

Abstract
Extracts prepared from the seeds of the medicinal plant milk thistle [Silybum marianum (L.) Gaertn. (Asteraceae)] are widely used as dietary supplements due to anti-inflammatory, antitumor, and hepatoprotective effects. Called silymarin, the main components of lipophilic extracts of milk thistle seeds are flavonoids and flavonolignans including silybin A, silybin B, isosilybin A, isosilybin B, silydianin, silychristin, taxifolin, and 2,3-dehydrosilybins. The aim of this study was to develop a method based on UHPLC-MS/MS for the chemical authentication and standardization of milk thistle silymarin. Validation included the method of standard addition to account for the lack of a blank matrix. Potential matrix effects were investigated by analyzing silymarin standards dissolved only in the initial UHPLC mobile phase. Measurements of six flavonolignans and taxifolin in the milk thistle extract using UHPLC-MS/MS with standard addition or external standard calibration produced similar results for all analytes except silydianin and 2,3-dehydrosilybin B, which showed significant peak enhancement during negative ion electrospray due to botanical matrix effects. The UHPLC-MS/MS-based method of standard addition requires <10 min per injection and is suitable for the standardization of silymarin from milk thistle in support of preclinical and clinical studies of safety and efficacy.
Introduction
Milk thistle (Silybum marianum L. Gaertn.) is native to the Mediterranean and is cultivated widely as a medicinal plant and dietary supplement.1 Extracts of milk thistle seeds have been used for over 2000 years, primarily for liver problems and hepatoprotection, and are under investigation for anti-inflammatory and antitumor activities.2−5 Milk thistle extract consists of >60% silymarin,6 which is a mixture of flavonolignans and flavonoids including silybin B, isosilybin A, isosilybin B, silychristin, isosilychristin, silydianin, 2,3-dehydrosilybin B, and taxifolin (Figure 1). Milk thistle is the 23rd most popular botanical dietary supplement in the United States,7 and the worldwide market for milk thistle extracts was $103 million in 2022.8
Figure 1.

Chemical structures of common milk thistle constituents in silymarin and deuterated internal standards.
Despite being the subject of over 70 clinical trials listed on clinicaltrials.gov alone, the therapeutic benefits of silymarin from milk thistle remain inconclusive, in part due to inadequately standardized test material.9 Without chemical standardization of the botanical being tested, dosages of the active compounds are unknown, results can vary from batch to batch, and preclinical as well as clinical studies cannot be reproduced.10,11 Variations in the composition of silybin from milk thistle results from different growth conditions12,13 and variations in extract preparation and handling.13−15 Therefore, a validated analytical method for the measurement and standardization of silymarin constituents in test materials is essential for the support of pharmacological and toxicological studies both in vitro and in vivo and to enable the correlation of activities with dosage.
Although there are many analytical methods for measuring silymarin constituents and metabolites in human serum16 and urine,17 mostly utilizing high-performance liquid chromatography-tandem mass spectrometry (HPLC-MS/MS), few methods have been reported for the standardization of silymarin compounds in milk thistle extracts. Analytical methods to measure constituents of silymarin have included thin layer chromatography, HPLC with UV absorbance detection, HPLC with electrochemical detection, HPLC-MS,18 and UHPLC-MS/MS.19
Due to the complexity of the botanical matrix, suppression or enhancement of MS signals can occur when using electrospray.20 To help compensate for these matrix effects, stable isotope-labeled analogues of each analyte (surrogate standards) can be added to the samples. Even if available, surrogate standards often do not completely correct for quantitation error caused by matrix effects during electrospray MS. One solution to this problem is to enhance the chromatographic separation such that all analytes are completely resolved from interfering matrix constituents using approaches such as ultrahigh-pressure liquid chromatography (UHPLC).21 Another solution to address this problem is to use a blank matrix when preparing standard curves that contain all components except for the analytes to be measured. Because surrogate standards for silymarin compounds and blank milk thistle extract matrix were unavailable, we used UHPLC instead of HPLC and the method of standard addition22 to validate a quantitative method based on UHPLC-MS/MS. In this approach, each milk thistle extract served as its own matrix for standard curve preparation. For comparison, external standard curves were also prepared using silymarin compounds dissolved in the initial UHPLC mobile phase. The method of standard addition has been used for the chemical standardization of other botanical extracts including extracts of licorice,23 garlic,24 and botanical mixtures.25 Here, we report the development and standardization of a quantitative assay for six milk thistle flavonolignans plus taxifolin in milk thistle extract using electrospray UHPLC-MS/MS that is fast, sensitive, and accurate.
