

Abstract
The actinide elements are attractive alternatives to transition metals or lanthanides for the design of exchange-coupled multinuclear single-molecule magnets. However, the synthesis of such compounds is challenging, as is unraveling any contributions from exchange coupling to the overall magnetism. To date, only a few actinide compounds have been shown to exhibit exchange coupling and single-molecule magnetism. Here, we report triangular uranium(III) clusters of the type (CpiPr5)3U3X (1-X; X = Cl, Br, I; CpiPr5 = pentaisopropylcyclopentadienyl), which are synthesized via reaction of the aryloxide-bridged precursor (CpiPr5)2U2(OPhtBu)4 with excess Me3SiX. Spectroscopic analysis suggests the presence of covalency in the uranium–halide interactions arising from 5f orbital participation in bonding. The dc magnetic susceptibility data reveal the presence of antiferromagnetic exchange coupling between the uranium(III) centers in these compounds, with the strength of the exchange decreasing down the halide series. Ac magnetic susceptibility data further reveal all compounds to exhibit slow magnetic relaxation under zero dc field. In 1-I, which exhibits particularly weak exchange, magnetic relaxation occurs via a Raman mechanism associated with the individual uranium(III) centers. In contrast, for 1-Br and 1-Cl, magnetic relaxation occurs via an Orbach mechanism, likely involving relaxation between ground and excited exchange-coupled states. Significantly, in the case of 1-Cl, magnetic relaxation is sufficiently slow such that open magnetic hysteresis is observed up to 2.75 K, and the compound exhibits a 100-s blocking temperature of 2.4 K. This compound provides the first example of magnetic blocking in a compound containing only actinide-based ions, as well as the first example involving the uranium(III) oxidation state.
Introduction
Single-molecule magnets possess a bistable magnetic ground state separated by an effective thermal energy barrier, Ueff, and as a result they can exhibit slow magnetic relaxation of molecular origin.1,2 Magnetic hysteresis can be observed in such systems below the magnetic blocking temperature, which has been defined as the temperature at which the relaxation time is 100 s (Tb,100s) or the temperature at which the zero-field cooled and field-cooled magnetic susceptibility curves diverge.1 Single-molecule magnets have garnered substantial interest for potential applications related to nanoscale information storage and spin-based computing.3−5 However, the vast majority of molecules studied to date exhibit slow magnetic relaxation only at very low temperatures. This is often due to “through-barrier” magnetic relaxation processes, such as quantum tunneling, that short-circuit the full relaxation barrier for a given molecule.6 As such, considerable research has focused on ways to suppress through-barrier relaxation processes.
One particularly effective strategy for suppressing quantum tunneling has been to design multinuclear complexes exhibiting strong magnetic exchange and high-spin ground states.7,8 Indeed, the first single-molecule magnet was the exchange-coupled cluster Mn12O12(CH3COO)16(H2O)4.2,9,10 However, the blocking temperatures of exchange-coupled transition metal clusters are relatively low,11 and it was found that systems with very high spin ground states—originally designed to maximize the barrier to magnetic relaxation—generally exhibit very low magnetic anisotropies and hence values of Ueff.12−14
In the last two decades, efforts have shifted to the design of single-molecule magnets based on the highly anisotropic lanthanide ions.11,13,15,16 In contrast to d-block metals, lanthanides engage in predominately electrostatic interactions with ligands, due to the limited radial extension of their valence 4f orbitals.17 Consequently, the lanthanide ions possess large spin–orbit coupled ground states, and with judicious choice of ligand field, lanthanide compounds can be designed that exhibit exceptionally large magnetic anisotropies.15,17,18 The study of lanthanide-based complexes has resulted in major breakthroughs in the design of single-molecule magnets with very large Ueff values.15,16,19−28 However, the majority of lanthanide single-molecule magnets display low blocking temperatures as a result of quantum tunneling.15,29 Strong exchange coupling in lanthanide compounds can suppress through barrier relaxation,8 although the lanthanides typically engage in only very weak magnetic exchange,13 with a few notable exceptions in which they are coupled to radical ligands27,30−33 or metal ions with diffuse spin orbitals.34
The actinide ions in principle combine advantageous properties of both the transition metal and lanthanide ions, although they remain heavily understudied in single-molecule magnetism.6,35−38 Like the lanthanides, the actinides possess unquenched orbital angular momenta and thus large magnetic anisotropies.6,36 At the same time, the 5f orbitals are more radially diffuse than the 4f orbitals,39 and thus actinide–ligand bonding can be more covalent.40−42 As such, strong magnetic exchange interactions are possible in actinide-based complexes, even in the absence of radical bridging ligands.43 Considering this, actinide-based exchange-coupled clusters represent desirable targets for single-molecule magnet research. This has perhaps been best exemplified by the multinuclear complexes {[UVO2(salen)]2MnII(py)3}6 (salenH2 = N,N′-ethylenebis(salicylimine); py = pyridine) and {[UVO2(Mesaldien)][MnII(TPA)I]2}+ (MesaldienH2 = N,N′-(2-aminomethyl)diethylenebis(salicylimine); TPA = tris(2-pyridyl-methyl)amine) (Figure 1, left and middle).44,45 Here, bridging oxygen atoms interact covalently with both the anisotropic UV (5f1, 2F5/2) and high-spin MnII (S = 5/2) ions, leading to significant ferromagnetic exchange, with an estimated coupling constant of JU–Mn = +7.5 cm–1 in the latter complex.45 These two molecules are the only actinide containing compounds known to exhibit molecular magnetic blocking, and they also exhibit large relaxation barriers of Ueff = 99(5) and 56.3(3) cm–1, respectively.
Figure 1.

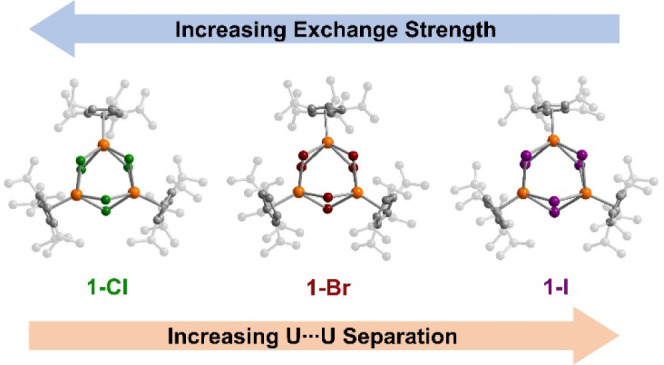
Molecular structures of reported exchange-coupled actinide-based single-molecule magnets known to exhibit magnetic blocking, the year they were reported, and their 100-s blocking temperatures: {[UO2(salen)]2Mn(Py)3}6 (left),44 {[UO2(Mesaldien)][Mn(TPA)I]2}I (middle),45 and, from this work, (CpiPr5)3U3Cl6 (right). Orange, teal, red, blue, gray, purple, and green spheres represent U, Mn, O, N, C, I, and Cl atoms, respectively; H atoms and an outer-sphere I– counterion are omitted for clarity.
In principle, even stronger magnetic exchange interactions should be possible in lower-valent actinide complexes. Here, uranium(III) (5f3, 4I9/2) is of particular interest because it possesses a larger total angular momentum ground state and more radially diffuse orbitals than uranium(V). Of note, the arene-bridged diuranium(III) complex (μ-toluene)U2(N[tBu]Ar)4 (Ar = 3,5-C6H3Me2)46 displays a maximum in the magnetic susceptibility, χM, indicative of antiferromagnetic coupling between uranium ions, at 125 K. This temperature is correlated with the strength of the exchange interaction,47 suggesting that exchange is far stronger in this complex than in actinyl-based single-molecule magnets44,45,48−50 or other uranium(V) complexes.51−55 However, to our knowledge there are only three reported examples of uranium(III)-based single-molecule magnets that exhibit exchange coupling (either between uranium(III) centers or uranium(III) and a radical ligand) and in these cases, the exchange is relatively weak.56−58
Herein, we report the synthesis and characterization of the halide-bridged uranium(III) clusters (CpiPr5)3UIII3X6 (1-X, X = Cl, Br, I). Trends in the UV–vis–NIR spectra suggest that the U–X bonds possess covalent character as a result of 5f orbital contributions to the bonding interactions. Static magnetic characterization reveals magnetic exchange coupling between UIII centers in all three triangular clusters, with the strength of the exchange increasing from 1-I to 1-Br to 1-Cl. Indeed, the exchange is sufficiently strong in 1-Cl that the molecule exhibits magnetic blocking at 2.4 K, providing a first example of such behavior for a uranium(III) compound.
Results and Discussion
Synthesis and Structural Characterization
We initially targeted the synthesis of dinuclear halide-bridged uranium complexes following a metathesis route analogous to that reported for the synthesis of the dinuclear lanthanide complexes (CpiPr5)2Ln2I4 (Ln = Y, Gd, Tb, Dy).26 In brief, the reaction of UBr3 or UI3 (or UI3(dioxane)1.5) with one equivalent of NaCpiPr5 in toluene at elevated temperature was allowed to proceed over the course of 8 days (see the Supporting Information for details). However, workup of the resulting products led only to isolation of the mononuclear metallocene complexes (CpiPr5)2UBr (Figure S34) and (CpiPr5)2UI59 as dark-green solids in low yields. We hypothesized that the larger ionic radius of UIII (1.025 Å) versus the trivalent lanthanides (for example, 0.938 Å for GdIII) may result in preferential coordination of two CpiPr5 ligands, rather than one as in the case of the lanthanides in (CpiPr5)2Ln2I4.60
We therefore sought to synthesize a monocyclopentadienyl uranium complex that could be used as a precursor to isolate halide-bridged complexes. Uranium borohydrides, legacy materials of the Manhattan project,61 have been used as starting materials to access monocyclopentadienyl uranium(III) and uranium(IV) compounds,62 and the monocyclopentadienyl dysprosium(III) complex, (CpiPr5)Dy(BH4)2(THF) has been synthesized via treatment of Dy(BH4)3(THF)3 with one equivalent of NaCpiPr5.23 Considering this, we turned to the versatile uranium(III) compound U(BH4)3(THF)263 and found that it reacts cleanly at room temperature in toluene with one equivalent of NaCpiPr5 to yield the trivalent monocyclopentadienyl complex (CpiPr5)U(BH4)2(THF) as a dark black-green solid in 52% crystalline yield. Single crystals of (CpiPr5)U(BH4)2(THF) were grown from a concentrated n-hexane solution at −25 °C. Analysis of single-crystal X-ray diffraction data collected at 100 K revealed that the compound crystallizes in the space group P21/c with two molecules in the asymmetric unit. Each complex adopts a pseudo three-legged piano-stool geometry in which each uranium ion is coordinated by two κ3-borohydride ligands (average U–H distances of 2.41(6) and 2.39(7) Å for each complex), a THF molecule (U–O bonds of 2.502(3) and 2.514(4) Å), and a capping η5-cyclopentadienyl ligand (U–Cp(cent) distances of 2.4971(6) and 2.4991(6) Å).
We next sought to synthesize an aryloxide precursor starting from
(CpiPr5)U(BH4)2(THF)
that could react with Me3SiX (X = halide) to generate halide-bridged
uranium complexes. Such reactivity would take advantage of the moderately
labile nature of the Si–X bond and the driving force for the
formation of a strong bond between silicon, a hard acid, and oxygen,
a hard base.64−66 Furthermore, there is precedent for the use of such
reagents to synthesize organouranium halides.67,68 We selected 4-tert-butylphenoxide (−OPhtBu) as a suitable ligand, given its
steric bulk and its ability to promote solubility in hydrocarbon solvents.
The reaction of (CpiPr5)U(BH4)2(THF) with two equivalents of KOPhtBu in toluene at room temperature resulted in the formation
of the aryloxide-bridged diuranium(III) complex (CpiPr5)2U2(OPhtBu)4 in high yield as a dark red-brown solid (Scheme 1). Single crystals suitable
for X-ray diffraction analysis were grown from a concentrated pentane
solution at −25 °C. The compound crystallizes in the space
group P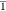 with
the asymmetric unit consisting of
one-half of the dimeric complex. The two U atoms in the complex are
related by inversion symmetry, with a resulting U···U
separation of 3.9735(3) Å. Each uranium(III) center adopts a
three-legged piano-stool geometry, with a U–Cp(cent) distance
of 2.5260(3) Å, a terminal U–OPhtBu distance
of 2.155(4) Å, bridging U–OPhtBu distances of 2.348(4) and 2.459(3) Å, and a U–O–U
angle of 111.5(1)°.
with
the asymmetric unit consisting of
one-half of the dimeric complex. The two U atoms in the complex are
related by inversion symmetry, with a resulting U···U
separation of 3.9735(3) Å. Each uranium(III) center adopts a
three-legged piano-stool geometry, with a U–Cp(cent) distance
of 2.5260(3) Å, a terminal U–OPhtBu distance
of 2.155(4) Å, bridging U–OPhtBu distances of 2.348(4) and 2.459(3) Å, and a U–O–U
angle of 111.5(1)°.
Scheme 1. Two-Step Synthesis of the Triangular Halide-Bridged Clusters (CpiPr5)3U3X6 (1-X, X = Cl, Br, I), Involving the Reaction of (CpiPr5)U(BH4)2(THF) with Potassium 4-tert-Butylphenoxide Followed by the Reaction of the Resulting Dinuclear UIII Precursor (CpiPr5)2U2(OPhtBu)4 with Me3SiX (X = Cl, Br, I).

The reaction of (CpiPr5)2U2(OPhtBu)4 with excess Me3SiX (X = Cl, Br, I) at room temperature yielded the halide-bridged uranium(III) complexes (CpiPr5)3U3X6 (1-X, X = Cl, Br, I) over the course of 2 to 7 days, as judged based on a change in the color of the reaction mixture from red-brown to dark blue (X = Cl, Br) or blue-green (X = I). The reaction time was found to correlate with the strength of the Si–X bond in the organosilane reagent, with the synthesis of 1-Cl taking the longest and the synthesis of 1-I taking the shortest amount of time. The compounds 1-Cl and 1-Br could be isolated as dark blue crystalline solids in moderate to good yields, and we suspect the isolated yield of 1-Cl to be limited by its high solubility in hydrocarbon solvents. Compound 1-I was isolated as a dark teal solid in very low yield (<10%), which we attribute to the concomitant formation of the mixed-valence uranium(III/IV) complex (CpiPr5)3U3I6(μ3-O), as verified by single-crystal X-ray diffraction analysis of olive-green crystals isolated from the reaction (Figure S40; note that shorter reaction times did not improve the yield of 1-I). Analogous mixed-valence compounds (CpMe4R)3U3I6(μ3-O) (R = Me, H, SiMe3)69 have been previously reported as resulting from activation of diethyl ether during the reaction of UI3 with KCpMe4R. In the toluene-based reaction described here, the μ3-O likely forms after activation of one of the aryloxide leaving groups. Nevertheless, 1-I can be readily separated from (CpiPr5)3U3I6(μ3-O) through successive washes with cold pentane. Analogous side-products (CpiPr5)3U3X6(μ3-O) (X = Cl and Br) were not isolated in the syntheses of 1-Cl and 1-Br.
