


Abstract
Interferons (IFNs) are a family of multifunctional proteins involved in immune activation, regulation of cell growth and antiviral response. They exert their functions by induction of several IFN-stimulated genes, including IFN regulatory factors (IRFs), a family of transcriptional regulators. One of these factors, IRF-2, was initially cloned as an antagonistic counterpart to IRF-1 with oncogenic potential. Here we describe a second isoform of IRF-2, termed IRF-2s, cloned from human and murine cells. This isoform lacks two amino acids located C-terminal of the DNA-binding domain, which is conserved in all IRF family members, leading to a change in the predicted secondary structure. Both isoforms have similar binding affinities to known target sequences in electrophoretic mobility shift assays. Using reporter gene constructs with the type IV promoter region of the MHC class II transactivator (CIITA), which is the essential factor for IFN-γ-induced MHC class II expression, we show that the short isoform IRF-2s exhibits a weaker activation ability compared to IRF-2. Thus, our data present the first evidence of two IRF-2 isoforms with different regulatory ability.
INTRODUCTION
Interferons (IFNs) are secreted cytokines with multiple functions in the immune system. Signalling by IFN-γ is mediated via successive phosphorylation events at the IFN-γ receptor by the associated tyrosine kinases JAK1 and JAK2. Subsequently, the signal transducer and activator of transcription 1α (STAT1α) becomes phosphorylated and dimerises (for reviews see 1–3). Homodimerised STAT1α translocates to the nucleus where it binds to the IFN-γ activation site (GAS) in promoters of IFN-inducible genes.
IFN-γ-induced up-regulation and subsequent phosphorylation of STAT1α leads to the induction of a family of transcription factors, the interferon regulatory factors (IRFs) (for reviews see 4,5). IRF-1 was detected through its ability to activate expression of interferon-inducible genes like IFN-β and MHC class I by binding to a sequence element named the interferon-stimulated response element (ISRE) located in the promoter region. An antagonistic factor termed IRF-2 was found to bind the same element but suppressed IRF-1 activation of IFN-inducible genes (6,7). The antagonistic effects are also represented in the oncogenic potential of IRF-2 and the anti-oncogenic potential of IRF-1 (8). Although the antagonistic role of the two factors prevails, IRF-2 has recently been shown to act as an activator of transcription for several genes (9–11). IRF-1 and IRF-2 can be detected in a wide variety of tissues. While the level of expression of IRF-1 in its constitutive form seems to be dependent on the cell cycle, the level of IRF-2 mRNA remains constant (6,7). Another member of the IRF family of transcription factors is the IFN consensus sequence binding protein (ICSBP), which represses transcription of target genes and is predominantly expressed in cells of the immune system (12–14).
The members of the IRF family share significant structural homologies. The N-terminal regions of the IRFs are highly conserved and confer DNA binding specificity. This region is called the DNA-binding domain (DBD) (4). In addition, an IRF-association domain (IAD) and several other regulatory domains are present in various IRFs, like IRF-1, IRF-2 and ICSBP (4,15,16). The binding affinities of IRF-1 and IRF-2 for specific sequences differ for each target sequence. The very low binding affinity of ICSBP to the ISRE is greatly enhanced by complex formation with IRF-1 or IRF-2, both in vivo and in vitro, in the presence or absence of DNA. The regulation of various target genes by IRFs is not only dependent on the formation of hetero-complexes with many different proteins, but also post-translational modifications (4,16–18), i.e. phosphorylation of ICSBP is necessary for binding DNA, which was also observed for IRF-1 and IRF-2 (4,15,19).
In addition to IFN-β and MHC class I, the MHC class II transactivator (CIITA) is another target for IRFs. CIITA was identified in studies of patients that suffered from primary immunodeficiency syndromes (20,21). Expression of CIITA, the key regulatory factor of IFN-γ-induced MHC class II transcription, is tightly regulated in humans and mice by different promoters preceding four alternatively used first exons (types I–IV) (22). The type IV promoter is predominantly responsible for induced expression of CIITA and subsequently MHC II following IFN-γ stimulation of target cells (22). Furthermore, an additional role of STAT1α for IFN-γ-induced regulation of CIITA expression has been described (23–27). These data can be explained by the presence of GAS and ISRE sequence elements in the CIITA promoter.