Methods
Materials and Reagents
Silychristin A (96.5% purity; DTXSID50187512), silydianin (96.2% purity; DTXSID70858696), and taxifolin (92.8% purity; DTXSID8022450) were purchased from ChromaDex (Los Angeles, CA). Isosilybin A (98.4% purity; DTXSID20453675), isosilybin B (≥88.7% purity; DTXSID80447055), and 2,3-dehydrosilybin B (99.3% purity; DTXSID601102493) were purchased from MilliporeSigma (St. Louis, MO). Silybin B (≥98.0% purity; DTXSID30858697), daidzein-d4, and genistein-d4 (≥99.0% purity) were purchased from Cayman Chemicals (Ann Arbor, MI). An ethanolic extract of milk thistle seed (90.6% silymarin compounds) was supplied by the Botanical Safety Consortium and had been purchased from Euromed S.A. (Barcelona, Spain). HPLC-MS grade acetonitrile, methanol, and formic acid were purchased from Thermo Fisher Scientific (Waltham, MA). Ultrapure water was prepared using a MilliporeSigma Milli-Q water purification system. All other reagents and solvents were reagent grade or better and were purchased from VWR (Visalia, CA).
Preparation of Calibration Standards and Internal Standards
Stock solutions of milk thistle flavonolignan and taxifolin standards (1 mg/mL each) were prepared in methanol, aliquoted, and stored at −20 °C until use. The working solutions for the calibration standards were prepared by diluting the stock solution in methanol/water (1:1, v/v). Internal standard stock solutions containing d4-daidzein and d4-genistein (1 mg/mL) were prepared by dissolving accurately weighed aliquots in dimethyl sulfoxide. Note that only d4-daidzein was used for data normalization. The stock solutions were diluted in methanol/water (1:1, v/v) to obtain working solutions (125 ng/mL) of each deuterium-labeled standard for addition during sample pretreatment. Since isotope-labeled milk thistle flavonolignans were not commercially available, d4-daizein and d4-genistein were used as internal standards to correct for variabilities during sample preparation and analysis as they displayed similar solubility and extraction efficiency.
Standard Addition Method and External Calibration Curve
Milk thistle extract was weighed and dissolved in methanol to produce a 1 mg/mL stock solution, which was diluted to 1 μg/mL with 50% aqueous methanol. Known amounts of flavonolignan and taxifolin standards were spiked into each sample to generate solutions for a standard addition calibration curve. For comparison, an external calibration curve was prepared by spiking different working solutions of flavonolignan and taxifolin standards in neat solvent to give a concentration range of 0.4–512 ng/mL for isosilybin A, isosilybin B, silybin B, 2,3-dehydrosilybin B, and taxifolin and 1.2–512 ng/mL for silychristin and silydianin.
UHPLC-MS and UHPLC-MS/MS of Milk Thistle Flavonolignans and Taxifolin
Chromatographic separation of the milk thistle flavonolignans and taxifolin was carried out using a Shimadzu (Kyoto, Japan) Nexera UHPLC system fitted with a Waters (Milford, MA) Cortecs UPLC C18 column (1.7 μm, 130 Å, 2.1 × 150 mm). The mobile phase consisted of a gradient from water (A) to methanol (B) both containing 0.01% formic acid as follows: 0–2 min 50% B, 2–4.5 min 50–55% B, 4.5–5.5 min 55–60% B, and 6–8 min 60–75% B. The column was reconditioned in 50% methanol for 2 min between injections. The flow rate was 0.3 mL/min, the injection volume was 5 μL, and the column and autosampler temperatures were 40 and 10 °C respectively. The total UHPLC analysis time per sample for the separation of milk thistle flavonolignans and taxifolin was 10 min.