The solid-state structures of 1-X were determined
by analysis of single-crystal X-ray diffraction data collected at
100 K (Figure 2; see Section 5 of the Supporting Information). The
compound 1-Cl crystallizes in the space group Pbca, while 1-Br and 1-I crystallize
in the space group P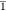 (1-I additionally crystallizes
with two toluene molecules in the asymmetric unit). Each structure
contains analogous trinuclear clusters featuring an edge-bridged triangular
[U3X6]3+ core, in contrast with the
dinuclear structures known for the lanthanide analogues that inspired
this work.26 As previously mentioned, this
difference reflects the larger ionic radius of uranium(III) compared
with the lanthanide(III) ions.60 Examples
of halide-bridged uranium clusters are rare,69−77 and 1-X represent the first examples of homotrivalent
uranium clusters containing a central U3X6 (X
= halide) core. While such M3X6 cores are commonly
observed for transition metals,78−90 the limited number of uranium clusters featuring this type of core
are either homotetravalent,75 or more commonly
possess a μ3-bridging atom,69,76,77 as a result of solvent- or ligand-activation
by uranium(III)—leading instead to mixed-valent species, such
as (CpMe4R)3U3I6(μ3-O)69 noted above. Notably, a mixed-valence
thorium(III/IV) cluster, (COT)3Th3Cl6 (COT2– = cyclooctatetraene dianion), was recently
reported that possesses an structurally analogous core to that in 1-X.91 While this cluster was reported
to feature direct Th–Th bonding interactions, there is no evidence
of direct U–U bonds in 1-X (see further discussion
below).
(1-I additionally crystallizes
with two toluene molecules in the asymmetric unit). Each structure
contains analogous trinuclear clusters featuring an edge-bridged triangular
[U3X6]3+ core, in contrast with the
dinuclear structures known for the lanthanide analogues that inspired
this work.26 As previously mentioned, this
difference reflects the larger ionic radius of uranium(III) compared
with the lanthanide(III) ions.60 Examples
of halide-bridged uranium clusters are rare,69−77 and 1-X represent the first examples of homotrivalent
uranium clusters containing a central U3X6 (X
= halide) core. While such M3X6 cores are commonly
observed for transition metals,78−90 the limited number of uranium clusters featuring this type of core
are either homotetravalent,75 or more commonly
possess a μ3-bridging atom,69,76,77 as a result of solvent- or ligand-activation
by uranium(III)—leading instead to mixed-valent species, such
as (CpMe4R)3U3I6(μ3-O)69 noted above. Notably, a mixed-valence
thorium(III/IV) cluster, (COT)3Th3Cl6 (COT2– = cyclooctatetraene dianion), was recently
reported that possesses an structurally analogous core to that in 1-X.91 While this cluster was reported
to feature direct Th–Th bonding interactions, there is no evidence
of direct U–U bonds in 1-X (see further discussion
below).
Figure 2.

Solid-state structures of (CpiPr5)3U3Cl6 (1-Cl, left), (CpiPr5)3U3Br6 (1-Br, middle), and (CpiPr5)3U3I6 (1-I, right). Orange, green, brown, purple, and gray spheres represent U, Cl, Br, I, and C atoms, respectively; H atoms are omitted for clarity.
All three uranium(III) centers in 1-X are crystallographically distinct and exhibit a four-legged piano-stool coordination geometry, defined by an η5-cyclopentandienyl ligand and four bridging halide ligands. Selected average distances and angles for these complexes are listed in Table 1 (see also Tables S7–S10). Progressing down the series from 1-Cl to 1-I, the main structural change is an increase of the average U···U separation, from 4.1946(7) Å in 1-Cl to 4.5700(5) Å in 1-I. This increase can be largely attributed to the increasing size of the halide ions and a corresponding increase in the U–X bond length, from an average of 2.812(2) Å in 1-Cl to 3.1914(8) Å in 1-I. This increase compensates for the decrease in the average X–U–X angle from 96.48(4)° in 1-Cl to 91.46(2)° in 1-I, which might otherwise favor shorter U···U separations. The U···U separations in 1-X are greater than the sum of covalent radii for two uranium atoms (3.92(7) Å) as tabulated by Alvarez et al.,92 suggesting no direct U–U bonding interactions are present. However, the average U–X bond length in each complex is less than the sum of the corresponding covalent radii (2.98 Å for U–Cl, 3.16 Å for U–Br, and 3.35 Å for U–I),92 suggesting the potential for covalent uranium–halide bonding in these clusters.
Table 1. Selected Average Bond Distances (Å) and Angles (Deg) for (CpiPr5)3U3Cl6 (1-Cl), (CpiPr5)3U3Br6 (1-Br), and (CpiPr5)3U3I6 (1-I)a.
| 1-Cl | 1-Br | 1-I | |
|---|---|---|---|
| U···U | 4.1946(7) | 4.3762(5) | 4.5700(5) |
| U–CpiPr5(cent) | 2.4836(5) | 2.5046(4) | 2.4806(4) |
| U–X | 2.812(1) | 2.9677(7) | 3.1914(8) |
| U–X–U | 96.48(4) | 95.01(2) | 91.46(2) |
| X–U–CpiPr5(cent) | 119.83(3) | 119.24(2) | 117.33(2) |
The standard
deviation of the
average value was estimated from 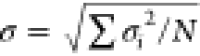 , where σi is the standard deviation of each bond distance (or angle) i and N is the number of distances (or
angles) averaged.
, where σi is the standard deviation of each bond distance (or angle) i and N is the number of distances (or
angles) averaged.
Electronic Absorption Spectra
Ultraviolet–visible–near-infrared (UV–vis–NIR) spectra were collected for all complexes (Figures S15–S30). The spectra for (CpiPr5)U(BH4)2(THF) and (CpiPr5)2U2(OPhtBu)4 are typical for 4I9/2 uranium(III) complexes (see Figures S15–S19), with many low- to medium-intensity features from λ = 400 to 900 nm that can be attributed to Laporte-allowed 5f3 → 5f26d1 transitions, as well as charge-transfer transitions involving metal and ligand orbitals.93 In the visible region, the spectra for 1-X are notably different (Figure 3, left). The dark blue and teal colors of these complexes come from a much more intense charge-transfer transition, most likely a ligand-to-metal charge transfer (LMCT), involving the bridging halide ligands and uranium. Interestingly, the energy of the most intense absorption feature for 1-Cl, 1-Br, and 1-I (located at λmax) decreases upon moving down the halide series, from 15,060 cm–1 (λmax = 664 nm) to 14,598 cm–1 (λmax = 685 nm) to 13,624 cm–1 (λmax = 734 nm), respectively. This trend likely arises as a result of the decreasing energy gap between the frontier orbitals of uranium and the bridging halide from Cl to Br to I. This trend is also evidence that this feature arises from LMCT, rather than metal-to-ligand charge transfer, which would likely show the opposite trend based on the relative energies of the uranium and halide frontier orbitals.40,41 The intensity of the band also decreases going down the series, concomitant with an increase in the intensity of the f → f transitions of 1-X, visible in the NIR region (Figure 3, right).
Figure 3.

UV–vis–NIR (left, 300–1500 nm) and NIR (right, 780–1500 nm) spectra for solutions of the clusters (CpiPr5)3U3X6 (1-X, X = Cl, Br, I) in hexane.
Owing to their electric dipole-forbidden nature, the f → f transitions of the actinides are typically very weak, with molar absorptivity values in the range 10 M–1 cm–1 ≤ ε ≤ 100 M–1 cm–1.93 However, the f → f transitions of 1-X are far more intense, with 100 M–1 cm–1 ≤ ε ≤ 1600 M–1 cm–1. Intense f → f transitions characterized for compounds of the f-elements can be the result of mixing of nf and (n + 1)d orbitals (n = 4 or 5).39,94 However, in many uranium-based complexes, such as the iodide-bridged diuranium(III) complex [U(N**)]2(μ–I)2 (N** = N(SiMe2tBu)2),95 such intense f → f transitions are proposed to result from intensity-stealing mechanisms96−98 involving vibronic coupling of these transitions to charge-transfer transitions. This coupling results in the mixing of some charge-transfer excited state character into the uranium-localized f → f states, which relaxes the electric dipole-forbidden nature of the latter. Such mixing is facilitated because there is common 5f orbital parentage in both the charge transfer and f → f states.99 This type of intensity-stealing mechanism is well-known for d → d transitions in d-metal complexes and reflects the extent of metal–ligand covalency.100 Covalency in f-element–ligand bonding has likewise been proposed to manifest in enhanced intensities of the f → f transitions by means of a similar coupling mechanism, and it has been suggested that the closer in energy the metal and ligand states, the stronger the coupling and intensity-stealing process.94
Together, the trends in the UV–vis–NIR spectra of 1-X suggest that the U–X bonding interactions have covalent character due to 5f orbital involvement. These trends also suggest the U–X bonding interactions may become more covalent upon progressing down the halide series. This could be ascribed to two factors: the extent of metal–ligand orbital overlap and the energy difference between the frontier atomic orbitals of the metal and halide ligands (see Figure S33 for details).40,42 The evidence for covalency in the U–X bonds, together with the bridging nature of the halide ligands, suggest the possibility of strong magnetic exchange interactions between the uranium(III) centers in the clusters.101 Supporting this, magnetic susceptibility data on the aforementioned complex [U(N**)]2(μ–I)2 are indicative of antiferromagnetic exchange between uranium centers at low temperatures,95 possibly as a result of uranium–halide covalency.
Static Magnetic Properties
Dc magnetic susceptibility data were collected on polycrystalline samples of 1-X (X = Cl, Br, I) and (CpiPr5)2U2(OPhtBu)4 between 2 and 300 K under applied fields of 0.1, 1.0, and 7.0 T (see Figures S45, S49, S70, and S81). At 1 T and 300 K, the χMT values are 1.93, 3.39, 3.78, and 3.96 cm3 K/mol for (CpiPr5)2U2(OPhtBu)4, 1-Cl, 1-Br, and 1-I, respectively (Figure 4, upper). The predicted χMT value for a single UIII ion is 1.64 cm3 K/mol at 300 K, and the experimental values for all complexes are therefore well below the values expected for complexes featuring two or three noninteracting uranium(III) centers (3.27 and 4.91 cm3 K/mol, respectively). This result is typical for uranium(III) complexes102 and can be ascribed to the presence of a large magnetic anisotropy, which results in incomplete thermal population of the MJ states in the ground 4I9/2 multiplet, even at room temperature. Within the 1-X series, the magnitude of χMT at 300 K increases upon moving from 1-Cl to 1-I, which is indicative of decreasing crystal-field splitting, and hence weaker total anisotropy, moving down the halide series.
Figure 4.
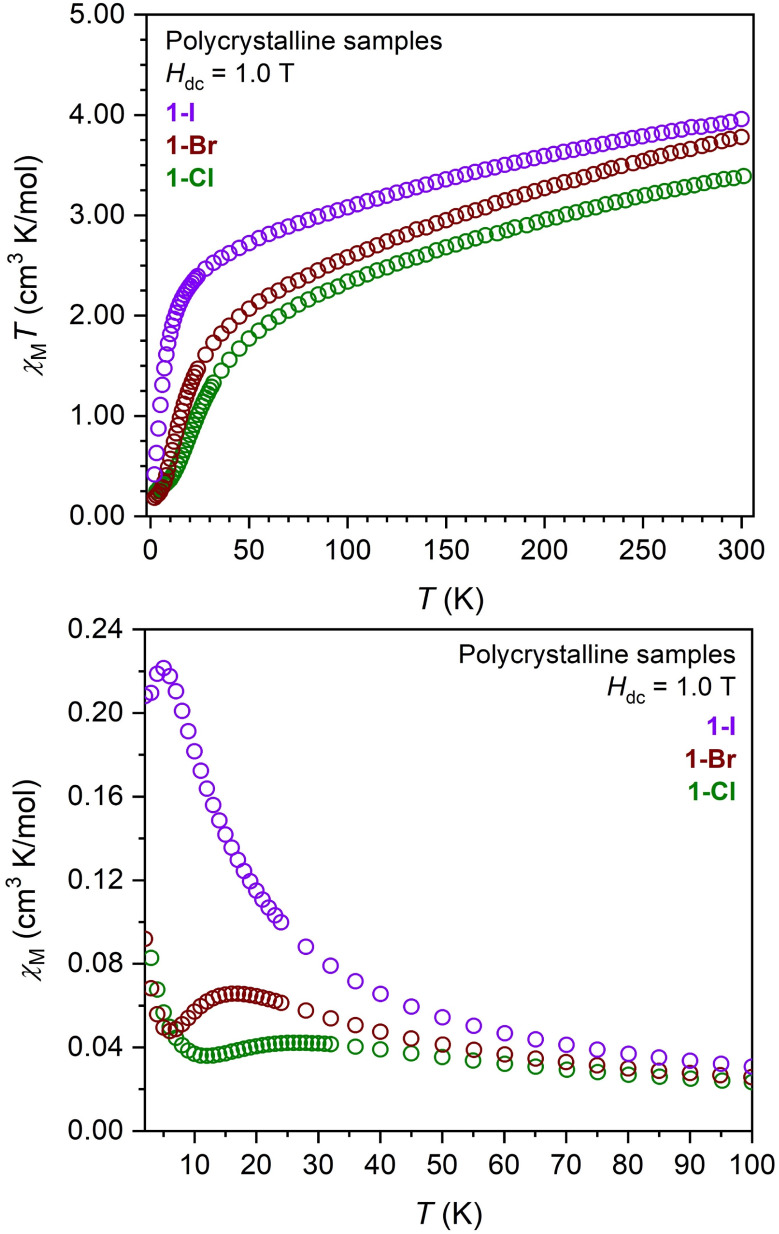
Plots of the molar magnetic susceptibility times temperature (χMT, upper) and molar magnetic susceptibility (χM, lower) versus temperature for polycrystalline samples of (CpiPr5)3U3X6 (1-X, X = Cl, Br, I) under a dc field of 1.0 T. The maxima in χM versus T at 28, 17, and 5 K, respectively, for 1-Cl, 1-Br, and 1-I are indicative of antiferromagnetic exchange interactions as discussed in the text. The subsequent increase in χM below these maxima as the temperature approaches 2 K likely originates from the presence of a minor paramagnetic impurity.47,55,85
As the temperature is decreased, χMT decreases for all complexes due to depopulation of excited MJ states; this decrease occurs in a linear fashion until approximately 45 K (for 1-X) and 24 K (for (CpiPr5)2U2(OPhtBu)4), below which χMT decreases more significantly, reaching values of 0.22, 0.18, 0.42, and 0.31 cm3 K/mol for 1-Cl, 1-Br, 1-I, and (CpiPr5)2U2(OPhtBu)4, respectively, at 2 K. This drop at low temperature can be ascribed to thermal depopulation of excited states and strong Zeeman splitting. A decrease in χMT at low temperatures can also result from the pairing of magnetic moments through magnetic exchange. However, in uranium compounds, competing single-ion effects can obscure evidence of magnetic exchange, particularly at low temperatures, an effect that will be exacerbated in plots of the χMT product versus T.43 On the other hand, changes in χM versus T resulting from, for example, antiferromagnetic exchange interactions, are more visually discernible than in plots of χMT.47 Notably, plots of χM versus temperature (Figure 4, lower) for 1-Cl, 1-Br, and 1-I feature maxima at 28, 17, and 5 K, respectively, which are indicative of antiferromagnetic interactions.46,95,103 Similarly, χM versus T data obtained for dilute frozen toluene solutions of 1-Cl (Figure S60) and 1-Br (Figure S74) also feature maxima at 18 and 11 K, respectively, establishing that the maxima in χM are not due to intermolecular interactions and instead are molecular in origin (by extension the same is likely true for 1-I). Of reported compounds with a M3X6 core, only a few have been shown to exhibit magnetic coupling. For example, the vanadium(II) complexes (CpMe4R)3V3Cl6 (R = Me, Et)80,85 exhibit very strong antiferromagnetic exchange (J = −113 and −119 cm–1, respectively), and (CpMe5)3U3I6(μ3-O) was reported to exhibit weak antiferromagnetic interactions between uranium centers mediated by the μ3-oxo bridge.69
The temperature at which χM approaches a maximum for a molecular cluster with antiferromagnetic coupling is correlated with the strength of the exchange interaction,47 suggesting that magnetic exchange interaction in 1-X becomes weaker progressing from 1-Cl to 1-Br to 1-I. Of note, no maximum is apparent in the χM versus T data for (CpiPr 52U2(OPhtBu)4 (Figure S46), indicating negligible, if any, magnetic exchange coupling in this complex. Significantly, complexes exhibiting unambiguous magnetic exchange between uranium(III) centers are exceedingly rare. To the best of our knowledge, other known examples include the recently reported triuranium cluster U3(iBuPOSS)3 (iBuPOSSH3 = (iBu)7Si7O9(OH)3),104 [Cs2{U(OSi(OtBu)3)3}2(μ-O)],105 [U(OSi(OtBu)3)2(μ-OSi(OtBu)3)]2,103 [U(N**)]2(μ–I)2,95 [K3{[U(OSi(OtBu)3)3]2(μ-N)}],52 and (μ-toluene)U2(N[tBu]Ar)4 (Ar = 3,5-C6H3Me2).46 Similar to 1-X, these compounds exhibit covalent uranium–bridging ligand bonding in tandem with antiferromagnetic exchange, with maxima in χM versus T at approximately 6, 10, 16, 17, 23, and 125 K, respectively.