In this work we describe the sequence and characterisation of a shorter isoform of IRF-2. To evaluate possible functional differences, we studied the binding affinities of both isoforms to known DNA target sequences by EMSA. We then analysed the regulatory activity of both isoforms using the CIITA type IV promoter in reporter gene assays.
MATERIALS AND METHODS
Cells and cell culture
The murine Leydig cell line TM3 was obtained from the ATCC (American Type Culture Collection, Rockville). Cells were cultured at 5% CO2 in DMEM/NUT-F12 medium (Gibco BRL, Eggenstein, Germany) supplemented with 10% foetal calf serum (Gibco BRL), 100 U/ml penicillin and 100 mg/ml streptomycin (Biochrom KG, Berlin, Germany). Stimulation experiments were performed with 200 U/ml IFN-γ (Boehringer Mannheim, Mannheim, Germany) for 24 h.
Primer extension assay
Since IRF-2 differs only in an additional 6 bp from IRF-2s, we employed a primer extension assay based on the PCR technique to analyse their expression. We used primers for IRF-2 spanning the variant region (sense, 5′-ATAGTACGGTGAACATCAT-3′; antisense, 5′-TTGCTGTCCAGATGGGACTG-3′) which were labelled with IRD-800 dye (MWG Biotech, Ebersberg, Germany). After a standard PCR (55°C annealing temperature, 25 cycles) the samples were loaded on a 30 cm acrylamide gel and run on an automated sequencer (LI-COR; MWG Biotech) as recommended by the manufacturer.
Generation of expression and reporter constructs
Murine cDNAs encoding IRF-2 and IRF-2s were synthesised from TM3 cell total RNA by reverse transcription (RT)–PCR using Superscript II reverse transcriptase (Gibco BRL) and the PCR system (Boehringer Mannheim) with the following primers: IRF-2 sense primer, ACCCAAGCTTGCGGGATTGTATTGGTAGCG; IRF-2 antisense primer, GCTGTCCAGATGGGACTGTCCTACAACT. The cDNA was then inserted into pcDNA3.1(+) (Invitrogen, San Diego, CA) between HindIII and BamHI restriction sites. The complete mICSBP cDNA clone was generously provided by Dr K. Ozato and the KpnI–BamHI backbone fragment was inserted into plasmid pcDNA3.
For the dual luciferase assays, the upstream 377 bp of the CIITA type IV promoter were PCR amplified from genomic DNA of TM3 cells. Primers containing specific restriction sites at their 5′-ends were used to insert the fragment into the pGL3-Basic firefly luciferase reporter vector (Promega, Madison, WI). Sequencing of the products was performed with either a LI-COR (MWG Biotech) or an ABI Prism 377 (PE Biosystems, Foster City, CA) automated sequencer as recommended by the manufacturer.
Calculation of secondary structure
Secondary structures were predicted using the PHDsec computer program (EMBL Laboratories) obtained via the Internet (http://www.embl-heidelberg.de/predictprotein/predictprotein.html ). This program calculates the secondary structure of proteins from their amino acid sequence using a system of neural networks.
In vitro transcription and translation
Plasmids containing IRF-2, IRF-2s and ICSBP under control of the T7 promoter were linearised downstream of the coding region with appropriate restriction enzymes. Aliquots of 3–5 mg of linearised plasmid were transcribed in vitro by T7 RNA polymerase using a commercial kit (Stratagene, La Jolla, CA). Proteins were translated in vitro using a rabbit reticulocyte (RRL) system (Promega) as recommended by the manufacturer. To monitor translation efficiency, small-scale reactions containing [35S]methionine were performed and the labelled protein was separated by 7% SDS–PAGE and subjected to autoradiography.
