


Abstract
Previous studies have shown that the repressive effect of thymidylate synthase (TS) mRNA translation is mediated by direct binding of TS itself to two cis-acting elements on its cognate mRNA. To identify the optimal RNA nucleotides that interact with TS, we in vitro synthesized a completely degenerate, linear RNA pool of 25 nt and employed in vitro selection to isolate high affinity RNA ligands that bind human TS protein. After 10 rounds of selection and amplification, a single RNA molecule was selected that bound TS protein with nearly 20-fold greater affinity than native, wild-type TS RNA sequences. Secondary structure analysis of this RNA sequence predicted it to possess a stem–loop structure. Deletion and/or modification of the UGU loop element within the RNA sequence decreased binding to TS by up to 1000-fold. In vivo transfection experiments revealed that the presence of the selected RNA sequence resulted in a significant increase in the expression of a heterologous luciferase reporter construct in human colon cancer H630 and TS-overexpressing HCT-C:His-TS+ cells, but not in HCT-C18 cells expressing a functionally inactive TS. In addition, the presence of this element in H630 cells leads to induced expression of TS protein. An immunoprecipitation method using RT–PCR confirmed a direct interaction between human TS protein and the selected RNA sequence in transfected human cancer H630 cells. This study identified a novel RNA sequence from a degenerate RNA library that specifically interacts with TS.
INTRODUCTION
The reaction catalyzed by thymidylate synthase (TS) provides the sole intracellular de novo source of synthesized dTMP (1). Thus, this folate-dependent enzyme is essential for regulating the balanced supply of the DNA precursors required for DNA replication. For this reason, significant research efforts have focused on TS as a critical target in cancer chemotherapy (2,3).
In addition to its essential role as a catalytic enzyme, TS also functions as an RNA binding protein (4–8). In this capacity, TS interacts directly with its own mRNA by binding to two different cis-acting elements. This interaction results in the translational repression of TS mRNA. The first site is a 30 nt sequence contained within the 5′-untranslated region (UTR) and includes the translational start site. The second element is encompassed within a 70 nt sequence in the protein coding region corresponding to nt 480–550 (9). An analysis of these two sequences has failed to identify a consensus nucleotide sequence. While the element in the 5′-upstream region adopts a stable stem–loop structure, the sequence in the protein coding region is predicted to form a different secondary structure, at least as suggested by preliminary secondary structure analyses. Significant efforts are on-going to more precisely elucidate the critical elements on TS mRNA that are required for protein binding.
Aside from interacting with its own mRNA, TS also interacts, with relatively high affinity, with several other cellular mRNAs, including p53 and members of the myc family (8,10–12). With regard to each of the characterized TS protein–cellular mRNA interactions, binding of TS to p53 and c-myc mRNAs results in translation repression. However, analysis of the various target cellular mRNA sequences has not yet identified a consensus nucleotide sequence and/or secondary structure that is recognized by TS protein.
In vitro selection methodology is a strategy to identify nucleic acid molecules with high affinity binding for a wide range of targets, including small molecules and proteins (13–15). This method has identified the optimal RNA sequences for several RNA binding proteins, including bacteriophage T4 DNA polymerase (13), iron regulatory factor (16), U1 small nuclear ribonucleoprotein A (17) and HIV Rev protein (18). The power of this approach derives from the fact that oligonucleotide libraries containing up to 1013–1015 RNA molecules can be readily synthesized in vitro. A process of reiterative in vitro amplification and selection can then be applied for the rapid isolation of high affinity ligands.
We employed this technique to further characterize the critical nucleotide elements required for RNA binding by TS protein. Using a completely random 25 nt oligoribonucleotide (ORN) library, we identified a single RNA ligand that bound TS protein with significantly higher affinity (up to 20-fold) than wild-type TS RNA sequences. RNA binding experiments suggest that the interaction between TS protein and this element requires the presence of a 3 nt stem–loop structure. Finally, transfection experiments in vivo confirm that this selected RNA sequence requires a functionally intact TS for its biological activity.