To acquire further support for the presence of antiferromagnetic exchange in 1-X, we collected variable-field magnetization data (M versus H). At 2 K and 7 T, M reaches values of 0.53 and 0.58 μB for 1-Cl and 1-Br, respectively (Figures S61 and S75). These values are quite small, suggesting that the magnetic moments of some of the uranium ions are aligned antiparallel with respect to each other. In contrast, for (CpiPr5)2U2(OPhtBu)4, M = 1.27 μB at 2 K and 7 T (Figure S48). Interestingly, in the case of 1-I, the exchange coupling is apparently weak enough such that there is an initial increase in M versus H to a value of 0.56 μB at 1.5 T, followed by a near plateau and then a more dramatic increase in the slope of the curve, with M reaching a value of 3.5 μB at 7 T (Figures S85–S87). We ascribe this behavior to a thermally broadened ground-state crossover upon reaching a critical magnetic field, HCF (based on the field at which dM/dH reaches a maximum, Figure S80),106 beyond which the excited ferromagnetically coupled state becomes lower in energy than the antiferromagnetically coupled ground state, leading to an increase in M. For 1-I, this occurs at magnetic fields greater than 3.5 T.
The magnetic exchange interaction, represented by the exchange coupling constant J, is a sum of an intramolecular dipolar interaction, Jdip, and an exchange-coupling interaction, Jex, namely superexchange.101 Based on the maxima in χM versus temperature for 1-X, the temperatures at which magnetic exchange influences the magnetic susceptibility are low enough that only the ground Kramers doublet (i.e., ± MJ states) of each uranium ion will be thermally populated.107,108 The dipolar interaction, in such a case, will involve through-space interactions between the magnetic moments of the ground Kramers doublets of each uranium ion, with the strength of the interaction decreasing with larger U···U separations, r, by the relation 1/r3. Dipolar interactions dominate magnetic exchange in many multilanthanide complexes (|Jdip| is typically on the order of ∼1–5 cm–1) and the magnitude of this exchange is strongly dependent on the orientation of the individual Kramers doublets with respect to each other.8,11,109−111 Each uranium(III) center in 1-X likely possesses strongly uniaxial magnetic anisotropy (i.e., gz > gx, gy) as a result of the strong U–CpiPr bonding interaction.59 In the simplest scenario, each uranium ion possesses a completely axial, and maximal, MJ = ± 9/2 ground state (gx, gy, gz = 0, 0, 6.55); assuming full parallel alignment of the magnetic moments, upper limits for |Jdip| for 1-Cl, 1-Br, and 1-I are 0.50, 0.44, and 0.39 cm–1, respectively (see Supporting Information for details). However, the temperature at which χM reaches a maximum, which is related to the strength of the exchange interaction as noted above,47 corresponds to thermal energies (kBT) of 19.5, 11.8, and 3.5 cm–1 for 1-Cl, 1-Br, and 1-I, respectively. Taking these energies to represent the upper limit of the exchange energy in each complex, then the total exchange interaction, J, in 1-X is approximately 1 to 2 orders of magnitude larger than the dipolar interaction energies. Thus, superexchange interactions facilitated by the overlap of uranium 5f and halide orbitals are likely the predominant source of magnetic exchange in 1-X.
Interestingly, the magnetic exchange strength decreases proceeding down the halide series, suggesting that other factors, beyond just degree of orbital overlap, play a role in the strength of the exchange interaction in these complexes.47,112−115 Nevertheless, the trend for 1-X is similar to that observed in the solid-state uranium trihalides UX3 (X = Cl, Br, I),116−119 namely in that the temperature at which χM reaches a maximum decreases moving from UCl3 (22 K), to UBr3 (15 K), to UI3 (3.4 K). This result has been attributed to both the increasing U···U separation, as well as the decreasing electronegativity of the bridging halide, down the series. Similar trends have also been suggested from computational studies on hypothetical halide-bridged uranium(III) and uranium(IV) complexes.120
Magnetic Hysteresis
A comparison of zero field-cooled and field-cooled dc magnetic susceptibility data collected for 1-Cl revealed a small divergence in the χMT values below 3 K (Figures S51 and S56), indicative of the onset of magnetic blocking that is a hallmark of single-molecule magnetism. Indeed, variable-field magnetization measurements revealed open magnetic hysteresis at 2 K (Figure 5, upper) and up to 2.75 K (Figure 5, lower). Hysteresis is also apparent in variable-field magnetization data collected for a 26.7 mM frozen toluene solution of 1-Cl, indicating that this behavior is molecular in origin (see Figures S66–S69). To the best of our knowledge, such slow magnetic relaxation behavior is unprecedented for uranium(III), and represents only the third example of open hysteresis in any actinide-based molecule.44,45 This is also the first example of magnetic blocking in a molecule containing only actinides as the metal ions. In contrast with these results, there are many examples of uranium48,56,95,121−124 and other actinide125 complexes that exhibit waist-restricted (or “butterfly”) hysteresis, in which the magnetization goes to zero upon removal of the applied magnetic field, as a result of fast, through-barrier magnetic relaxation. With a few exceptions,48 the magnetic hysteresis exhibited by these systems is not molecular in origin and instead arises from phonon bottleneck effects6,126 or long-range ordering as a result of intermolecular interactions.36,127
Figure 5.
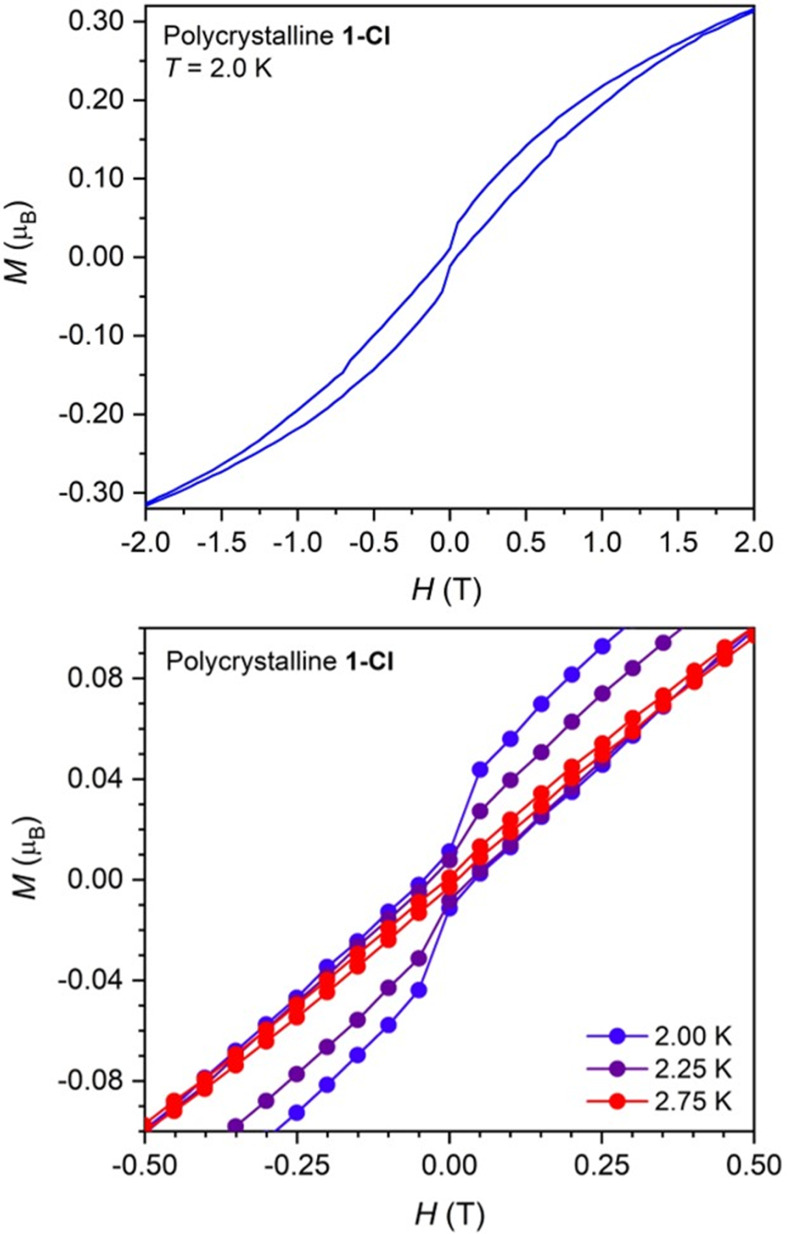
Variable-field magnetization, M, versus field, H, plots for polycrystalline 1-Cl, showing magnetic hysteresis at 2 K (upper) and between 2 and 2.75 K (lower). Sweep rates of 140(4) Oe/s and 33(1) Oe/s were used for |Hdc| > 20 kOe and |Hdc| < 20 kOe, respectively. The sharp drop in the magnetization at zero field is ascribed to quantum tunneling.
Interestingly, hysteresis evident for the solution sample of 1-Cl is narrower than that measured in the solid state and persists over a slightly smaller temperature range of 2–2.5 K (Figures S68 and S69), indicating that the relaxation is slightly faster in the magnetically dilute solution phase. In contrast, magnetic dilution can often enhance magnetic relaxation times relative to those measured for the bulk samples, particularly at low temperature, as the removal of neighboring magnetic moments minimizes transverse fields that can give rise to quantum tunneling.6 The faster relaxation observed for 1-Cl in solution might be explained by a slight structural change upon dissolution in toluene; indeed, the measured maximum in a plot of χM versus T for 1-Cl changes from 28 K in the solid state to about 18 K in solution (both complexes measured at 7 T; Figure S60). This change suggests a weakening of the exchange interaction in solution, and together with the change in the variable-field magnetization data with dilution, is strongly suggestive that the observed hysteresis for 1-Cl is correlated with the strength of exchange coupling. In support of this hypothesis, no magnetic hysteresis was apparent in variable-field magnetization data collected for 1-Br or 1-I (Figures S75, S76, and S85), which exhibit weaker exchange coupling than 1-Cl.
Dynamic Magnetic Properties
To further explore the magnetization relaxation dynamics in 1-Cl, we collected temperature-dependent alternating current (ac) magnetic susceptibility data using a 4 Oe ac field at frequencies ranging from 1 to 1500 Hz. Ac magnetic susceptibility measurements can probe magnetic relaxation time scales that are too fast to observe on the time scale of variable-field magnetization measurements, and therefore we also collected ac susceptibility data for 1-Br and 1-I to probe for evidence of slow magnetic relaxation in these compounds (data were also collected for (CpiPr5)2U2(OPhtBu)4; see the Supporting Information for details and Figures S93–S97 and S126). Both 1-Cl and 1-Br exhibit temperature-dependent maxima in the molar out-of-phase magnetic susceptibility (χM″) versus frequency data collected under zero dc field, from 3 to 4 K and 1.9 to 3 K, respectively (see Figures S98 and S108), indicative of slow magnetic relaxation. This result is noteworthy, as slow magnetic relaxation under zero applied field is extremely rare for actinide-based complexes because of efficient quantum tunneling.36,37 The zero-field slow magnetic relaxation characterized for 1-Cl and 1-Br—and the open magnetic hysteresis measured for 1-Cl—suggest that exchange interactions in these complexes serve to diminish this through-barrier process.7,8 In the case of 1-I, a nonzero signal in χM″ was also measured at 2 K under zero dc field at the highest frequency, although there is no clear maximum under these conditions (Figure S115). The presence of a χM″ signal at zero-field suggests that magnetic exchange may also be suppressing fast zero-field relaxation in 1-I, but relaxation in this compound is much faster than in 1-Cl and 1-Br. Below, we discuss in more detail the zero-field temperature-dependent slow magnetic relaxation in 1-Cl and 1-Br, and, separately, temperature-dependent magnetic relaxation in 1-I characterized under a small dc field.
Temperature-dependent relaxation times, τ, were extracted for 1-Cl and 1-Br from simultaneous fits of the molar in-phase (χM′) and χM″ data for both compounds using a generalized Debye model1 (see Figures S98 and S108). In the case of 1-Cl, an adequate model of the ac susceptibility data required fitting with two relaxation processes, a “slow” process occurring at low frequencies, and a “fast” process occurring at higher frequencies (see Figures S99–S101). In contrast, the data for 1-Br are more uniform and could be fit with a single relaxation process (Figures S108 and S109). We also collected dc relaxation data for 1-Cl at 1.9, 2.25, and 2.5 K, which were fit with a stretched exponential to extract corresponding relaxation times (Figure S121).
Plots of ln(τ) versus 1/T generated for 1-Cl and 1-Br using relaxation times extracted from ac and dc data are shown in Figure 6. For 1-Cl, the data at high temperatures corresponding to the fast and slow relaxation processes are relatively linear, suggesting that magnetic relaxation is occurring via an Orbach mechanism,128 rather than the through-barrier Raman relaxation typically observed for actinide complexes (open green and red symbols, Figure 6, upper; see also Figure S127). In contrast, the relaxation times extracted from dc relaxation data are relatively invariant with temperature (Figure 6, upper, open purple symbols), consistent with tunneling of the magnetization. At high temperatures, the plot of ln(τ) versus 1/T for 1-Br also trends toward linear, although there is more pronounced curvature in the data at the lowest temperatures (Figure 6 middle, open blue circles). The zero-field relaxation times for both compounds were fit to a sum of quantum tunneling129 and Orbach relaxation mechanisms, according to the equation, τ–1 = τtunnel–1 + τ0–1 exp(−Ueff/kBT), where τtunnel is the relaxation time for quantum tunneling, τ0 is the attempt time, Ueff is the effective thermal barrier to magnetic relaxation, and kB is the Boltzmann constant (0.695 cm–1).126
Figure 6.
