorectal cancer: pathogenic role and therapeutic target. Mol. Cancer 21, 144. 10.1186/s12943-022-01616-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schatoff EM, Leach BI, and Dow LE (2017). Wnt Signaling and Colorectal Cancer. Curr. Colorectal Cancer Rep 13, 101–110. 10.1007/s11888-017-0354-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Scully MA, Wilhelm R, Wilkins DE, and Day ES (2024). Membrane-Cloaked Nanoparticles for RNA Interference of beta-Catenin in Triple-Negative Breast Cancer. ACS Biomater. Sci. Eng 10, 1355–1363. 10.1021/acsbiomaterials.4c00160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cadigan KM (2012). TCFs and Wnt/β-catenin signaling: more than one way to throw the switch. Curr. Top. Dev. Biol 98, 1–34. 10.1016/B978-0-12-386499-4.00001-X. [DOI] [PubMed] [Google Scholar]
- 15.Behrens J, von Kries JP, Kühl M, Bruhn L, Wedlich D, Grosschedl R, and Birchmeier W. (1996). Functional interaction of beta-catenin with the transcription factor LEF-1. Nature 382, 638–642. 10.1038/382638a0. [DOI] [PubMed] [Google Scholar]
- 16.Krieghoff E, Behrens J, and Mayr B. (2006). Nucleo-cytoplasmic distribution of beta-catenin is regulated by retention. J. Cell Sci 119, 1453–1463. 10.1242/jcs.02864. [DOI] [PubMed] [Google Scholar]
- 17.Lawrence PA, Casal J, and Struhl G. (2002). Towards a model of the organisation of planar polarity and pattern in the Drosophila abdomen. Development 129, 2749–2760. [DOI] [PubMed] [Google Scholar]
- 18.Cliffe A, Hamada F, and Bienz M. (2003). A role of Dishevelled in relocating Axin to the plasma membrane during wingless signaling. Curr. Biol 13, 960–966. [DOI] [PubMed] [Google Scholar]
- 19.Molenaar M, van de Wetering M, Oosterwegel M, Peterson-Maduro J, Godsave S, Korinek V, Roose J, Destrée O, and Clevers H. (1996). XTcf-3 transcription factor mediates beta-catenin-induced axis formation in Xenopus embryos. Cell 86, 391–399. 10.1016/s0092-8674(00)80112-9. [DOI] [PubMed] [Google Scholar]
- 20.Zecca M, and Struhl G. (2002). Subdivision of the Drosophila wing imaginal disc by EGFR-mediated signaling. Development 129, 1357–1368. 10.1242/dev.129.6.1357. [DOI] [PubMed] [Google Scholar]
- 21.Orsulic S, and Peifer M. (1996). An in vivo structure-function study of armadillo, the beta-catenin homologue, reveals both separate and overlapping regions of the protein required for cell adhesion and for wingless signaling. J. Cell Biol 134, 1283–1300. 10.1083/jcb.134.5.1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Coates JC (2003). Armadillo repeat proteins: beyond the animal kingdom. Trends Cell Biol. 13, 463–471. 10.1016/s0962-8924(03)00167-3. [DOI] [PubMed] [Google Scholar]
- 23.Huber AH, Nelson WJ, and Weis WI (1997). Three-dimensional structure of the armadillo repeat region of beta-catenin. Cell 90, 871–882. 10.1016/s0092-8674(00)80352-9. [DOI] [PubMed] [Google Scholar]
- 24.Gul IS, Hulpiau P, Saeys Y, and van Roy F. (2017). Metazoan evolution of the armadillo repeat superfamily. Cell. Mol. Life Sci 74, 525–541. 10.1007/s00018-016-2319-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ha NC, Tonozuka T, Stamos JL, Choi HJ, and Weis WI (2004). Mechanism of phosphorylation-dependent binding of APC to beta-catenin and its role in beta-catenin degradation. Mol. Cell 15, 511–521. 10.1016/j.molcel.2004.08.010. [DOI] [PubMed] [Google Scholar]
- 26.Xing Y, Takemaru KI, Liu J, Berndt JD, Zheng JJ, Moon RT, and Xu W. (2008). Crystal structure of a full-length beta-catenin. Structure 16, 478–487. 10.1016/j.str.2007.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Blauwkamp TA, Chang MV, and Cadigan KM (2008). Novel TCF-binding sites specify transcriptional repression by Wnt signalling. Embo J 27, 1436–1446. 10.1038/emboj.2008.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vuong LT, Mukhopadhyay B, and Choi KW (2014). Kinesin-II recruits Armadillo and Dishevelled for Wingless signaling in Drosophila. Development 141, 3222–3232. 10.1242/dev.106229. [DOI] [PubMed] [Google Scholar]