Electrophoretic mobility shift assay (EMSA)
In vitro translated proteins were prepared as described above. Nuclear extracts were prepared according to Dignam et al. (28). The oligonucleotides used for gel shift assays were as follows. PRDI trimer: forward, GATCAAGTGAAAGTGAAAGTGA; reverse complementary, GATCTCACTTTCACTTTCACTT. λB: forward, gatctcttggtttcacttccttttatttctg; reverse complementary, gatccagaaataaaaggaagtgaaaccaaga. ISG-15: forward, GATCCTCGGGAAAGGGAAACCGAAACTGAAGCC; reverse complementary, GATCGGCTTCAGTTTCGGTTTCCCTTTCCCGAG. Gel shift assays were performed as described elsewhere (15). A typical reaction contained up to 6 µl of RRL with in vitro translated proteins or nuclear extracts containing 2–5 µg protein. The samples were incubated with radiolabelled oligonucleotide in binding buffer [10 mM HEPES, pH 8.0, 5 mM MgCl2, 50 mM KCl, 1 mM dithiothreitol, 10% Ficoll, 3% glycerol, 0.025% Bromophenol blue, 0.005% xylene cyanol, 1 mg sonicated poly(dI·dC), 1 mg sheared salmon sperm DNA] for 10 min on ice. For supershift experiments the samples were incubated with 2–4 µl of antiserum or pre-immune serum for 1 h on ice before adding the oligonucleotide. The antisera used against IRF2 (Ab 311, against peptide 205–226; Ab 313, against peptide 250–268) were a generous gift from Dr H. Hauser. For competition experiments, the samples were incubated with the indicated amount of unlabelled competitor for 10 min before the labelled probe was added. The samples were then applied to a pre-run 7% polyacrylamide gel. The dried gel was subsequently autoradiographed.
Transfection of TM3 cells using a ballistic vector system
Transfection for transient expression was performed with a ballistic vector system (BVS). Since BVS transfers large numbers of plasmid vectors directly into the nucleus, efficient co-transfection of the same cell with different constructs can be achieved (29). TM3 cells were grown to confluency in 3 cm Petri dishes and the culture medium was removed prior to transfection. The transfection was performed using BVS with the Hydra extension module (Bio-Rad, Richmond, CA). This allows simultaneous transfection of up to six culture dishes. In all experiments the plasmid concentration was as follows: 400 nM reporter gene construct (CIITA type IV promoter/firefly luciferase gene), 110 nM transfection control construct (TK promoter/renilla luciferase gene) and 8 nM IRF expression vector (CMV promoter/IRF-2 or IRF-2s).
Dual luciferase assay
Promoter function was measured using the dual luciferase assay (Promega). In this system a co-transfected expression plasmid for renilla luciferase provides an internal reference for efficiency of transfection and expression. The promoter-dependent signal was generated from an expression plasmid carrying the CIITA type IV promoter controlling expression of the firefly luciferase gene. Twenty-four hours after transfection, culture medium was removed and replaced with medium containing 200 U/ml IFN-γ. Cells were harvested for determination of luciferase activity 24 h later. Luciferase assays were performed with a LB 96 P microlumat (E.G. & G. Berthold, Bad Wildbad, Germany). The results are presented as the ratio of measured firefly relative light units (r.l.u.) to renilla r.l.u.
RESULTS
A naturally occurring isoform of IRF-2, termed IRF-2s
During cloning experiments with TM3 cells, a murine Leydig cell line, we found an isoform of IRF-2 which lacked six bases in the mRNA sequence as compared to the database sequence of the originally described IRF-2 (Fig. 1). These six bases are located at the exon 5/exon 6 boundary and their absence leads to a shorter IRF-2 protein lacking two valines at that position (Val177 and Val178). We have termed this isoform IRF-2s (short). We tested whether the isoforms were also expressed in other species and found the same shortened isoform in J774.A1 cells, a murine monocytic/macrophage line. Primer extension analysis showed expression of both isoforms in these cells (Fig. 2). Interestingly, the ratio of IRF-2 to IRF-2s varied considerably: in TM3 cells both isoforms were expressed similarly, whereas IRF-2 dominated in J774.A1 cells (Fig. 2). The second isoform, IRF-2s, was also isolated from human peripheral blood and lacked six bases at a comparable site (Val177 and Val178) (Fig. 1) (30). Using the PHDsec computer program (EMBL Laboratories) the secondary structure of both isoforms was predicted (Fig. 3). The missing two valines in the IRF-2s isoform are located in a β-sheet, which becomes shorter and thus may be destabilised. Furthermore, the formation of a new β-sheet domain is predicted approximately 15 amino acids N-terminal of the original β-sheet (Fig. 3).
Figure 1.