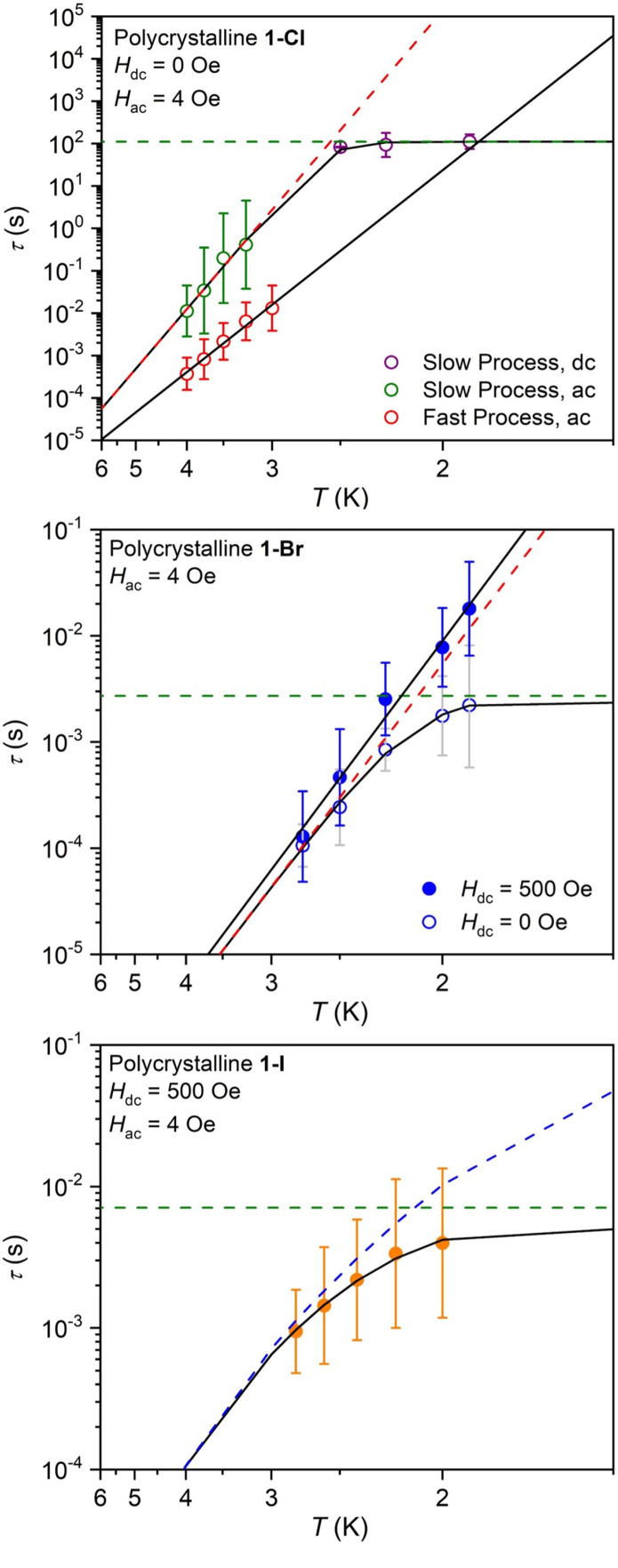
Plots of the magnetic relaxation time (τ, log scale) versus T (inverse scale) for 1-Cl (upper), 1-Br (middle), and 1-I (lower). Black lines represent total fits of the data as discussed in the text. Green, red, and blue dashed lines represent quantum tunneling, Orbach, and Raman processes for 1-Cl (slow process, Hdc = 0 Oe), 1-Br (Hdc = 0 Oe), and 1-I (Hdc = 500) Oe. The fast process for 1-Cl and the data for 1-Br at 500 Oe were also fit to an Orbach process.
The resulting fit parameters for both compounds are listed in Table 2. From the fit of the slower process for 1-Cl, we were also able to estimate a 100-s blocking temperature of Tb,100s = 2.4 K, which is slightly lower than the temperature at which magnetic hysteresis loops for the compound begin to close. The values of Ueff extracted for 1-Cl are 45.1(1) and 30.49(9) cm–1, which are among the largest reported for uranium single-molecule magnets. The Ueff and Tb,100s values are smaller than those reported for {[UO2(salen)]2Mn(py)3}6 (Ueff = 99 cm–1, Tb,100s = 4.6 K)44 and {[UO2(Mesaldien)][Mn(TPA)I]2}I (Ueff = 56.3 cm–1, Tb,100s = 3.1 K),45 while Ueff value for the slow process in 1-Cl is larger than the values reported for the exchange-coupled complexes [{UO2(Mesaldien)}{Fe(TPA)Cl}2]I (Ueff = 37.5 cm–1) and [{UO2(Mesaldien)}{Ni(BPPA)(py)}2]I (Ueff = 19.0 cm–1; BPPAH = bis(2-poclyl)(2-hydroxybenzyl)amine),48 which do not exhibit magnetic blocking. More generally, it is noteworthy that 1-Cl is the first all-uranium cluster compound to exhibit magnetic exchange giving rise to magnetic blocking and Orbach relaxation under zero applied field.
Table 2. Fit Parameters of the Magnetic Relaxation Times (from AC Magnetic Susceptibility Measurements) for 1-Xa.
| compound | Hdc (Oe) | τtunnel (s) | C (K–n s–1) | n | τ0 (s) | Ueff (cm–1) |
|---|---|---|---|---|---|---|
| 1-Cl | 0 (slow process) | 111(9) | 10–8.94(3) | 45.1(1) | ||
| 0 (fast process) | 10–8.15(2) | 30.49(9) | ||||
| 1-Br | 0 | 0.0027(1) | 10–8.56(2) | 20.1(1) | ||
| 500 | 10–8.50(4) | 20.7(1) | ||||
| 1-I | 500 (slow process) | 0.0071(2) | 1.01(3) | 6.59(2) |
The quantum tunneling contribution to the magnetic relaxation of 1-Cl was determined from dc relaxation measurements (Figures S121 and S122), while values of τ0 and Ueff were obtained from fits of only the ac data. Two relaxation processes were characterized for 1-I on the ac time scale, but only the temperature dependence of the slower process (i.e., process 1, see Figure S119) could be determined.
The value of Ueff extracted for 1-Br is 20.1(1) cm–1 (τ0 = 10–8.50(4) s), which is similar in magnitude to relaxation barriers reported for many mononuclear actinide single-molecule magnets,37 although for the latter systems, magnetic relaxation likely occurs via through-barrier Raman processes involving vibronically accessible “virtual” excited states, often much lower in energy than excited single-ion or exchange-coupled states.6,36 To examine the possibility that the zero-field relaxation observed for 1-Br arises due to Raman relaxation, we also collected ac susceptibility data under a 500 Oe applied field from 1.9 to 3 K (see Figures S110 and S111) and fit both sets of data using the equation τ–1 = CTn, where C and n are fit parameters that describe Raman relaxation. For a Kramers system, it has been shown that the Raman process is independent of the magnitude of the applied field.130 However, we obtained dramatically different fit parameters under zero and applied field (C = 1.7(1) s–1 K–n and n = 8.45(4) versus C = 0.0119(8) s–1 K–n and n = 13.17(5), respectively), which indicates the Raman mechanism is not valid in this case (see Figures S131 and S132 for these fits and additional discussion).
As further support for Orbach relaxation in 1-Br, the plot of the natural log of τ (under an applied field) versus 1/T is linear across the examined temperature range (Figure 6, middle, filled blue circles), indicating that quantum tunneling has effectively been quenched by the applied field. The data could be fit to an Orbach relaxation process, and the resulting values of Ueff and τ0 are consistent with those determined from the fit of the zero-field data, as well as those determined from fits of the data for 1-Cl. These values are also reasonable when compared with other uranium single-molecule magnets that undergo Orbach relaxation.44,45,48,50 Altogether, these results support the relevance of the Orbach relaxation mechanism for 1-Br under zero field. We note that application of a magnetic field also leads to longer measured relaxation times for 1-Cl, this time on the on the dc time scale (Figures S122 and S128), with τ increasing from 111 s (under zero field) to 335 s (under Hdc = 500 Oe) at 1.9 K. However, unlike for 1-Br, quantum tunneling is not completely suppressed by the applied field (Figure 6).
Thermally activated slow magnetic relaxation under zero applied field is exceedingly rare for uranium single-molecule magnets, and to our knowledge, 1-Cl and 1-Br are the first uranium-only molecules to exhibit zero-field Orbach relaxation. We propose that the magnetic exchange coupling in these compounds is effective at suppressing fast zero-field relaxation and promoting relaxation via an Orbach mechanism. Further, the decrease in Ueff upon going from 1-Cl to 1-Br suggests that the relaxation barriers are correlated with the strength of exchange coupling in these compounds (refer to further discussion in the following section).
In contrast to what is seen for 1-Cl and 1-Br, a dc field is required to observe slow magnetic relaxation in 1-I at temperatures above 2 K on the ac time scale (Figures S114–S116), which is more typical for mononuclear actinide-based single-molecule magnets. Ac susceptibility data collected under an applied dc field of 500 Oe revealed temperature-dependent χM″ versus frequency data between 2 and 2.8 K (Figures S118–S120), with two distinguishable relaxation processes observable between 1 and 1500 Hz. Maxima were only evident in χM″ for the slower relaxation process, and τ values extracted for this process are plotted versus T in the lower part of Figure 6. Fitting these data to a sum of quantum tunneling and Raman relaxation processes129,131 according to the equation τ–1 = τtunnel–1 + CTn yielded τtunnel = 0.0071(2) s, n = 6.59(2), and C = 1.01(3) K–n s–1, the latter two of which are typical for actinide single-molecule magnets (Table 2).36,37
Rationalizing Magnetic Exchange and Relaxation Using the Ising Model
As alluded to above, the dynamic magnetic properties of 1-X appear to correlate with the magnitude of the exchange coupling strength in each compound, as estimated based on the maximum in plots of χM versus T. In particular, strong exchange coupling in 1-Cl and 1-Br promotes Orbach relaxation, as well as magnetic blocking in the case of 1-Cl, while through-barrier processes appear to dominate for 1-I in the examined temperature and frequency range. Furthermore, hysteresis and ac susceptibility data collected for dilute, frozen solutions of 1-Cl and 1-Br, respectively, revealed that relaxation is faster than measured for the polycrystalline compounds (see Figures S68, S69, S112, and S133). We attribute these results to slight structural changes in solution that give rise to weaker exchange interactions in both complexes, compared with their solid-state structures, based on the decrease in the temperature at which χM reaches a maximum in both solution samples (Figures S60 and S74). A more rigorous quantification of this exchange interaction is thus highly desirable. However, this would require knowledge of the anisotropy tensors (gx, gy, gz) of each of the three individual uranium ions in each cluster, and therefore the use of ab initio calculations that can be both computationally and time intensive to execute, and are associated with a large margin of error.108,132−134 To the best of our knowledge, no such methods have yet been used to model the exchange interactions in such complicated systems as actinide-based cluster compounds.
One approach that has been used in the literature for estimating values of J in uranium-based, heterometallic complexes is the so-called subtraction method.45,135,136 Here, the paramagnetic transition metal ion in the compound of interest is replaced with a diamagnetic metal ion, and the magnetic susceptibility of the resulting compound is subtracted from the data of the original compound to “eliminate” ligand-field and spin–orbit coupling effects from uranium, leaving behind only the part of the magnetic susceptibility that is due to magnetic exchange. The residual magnetic susceptibility can then be fit to a Hamiltonian describing an isotropic exchange interaction.43,137 This approach has been used to estimate the magnitude of the ferromagnetic exchange in {[UVO2(Mesaldien)][MnII(TPA)I]2}I (J = +7.5 cm–1).45 In the case of 1-X, the most suitable substitution would be with a similarly sized diamagnetic ion, such as LaIII. However, the synthesis of substitutionally pure complexes of form (CpiPr5)3U3–nLanX6 (n = 1, 2), which could enable the use of the subtraction method, would be prohibitively challenging.
In considering an alternative approach to evaluating exchange coupling in 1-X, we turned to the literature on strongly coupled dilanthanide compounds, wherein the exchange can be modeled by an Ising-type interaction.108 In the case of the N23–-bridged compounds [K(18-crown-6)][{(Me3Si)2N)2LnIII(THF)}2(μ-N2·)] (Ln = Tb, Dy, Ho),30 modeling and computational analyses suggested that the corresponding Orbach relaxation process involves the ground and first excited exchange-coupled states.138 These results were corroborated by an inelastic neutron scattering study of [K(18-crown-6)][{(Me3Si)2N)2TbIII(THF)}2(μ-N2·)],139 and a recent investigation of [(Cp*2LnIII)2(μ-5,5′-R2bpym)](BPh4) (Ln = Gd, Dy; Cp* = pentamethylcyclopentadienyl; bpym = 2,2′-bipyrimidine; R = NMe2, OEt, Me, F)33 suggested that this behavior might be a more common phenomenon in dilanthanide radical-bridged compounds. In such cases, the magnitude of Ueff is directly proportional to the magnitude of the exchange-coupling, Jex, which establishes the energetic separation between the exchange-coupled states.138,139 Knowledge of Ueff can therefore be used to estimate Jex, and vice versa (see ref. (33) and the corresponding Supporting Information for more details).33
Here, we endeavored to qualitatively describe the exchange interactions in 1-X by employing a variation of the Ising model that has been adopted extensively to model magnetic exchange in the above-mentioned lanthanide complexes and many others,8,132,140−144 and even some actinide43,49,51,54,55 compounds. Our rationale for this approach in the evaluation of 1-X is that the U–CpiPr5 bonding interaction (defined as along the unique z axis for each ion) will be much stronger than the U–X bonding interactions, resulting in a largely uniaxial magnetic anisotropy for the ground Kramers doublet of each uranium ion (gz > gx, gy).18,59 Indeed, X-band electron paramagnetic resonance (EPR) spectra collected at 6 K for solid samples of 1-X and a solution of 1-Cl in C6D6 do not feature any signals attributable to the clusters, which indicates that gx and gy are negligible in the ground state (see Figures S88 and S89 and Section 6.3 of the Supporting Information for details). In the limit that gx and gy are negligible compared with gz (with the z axis largely defined by the U–CpiPr5 bond axis for each uranium ion), the Ising model focuses only on the magnetic exchange between each Kramers doublet (more specifically, the z-component), and results in three quasi-degenerate antiferromagnetic Kramers doublets, which differ in energy with respect to each other by differences in the exchange interactions between each pair of uranium ions (see Supporting Information for more details). The ferromagnetic excited Kramers doublet would then reside above these states by an energy equivalent to 2Javg, where Javg is the average exchange interaction (Figure 7), when formulated using a pseudospin Hamiltonian (−2J formalism). The use of pseudospins is justified when the Kramers doublets involved in the exchange interaction are well-isolated, and they have previously been used to model magnetic exchange in uranium(V) complexes.51,54,55 However, the MJ states in 1-X are likely admixed, as such, a more accurate formulation of the exchange would consider the z-component of the total angular momentum (i.e., the MJ value).49,108 Such a detailed analysis is beyond the scope of this paper, and the complexity of the electronic structure of uranium(III) likely precludes a more accurate modeling of the magnetic exchange in such a fashion; namely, the ground state, and its composition, is unknown for 1-X. It should also be noted that as a consequence of this admixture of MJ states, the uranium centers in 1-X are not likely truly axial (only a pure, maximal MJ = ± 9/2 ground state would possess gx, gy, gz = 0, 0, 6.55, with the z axis as defined above). This is evidenced by the presence of quantum tunneling in 1-Cl and 1-Br at zero field, which indicates that there are nonaxial components in the ground Kramers doublets of each uranium ion.1 Nevertheless, as noted above, it is likely that the ground states in 1-X are predominantly axial.