- 29.Vuong LT, Iomini C, Balmer S, Esposito D, Aaronson SA, and Mlodzik M. (2018). Kinesin-2 and IFT-A act as a complex promoting nuclear localization of beta-catenin during Wnt signalling. Nat. Commun 9, 5304. 10.1038/s41467-018-07605-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu TH, Li L, and Vaessin H. (2002). Transcription of the Drosophila CKI gene dacapo is regulated by a modular array of cis-regulatory sequences. Mech. Dev 112, 25–36. 10.1016/s0925-4773(01)00626-8. [DOI] [PubMed] [Google Scholar]
- 31.Pai LM, Orsulic S, Bejsovec A, and Peifer M. (1997). Negative regulation of Armadillo, a Wingless effector in Drosophila. Development 124, 2255–2266. [DOI] [PubMed] [Google Scholar]
- 32.Balmer S, Dussert A, Collu GM, Benitez E, Iomini C, and Mlodzik M. (2015). Components of Intraflagellar Transport Complex A Function Independently of the Cilium to Regulate Canonical Wnt Signaling in Drosophila. Dev. Cell 34, 705–718. 10.1016/j.devcel.2015.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Williams JA, Paddock SW, Vorwerk K, and Carroll SB (1994). Organization of wing formation and induction of a wing-patterning gene at the dorsal/ventral compartment boundary. Nature 368, 299–305. 10.1038/368299a0. [DOI] [PubMed] [Google Scholar]
- 34.Groth AC, Fish M, Nusse R, and Calos MP (2004). Construction of transgenic Drosophila by using the site-specific integrase from phage phiC31. Genetics 166, 1775–1782. 10.1534/genetics.166.4.1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gustafson K, and Boulianne GL (1996). Distinct expression patterns detected within individual tissues by the GAL4 enhancer trap technique. Genome 39, 174–182. 10.1139/g96-023. [DOI] [PubMed] [Google Scholar]
- 36.Spencer ZT, Ng VH, Benchabane H, Siddiqui GS, Duwadi D, Maines B, Bryant JM, Schwarzkopf A, Yuan K, Kassel SN, et al. (2023). The USP46 deubiquitylase complex increases Wingless/Wnt signaling strength by stabilizing Arrow/LRP6. Nat. Commun 14, 6174. 10.1038/s41467-023-41843-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Diaz-Benjumea FJ, and Cohen SM (1995). Serrate signals through Notch to establish a Wingless-dependent organizer at the dorsal/ventral compartment boundary of the Drosophila wing. Development 121, 4215–4225. 10.1242/dev.121.12.4215. [DOI] [PubMed] [Google Scholar]
- 38.Neumann CJ, and Cohen SM (1997). Long-range action of Wingless organizes the dorsal-ventral axis of the Drosophila wing. Development 124, 871–880. 10.1242/dev.124.4.871. [DOI] [PubMed] [Google Scholar]
- 39.Zecca M, Basler K, and Struhl G. (1996). Direct and long-range action of a wingless morphogen gradient. Cell 87, 833–844. 10.1016/s0092-8674(00)81991-1. [DOI] [PubMed] [Google Scholar]
- 40.Campbell G, and Tomlinson A. (1998). The roles of the homeobox genes aristaless and Distal-less in patterning the legs and wings of Drosophila. Development 125, 4483–4493. 10.1242/dev.125.22.4483. [DOI] [PubMed] [Google Scholar]
- 41.Hay BA, Wolff T, and Rubin GM (1994). Expression of baculovirus P35 prevents cell death in Drosophila. Development 120, 2121–2129. 10.1242/dev.120.8.2121. [DOI] [PubMed] [Google Scholar]
- 42.Hrdlicka L, Gibson M, Kiger A, Micchelli C, Schober M, Schöck F, and Perrimon N. (2002). Analysis of twenty-four Gal4 lines in Drosophila melanogaster. Genesis 34, 51–57. 10.1002/gene.10125. [DOI] [PubMed] [Google Scholar]
- 43.Abu-Shaar M, Ryoo HD, and Mann RS (1999). Control of the nuclear localization of Extradenticle by competing nuclear import and export signals. Genes Dev. 13, 935–945. 10.1101/gad.13.8.935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, and Sunyaev SR (2010). A method and server for predicting damaging missense mutations. Nat. Methods 7, 248–249. 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sim NL, Kumar P, Hu J, Henikoff S, Schneider G, and Ng PC (2012). SIFT web server: predicting effects of amino acid substitutions on proteins. Nucleic Acids Res. 40, W452–W457. 10.1093/nar/gks539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kim S, and Jeong S. (2019). Mutation Hotspots in the beta-Catenin Gene: Lessons from the Human Cancer Genome Databases. Mol. Cells 42, 8–16. 10.14348/molcells.2018.0436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Oh JH, Ku JL, Yoon KA, Kwon HJ, Kim WH, Park HS, Yeo KS,Song SY, Chung JK, and Park JG (1999). Establishment and characterization of 12 human colorectal-carcinoma cell lines. Int. J. Cancer 81, 902–910. . [DOI] [PubMed] [Google Scholar]