The different IRF-2 isoforms. (A) Protein composition of human IRF-2. The arrow denotes the location of the two amino acids missing in IRF-2s. (B and C) Alignment of IRF-2 and IRF-2s mRNA and protein sequences, respectively (note that the human and murine sequences in this region are identical). DBD, DNA-binding domain; IAD, IRF-association domain; RP, repressor domain (4,16,31).
Figure 2.

Expression of IRF-2 isoforms in cell lines. IRF-2/IRF-2s mRNA levels (arrows) were determined by primer extension assay. (A) Three experiments for TM3 (left) and J774.A1 (right) were performed. (B) As controls, plasmids with IRF-2s (lane 1), IRF-2 (lane 2) and both (lane 3) were studied.
Figure 3.

Predicted secondary structure of IRF-2 isoforms. The amino acid (AA) sequence is shown on the upper lane starting from amino acid 125 to 180 or 182, respectively. The region corresponding to the 6 bp difference in the RNA sequence is shown by arrows. The calculated secondary structure motifs (with the PHDsec program) are shown in the second lane (PHD sec). H, α-helices; E, β-sheets. The third lane (Rel sec) shows the reliability index of the prediction (9 is highest). Differences in secondary structure motifs are marked with a star.
IRF-2 and IRF-2s have comparable binding affinities to target DNA elements in vitro
To investigate possible functional differences of the two isoforms, we first asked whether they differ in their ability to bind certain target DNA sequences in EMSA. Three different target probes of standard consensus sequences were used: a positive regulatory domain I trimer (PRDI) from the IFN-β promoter (15,32); the ISRE of the IFN-stimulated gene 15 promoter (ISG15) (14,33); the λB element of the immunoglobulin light chain gene enhancer (34). We used in vitro translated proteins of IRF-2 and IRF-2s and ensured equal amounts of protein by calibrating translation via incorporation of radiolabelled methionine. We found no significant difference between binding of IRF-2 and IRF-2s to the above elements (Fig. 4). To investigate the DNA-binding affinity of IRF-2 and IRF-2s in more detail, we performed competitive EMSA with a labelled PRDI probe. The binding of both complexes was comparably diminished by increasing amounts of unlabelled PRDI competitor probe (Fig. 5). The same was found for competition with a probe comprising the λB element (data not shown). We then performed supershift experiments with in vitro translated proteins and two different antibodies against IRF-2, Ab 311 (against peptide 205–226) and Ab 313 (against peptide 250–268). Both complexes, IRF-2–DNA and IRF-2s–DNA, were supershifted to a similar extent (data not shown). In addition, both isoforms were able to form hetero-complexes with ICSBP. No significant difference in the strength of binding to the PRDI probe was observed (data not shown). These data argue that the DNA-binding affinity of both proteins is similar, if not identical. This can be rationalised by the location of the two missing amino acids C-terminal of the DBD.
Figure 4.
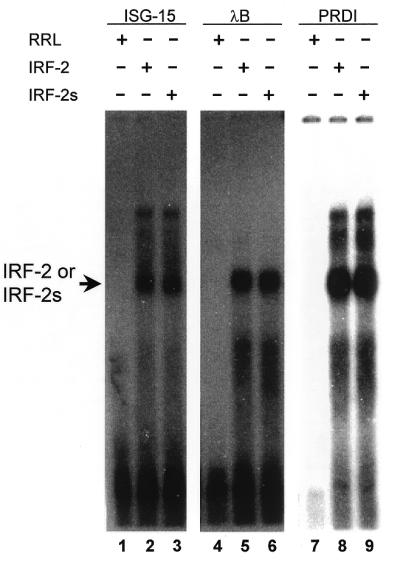
DNA-binding ability of IRF-2/IRF-2s to various target sequences. Binding of IRF-2 and IRF-2s to ISG15, λB and PRDI probes in EMSA. No significant difference in binding ability between IRF-2 (lanes 2, 5 and 8) and IRF-2s (lanes 3, 6 and 9) was detected. RRL, rabbit reticulocyte lysate.
Figure 5.