Figure 7.
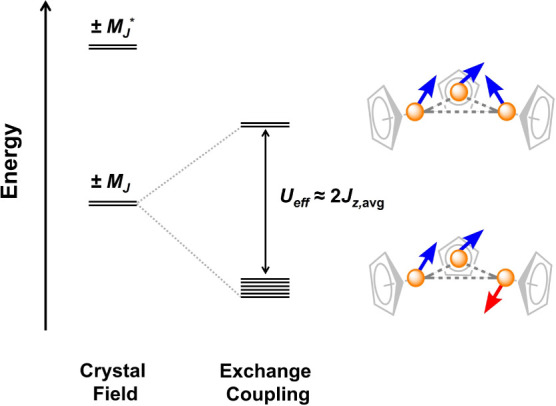
Simplified qualitative depiction of proposed exchange-coupled states in 1-X generated using the Ising model. Differences between pairs of uranium ions are assumed to be negligible, leading to three quasi-degenerate antiferromagnetic states separated in energy from the excited, doubly degenerate, ferromagnetic state by 2Jz, avg (≈ 2Javg; −2J formalism). Here, Javg is the average exchange interaction (see Section 6.4 of the Supporting Information for more details). As discussed in the text, this model was used to estimate Javg for 1-Cl and 1-Br based on their Ueff values. Weaker exchange in the case of 1-I is proposed to give rise to the diagram depicted in Figure S124, wherein there is thermal population of the exchange-coupled excited state, and relaxation of individual uranium centers occurs via a Raman mechanism.
Analogous to the situation described above for radical-bridged lanthanide complexes,33,139 if magnetic relaxation is indeed occurring primarily between the ground antiferromagnetic and excited ferromagnetic states of 1-X (Figure S125), then Ueff is related to the energy required to invert one of the magnetic moments such that all three are then aligned in a near-parallel fashion. This reorientation in the direction of the magnetic moment will occur along the magnetic anisotropy axis1 of the corresponding uranium ion in 1-X. Consequently, while the z axes of the individual uranium ions in 1-X are likely not colinear (i.e., nonparallel), the relation Ueff ≈ 2Jz,avg allows for the estimate of the average exchange strength (Javg) in these complexes, assuming that Jx and Jy are relatively small. In the case of 1-Br, this implies Javg ≈ −10.1 cm–1, which is reasonable given that the temperature at which χM reaches a maximum for 1-Br is 17 K (corresponding thermal energy of kBT = 11.8 cm–1). However, for 1-Cl, this analysis is complicated by the presence of two relaxation processes on the ac time scale (Figures S98–S100), suggesting, possibly, a more complicated electronic structure, and again suggesting that the Ising model is not entirely valid. Nevertheless, adopting the same analysis as for 1-Br, the Ueff values for the fast and slow processes in 1-Cl suggest that Javg is within the range of −15.3 to −22.5 cm–1. These values provide upper estimates to the values of the exchange interactions in 1-Br and 1-Cl, and indicate that the exchange interactions here are on par with (and possibly stronger than) those reported for other exchange-coupled actinide-based single-molecule magnets (J = +7.5 cm–1).45,49
In contrast to what is seen for 1-Cl and 1-Br, the magnetic exchange interaction in 1-I is apparently weak enough to allow thermal population of the excited exchange-coupled state, such that observed magnetic relaxation is associated with the individual uranium ions and not the exchange-coupled system (Figure S124). The uranium(III) centers in 1-I are inequivalent by symmetry, and the multiple relaxation processes observed for this compound may reflect the magnetization dynamics of the individual ions (see Figures S118–S120). Indeed, in several low-symmetry multinuclear lanthanide complexes possessing negligible, or weak, exchange coupling, the lanthanide ions have been shown to relax independently, giving rise to multiple processes on the ac time scale.145−147
Lastly, regarding the observation of two relaxation processes in 1-Cl, we note that multiple magnetic relaxation processes have been reported in exchange-coupled f-element complexes.27,30 For example, magnetic relaxation in [(Cp*2Dy)2(μ-5,5′-R2bpym)](BPh4) (R = NMe2, OEt)33 occurs via both the first and second exchange-coupled excited states. The experimentally determined relaxation barriers associated with these two relaxation processes are similar, resulting in broad, poorly resolved features in the ac susceptibility data, such as is observed for 1-Cl. Another interesting literature compound in this regard is the mixed-valence neptunyl(V/VI) cluster (NpVIO2Cl2)[NpVO2Cl(THF)3]2.49 Here, a form of the Ising model was used to estimated JNp(V)–Np(VI) = +7.5 cm–1, while dynamic magnetic susceptibility data were fit to extract a thermal barrier to relaxation of Ueff = 97.3 cm–1. In this system, magnetic relaxation is believed to occur via a predominately MJ = ± 5/2 excited state of the Np(VI) ion. We propose that a similar situation might hold true for 1-Cl: as a result of the strong exchange coupling in this complex, the excited ferromagnetic state may be close in energy to a thermally accessible excited crystal field (i.e., MJ*) state of one of the uranium ions, and magnetic relaxation via either the ferromagnetic excited state, or a single-ion state, should be energetically feasible. Considering this, one approach to enhancing Ueff and the Tb,100s in such complexes may be to increase the single-ion anisotropy of the individual uranium ions, such as by modifying or replacing the supporting cyclopentadienyl ligand.20,23−25,27,30,148
Conclusions
We have reported the synthesis and detailed spectroscopic and magnetic characterization of the isostructural series of halide-bridged triuranium(III) complexes (CpiPr5)3U3X6 (1-X; X = Cl, Br, I). Spectroscopic analysis suggests the presence of covalent U–X bonding interactions in these compounds, while static magnetic susceptibility data reveal evidence for magnetic exchange coupling that becomes stronger moving from 1-I to 1-Br to 1-Cl. Notably, while all three molecules exhibit features of slow magnetic relaxation, as the exchange interaction becomes stronger, the single-molecule magnet properties are clearly enhanced. In the case of 1-I, slow magnetic relaxation via a Raman process is observed under an applied field and appears to be single-ion in origin. In contrast, 1-Br and 1-Cl exhibit stronger magnetic exchange interactions and both undergo slow magnetic relaxation under zero field via an Orbach process. In the case of 1-Cl, magnetic exchange is strong enough to observe open magnetic hysteresis, with a corresponding 100-s blocking temperature of Tb,100s = 2.4 K, the first example of such behavior in a uranium(III) complex, as well as the first example of such behavior in a complex containing only actinide ions.
These results demonstrate that strong magnetic exchange sufficient to engender Orbach relaxation can be achieved in uranium(III) compounds. Finally, the chemistry developed here is likely to be generalizable such that, with judicious choice of bridging ligands, it should be possible to obtain uranium complexes with different nuclearity, greater uniaxial magnetic anisotropy, and stronger exchange-coupling interactions, with potential relevance to the discovery of new design parameters for creating high-performance actinide-based single-molecule magnets.
Acknowledgments
This work was supported by the Director, Office of Science, Office of Basic Energy Sciences, Division of Chemical Sciences, Geosciences, and Biosciences Heavy Element Chemistry Program of the U.S. Department of Energy (DOE) at LBNL under Contract DE-AC02-05CH11231 (D.J.L. and D.K.S). Magnetic characterization by SQUID magnetometry was supported by NSF grant CHE-2102603 awarded to J.R.L. Additional support was provided by the Naval Air Warfare Center Weapons Division ILIR program (K.R.M. and B.G.H.) and well as the Funai Overseas Scholarship (E.I.). We also thank the ILJU Academy and Culture Foundation for the support of H.K. through an Overseas Ph.D. Scholarship. We are grateful to Dr. Hasan Celik and UC Berkeley’s NMR facility in the College of Chemistry (CoC-NMR) for spectroscopic assistance. Instruments in the CoC-NMR are supported in part by NIH S10OD024998. We further thank Dr. Nicholas Settineri and UC Berkeley’s Small Molecule X-ray Crystallography Facility (CheXray) for crystallographic assistance, Dr. Wayne W. Lukens, Dr. Hossein Taghinejad, and Dr. T. David Harris for helpful discussions, and Dr. Katie R. Meihaus for helpful discussions and assistance with writing and editing.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.3c11678.
Additional experimental details, NMR spectroscopy, IR spectroscopy, additional UV–vis–NIR spectra, X-ray crystallographic information and refinement details, and additional magnetic data (PDF)
Author Contributions
# E.I. and K.R.M. contributed equally to this work.
The authors declare no competing financial interest.
Supplementary Material
References
- Gatteschi D.; Sessoli R.; Villain J.. Molecular Nanomagnets, Mesosc. Phys. Nanotechnol.; Oxford University Press: Oxford; New York, 2006. [Google Scholar]
- Sessoli R.; Gatteschi D.; Caneschi A.; Novak M. A. Magnetic Bistability in a Metal-Ion Cluster. Nature 1993, 365 (6442), 141–143. 10.1038/365141a0. [DOI] [Google Scholar]
- Bogani L.; Wernsdorfer W. Molecular Spintronics Using Single-Molecule Magnets. Nat. Mater. 2008, 7 (3), 179–186. 10.1038/nmat2133. [DOI] [PubMed] [Google Scholar]
- Vincent R.; Klyatskaya S.; Ruben M.; Wernsdorfer W.; Balestro F. Electronic Read-out of a Single Nuclear Spin Using a Molecular Spin Transistor. Nature 2012, 488 (7411), 357–360. 10.1038/nature11341. [DOI] [PubMed] [Google Scholar]
- Thiele S.; Balestro F.; Ballou R.; Klyatskaya S.; Ruben M.; Wernsdorfer W. Electrically Driven Nuclear Spin Resonance in Single-Molecule Magnets. Science 2014, 344 (6188), 1135–1138. 10.1126/science.1249802. [DOI] [PubMed] [Google Scholar]
- Liddle S. T.; van Slageren J. Improving F-Element Single Molecule Magnets. Chem. Soc. Rev. 2015, 44 (19), 6655–6669. 10.1039/C5CS00222B. [DOI] [PubMed] [Google Scholar]
- Wernsdorfer W.; Aliaga-Alcalde N.; Hendrickson D. N.; Christou G. Exchange-Biased Quantum Tunnelling in a Supramolecular Dimer of Single-Molecule Magnets. Nature 2002, 416 (6879), 406–409. 10.1038/416406a. [DOI] [PubMed] [Google Scholar]
- Guo Y.-N.; Xu G.-F.; Wernsdorfer W.; Ungur L.; Guo Y.; Tang J.; Zhang H.-J.; Chibotaru L. F.; Powell A. K. Strong Axiality and Ising Exchange Interaction Suppress Zero-Field Tunneling of Magnetization of an Asymmetric Dy2 Single-Molecule Magnet. J. Am. Chem. Soc. 2011, 133 (31), 11948–11951. 10.1021/ja205035g. [DOI] [PubMed] [Google Scholar]
- Caneschi A.; Gatteschi D.; Sessoli R.; Barra A. L.; Brunel L. C.; Guillot M. Alternating Current Susceptibility, High Field Magnetization, and Millimeter Band EPR Evidence for a Ground S = 10 State in [Mn12O12(CH3COO)16(H2O)4]·2CH3COOH·4H2O. J. Am. Chem. Soc. 1991, 113 (15), 5873–5874. 10.1021/ja00015a057. [DOI] [Google Scholar]
- Sessoli R.; Tsai H. L.; Schake A. R.; Wang S.; Vincent J. B.; Folting K.; Gatteschi D.; Christou G.; Hendrickson D. N. High-Spin Molecules: [Mn12O12(O2CR)16(H2O)4]. J. Am. Chem. Soc. 1993, 115 (5), 1804–1816. 10.1021/ja00058a027. [DOI] [Google Scholar]
- Zabala-Lekuona A.; Seco J. M.; Colacio E. Single-Molecule Magnets: From Mn12-Ac to Dysprosium Metallocenes, a Travel in Time. Coord. Chem. Rev. 2021, 441, 213984. 10.1016/j.ccr.2021.213984. [DOI] [Google Scholar]
- Abbasi P.; Quinn K.; Alexandropoulos D. I.; Damjanović M.; Wernsdorfer W.; Escuer A.; Mayans J.; Pilkington M.; Stamatatos T. C. Transition Metal Single-Molecule Magnets: A {Mn31} Nanosized Cluster with a Large Energy Barrier of ∼ 60 K and Magnetic Hysteresis at ∼ 5 K. J. Am. Chem. Soc. 2017, 139 (44), 15644–15647. 10.1021/jacs.7b10130. [DOI] [PubMed] [Google Scholar]
- Benelli C.; Gatteschi D.. Introduction to Molecular Magnetism: from Transition Metals to Lanthanides; Wiley-VCH: Weinheim, 2015. [Google Scholar]
- Neese F.; Pantazis D. A. What Is Not Required to Make a Single Molecule Magnet. Faraday Discuss. 2011, 148, 229–238. 10.1039/C005256F. [DOI] [PubMed] [Google Scholar]