High-resolution mass spectra and tandem mass spectra (using data-dependent acquisition to acquire tandem mass spectra of all major constituents) of flavolignans and taxifolin in milk thistle extract were obtained using a Shimadzu (Kyoto, Japan) 9030 Q-ToF tandem mass spectrometer interfaced with a Shimadzu Nexera UHPLC system. The electrospray ionization interface temperature was 300 °C, and the voltage was −3.5 kV for negative ion mode. The heat block and desolvation line temperatures were 400 and 250 °C, respectively. Nitrogen was used as a drying gas at a flow rate of 10 L/min, for nebulization at 3 L/min, and as a heating gas at 10 L/min. Mass spectra and product ion tandem mass spectra were acquired every 100 ms over the mass range m/z 70–700. During data-dependent acquisition, six dependent events were chosen at an intensity threshold of 3000, and the product ion tandem mass spectra were obtained using a collision energy of 35 V with an energy spread of 17 V.
For quantitative analysis, the UHPLC system was coupled to a Shimadzu LCMS-8060 triple quadruple mass spectrometer equipped with negative ion electrospray. Nitrogen was used as drying gas at a flow rate of 5 L/min and for nebulization at 3 L/min. The interface and desolvation line temperatures were set to 400 and 300 °C, respectively. The milk thistle flavonolignans and taxifolin were measured using collision-induced dissociation with selected reaction monitoring (SRM). The SRM dwell time for each transition was 15 ms, and the collision gas pressure was 230 kPa. Data acquisition, integration, and linear standard curves fitting were carried out using Shimadzu Lab Solutions software version 5.7. The concentrations of flavonolignans and taxifolin in the milk thistle extract were calculated using Microsoft Excel software (Seattle, WA).
Results and Discussion
Chemical Authentication of Milk Thistle Extract
The goal of this investigation was to develop and validate an accurate and efficient assay for the chemical authentication and standardization of milk thistle extracts for use in preclinical and clinical studies of safety and efficacy.26,27 This assay helped support the authentication and standardization of the milk thistle extract used by the Botanical Safety Consortium,28 which is an international group of experts working to identify fit-for-purpose assays for the evaluation of the safety of botanicals used as dietary supplements or medicinal plants.29,30
As a first step in this process, reversed phase UHPLC with high-resolution mass spectrometry was used to separate the major constituents in silymarin (Figure 2 and Supplemental Figures 1 and 2) and confirm their elemental compositions (Supplemental Table 1). Most flavonolignans in silymarin are constitutional isomers with the molecular formulas C25H22O10 (Figure 1), and the measured masses of these compounds in this silymarin preparation were within 10 ppm of the expected values (Supplemental Table 1). The major flavanonol taxifolin (molecular formula C15H12O7) was also detected and measured in this extract. Data-dependent high-resolution tandem mass spectra of the major silymarin constituents were acquired and compared with those of standards. Based on identical tandem mass spectra (Supplemental Figures 3–21), identical accurate mass measurements, and identical retention times during UHPLC as standards (Supplemental Table 1), the milk thistle constituents silychristin, silydianin, silybin A, silybin B, isosilybin A, isosilybin B, 2,3-dehydrosilybin B, and taxifolin were identified, additional constituents were characterized, and the extract was chemically authenticated as silymarin from milk thistle. Although no pure commercial standard was available for silybin A, a commercially available equimolar silybin A and silybin B mixture was used to isolate silybin A as described previously.16 The abundant peak eluting at 19.6 min had an elemental composition (C25H22O10), tandem mass spectrum (Supplemental Figure 10), and retention time that were consistent with silybin A. Also, the peak at 12.8 min in the UHPLC-MS/MS chromatogram (Figure 2) had an elemental composition (C25H22O10), tandem mass spectrum, and retention time consistent with neusilychristin, a constitutional isomer of silychristin18 (Supplemental Figure 6).
Figure 2.

High-resolution negative ion electrospray UHPLC-MS total ion chromatogram survey of milk thistle extract (silymarin) showing separation and detection of major silymarin constituents plus the internal standard daidzein-d4. The corresponding high-resolution Q-ToF positive ion electrospray chromatogram is shown in Supplemental Figure 2. The most abundant milk thistle constituents (labeled in red font) were then measured using a faster UHPLC-MS/MS method on a triple quadrupole mass spectrometer.