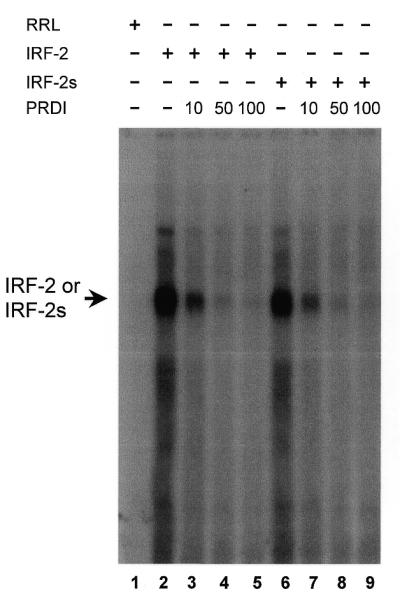
DNA-binding ability of IRF-2/IRF-2s in competition EMSA. Binding of IRF-2 (lanes 2–5) and IRF-2s (lanes 6–9) to a labelled PRDI trimer probe was comparably diminished by increasing amounts of unlabelled PRDI competitor (10, 50 and 100 times excess). RRL, rabbit reticulocyte lysate.
IRF-2s is a weaker transcriptional activator in vivo than IRF-2
Since both isoforms are present in several cell types, we addressed whether the variation in primary structure of IRF-2s leads to a differential ability to regulate target genes. We chose the CIITA type IV promoter, which is known to be activated by IFN-γ and IRF-2 (11). The coding sequences of both isoforms were inserted into identical expression vectors, which were co-transfected with the CIITA type IV promoter–reporter gene construct. The reporter activity was measured in unstimulated and IFN-γ-stimulated TM3 cells. The basal reporter activity without the IRF expression vector was enhanced two to three times by IFN-γ (Fig. 6A). Co-transfection with IRF-2 led to an increase in reporter activity to nearly three times the basal level, which was further increased by IFN-γ to >4-fold above the basal level (Fig. 6A). Co-transfection of IRF-2s also enhanced basal and IFN-γ-induced reporter activity, but the level of activation was 0.4–0.7 times lower compared to that of IRF-2 (Fig. 6A). Furthermore, these differences persisted with IFN-α treatment, which reduced IRF-2/IRF-2s-mediated reporter gene activation (Fig. 6B). These data clearly indicate that IRF-2s has a weaker activating ability on transcription through the CIITA type IV promoter than IRF-2 in TM3 cells.
Figure 6.

Activation ability of IRF-2/IRF-2s in reporter gene assays. IRF-2 or IRF-2s expression plasmid or no IRF was transfected into TM3 cells with the CIITA type IV promoter/luciferase vector and the TK promoter/renilla luciferase vector (as an internal control). Effect of IFN-γ (A) and IFN-α (B) on IRF-2/IRF-2s-induced reporter gene expression. Open boxes denote no stimulation and dark grey boxes IFN stimulation (24 h). The relative promoter activity is calculated as the ratio of firefly luciferase to renilla luciferase activity. The mean of three experiments is shown (± SE).
DISCUSSION
We cloned a naturally expressed IRF-2 isotype, IRF-2s, from both murine and human cells. This factor, which is two amino acids shorter than the previously described IRF-2, exhibits a comparable binding affinity to typical target promoter elements. Both isotypes are identical except for 6 bp, arguing against two genes coding for IRF-2. Alternative splicing is the most likely explanation for the existance of the two IRF-2 isoforms. There is growing evidence that alternative splicing, the use of different splice sites by the spliceosome, may be a major mechanism for controlling the expression of eukaryotic genes. Indeed, alternative splicing has been shown to be involved in the generation of various immunomodulatory proteins. These include the cytokines EGF, TGF-β1, IL-2, IL-4 and others (35). Alternative splicing has also been described for a variety of other proteins, including kinases, receptors like MHC class I, whose alternatively spliced form leads to a soluble protein, and transcription factors (36–38). The functional correlation of alternative splicing may include: (i) protein localisation; (ii) deletion, modulation or change of function; (iii) participation in developmental pathways.