- Ungur L.; Chibotaru L. F. Strategies toward High-Temperature Lanthanide-Based Single-Molecule Magnets. Inorg. Chem. 2016, 55 (20), 10043–10056. 10.1021/acs.inorgchem.6b01353. [DOI] [PubMed] [Google Scholar]
- Woodruff D. N.; Winpenny R. E. P.; Layfield R. A. Lanthanide Single-Molecule Magnets. Chem. Rev. 2013, 113 (7), 5110–5148. 10.1021/cr400018q. [DOI] [PubMed] [Google Scholar]
- Parker D.; Suturina E. A.; Kuprov I.; Chilton N. F. How the Ligand Field in Lanthanide Coordination Complexes Determines Magnetic Susceptibility Anisotropy, Paramagnetic NMR Shift, and Relaxation Behavior. Acc. Chem. Res. 2020, 53 (8), 1520–1534. 10.1021/acs.accounts.0c00275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinehart J. D.; Long J. R. Exploiting Single-Ion Anisotropy in the Design of f-Element Single-Molecule Magnets. Chem. Sci. 2011, 2 (11), 2078–2085. 10.1039/c1sc00513h. [DOI] [Google Scholar]
- Goodwin C. A. P.; Ortu F.; Reta D.; Chilton N. F.; Mills D. P. Molecular Magnetic Hysteresis at 60 K in Dysprosocenium. Nature 2017, 548 (7668), 439–442. 10.1038/nature23447. [DOI] [PubMed] [Google Scholar]
- McClain K. R.; Gould C. A.; Chakarawet K.; Teat S. J.; Groshens T. J.; Long J. R.; Harvey B. G. High-Temperature Magnetic Blocking and Magneto-Structural Correlations in a Series of Dysprosium(III) Metallocenium Single-Molecule Magnets. Chem. Sci. 2018, 9 (45), 8492–8503. 10.1039/C8SC03907K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gould C. A.; McClain K. R.; Yu J. M.; Groshens T. J.; Furche F.; Harvey B. G.; Long J. R. Synthesis and Magnetism of Neutral, Linear Metallocene Complexes of Terbium(II) and Dysprosium(II). J. Am. Chem. Soc. 2019, 141 (33), 12967–12973. 10.1021/jacs.9b05816. [DOI] [PubMed] [Google Scholar]
- Guo F.-S.; Day B. M.; Chen Y.-C.; Tong M.-L.; Mansikkamäki A.; Layfield R. A. A Dysprosium Metallocene Single-Molecule Magnet Functioning at the Axial Limit. Angew. Chem., Int. Ed. 2017, 56 (38), 11445–11449. 10.1002/anie.201705426. [DOI] [PubMed] [Google Scholar]
- Guo F.-S.; Day B. M.; Chen Y.-C.; Tong M.-L.; Mansikkamäki A.; Layfield R. A. Magnetic Hysteresis up to 80 K in a Dysprosium Metallocene Single-Molecule Magnet. Science 2018, 362 (6421), 1400–1403. 10.1126/science.aav0652. [DOI] [PubMed] [Google Scholar]
- Vincent A. H.; Whyatt Y. L.; Chilton N. F.; Long J. R. Strong Axiality in a Dysprosium(III) Bis(Borolide) Complex Leads to Magnetic Blocking at 65 K. J. Am. Chem. Soc. 2023, 145 (3), 1572–1579. 10.1021/jacs.2c08568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanjak J. C.; Wilkins B. O.; Vieru V.; Bhuvanesh N. S.; Reibenspies J. H.; Martin C. D.; Chibotaru L. F.; Nippe M. A High-Performance Single-Molecule Magnet Utilizing Dianionic Aminoborolide Ligands. J. Am. Chem. Soc. 2022, 144 (39), 17743–17747. 10.1021/jacs.2c06698. [DOI] [PubMed] [Google Scholar]
- Gould C. A.; McClain K. R.; Reta D.; Kragskow J. G. C.; Marchiori D. A.; Lachman E.; Choi E.-S.; Analytis J. G.; Britt R. D.; Chilton N. F.; Harvey B. G.; Long J. R. Ultrahard Magnetism from Mixed-Valence Dilanthanide Complexes with Metal-Metal Bonding. Science 2022, 375 (6577), 198–202. 10.1126/science.abl5470. [DOI] [PubMed] [Google Scholar]
- Demir S.; Gonzalez M. I.; Darago L. E.; Evans W. J.; Long J. R. Giant Coercivity and High Magnetic Blocking Temperatures for N23– Radical-Bridged Dilanthanide Complexes upon Ligand Dissociation. Nat. Commun. 2017, 8 (1), 2144. 10.1038/s41467-017-01553-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Z.; Zhao C.; Feng T.; Liu X.; Ying X.; Li X.-L.; Zhang Y.-Q.; Tang J. Air-Stable Chiral Single-Molecule Magnets with Record Anisotropy Barrier Exceeding 1800 K. J. Am. Chem. Soc. 2021, 143 (27), 10077–10082. 10.1021/jacs.1c05279. [DOI] [PubMed] [Google Scholar]
- Ding Y.-S.; Chilton N. F.; Winpenny R. E. P.; Zheng Y.-Z. On Approaching the Limit of Molecular Magnetic Anisotropy: A Near-Perfect Pentagonal Bipyramidal Dysprosium(III) Single-Molecule Magnet. Angew. Chem., Int. Ed. 2016, 55 (52), 16071–16074. 10.1002/anie.201609685. [DOI] [PubMed] [Google Scholar]
- Rinehart J. D.; Fang M.; Evans W. J.; Long J. R. A N23– Radical-Bridged Terbium Complex Exhibiting Magnetic Hysteresis at 14 K. J. Am. Chem. Soc. 2011, 133 (36), 14236–14239. 10.1021/ja206286h. [DOI] [PubMed] [Google Scholar]
- Demir S.; Jeon I.-R.; Long J. R.; Harris T. D. Radical Ligand-Containing Single-Molecule Magnets. Coord. Chem. Rev. 2015, 289–290, 149–176. 10.1016/j.ccr.2014.10.012. [DOI] [Google Scholar]
- Demir S.; Zadrozny J. M.; Nippe M.; Long J. R. Exchange Coupling and Magnetic Blocking in Bipyrimidyl Radical-Bridged Dilanthanide Complexes. J. Am. Chem. Soc. 2012, 134 (45), 18546–18549. 10.1021/ja308945d. [DOI] [PubMed] [Google Scholar]
- Gould C. A.; Mu E.; Vieru V.; Darago L. E.; Chakarawet K.; Gonzalez M. I.; Demir S.; Long J. R. Substituent Effects on Exchange Coupling and Magnetic Relaxation in 2,2′-Bipyrimidine Radical-Bridged Dilanthanide Complexes. J. Am. Chem. Soc. 2020, 142 (50), 21197–22120. 10.1021/jacs.0c10612. [DOI] [PubMed] [Google Scholar]
- Darago L. E.; Boshart M. D.; Nguyen B. D.; Perlt E.; Ziller J. W.; Lukens W. W.; Furche F.; Evans W. J.; Long J. R. Strong Ferromagnetic Exchange Coupling and Single-Molecule Magnetism in MoS43–-Bridged Dilanthanide Complexes. J. Am. Chem. Soc. 2021, 143 (22), 8465–8475. 10.1021/jacs.1c03098. [DOI] [PubMed] [Google Scholar]
- McAdams S. G.; Ariciu A.-M.; Kostopoulos A. K.; Walsh J. P. S.; Tuna F. Molecular Single-Ion Magnets Based on Lanthanides and Actinides: Design Considerations and New Advances in the Context of Quantum Technologies. Coord. Chem. Rev. 2017, 346, 216–239. 10.1016/j.ccr.2017.03.015. [DOI] [Google Scholar]
- Meihaus K. R.; Long J. R. Actinide-Based Single-Molecule Magnets. Dalton Trans. 2015, 44 (6), 2517–2528. 10.1039/C4DT02391A. [DOI] [PubMed] [Google Scholar]
- Liu M.; Peng X.-H.; Guo F.-S.; Tong M.-L. Actinide-Based Single-Molecule Magnets: Alone or in a Group?. Inorg. Chem. Front. 2023, 10, 3742–3755. 10.1039/D3QI00523B. [DOI] [Google Scholar]
- Magnani N.; Caciuffo R. Future Directions for Transuranic Single Molecule Magnets. Inorganics 2018, 6 (1), 26. 10.3390/inorganics6010026. [DOI] [Google Scholar]
- The Chemistry of the Actinide and Transactinide Elements, Morss L. R.; Edelstein N. M.; Fuger J., Ed.; Springer: Dordrecht, Netherlands, 2006. [Google Scholar]
- Neidig M. L.; Clark D. L.; Martin R. L. Covalency in F-Element Complexes. Coord. Chem. Rev. 2013, 257 (2), 394–406. 10.1016/j.ccr.2012.04.029. [DOI] [Google Scholar]
- Choppin G. R. Covalency in f-Element Bonds. J. Alloys Compd 2002, 344 (1), 55–59. 10.1016/S0925-8388(02)00305-5. [DOI] [Google Scholar]
- Pace K. A.; Klepov V. V.; Berseneva A. A.; zur Loye H.-C. Covalency in Actinide Compounds. Chemistry 2021, 27 (19), 5835–5841. 10.1002/chem.202004632. [DOI] [PubMed] [Google Scholar]
- Rinehart J. D.; Harris T. D.; Kozimor S. A.; Bartlett B. M.; Long J. R. Magnetic Exchange Coupling in Actinide-Containing Molecules. Inorg. Chem. 2009, 48 (8), 3382–3395. 10.1021/ic801303w. [DOI] [PubMed] [Google Scholar]
- Mougel V.; Chatelain L.; Pécaut J.; Caciuffo R.; Colineau E.; Griveau J.-C.; Mazzanti M. Uranium and Manganese Assembled in a Wheel-Shaped Nanoscale Single-Molecule Magnet with High Spin-Reversal Barrier. Nat. Chem. 2012, 4 (12), 1011–1017. 10.1038/nchem.1494. [DOI] [PubMed] [Google Scholar]
- Chatelain L.; Walsh J. P. S.; Pécaut J.; Tuna F.; Mazzanti M. Self-Assembly of a 3d–5f Trinuclear Single-Molecule Magnet from a Pentavalent Uranyl Complex. Angew. Chem., Int. Ed. 2014, 53 (49), 13434–13438. 10.1002/anie.201407334. [DOI] [PubMed] [Google Scholar]
- Vlaisavljevich B.; Diaconescu P. L.; Lukens W. L. Jr; Gagliardi L.; Cummins C. C. Investigations of the Electronic Structure of Arene-Bridged Diuranium Complexes. Organometallics 2013, 32 (5), 1341–1352. 10.1021/om3010367. [DOI] [Google Scholar]
- Kahn O.Molecular Magnetism; VCH, 1993. [Google Scholar]
- Chatelain L.; Pécaut J.; Tuna F.; Mazzanti M. Heterometallic Fe2II–UV and Ni2II–UV Exchange-Coupled Single-Molecule Magnets: Effect of the 3 d Ion on the Magnetic Properties. Chem. – Eur. J. 2015, 21 (50), 18038–18042. 10.1002/chem.201503637. [DOI] [PubMed] [Google Scholar]
- Magnani N.; Colineau E.; Eloirdi R.; Griveau J.-C.; Caciuffo R.; Cornet S. M.; May I.; Sharrad C. A.; Collison D.; Winpenny R. E. P. Superexchange Coupling and Slow Magnetic Relaxation in a Transuranium Polymetallic Complex. Phys. Rev. Lett. 2010, 104 (19), 197202. 10.1103/PhysRevLett.104.197202. [DOI] [PubMed] [Google Scholar]
- Chatelain L.; Tuna F.; Pécaut J.; Mazzanti M. Synthesis and SMM Behaviour of Trinuclear versus Dinuclear 3d–5f Uranyl(V)–Cobalt(II) Cation–Cation Complexes. Dalton Trans. 2017, 46 (17), 5498–5502. 10.1039/C6DT04558H. [DOI] [PubMed] [Google Scholar]
- Spencer L. P.; Schelter E. J.; Yang P.; Gdula R. L.; Scott B. L.; Thompson J. D.; Kiplinger J. L.; Batista E. R.; Boncella J. M. Cation–Cation Interactions, Magnetic Communication, and Reactivity of the Pentavalent Uranium Ion [U(NtBu)2]+. Angew. Chem. 2009, 121 (21), 3853–3856. 10.1002/ange.200806190. [DOI] [PubMed] [Google Scholar]
- Falcone M.; Barluzzi L.; Andrez J.; Fadaei Tirani F.; Zivkovic I.; Fabrizio A.; Corminboeuf C.; Severin K.; Mazzanti M. The Role of Bridging Ligands in Dinitrogen Reduction and Functionalization by Uranium Multimetallic Complexes. Nat. Chem. 2019, 11 (2), 154–160. 10.1038/s41557-018-0167-8. [DOI] [PubMed] [Google Scholar]
- Barluzzi L.; Chatelain L.; Fadaei-Tirani F.; Zivkovic I.; Mazzanti M. Facile N-Functionalization and Strong Magnetic Communication in a Diuranium(V) Bis-Nitride Complex. Chem. Sci. 2019, 10 (12), 3543–3555. 10.1039/C8SC05721D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt A.-C.; Heinemann F. W.; Lukens W. W. Jr; Meyer K. Molecular and Electronic Structure of Dinuclear Uranium Bis-μ-Oxo Complexes with Diamond Core Structural Motifs. J. Am. Chem. Soc. 2014, 136 (34), 11980–11993. 10.1021/ja504528n. [DOI] [PubMed] [Google Scholar]
- Rosen R. K.; Andersen R. A.; Edelstein N. M. [(MeC5H4)3U]2[μ.-1,4-N2C6H4]: A Bimetallic Molecule with Antiferromagnetic Coupling between the Uranium Centers. J. Am. Chem. Soc. 1990, 112 (11), 4588–4590. 10.1021/ja00167a092. [DOI] [Google Scholar]
- Mills D. P.; Moro F.; McMaster J.; van Slageren J.; Lewis W.; Blake A. J.; Liddle S. T. A Delocalized Arene-Bridged Diuranium Single-Molecule Magnet. Nat. Chem. 2011, 3 (6), 454–460. 10.1038/nchem.1028. [DOI] [PubMed] [Google Scholar]
- Modder D. K.; Batov M. S.; Rajeshkumar T.; Sienkiewicz A.; Zivkovic I.; Scopelliti R.; Maron L.; Mazzanti M. Assembling Diuranium Complexes in Different States of Charge with a Bridging Redox-Active Ligand. Chem. Sci. 2022, 13 (38), 11294–11303. 10.1039/D2SC03592H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coutinho J. T.; Antunes M. A.; Pereira L. C. J.; Marçalo J.; Almeida M. Zero-Field Slow Magnetic Relaxation in a Uranium(III) Complex with a Radical Ligand. Chem. Commun. 2014, 50 (71), 10262–10264. 10.1039/C4CC04486J. [DOI] [PubMed] [Google Scholar]
- Guo F.-S.; Chen Y.-C.; Tong M.-L.; Mansikkamäki A.; Layfield R. A. Uranocenium: Synthesis, Structure, and Chemical Bonding. Angew. Chem., Int. Ed. 2019, 58 (30), 10163–10167. 10.1002/anie.201903681. [DOI] [PubMed] [Google Scholar]
- Shannon R. D. Revised Effective Ionic Radii and Systematic Studies of Interatomic Distances in Halides and Chalcogenides. Acta Crystallogr., Sect. A 1976, 32 (5), 751–767. 10.1107/S0567739476001551. [DOI] [Google Scholar]