Chemical Standardization of Milk Thistle Extract
A UHPLC-MS/MS quantitative assay was developed for the measurement of six flavonolignans and taxifolin in milk thistle extract. The SRM transitions used for MS/MS quantitative analysis of each analyte are shown in Table 1. Optimized for speed, baseline separation of each analyte was achieved in <10 min using a C18 UHPLC column (Figure 3). This separation is comparable in speed to a previous UHPLC-MS/MS method19 for the measurement of flavonolignans in milk thistle extract but considerably faster than an HPLC-MS method that required 40 min per analysis.31 Because no blank matrix is available for milk thistle extract, standards for external standard curves were prepared in 50% methanol. The external calibration curves for silybin B, isosilybin A, isosilybin B, and taxifolin were linear over 3 orders of magnitude (0.4–512 mg/g dry extract), and the calibration curves for silychristin and silydianin were linear from 1.2 to 512 mg/g dry extract (Table 1). Note that the dynamic range of this method is approximately 10-fold greater than that of a previous UHPLC-MS/MS method.19
Table 1. UHPLC-MS/MS Negative Ion Electrospray Retention Times (RT), MS/MS SRM Transitions, Collision Energies (CE), Concentrations (ng/mL) of Standards Spiked into Dilute Milk Thistle Extract, and Coefficients of Determination (R2) for External Calibrants in Neat Solvent and for the Method of Standard Addition.
| analyte | RT (min) | SRM transitions m/z | CE (V) | spiked concentrations (ng/mL) | R2 neat solvent | R2 std addn | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| taxifolin | 1.7 | 303 → 285a | 12 | 0.4 | 2 | 4 | 8 | 16 | 32 | 64 | 128 | 256 | 512 | 0.9991 | 0.9999 |
| 303 → 125 | 22 | ||||||||||||||
| silychristin | 2.0 | 481 → 125 | 31 | 1.2 | 2 | 4 | 8 | 16 | 32 | 64 | 128 | 256 | 512 | 0.9984 | 0.9988 |
| 481 → 325 | 21 | ||||||||||||||
| silydianin | 2.5 | 481 → 179 | 29 | 1.2 | 2 | 4 | 8 | 16 | 32 | 64 | 128 | 256 | 512 | 0.9963 | 0.9956 |
| 481 → 125 | 26 | ||||||||||||||
| silybin B | 4.4 | 481 → 125 | 28 | 0.4 | 2 | 4 | 8 | 16 | 32 | 64 | 128 | 256 | 512 | 0.9992 | 0.9996 |
| 481 → 301 | 20 | ||||||||||||||
| isosilybin A | 5.4 | 481 → 125 | 28 | 0.4 | 2 | 4 | 8 | 16 | 32 | 64 | 128 | 256 | 512 | 0.9994 | 0.9997 |
| 481 → 453 | 17 | ||||||||||||||
| isosilybin B | 5.7 | 481 → 125 | 27 | 0.4 | 2 | 4 | 8 | 16 | 32 | 64 | 128 | 256 | 512 | 0.9991 | 0.9998 |
| 481 → 453 | 19 | ||||||||||||||
| 2,3-dehydrosilybin B | 8.4 | 479 → 299 | 18 | 0.4 | 2 | 4 | 8 | 16 | 32 | 64 | 128 | 256 | 512 | 0.9968 | 0.9959 |
| 479 → 271 | 35 | ||||||||||||||
| d4-daidzein | 3.5 | 257 → 212 | 29 | ||||||||||||
| 257 → 136 | 39 | ||||||||||||||
| d4-genistein | 5.0 | 273 → 163 | 30 | ||||||||||||
| 273 → 137 | 31 | ||||||||||||||
The first SRM transition for each analyte was used as the quantifier while the second SRM transition was used as a qualifier in the UHPLC-MS/MS assay.
Figure 3.

Negative ion electrospray UHPLC-MS/MS selected reaction monitoring chromatograms of flavonolignans, flavanonol taxifolin, and deuterated internal standards daidzein and genistein. (A) Standards at 256 ng/mL in 50% aqueous methanol; (B) internal standards at 100 ng/mL; and (C) silymarin (10 μg/mL) extracted from milk thistle seed.