The difference of 6 bp between the two IRF-2 isotypes most likely accounts for the differences in activation activity. The predicted secondary structure of the two isotypes indicates a putative destabilisation of a β-sheet inside the IRF-2s protein and a new β-sheet further N-terminal. The reduction of IRF-2-induced transcriptional activity at the CIITA type IV promoter without affecting the binding ability may be explained by a change in protein–protein interactions which is essential for the function of IRF family members (15,16,39). It is also possible that a conformational change of the protein may lead to stabilisation of its repression domains (31,40,41). Another possibility may be disruption of the latent activation domain located at amino acids 160–220 (31) by the missing amino acids at positions 177 and 178. A pivotal element called the IAD, which was shown to be essential for formation of hetero-complexes (4,15,16), was not directly affected by absence of the two amino acids (Fig. 1A). It seems unlikely that the lack of just 6 bp accounts for a reduced RNA stability or translation efficiency which would then lead to decreased promoter activation.
As previously described, the expression of IFN-γ target genes is regulated by various IRFs, like the activators IRF-1, IRF-2 and IRF-2s and the repressor ICSBP. The exact combination of these transcription factors, comprised of activators of different strengths as well as inhibitors, may be important in fine tuning expression of several crucial genes, such as control of T cell activation by MHC class II expression through its key regulator CIITA. Differential usage of the IRF-2 isotypes may thus be a novel mechanism for precise regulation of target gene expression by the same IRF protein. These data are substantiated by our results that the two isoforms are expressed in different ratios in the two cell types, TM3 Leydig and J774.A1 monocytic/macrophage cells. Thus it may be interesting in future studies with IRF-2/IRF-2s to reveal the differences between the two isotypes in a system in which IRF-2 acts as repressor (e.g. MHC class I).
Previous studies on IRF-2 have contributed to a more complicated picture. It has been shown that upon induction with double-stranded RNA or viruses, 185 amino acids at the C-terminal end of IRF-2 are proteolytically removed (42–44). This modification can alter IRF-2 repression of the IFN-β gene (42–44). Other truncated forms of IRF-2 have been found in induced complexes, such as I-Bfi and TH3 (42,45). Formation of hetero-complexes is an important regulatory mechanism used by IRF-2 (44) and other IRFs (16,18). Moreover, it has been shown that phosphorylation of IRF-2 is important for binding to ICSBP (15,19). This complex network of interactions has been further complicated by our findings that IRF-2 exists in two different isoforms. Although IRF-2s exhibited a reduced activation ability, no obvious alteration in binding target sequences and in forming hetero-complexes with ICSBP was detected in EMSAs. Another IRF family member, IRF4 (LSIRF/Pip/ICSAT/MUM1), is known to express two splice forms with a difference of one amino acid (Gln164), which is also not located in the DBD or IAD (4,46). Thus, the existence of splice variants of other IRFs than IRF-2 with differential regulatory functions is possible.
Our experiments imply a putative new regulatory mechanism, namely the generation of splice variants of the same IRF transcription factor, which results in proteins with different regulatory abilities.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Katrin Büttner, Susanne Brendel and Iris Ziglowsky for excellent technical assistance. We are grateful to Dr Keiko Ozato for providing the mICSBP cDNA clone and to Dr Hansjörg Hauser for providing IRF-2 antisera. Our work was enabled by generous funds provided through Centrum für Somatische Gentherapie e.V., the Deutsche Forschungsgemeinschaft (SFB366 to B.W. and A.N., SFB506 to B.W. and Ne310/6-3 to A.N.), the H.W. & J. Hector Stiftung (to M.S. and A.N.) and the German–Israeli Foundation for Scientific Research and Development (to B.-Z.L.). S.A.K.M. was funded by the Deutsche Forschungsgemeinschaft as a fellow of the Graduiertenkolleg Signaltransduction. M.S. was funded in part by a short-term fellowship from the European Molecular Biology Organization (ASTF-9095).