- Schlesinger H. I.; Brown H. C. Uranium(IV) Borohydride. J. Am. Chem. Soc. 1953, 75 (1), 219–221. 10.1021/ja01097a058. [DOI] [Google Scholar]
- Ephritikhine M. Synthesis, Structure, and Reactions of Hydride, Borohydride, and Aluminohydride Compounds of the f-Elements. Chem. Rev. 1997, 97 (6), 2193–2242. 10.1021/cr960366n. [DOI] [PubMed] [Google Scholar]
- Arnold P. L.; Stevens C. J.; Farnaby J. H.; Gardiner M. G.; Nichol G. S.; Love J. B. New Chemistry from an Old Reagent: Mono- and Dinuclear Macrocyclic Uranium(III) Complexes from [U(BH4)3(THF)2]. J. Am. Chem. Soc. 2014, 136 (29), 10218–10221. 10.1021/ja504835a. [DOI] [PubMed] [Google Scholar]
- Olah G. A.; Narang S. C. Iodotrimethylsilane—a Versatile Synthetic Reagent. Tetrahedron 1982, 38 (15), 2225–2277. 10.1016/0040-4020(82)87002-6. [DOI] [Google Scholar]
- Jung M. E.; Lyster M. A. Quantitative Dealkylation of Alkyl Ethers via Treatment with Trimethylsilyl Iodide. A New Method for Ether Hydrolysis. J. Org. Chem. 1977, 42 (23), 3761–3764. 10.1021/jo00443a033. [DOI] [Google Scholar]
- Morita T.; Okamoto Y.; Sakurai H. Novel Method for Dealkylation of Esters, Ethers, and Acetals by Chlorotrimethylsilane–Sodium Iodide. J. Chem. Soc., Chem. Commun. 1978, 20, 874–875. 10.1039/C39780000874. [DOI] [Google Scholar]
- Gardner B. M.; Stewart J. C.; Davis A. L.; McMaster J.; Lewis W.; Blake A. J.; Liddle S. T. Homologation and Functionalization of Carbon Monoxide by a Recyclable Uranium Complex. Proc. Natl. Acad. Sci. U. S. A. 2012, 109 (24), 9265–9270. 10.1073/pnas.1203417109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matson E. M.; Fanwick P. E.; Bart S. C. Formation of Trivalent U–C, U–N, and U–S Bonds and Their Reactivity toward Carbon Dioxide and Acetone. Organometallics 2011, 30 (21), 5753–5762. 10.1021/om200612h. [DOI] [Google Scholar]
- Larch C. P.; Cloke F. G. N.; Hitchcock P. B. Activation and Reduction of Diethyl Ether by Low Valent Uranium: Formation of the Trimetallic, Mixed Valence Uranium Oxo Species [U(CpRR′)(μ-I)2]3(μ3-O) (CpRR′= C5Me5, C5Me4H, C5H4SiMe3). Chem. Commun. 2008, 1, 82–84. 10.1039/B714211K. [DOI] [PubMed] [Google Scholar]
- Franolic J. D.; Long J. R.; Holm R. H. Comprehensive Tungsten-Iodine Cluster Chemistry: Isolated Intermediates in the Solid-State Nucleation of [W6I14]2–. J. Am. Chem. Soc. 1995, 117 (31), 8139–8153. 10.1021/ja00136a011. [DOI] [Google Scholar]
- Manriquez J. M.; Fagan P. J.; Marks T. J.; Vollmer S. H.; Day C. S.; Day V. W. Pentamethylcyclopentadienyl Organoactinides, Trivalent Uranium Organometallic Chemistry and the Unusual Structure of Bis(Pentamethylcyclopentadienyl)Uranium Monochloride. J. Am. Chem. Soc. 1979, 101 (17), 5075–5078. 10.1021/ja00511a054. [DOI] [Google Scholar]
- Clark D. L.; Gordon J. C.; Huffman J. C.; Watkin J. G.; Zwick B. D. Preparation of Mono-Pentamethylcyclopentadienyl Uranium(IV) Sulfido Clusters through Oxidation of (η-C5Me5)3U3(μ3-S)(μ2-I)3I3. New J. Chem. 1995, 19 (5–6), 495–502. [Google Scholar]
- Korobkov I.; Gambarotta S. Ligand Metalation in the Reactivity of a Tetravalent Uranium Amides. Inorg. Chem. 2010, 49 (7), 3409–3418. 10.1021/ic902567m. [DOI] [PubMed] [Google Scholar]
- Arliguie T.; Ephritikhine M.; Lance M.; Vigner J.; Nierlich M. Reaction of Uranium Tetrachloride with Trimethylsilylcyclopentadiene: Crystal Structure of a Dichloromethane Adduct of [{U(Cp)2(μ-Cl)}3 (μ3-Cl)2][{U(Cp)Cl2}2(μ-Cl)3] (Cp = η = C5H5). J. Organomet. Chem. 1994, 484 (1), 195–201. 10.1016/0022-328X(94)87205-8. [DOI] [Google Scholar]
- Old J.; Danopoulos A. A.; Winston S. Uranium Complexes with Dianionic O-Methylated Calix[4]Arene Ligands. New J. Chem. 2003, 27 (4), 672–674. 10.1039/b212092e. [DOI] [Google Scholar]
- Boronski J. T.; Doyle L. R.; Wooles A. J.; Seed J. A.; Liddle S. T. Synthesis and Characterization of an Oxo-Centered Homotrimetallic Uranium(IV)–Cyclobutadienyl Dianion Complex. Organometallics 2020, 39 (10), 1824–1831. 10.1021/acs.organomet.0c00104. [DOI] [Google Scholar]
- Evans W. J.; Miller K. A.; Ziller J. W.; Greaves J. Analysis of Uranium Azide and Nitride Complexes by Atmospheric Pressure Chemical Ionization Mass Spectrometry. Inorg. Chem. 2007, 46 (19), 8008–8018. 10.1021/ic7006099. [DOI] [PubMed] [Google Scholar]
- Franolic J. D.; Long J. R.; Holm R. H. Comprehensive Tungsten-Iodine Cluster Chemistry: Isolated Intermediates in the Solid-State Nucleation of [W6I14]2–. J. Am. Chem. Soc. 1995, 117 (31), 8139–8153. 10.1021/ja00136a011. [DOI] [Google Scholar]
- Schäfer S.; Bauer H.; Becker J.; Sun Y.; Sitzmann H. Cyclononatetraenyl-Indenyl Transformation and a Zirconium(III) Trimer from Bulky Alkylcyclopentadienylzirconium Chlorides. Eur. J. Inorg. Chem. 2013, 2013 (33), 5694–5700. 10.1002/ejic.201300885. [DOI] [Google Scholar]
- Ting C.; Hammer M. S.; Baenziger N. C.; Messerle L.; Deak J.; Li S.; McElfresh M. Dimeric and Cyclotrimeric Piano-Stool Vanadium(III) Dihalides with Unusual Differences in V–V Distance and Magnetochemistry. Syntheses, Structures, and Reactivities of (η-C5Me4R)2V2(μ-Br)4 and the Trivanadium Cluster (η-C5Me4R)3V3(μ-Cl)6, New Mid-Valent Organovanadium Synthons. Organometallics 1997, 16 (9), 1816–1818. 10.1021/om970045h. [DOI] [Google Scholar]
- Lee T.-Y.; Wooten A. J.; Luci J. J.; Swenson D. C.; Messerle L. Four-Electron Reduction of Dinitrogen during Solution Disproportionation of the Organodimetallic (η-C5Me4R)2Ta2(μ-Cl)4 (R = Me, Et) to a New μ- η1, η1-N2 Complex and Odd-Electron Organotrimetallic Cluster. Chem. Commun. 2005, 43, 5444–5446. 10.1039/b508148c. [DOI] [PubMed] [Google Scholar]
- Stollmaier F.; Thewalt U. Isolierung und struktur des dreikernigen hexamethylbenzolzirkonium-komplexes [(Me6C6)3Zr3Cl6]2+[(Al2Cl7)−]2. J. Organomet. Chem. 1981, 208 (3), 327–334. 10.1016/S0022-328X(00)86719-4. [DOI] [Google Scholar]
- Solari E.; Gauthier S.; Scopelliti R.; Severin K. Multifaceted Chemistry of [(Cymene)RuCl2]2 and PCy3. Organometallics 2009, 28 (15), 4519–4526. 10.1021/om900374e. [DOI] [Google Scholar]
- Abernethy C. D.; Bottomley F.; Decken A.; Thompson R. C. Organometallic Halides: Preparation and Physical and Chemical Properties of Tris[(η-Pentamethylcyclopentadienyl)Dichlorovanadium], [(η-C5Me5)V(μ-Cl)2]3. Organometallics 1997, 16 (9), 1865–1869. 10.1021/om960482g. [DOI] [Google Scholar]
- Abernethy C. D.; Bottomley F.; Decken A.; Summers D. A.; Thompson R. C. Organometallic Halides: Structure and Magnetism of Tris[(η-Pentamethylcyclopentadienyl)Dichlorovanadium], [(η-C5Me5)V(μ-Cl)2]3. Organometallics 1998, 17 (6), 1248–1250. 10.1021/om970903+. [DOI] [Google Scholar]
- Abernethy C. D.; Bottomley F.; Day R. W.; Decken A.; Summers D. A.; Thompson R. C. Organometallic Oxides: Preparation and Properties of the Clusters [(η-C5Me5)V(μ3-O)]4 and [(η-C5Me5)V]4(μ-O)6 by Reductive Aggregation of (η-C5Me5)VCl2(O). Organometallics 1999, 18 (5), 870–879. 10.1021/om9807809. [DOI] [Google Scholar]
- Gauthier S.; Scopelliti R.; Severin K. Syntheses and Structures of the Trinuclear Ruthenium Complexes [RuCl2(PAd2Bu)]3 and [RuCl2(PtBu2Cy)]3. Organometallics 2005, 24 (24), 5792–5794. 10.1021/om050649i. [DOI] [Google Scholar]
- Goldberg S. Z.; Spivack B.; Stanley G.; Eisenberg R.; Braitsch D. M.; Miller J. S.; Abkowitz M. Synthesis, Structure, and Physical Properties of the Bis(7,7,8,8-Tetracyano-p-Quinodimethane) Salt of the Paramagnetic Cluster Tris[(Di-μ-Chloro)(Hexamethylbenzene)Niobium], [Nb3(μ-Cl)6(C6Me6)3]2+(TCNQ)22–. J. Am. Chem. Soc. 1977, 99 (1), 110–117. 10.1021/ja00443a021. [DOI] [Google Scholar]
- García-Castro M.; García-Iriepa C.; Del Horno E.; Martín A.; Mena M.; Pérez-Redondo A.; Temprado M.; Yélamos C. The Puzzling Monopentamethylcyclopentadienyltitanium(III) Dichloride Reagent: Structure and Properties. Inorg. Chem. 2019, 58 (8), 5314–5324. 10.1021/acs.inorgchem.9b00437. [DOI] [PubMed] [Google Scholar]
- Churchill M. R.; Chang S. W.-Y. X-Ray Crystallographic Evidence for a Trinuclear Formulation for the ‘Non-Oxidized’ Hexamethylbenzene–Niobium Chloride Cluster Complex, [(Me6C6)3Nb3Cl6]Cl. J. Chem. Soc., Chem. Commun. 1974, 7, 248–249. 10.1039/C39740000248. [DOI] [Google Scholar]
- Boronski J. T.; Seed J. A.; Hunger D.; Woodward A. W.; van Slageren J.; Wooles A. J.; Natrajan L. S.; Kaltsoyannis N.; Liddle S. T. A Crystalline Tri-Thorium Cluster with σ-Aromatic Metal–Metal Bonding. Nature 2021, 598 (7879), 72–75. 10.1038/s41586-021-03888-3. [DOI] [PubMed] [Google Scholar]
- Cordero B.; Gómez V.; Platero-Prats A. E.; Revés M.; Echeverría J.; Cremades E.; Barragán F.; Alvarez S. Covalent Radii Revisited. Dalton Trans. 2008, 21, 2832–2838. 10.1039/b801115j. [DOI] [PubMed] [Google Scholar]
- Liddle S. T. The Renaissance of Non-Aqueous Uranium Chemistry. Angew. Chem., Int. Ed. 2015, 54 (30), 8604–8641. 10.1002/anie.201412168. [DOI] [PubMed] [Google Scholar]
- Henrie D. E.; Fellows R. L.; Choppin G. R. Hypersensitivity in the Electronic Transitions of Lanthanide and Actinide Complexes. Coord. Chem. Rev. 1976, 18 (2), 199–224. 10.1016/S0010-8545(00)82044-5. [DOI] [Google Scholar]
- Goodwin C. A. P.; Tuna F.; McInnes E. J. L.; Mills D. P. Exploring Synthetic Routes to Heteroleptic UIII, UIV, and ThIV Bulky Bis(Silyl)Amide Complexes. Eur. J. Inorg. Chem. 2018, 2018 (20–21), 2356–2362. 10.1002/ejic.201800036. [DOI] [Google Scholar]
- Evans W. J.; Champagne T. M.; Davis B. L.; Allen N. T.; Nyce G. W.; Johnston M. A.; Lin Y.-C.; Khvostov A.; Ziller J. W. Structural Studies of Mono(Pentamethylcyclopentadienyl)Lanthanide Complexes. J. Coord. Chem. 2006, 59 (10), 1069–1087. 10.1080/00958970500512831. [DOI] [Google Scholar]
- Graves C. R.; Yang P.; Kozimor S. A.; Vaughn A. E.; Clark D. L.; Conradson S. D.; Schelter E. J.; Scott B. L.; Thompson J. D.; Hay P. J.; Morris D. E.; Kiplinger J. L. Organometallic Uranium(V)–Imido Halide Complexes: From Synthesis to Electronic Structure and Bonding. J. Am. Chem. Soc. 2008, 130 (15), 5272–5285. 10.1021/ja711010h. [DOI] [PubMed] [Google Scholar]
- Morris D. E.; Da Re R. E.; Jantunen K. C.; Castro-Rodriguez I.; Kiplinger J. L. Trends in Electronic Structure and Redox Energetics for Early-Actinide Pentamethylcyclopentadienyl Complexes. Organometallics 2004, 23 (22), 5142–5153. 10.1021/om049634v. [DOI] [Google Scholar]
- Graves C. R.; Vaughn A. E.; Schelter E. J.; Scott B. L.; Thompson J. D.; Morris D. E.; Kiplinger J. L. Probing the Chemistry, Electronic Structure and Redox Energetics in Organometallic Pentavalent Uranium Complexes. Inorg. Chem. 2008, 47 (24), 11879–11891. 10.1021/ic8017375. [DOI] [PubMed] [Google Scholar]
- Carlin R. L. Inorganic Electronic Spectroscopy. J. Chem. Educ. 1969, 46, A628. 10.1021/ed046pA628.1. [DOI] [Google Scholar]
- Anderson P. W. New Approach to the Theory of Superexchange Interactions. Phys. Rev. 1959, 115 (1), 2–13. 10.1103/PhysRev.115.2. [DOI] [Google Scholar]
- Kindra D. R.; Evans W. J. Magnetic Susceptibility of Uranium Complexes. Chem. Rev. 2014, 114 (18), 8865–8882. 10.1021/cr500242w. [DOI] [PubMed] [Google Scholar]
- Camp C.; Mougel V.; Pécaut J.; Maron L.; Mazzanti M. Cation-Mediated Conversion of the State of Charge in Uranium Arene Inverted-Sandwich Complexes. Chemistry 2013, 19 (51), 17528–17540. 10.1002/chem.201302752. [DOI] [PubMed] [Google Scholar]
- Tricoire M.; Jori N.; Tirani F. F.; Scopelliti R.; Ivković I.; Natrajan L. S.; Mazzanti M. A Trinuclear Metallasilsesquioxane of Uranium(III). Chem. Commun. 2023, 60 (1), 55–58. 10.1039/D3CC05390C. [DOI] [PubMed] [Google Scholar]