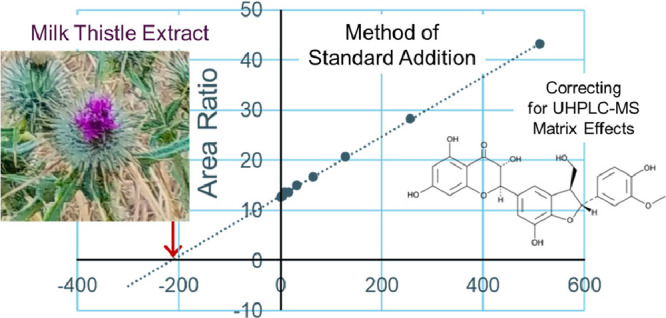
To test for matrix effects during UHPLC-MS/MS, which can interfere with quantitative measurements of constituents in complex botanical extracts, the standard addition method of quantitation was also used for the determination of levels of constituents in the milk thistle extract. An aliquot of milk thistle extract was diluted to produce UHPLC-MS/MS signals for the endogenous flavonolignans and taxifolin with signal-to-noise ratios of ∼10:1. Different concentrations of milk thistle standards were spiked into this extract as indicated in Table 1. After UHPLC-MS/MS with SRM analysis, the internal standard normalized peak area for each flavonolignan or taxifolin spiked sample was plotted on the y-axis while the known concentration of each spiked standard was plotted on the x-axis. The resulting linear trendline was extrapolated to the zero-peak area, and the absolute point of interception of the abscissa was the endogenous concentration of the flavonolignan or taxifolin in the sample (as an example, see the method of standard addition curve for silychristin in Figure 4).
Figure 4.

Using the method of standard addition, the concentration of the analyte silychristin was determined by extrapolating the calibration curve to 0 on the negative side of the x-axis (in the equation, solve for x when y = 0). A constant amount of internal standard was added to each aliquot of milk thistle extract immediately before UHPLC-MS/MS analysis. The peak area of silychristin, spiked with different amounts of silychristin standard, was normalized by dividing with the internal standard peak area. This area ratio was plotted versus the amount of silychristin spiked into the extract.
Peak suppression or enhancement during electrospray mass spectrometry that might be caused by milk thistle matrix was evaluated by comparing the concentrations obtained using the standard addition method to that of an external calibration curve prepared in neat solvent. Ion suppression can occur when coeluting compounds compete for ionization during the saturable electrospray process. Peak enhancement can occur when coeluting compounds, such as isomers of the analyte, contribute to the signal or when coeluting compounds potentiate the ionization of the analyte, perhaps through charge exchange. Although both analytical methods produced linear standard curves (Table 1) and nearly identical values for most analytes, matrix effects produced significant ion suppression that was observed for the milk thistle compounds silydianin (10.8%) and 2,3-dehydrosilybin B (28%) (Table 2). Therefore, only the method of standard addition provided accurate measurement for the chemical standardization of milk thistle extracts in this investigation.
Table 2. Quantitative Analysis of Flavonolignans in Milk Thistle Extract Using UHPLC-MS/MS with the Method of Standard Addition or an External Standard Curve Prepared Using Neat Solvent.
| milk
thistle extract (mg/g extract) |
||
|---|---|---|
| milk thistle compound | standard addition (n = 3) | neat solvent standard curve (n = 3) |
| taxifolin | 37.48 ± 2.51 | 35.04 ± 0.64 |
| silychristin | 158.7 ± 7.6 | 159.0 ± 4.5 |
| silydianin | 13.95 ± 3.10 | 12.45 ± 0.63 |
| silybin B | 263.1 ± 13.3 | 242.3 ± 8.3 |
| isosilybin A | 35.17 ± 1.34 | 35.65 ± 0.75 |
| isosilybin B | 19.75 ± 1.13 | 20.18 ± 1.91 |
| 2,3-dehydrosilybin B | 3.060 ± 0.510 | 2.210 ± 0.065 |
Due to superior speed, sensitivity, and selectivity, UHPLC-MS/MS-based assays are widely used for the quantitative analysis of natural products from foods, dietary supplements, and medicinal plants in serum, plasma, urine, and tissues. For those matrices, blank specimens are readily available that contain possible interfering substances but not the natural product analytes. Therefore, during the development of assays for these matrices utilizing electrospray MS, which is highly susceptible to matrix effects like peak enhancement or suppression, potential matrix interference can be detected. Once identified, the analytical method can be changed to mitigate or eliminate matrix interference. However, measurement of endogenous natural products in samples such as botanicals and botanical extracts poses a special problem due to the absence of blank botanical material.