REFERENCES
- 1.Schindler C. and Darnell,J.E.,Jr (1995) Annu. Rev. Biochem., 64, 621–651. [DOI] [PubMed] [Google Scholar]
- 2.Leonard W.J. and O’Shea,J.J.O. (1998) Annu. Rev. Immunol., 16, 293–322. [DOI] [PubMed] [Google Scholar]
- 3.Ward A.C., Touw,I. and Yoshimura,A. (2000) Blood, 95, 19–29. [PubMed] [Google Scholar]
- 4.Nguyen H.,Hiscott,J. and Pitha,P.M. (1997) Cytokine Growth Factor Rev., 8, 293–312. [DOI] [PubMed] [Google Scholar]
- 5.Taniguchi T. (1997) J. Cell. Physiol., 173, 128–130. [DOI] [PubMed] [Google Scholar]
- 6.Harada H., Fujita,T., Miyamoto,M., Kimura,Y., Maruyama,M., Furia,A., Miyata,T. and Taniguchi,T. (1989) Cell, 58, 729–739. [DOI] [PubMed] [Google Scholar]
- 7.Miyamoto M., Fujita,T., Kimura,Y., Maruyama,M., Harada,H., Sudo,Y., Miyata,T. and Taniguchi,T. (1988) Cell, 54, 903–913. [DOI] [PubMed] [Google Scholar]
- 8.Harada H., Kitagawa,M., Tanaka,N., Yamamoto,H., Harada,K., Ishihara,M. and Taniguchi,T. (1993) Science, 259, 971–974. [DOI] [PubMed] [Google Scholar]
- 9.Vaughan P., Aziz,F., van Wijnen,A., Wu,S., Harada,H., Taniguchi,T., Soprano,K., Stein,J. and Stein,G. (1995) Nature, 377, 362–365. [DOI] [PubMed] [Google Scholar]
- 10.Nonkwelo C., Ruf,I.K. and Sample,J. (1997) J. Virol., 71, 6887–6897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xi H., Eason,D.D., Ghosh,D., Dovhey,S., Wright,K.L. and Blanck,G. (1999) Oncogene, 18, 5889–5903. [DOI] [PubMed] [Google Scholar]
- 12.Driggers P.H., Ennist,D.L., Gleason,S.L., Mak,W.H., Marks,M.S., Levi,B.Z., Flanagan,J.R., Appella,E. and Ozato,K. (1990) Proc. Natl Acad. Sci. USA, 87, 3743–3747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Weisz A., Marx,P., Sharf,R., Appella,E., Driggers,P.H., Ozato,K. and Levi,B.Z. (1992) J. Biol. Chem., 267, 25589–25596. [PubMed] [Google Scholar]
- 14.Nelson N., Marks,M.S., Driggers,P.H. and Ozato,K. (1993) Mol. Cell. Biol., 13, 588–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sharf R., Meraro,D., Azriel,A., Thornton,A.M., Ozato,K., Petricoin,E.F., Larner,A.C., Schaper,F., Hauser,H. and Levi,B.Z. (1997) J. Biol. Chem., 272, 9785–9792. [DOI] [PubMed] [Google Scholar]
- 16.Meraro D., Hashmueli,S., Koren,B., Azriel,A., Oumard,A., Kirchhoff,S., Hauser,H., Nagulapalli,S., Atchison,M.L. and Levi,B.Z. (1999) J. Immunol., 163, 6468–6478. [PubMed] [Google Scholar]
- 17.Schaper F., Kirchhoff,S., Posern,G., Köster,M., Oumard,A., Sharf,R., Levi,B.Z. and Hauser,H. (1998) Biochem. J., 335, 147–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Eklund E.A. and Kakar,R. (1999) J. Immunol., 163, 6095–6105. [PubMed] [Google Scholar]
- 19.Birnbaum M.J., van Zundert,B., Vaughan,P.S., Whitmarsh,A.J., van Wijnen,A.J., Davis,R.J., Stein,G.S. and Stein,J.L. (1997) J. Cell. Biochem., 66, 175–183. [PubMed] [Google Scholar]
- 20.Steimle V., Siegrist,C.A., Mottet,A., Lisowska-Grospierre,B. and Mach,B. (1994) Science, 265, 106–109. [DOI] [PubMed] [Google Scholar]
- 21.Mach B., Steimle,V., Martinez Soria,E. and Reith,W. (1996) Annu. Rev. Immunol., 14, 301–331. [DOI] [PubMed] [Google Scholar]
- 22.Muhlethaler-Mottet A., Otten,L.A., Steimle,V. and Mach,B. (1997) EMBO J., 16, 2851–2860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee Y.J. and Benveniste,E.N. (1996) J. Immunol., 157, 1559–1568. [PubMed] [Google Scholar]