- Jori N.; Rajeshkumar T.; Scopelliti R.; Ivković I.; Sienkiewicz A.; Maron L.; Mazzanti M. Cation Assisted Binding and Cleavage of Dinitrogen by Uranium Complexes. Chem. Sci. 2022, 13 (32), 9232–9242. 10.1039/D2SC02530B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapira Y.; Bindilatti V. Magnetization-Step Studies of Antiferromagnetic Clusters and Single Ions: Exchange, Anisotropy, and Statistics. J. Appl. Phys. 2002, 92 (8), 4155–4185. 10.1063/1.1507808. [DOI] [Google Scholar]
- Lines M. E. Orbital Angular Momentum in the Theory of Paramagnetic Clusters. J. Chem. Phys. 1971, 55 (6), 2977–2984. 10.1063/1.1676524. [DOI] [Google Scholar]
- Chibotaru L. F.; Iwahara N. Ising Exchange Interaction in Lanthanides and Actinides. New J. Phys. 2015, 17 (10), 103028. 10.1088/1367-2630/17/10/103028. [DOI] [Google Scholar]
- Habib F.; Murugesu M. Lessons Learned from Dinuclear Lanthanide Nano-Magnets. Chem. Soc. Rev. 2013, 42 (8), 3278–3288. 10.1039/c2cs35361j. [DOI] [PubMed] [Google Scholar]
- Meng Y.-S.; Xiong J.; Yang M.-W.; Qiao Y.-S.; Zhong Z.-Q.; Sun H.-L.; Han J.-B.; Liu T.; Wang B.-W.; Gao S. Experimental Determination of Magnetic Anisotropy in Exchange-Bias Dysprosium Metallocene Single-Molecule Magnets. Angew. Chem., Int. Ed. 2020, 59 (31), 13037–13043. 10.1002/anie.202004537. [DOI] [PubMed] [Google Scholar]
- Han T.; Ding Y.-S.; Giansiracusa M. J.; Chilton N. F.; Winpenny R. E. P.; Zheng Y.-Z. Determinative Effect of Axial Linearity on Single-Molecule Magnet Performance in Dinuclear Dysprosium Complexes. Chem. – Eur. J. 2023, 29 (29), e202300256 10.1002/chem.202300256. [DOI] [PubMed] [Google Scholar]
- Palumbo C. T.; Barluzzi L.; Scopelliti R.; Zivkovic I.; Fabrizio A.; Corminboeuf C.; Mazzanti M. Tuning the Structure, Reactivity and Magnetic Communication of Nitride-Bridged Uranium Complexes with the Ancillary Ligands. Chem. Sci. 2019, 10 (38), 8840–8849. 10.1039/C9SC02149C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coetzee S.; Turnbull M. M.; Landee C. P.; Monroe J. C.; Deumal M.; Novoa J. J.; Rademeyer M. Satellite Ligand Effects on Magnetic Exchange in Dimers. A Structural, Magnetic and Theoretical Investigation of Cu2L2X4 (L = Methylisothiazolinone and X = Cl–, Br–). Phys. Chem. Chem. Phys. 2023, 25 (13), 9176–9187. 10.1039/D2CP05629A. [DOI] [PubMed] [Google Scholar]
- Landee C. P.; Greeney R. E. Strong Antiferromagnetic Exchange in a Bromide-Bridged Copper Dimer, Bis(μ-Bromo)Bis[Bromobis(Tetramethylene Sulfoxide)Copper(II)]. Structure, Susceptibility, and Magnetostructural Correlations. Inorg. Chem. 1986, 25 (21), 3771–3775. 10.1021/ic00241a013. [DOI] [Google Scholar]
- Felthouse T. R.; Laskowski E. J.; Hendrickson D. N. Magnetic Exchange Interactions in Transition Metal Dimers. 10. Structural and Magnetic Characterization of Oxalate-Bridged, Bis(1,1,4,7,7-Pentaethyldiethylene Triamine)Oxalatodicopper Tetraphenylborate and Related Dimers. Effects of Nonbridging Ligands and Counterions on Exchange Interactions. Inorg. Chem. 1977, 16 (5), 1077–1089. 10.1021/ic50171a023. [DOI] [Google Scholar]
- Jones E. R. Jr; Hendricks M. E.; Stone J. A.; Karraker D. G. Magnetic Properties of the Trichlorides, Tribromides, and Triiodides of U(III), Np(III), and Pu(III). J. Chem. Phys. 2003, 60 (5), 2088–2094. 10.1063/1.1681318. [DOI] [Google Scholar]
- Hinatsu Y.; Miyake C.; Imoto S. Antiferromagnetic Transition of Uranium Trichloride. J. Nucl. Sci. Technol. 1980, 17 (12), 929–934. 10.1080/18811248.1980.9732676. [DOI] [Google Scholar]
- Murasi A.; Fischer P.; Furrer A.; Schmid B.; Svcvepaniak W. Magnetic Phase Transitions and Crystal Field Splitting of UX3 Compounds (X = Cl, Br, I) Investigated by Neutron Scattering. J. Common Met. 1986, 121, 151–155. 10.1016/0022-5088(86)90523-0. [DOI] [Google Scholar]
- Schmid B.; Murasik A.; Fischer P.; Furrer A.; Kanellakopulos B. Ningeutron Scatter Investigation of Magnetic Phase Transitions and Crystal-Field Splitting in UX3 (X ≡ Br, Cl). J. Phys.: Condens. Matter 1990, 2 (14), 3369. 10.1088/0953-8984/2/14/021. [DOI] [Google Scholar]
- Wang C.-Z.; Wu Q.-Y.; Lan J.-H.; Chai Z.-F.; Gibson J. K.; Shi W.-Q. Binuclear Trivalent and Tetravalent Uranium Halides and Cyanides Supported by Cyclooctatetraene Ligands. Radiochim. Acta 2017, 105 (1), 21–32. 10.1515/ract-2016-2615. [DOI] [Google Scholar]
- Goodwin C. A. P.; Tuna F.; McInnes E. J. L.; Liddle S. T.; McMaster J.; Vitorica-Yrezabal I. J.; Mills D. P. [UIII{N(SiMe2tBu)2}3]: A Structurally Authenticated Trigonal Planar Actinide Complex. Chem. – Eur. J. 2014, 20 (45), 14579–14583. 10.1002/chem.201404864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King D. M.; Tuna F.; McMaster J.; Lewis W.; Blake A. J.; McInnes E. J. L.; Liddle S. T. Single-Molecule Magnetism in a Single-Ion Triamidoamine Uranium(V) Terminal Mono-Oxo Complex. Angew. Chem., Int. Ed. 2013, 52 (18), 4921–4924. 10.1002/anie.201301007. [DOI] [PubMed] [Google Scholar]
- Barluzzi L.; Giblin S. R.; Mansikkamäki A.; Layfield R. A. Identification of Oxidation State + 1 in a Molecular Uranium Complex. J. Am. Chem. Soc. 2022, 144 (40), 18229–18233. 10.1021/jacs.2c06519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coutinho J. T.; Antunes M. A.; Pereira L. C. J.; Bolvin H.; Marçalo J.; Mazzanti M.; Almeida M. Single-Ion Magnet Behaviour in [U(TpMe2)2I]. Dalton Trans. 2012, 41 (44), 13568–13571. 10.1039/c2dt31421e. [DOI] [PubMed] [Google Scholar]
- Magnani N.; Apostolidis C.; Morgenstern A.; Colineau E.; Griveau J.-C.; Bolvin H.; Walter O.; Caciuffo R. Magnetic Memory Effect in a Transuranic Mononuclear Complex. Angew. Chem., Int. Ed. 2011, 50 (7), 1696–1698. 10.1002/anie.201006619. [DOI] [PubMed] [Google Scholar]
- Abragam A.; Bleaney B.. Electron Paramagnetic Resonance of Transition Ions; Oxford Classic Texts in the Physical Sciences; OUP Oxford, 2012. [Google Scholar]
- Meihaus K. R.; Rinehart J. D.; Long J. R. Dilution-Induced Slow Magnetic Relaxation and Anomalous Hysteresis in Trigonal Prismatic Dysprosium(III) and Uranium(III) Complexes. Inorg. Chem. 2011, 50 (17), 8484–8489. 10.1021/ic201078r. [DOI] [PubMed] [Google Scholar]
- Finn C. B. P.; Orbach R.; Wolf W. P. Spin-Lattice Relaxation in Cerium Magnesium Nitrate at Liquid Helium Temperature: A New Process. Proc. Phys. Soc. 1961, 77 (2), 261. 10.1088/0370-1328/77/2/305. [DOI] [Google Scholar]
- Shrivastava K. N. Theory of Spin–Lattice Relaxation. Phys. Status Solidi B 1983, 117 (2), 437–458. 10.1002/pssb.2221170202. [DOI] [Google Scholar]
- Ho L. T. A.; Chibotaru L. F. Spin-Lattice Relaxation of Magnetic Centers in Molecular Crystals at Low Temperature. Phys. Rev. B 2018, 97 (2), 024427. 10.1103/PhysRevB.97.024427. [DOI] [Google Scholar]
- Van Vleck J. H. Paramagnetic Relaxation Times for Titanium and Chrome Alum. Phys. Rev. 1940, 57 (5), 426–447. 10.1103/PhysRev.57.426. [DOI] [Google Scholar]
- Ungur L.; van den Heuvel W.; Chibotaru L. F. Ab Initio Investigation of the Non-Collinear Magnetic Structure and the Lowest Magnetic Excitations in Dysprosium Triangles. New J. Chem. 2009, 33 (6), 1224–1230. 10.1039/b903126j. [DOI] [Google Scholar]
- Gao S.Molecular Nanomagnets and Related Phenomena; Springer: Berlin, 2015. [Google Scholar]
- Giansiracusa M. J.; Moreno-Pineda E.; Hussain R.; Marx R.; Martínez Prada M.; Neugebauer P.; Al-Badran S.; Collison D.; Tuna F.; van Slageren J.; Carretta S.; Guidi T.; McInnes E. J. L.; Winpenny R. E. P.; Chilton N. F. Measurement of Magnetic Exchange in Asymmetric Lanthanide Dimetallics: Toward a Transferable Theoretical Framework. J. Am. Chem. Soc. 2018, 140 (7), 2504–2513. 10.1021/jacs.7b10714. [DOI] [PubMed] [Google Scholar]
- Kozimor S. A.; Bartlett B. M.; Rinehart J. D.; Long J. R. Magnetic Exchange Coupling in Chloride-Bridged 5f–3d Heterometallic Complexes Generated via Insertion into a Uranium(IV) Dimethylpyrazolate Dimer. J. Am. Chem. Soc. 2007, 129 (35), 10672–10674. 10.1021/ja0725044. [DOI] [PubMed] [Google Scholar]
- Rinehart J. D.; Bartlett B. M.; Kozimor S. A.; Long J. R. Ferromagnetic Exchange Coupling in the Linear, Chloride-Bridged Cluster (Cyclam)CoII[(μ-Cl)UIV(Me2Pz)4]2. Inorg. Chim. Acta 2008, 361 (12), 3534–3538. 10.1016/j.ica.2008.03.006. [DOI] [Google Scholar]
- Le Borgne T.; Rivière E.; Marrot J.; Thuéry P.; Girerd J.-J.; Ephritikhine M. Syntheses, X-Ray Crystal Structures, and Magnetic Properties of Novel Linear M2IIUIV Complexes (M = Co, Ni, Cu, Zn). Chem. – Eur. J. 2002, 8 (4), 773–783. . [DOI] [PubMed] [Google Scholar]
- Vieru V.; Iwahara N.; Ungur L.; Chibotaru L. F. Giant Exchange Interaction in Mixed Lanthanides. Sci. Rep. 2016, 6 (1), 24046. 10.1038/srep24046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prša K.; Nehrkorn J.; Corbey J. F.; Evans W. J.; Demir S.; Long J. R.; Guidi T.; Waldmann O. Perspectives on Neutron Scattering in Lanthanide-Based Single-Molecule Magnets and a Case Study of the Tb2(μ-N2) System. Magnetochemistry 2016, 2, 45. 10.3390/magnetochemistry2040045. [DOI] [Google Scholar]
- Tang J.; Hewitt I.; Madhu N. T.; Chastanet G.; Wernsdorfer W.; Anson C. E.; Benelli C.; Sessoli R.; Powell A. K. Dysprosium Triangles Showing Single-Molecule Magnet Behavior of Thermally Excited Spin States. Angew. Chem., Int. Ed. 2006, 45 (11), 1729–1733. 10.1002/anie.200503564. [DOI] [PubMed] [Google Scholar]
- Chibotaru L. F.; Ungur L.; Soncini A. The Origin of Nonmagnetic Kramers Doublets in the Ground State of Dysprosium Triangles: Evidence for a Toroidal Magnetic Moment. Angew. Chem., Int. Ed. 2008, 47 (22), 4126–4129. 10.1002/anie.200800283. [DOI] [PubMed] [Google Scholar]
- Luzon J.; Bernot K.; Hewitt I. J.; Anson C. E.; Powell A. K.; Sessoli R. Spin Chirality in a Molecular Dysprosium Triangle: The Archetype of the Noncollinear Ising Model. Phys. Rev. Lett. 2008, 100 (24), 247205. 10.1103/PhysRevLett.100.247205. [DOI] [PubMed] [Google Scholar]
- Bernot K.; Luzon J.; Bogani L.; Etienne M.; Sangregorio C.; Shanmugam M.; Caneschi A.; Sessoli R.; Gatteschi D. Magnetic Anisotropy of Dysprosium(III) in a Low-Symmetry Environment: A Theoretical and Experimental Investigation. J. Am. Chem. Soc. 2009, 131 (15), 5573–5579. 10.1021/ja8100038. [DOI] [PubMed] [Google Scholar]
- Long J.; Habib F.; Lin P.-H.; Korobkov I.; Enright G.; Ungur L.; Wernsdorfer W.; Chibotaru L. F.; Murugesu M. Single-Molecule Magnet Behavior for an Antiferromagnetically Superexchange-Coupled Dinuclear Dysprosium(III) Complex. J. Am. Chem. Soc. 2011, 133 (14), 5319–5328. 10.1021/ja109706y. [DOI] [PubMed] [Google Scholar]
- Meng Y.-S.; Qiao Y.-S.; Yang M.-W.; Xiong J.; Liu T.; Zhang Y.-Q.; Jiang S.-D.; Wang B.-W.; Gao S. Weak Exchange Coupling Effects Leading to Fast Magnetic Relaxations in a Trinuclear Dysprosium Single-Molecule Magnet. Inorg. Chem. Front. 2020, 7 (2), 447–454. 10.1039/C9QI01252D. [DOI] [Google Scholar]
- Chilton N. F.; Deacon G. B.; Gazukin O.; Junk P. C.; Kersting B.; Langley S. K.; Moubaraki B.; Murray K. S.; Schleife F.; Shome M.; Turner D. R.; Walker J. A. Structure, Magnetic Behavior, and Anisotropy of Homoleptic Trinuclear Lanthanoid 8-Quinolinolate Complexes. Inorg. Chem. 2014, 53 (5), 2528–2534. 10.1021/ic402672m. [DOI] [PubMed] [Google Scholar]
- Su S.-D.; Li J.-X.; Xu F.; Wang C.-X.; Wang K.; Li Y.; Zhang S.-H.; Zhang X.-Q.; Zhang Y.-Q.; Liang F.-P. DyIII Single-Molecule Magnets from Ligands Incorporating Both Amine and Acylhydrazine Schiff Base Groups: The Centrosymmetric {Dy2} Displaying Dual Magnetic Relaxation Behaviors. Dalton Trans. 2020, 49 (44), 15739–15749. 10.1039/C9DT04434E. [DOI] [PubMed] [Google Scholar]
- Durrant J. P.; Day B. M.; Tang J.; Mansikkamäki A.; Layfield R. A. Dominance of Cyclobutadienyl Over Cyclopentadienyl in the Crystal Field Splitting in Dysprosium Single-Molecule Magnets. Angew. Chem., Int. Ed. 2022, 61 (17), e202200525 10.1002/anie.202200525. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.