Matrix effects resulting in signal enhancement or suppression can be mitigated but not always eliminated by using stable isotope-labeled surrogate standards of each analyte. However, surrogate standards are expensive and unavailable for many natural products such as the flavonolignans in milk thistle. Another approach used to minimize matrix effects is to optimize chromatographic separation of analytes from interfering matrix compounds. In this investigation, UHPLC was used instead of HPLC for improved analyte separation, but matrix interference still occurred. Slower gradient separations might have overcome these matrix effects but at the cost of longer analysis times and possibly loss of sensitivity due to broader peak shape. Note that a previous UHPLC-MS/MS method did not detect any matrix effects but did not test for them using the method of standard addition.19
Conclusions
By comparing electrospray UHPLC-MS/MS measurements of constituents in the milk thistle extract silymarin using the method of standard addition to those obtained using an external standard curve with standard dissolved in neat solvent, matrix effects were observed that suppressed the signals for two flavonolignans, silydianin and 2,3-dehydrosilybin B. Although both methods were fast (<10 min per UHPLC-MS analysis) and produced linear standard curves over a wider dynamic range than previous methods, only the method of standard addition corrected for the matrix effects and produced accurate values for all analytes. This method of standard addition was determined to be fit-for-purpose for use in the chemical standardization of milk thistle extracts intended for preclinical and clinical investigation.
Acknowledgments
The authors would like to thank Shimadzu for providing the LCMS-9030 Q-ToF system used in this study. This work was supported by the Health and Environmental Sciences Institute (HESI) Botanical Safety Consortium, funded by the U.S. FDA, HHS, NIEHS, and HESI via the DOI FCG under Blanket Purchase Agreement Order 140D0421F0068. Additional support came from the NIH Intramural Research Program, NIEHS projects ZIC ES103391-01 and ZIC ES103373-02, and contracts HHSN273201400027C (Battelle) and HHSN273201400020C (MRIGlobal). The HESI BSC initiative is primarily funded by in-kind contributions of time, expertise, and experimental effort from public and private sector participants, supplemented by direct support for program infrastructure and management from HESI’s corporate supporters. A list of supporting organizations is available at https://botanicalsafetyconsortium.org/.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jasms.4c00125.
Peak assignments for the high-resolution UHPLC-MS analyses of milk thistle extract; tandem mass spectra of all milk thistle analytes measured in this study (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Marceddu R.; Dinolfo L.; Carrubba A.; Sarno M.; Di Miceli G. Milk Thistle (Silybum marianum L.) as a Novel Multipurpose Crop for Agriculture in Marginal Environments: A Review. Agronomy. 2022, 12 (3), 729. 10.3390/agronomy12030729. [DOI] [Google Scholar]
- Hamilton W. R.; Stohs S.. Hepatic Effects of Herbal Remedies, Chapter 3. In Herbal Medicinals: A Clinician’s Guide; Miller L. G., Murray W. J., Eds.; Pharmaceutical Products Press: New York, 1998; p 37. [Google Scholar]
- Rainone F. Milk Thistle. Am. Fam. Physician 2005, 72, 1285–1292. [PubMed] [Google Scholar]
- Polyak S. J.; Morishima C.; Lohmann V.; Pal S.; Lee D. Y. W.; Liu Y.; Graf T. N.; Oberlies N. H. Identification of Hepatoprotective Flavonolignans from Silymarin. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 5995–5999. 10.1073/pnas.0914009107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saller R.; Brignoli R.; Melzer J.; Meier R. An Updated Systematic Review with Meta-Analysis for the Clinical Evidence of Silymarin. Forsch. Komplementärmedizin 2008, 15, 9–20. 10.1159/000113648. [DOI] [PubMed] [Google Scholar]
- Berman J.50-Complementary and Alternative Medicines for Infectious Diseases. In Mandell, Douglas, and Bennett’s Principles and Practice of Infectious Diseases, 8th ed.; Bennett J. E., Dolin R., Blaser M. J., Eds.; W. B. Saunders: Philadelphia, PA, 2015; p 597. [Google Scholar]
- Smith T.; Bauman H.; Resetar H. US Sales of Herbal Supplements Decline Slightly in 2022. HerbalGram 2024, 139, 52–69. [Google Scholar]
- Milk Thistle Supplements Market. https://www.factmr.com/report/662/milk-thistle-supplements-market (accessed 2024–05–28).