- 24.Meraz M.A., White,J.M., Sheehan,K.C., Bach,E.A., Rodig,S.J., Dighe,A.S., Kaplan,D.H., Riley,J.K., Greenlund,A.C., Campbell,D., Carver-Moore,K., DuBois,R.N., Clark,R., Aguet,M. and Schreiber,R.D. (1996) Cell, 84, 431–442. [DOI] [PubMed] [Google Scholar]
- 25.Piskurich J.F., Linhoff,M.W., Wang,Y. and Ting,J.P. (1999) Mol. Cell. Biol., 19, 431–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Muhlethaler-Mottet A., Di Berardino,W., Otten,L.A. and Mach,B. (1998) Immunity, 8, 157–166. [DOI] [PubMed] [Google Scholar]
- 27.Dong Z., Kumar,R., Yang,X. and Fidler,I.J. (1997) Cell, 88, 801–810. [DOI] [PubMed] [Google Scholar]
- 28.Dignam J.D., Lebovitz,R.M. and Roeder,R.G. (1983) Nucleic Acids Res., 11, 1475–1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sun Y., Jurgovsky,K., Möller,P., Alijagic,S., Dorbic,T., Georgieva,J., Wittig,B. and Schadendorf,D. (1998) Gene Ther., 5, 481–490. [DOI] [PubMed] [Google Scholar]
- 30.Cha Y. and Deisseroth,A.B. (1994) J. Biol. Chem., 269, 5279–5287. [PubMed] [Google Scholar]
- 31.Yamamoto H., Lamphier,M.S., Fujita,T., Taniguchi,T. and Harada,H. (1994) Oncogene, 9, 1423–1428. [PubMed] [Google Scholar]
- 32.Kirchhoff S., Schaper,F. and Hauser,H. (1993) Nucleic Acids Res., 21, 2881–2889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bovolenta C., Driggers,P.H., Marks,M.S., Medin,J.A., Politis,A.D., Vogel,S.N., Levy,D.E., Sakaguchi,K., Appella,E., Coligan,J.E. and Ozato,K. (1994) Proc. Natl Acad. Sci. USA, 91, 5046–5050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Eisenbeis C.F., Singh,H. and Storb,U. (1995) Genes Dev., 9, 1377–1387. [DOI] [PubMed] [Google Scholar]
- 35.Atamas S.P. (1997) Life Sci., 61, 1105–1112. [DOI] [PubMed] [Google Scholar]
- 36.Thomis D.C., Floyd-Smith,G. and Samuel,C.E. (1992) J. Biol. Chem., 267, 10723–10728. [PubMed] [Google Scholar]
- 37.He X. and Le,J. (1995) Cell. Immunol., 162, 159–168. [DOI] [PubMed] [Google Scholar]
- 38.Hsu T., Gogos,J.A., Kirsh,S.A. and Kafatos,F.C. (1992) Science, 257, 1946–1950. [DOI] [PubMed] [Google Scholar]
- 39.Sharf R., Azriel,A., Lejbkowicz,F., Winograd,S.S., Ehrlich,R. and Levi,B.Z. (1995) J. Biol. Chem., 270, 13063–13069. [DOI] [PubMed] [Google Scholar]
- 40.Lin R., Mustafa,A., Nguyen,H. and Hiscott,J. (1994) J. Biol. Chem., 269, 17542–17549. [PubMed] [Google Scholar]
- 41.Nguyen H., Mustafa,A., Hiscott,J. and Lin,R. (1995) Oncogene, 11, 537–544. [PubMed] [Google Scholar]
- 42.Palombella V.J. and Maniatis,T. (1992) Mol. Cell. Biol., 12, 3325–3336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yoneyama M., Suhara,W., Fukuhara,Y., Fukuda,M., Nishida,E. and Fujita,T. (1998) EMBO J., 17, 1087–1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Whiteside S.T., King,P. and Goodbourn,S. (1994) J. Biol. Chem., 269, 27059–27065. [PubMed] [Google Scholar]
- 45.Cohen L. and Hiscott,J. (1992) Virology, 191, 589–599. [DOI] [PubMed] [Google Scholar]
- 46.Grossman A., Mittrücker,H.W., Nicholl,J., Suzuki,A., Chung,S., Antonio,L., Suggs,S., Sutherland,G.R., Siderovski,D.P. and Mak,T.W. (1996) Genomics, 37, 229–233. [DOI] [PubMed] [Google Scholar]