- 48.Koura M, and Isaka H. (1980). Establishment of a human colon adenocarcinoma cell line producing carcinoembryonic antigen. Gan 71, 313–318. [PubMed] [Google Scholar]
- 49.Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, Wilson CJ, Lehár J, Kryukov GV, Sonkin D, et al. (2012). The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 483, 603–607. 10.1038/nature11003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bignell GR, Greenman CD, Davies H, Butler AP, Edkins S, Andrews JM, Buck G, Chen L, Beare D, Latimer C, et al. (2010). Signatures of mutation and selection in the cancer genome. Nature 463, 893–898. 10.1038/nature08768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kitamura T, Koshino Y, Shibata F, Oki T, Nakajima H, Nosaka T, and Kumagai H. (2003). Retrovirus-mediated gene transfer and expression cloning: powerful tools in functional genomics. Exp. Hematol 31, 1007–1014. [PubMed] [Google Scholar]
- 52.Crowley-Weber CL, Payne CM, Gleason-Guzman M, Watts GS, Futscher B, Waltmire CN, Crowley C, Dvorakova K, Bernstein C, Craven M, et al. (2002). Development and molecular characterization of HCT-116 cell lines resistant to the tumor promoter and multiple stress-inducer, deoxycholate. Carcinogenesis 23, 2063–2080. 10.1093/carcin/23.12.2063. [DOI] [PubMed] [Google Scholar]
- 53.Yeung TM, Gandhi SC, Wilding JL, Muschel R, and Bodmer WF (2010). Cancer stem cells from colorectal cancer-derived cell lines. Proc. Natl. Acad. Sci. USA 107, 3722–3727. 10.1073/pnas.0915135107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ahmed D, Eide PW, Eilertsen IA, Danielsen SA, Eknæs M, Hektoen M, Lind GE, and Lothe RA (2013). Epigenetic and genetic features of 24 colon cancer cell lines. Oncogenesis 2, e71. 10.1038/oncsis.2013.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Berg KCG, Eide PW, Eilertsen IA, Johannessen B, Bruun J, Danielsen SA, Bjørnslett M, Meza-Zepeda LA, Eknæs M, Lind GE, et al. (2017). Multi-omics of 34 colorectal cancer cell lines - a resource for biomedical studies. Mol. Cancer 16, 116. 10.1186/s12943-017-0691-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ilyas M, Tomlinson IP, Rowan A, Pignatelli M, and Bodmer WF (1997). Beta-catenin mutations in cell lines established from human colorectal cancers. Proc. Natl. Acad. Sci. USA 94, 10330–10334. 10.1073/pnas.94.19.10330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bhattacharyya NP, Skandalis A, Ganesh A, Groden J, and Meuth M. (1994). Mutator phenotypes in human colorectal carcinoma cell lines. Proc. Natl. Acad. Sci. USA 91, 6319–6323. 10.1073/pnas.91.14.6319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chandra SHV, Wacker I, Appelt UK, Behrens J, and Schneikert J. (2012). A common role for various human truncated adenomatous polyposis coli isoforms in the control of beta-catenin activity and cell proliferation. PLoS One 7, e34479. 10.1371/journal.pone.0034479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Liu J, Pan S, Hsieh MH, Ng N, Sun F, Wang T, Kasibhatla S, Schuller AG, Li AG, Cheng D, et al. (2013). Targeting Wnt-driven cancer through the inhibition of Porcupine by LGK974. Proc. Natl. Acad. Sci. USA 110, 20224–20229. 10.1073/pnas.1314239110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Vuong LT, and Mlodzik M. (2022). Different strategies by distinct Wnt-signaling pathways in activating a nuclear transcriptional response. Curr. Top. Dev. Biol 149, 59–89. 10.1016/bs.ctdb.2022.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hwang WY, Kostiuk V, Gonzalez DP, Lusk CP, and Khokha MK (2022). Kap-beta2/Transportin mediates beta-catenin nuclear transport in Wnt signaling. Elife 11, e70495. 10.7554/eLife.70495. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data reported in this paper will be shared by the lead contact upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.