- Ball K. R.; Kowdley K. V. A Review of Silybum marianum (Milk Thistle) as a Treatment for Alcoholic Liver Disease. J. Clin. Gastroenterol. 2005, 39, 520–528. 10.1097/01.mcg.0000165668.79530.a0. [DOI] [PubMed] [Google Scholar]
- Tamayo C.; Diamond S. Review of Clinical Trials Evaluating Safety and Efficacy of Milk Thistle (Silybum marianum [L.] Gaertn.). Integr. Cancer Ther. 2007, 6, 146–157. 10.1177/1534735407301942. [DOI] [PubMed] [Google Scholar]
- Fenclova M.; Novakova A.; Viktorova J.; Jonatova P.; Dzuman Z.; Ruml T.; Kren V.; Hajslova J.; Vitek L.; Stranska-Zachariasova M. Poor Chemical and Microbiological Quality of the Commercial Milk Thistle-Based Dietary Supplements May Account for Their Reported Unsatisfactory and Non-Reproducible Clinical Outcomes. Sci. Rep. 2019, 9 (1), 11118. 10.1038/s41598-019-47250-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambers C. S.; Holečková V.; Petrásková L.; Biedermann D.; Valentová K.; Buchta M.; Křen V. The Silymarin Composition. . . and Why Does It Matter???. Food Res. Int. 2017, 100, 339–353. 10.1016/j.foodres.2017.07.017. [DOI] [PubMed] [Google Scholar]
- Booker A.; Suter A.; Krnjic A.; Strassel B.; Zloh M.; Said M.; Heinrich M. A Phytochemical Comparison of Saw Palmetto Products Using Gas Chromatography and 1H Nuclear Magnetic Resonance Spectroscopy Metabolomic Profiling. J. Pharm. Pharmacol. 2014, 66, 811–822. 10.1111/jphp.12198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harkey M. R.; Henderson G. L.; Gershwin M. E.; Stern J. S.; Hackman R. M. Variability in Commercial Ginseng Products: An Analysis of 25 Preparations. Am. J. Clin. Nutr. 2001, 73, 1101–1106. 10.1093/ajcn/73.6.1101. [DOI] [PubMed] [Google Scholar]
- Duan L.; Carrier D. J.; Clausen E. C. Silymarin Extraction from Milk Thistle Using Hot Water. Appl. Biochem. Biotechnol. 2004, 114, 559–568. 10.1385/ABAB:114:1-3:559. [DOI] [PubMed] [Google Scholar]
- Muchiri R. N.; van Breemen R. B. Single Laboratory Validation of UHPLC-MS/MS Assays for Six Milk Thistle Flavonolignans in Human Serum. J. AOAC Int. 2021, 104, 232–238. 10.1093/jaoacint/qsaa110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazzeroni M.; Petrangolini G.; Legarreta Iriberri J. A.; Pascual Avellana J.; Tost Robusté D.; Cagnacci S.; Macis D.; Aristarco V.; Bonanni B.; Morazzoni P.; Johansson H.; Riva A. Development of an HPLC-MS/MS Method for the Determination of Silybin in Human Plasma, Urine and Breast Tissue. Molecules. 2020, 25 (12), 2918. 10.3390/molecules25122918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csupor D.; Csorba A.; Hohmann J. Recent Advances in the Analysis of Flavonolignans of Silybum marianum. J. Pharm. Biomed. Anal. 2016, 130, 301–317. 10.1016/j.jpba.2016.05.034. [DOI] [PubMed] [Google Scholar]
- Graf T. N.; Cech N. B.; Polyak S. J.; Oberlies N. H. A Validated UHPLC-Tandem Mass Spectrometry Method for Quantitative Analysis of Flavonolignans in Milk Thistle (Silybum marianum) Extracts. J. Pharm. Biomed. Anal. 2016, 126, 26–33. 10.1016/j.jpba.2016.04.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furey A.; Moriarty M.; Bane V.; Kinsella B.; Lehane M. Ion Suppression; a Critical Review on Causes, Evaluation, Prevention and Applications. Talanta. 2013, 115, 104–122. 10.1016/j.talanta.2013.03.048. [DOI] [PubMed] [Google Scholar]
- Van De Steene J. C.; Lambert W. E. Comparison of Matrix Effects in HPLC-MS/MS and UPLC-MS/MS Analysis of Nine Basic Pharmaceuticals in Surface Waters. J. Am. Soc. Mass Spectrom. 2008, 19, 713–718. 10.1016/j.jasms.2008.01.013. [DOI] [PubMed] [Google Scholar]
- Guidelines for Standard Method Performance Requirements AOAC Official Methods of Analysis. Appendix F. 2016; https://www.aoac.org/wp-content/uploads/2019/08/app_f.pdf (accessed 2024–05–28).
- Li G.; Nikolic D.; van Breemen R. B. Identification and Chemical Standardization of Licorice Raw Materials and Dietary Supplements Using UHPLC-MS/MS. J. Agric. Food Chem. 2016, 64, 8062–8070. 10.1021/acs.jafc.6b02954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rybak M. E.; Calvey E. M.; Harnly J. M. Quantitative Determination of Allicin in Garlic: Supercritical Fluid Extraction and Standard Addition of Alliin. J. Agric. Food Chem. 2004, 52, 682–687. 10.1021/jf034853x. [DOI] [PubMed] [Google Scholar]
- Wen D.; Li C.; Di H.; Liao Y.; Liu H. A Universal HPLC Method for the Determination of Phenolic Acids in Compound Herbal Medicines. J. Agric. Food Chem. 2005, 53, 6624–6629. 10.1021/jf0511291. [DOI] [PubMed] [Google Scholar]
- Patel D.; Sorkin B. C.; Mitchell C. A.; Embry M. R.; Rina-Kong S.; Adams R. E.; DeTemple E. R.; Reddam A.; Gafner S.; Kelber O.; Rider C. V.; Oketch-Rabah H.; Roe A. L.; Marles R. J.; Dever J.; Dentali S. Improving the Rigor and Utility of Botanical Toxicity Studies: Recommended Resources. Regul. Toxicol. Pharmacol. 2023, 144, 105471. 10.1016/j.yrtph.2023.105471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Breemen R. B.; Fong H. H. S.; Farnsworth N. R. The Role of Quality Assurance and Standardization in the Safety of Botanical Dietary Supplements. Chem. Res. Toxicol. 2007, 20, 577–582. 10.1021/tx7000493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- National Toxicology Program. Botanical Safety Consortium–Chemical Analysis. 10.22427/NTP-DATA-500-007-001-000-3 (accessed 2014–05–28). [DOI]
- Mitchell C. A.; Dever J. T.; Gafner S.; Griffiths J. C.; Marsman D. S.; Rider C.; Welch C.; Embry M. R. The Botanical Safety Consortium: A Public-Private Partnership to Enhance the Botanical Safety Toolkit. Regul. Toxicol. Pharmacol. 2022, 128, 105090. 10.1016/j.yrtph.2021.105090. [DOI] [PubMed] [Google Scholar]
- Waidyanatha S.; Collins B. J.; Cristy T.; Embry M.; Gafner S.; Johnson H.; Kellogg J.; Krzykwa J.; Li S.; Mitchell C. A.; Mutlu E.; Pickett S.; You H.; van Breemen R.; Baker T. R. Advancing Botanical Safety: A Strategy for Selecting, Sourcing, and Characterizing Botanicals for Developing Toxicological Tools. Food Chem. Toxicol. 2024, 186 (3), 114537. 10.1016/j.fct.2024.114537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J. I.; Narayan M.; Barrett J. S. Analysis and Comparison of Active Constituents in Commercial Standardized Silymarin Extracts by Liquid Chromatography–Electrospray Ionization Mass Spectrometry. J. Chromatogr. B 2007, 845, 95–103. 10.1016/j.jchromb.2006.07.063. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.