MATERIALS AND METHODS
Cell culture
The human colon cancer cell line H630 has been previously described (19). Cells were grown in 75 cm2 plastic tissue culture flasks in RPMI 1640 growth medium with 10% dialyzed fetal bovine serum (Gibco BRL, Grand Island, NY).
The human colon cancer HCT-C18 cell line has been previously described (20) and was a kind gift from Dr Sondra Berger. HCT-C18 cells were maintained in RPMI 1640 growth medium containing 10% dialyzed fetal bovine serum and supplemented with 10 µM thymidine. The HCT-C:His-TS(+) cell line was established by stable transfection of HCT-C18 cells with human His-tagged TS cDNA, as previously described (10).
Purification of human TS protein
Human recombinant TS protein was purified according to previously described methods (21–23). The specific activity of this protein preparation was 0.05 U/mg. The protein was further purified by DE-52 anion exchange chromotography and the specific activity of the DE-52 purified protein was 0.08 U/mg. Human recombinant, His-tagged TS protein was purified as previously described (23) and the specific activity of this protein was 1.5 U/mg.
RNA gel mobility shift assay
The RNA mobility shift assay was performed as previously described (4,5,24). Competition experiments were performed with 32P-radiolabeled TS-30 RNA (2.2 fmol, 100 000 c.p.m.) and recombinant human TS protein (3 pmol). TS-30 RNA represents the 5′-upstream binding site on TS mRNA, with the sequence 5′-CCG CCC GCC GCG CCA UGC CUG UGG CCG GCU-3′. The conditions selected were based on control experiments using a fixed amount of radiolabeled TS-30 RNA with increasing concentrations of TS protein to determine linearity of binding.
Synthesis of the randomized RNA pool and in vitro RNA selection
Double-stranded transcription template was prepared as previously described (15), by subjecting 5 ng linear N25 to PCR amplification using 100 ng T7Univ and RevUniv oligodeoxyribonucleotide primers, respectively. The respective sequences for these oligonucleotides were (underlined bases represent the EcoRI and HindIII restriction sites): Linear 25, d(TAATACGACTCACTATAGGGN24AAGCTTGGC); T7Univ, d(GGAATTCTAATACGACTCACTATAGGG); RevUniv, d(GCCAAGCTTN9).
PCR reactions were performed as previously described (6,11). Reactions were incubated at 96°C for 1 min, 55°C for 1 min and 72°C for 1.5 min for 35 cycles. At the end of the reaction, samples were incubated for an additional 10 min and then cooled to 4°C.
In vitro RNA selection
The randomized, radiolabeled RNA pool was subjected to up to 10 rounds of in vitro selection and amplification as outlined in Figure 1. In brief, 100 ng of pool RNA was incubated with pure human recombinant His-tagged TS protein (1 µg) in the RNA gel shift binding assay. RNA–protein complexes were detected by means of autoradiography, excised and isolated from the 5% polyacrylamide gel in a buffer containing 0.5 M sodium acetate, 0.1% SDS and 1 mM EDTA overnight at 37°C. Eluted RNAs were then purified by phenol/chloroform extraction and ethanol precipitation. The selected RNA was subsequently converted to single-strand cDNA with AMV reverse transcriptase (Gibco BRL) in a 1 h reaction at 42°C. RNA was removed by treatment of the reaction mixture with RNase A for 10 min at 37°C. The resulting cDNA was amplified by PCR and the product transcribed in vitro and used for the next round of selection. After 10 rounds of selection and amplification, the PCR-amplified DNA was cloned into the EcoR1 and HindIII restriction sites of plasmid pGEM-4Z (Promega, Madison, WI). Positive clones were isolated and the DNA was subsequently purified and sequenced.
Figure 1.

Strategy for RNA selection in vitro. The specific steps of selection and amplification are detailed in Materials and Methods. The T7 promoter region is marked by T7 primer. DNA and RNA are represented by double lines and single lines, respectively. The linear N25 template is shown and the degenerate nucleotides are represented by N.
Synthesis of recombinant plasmid constructs
Luciferase reporter plasmid p644 was constructed, as previously described (10), by inserting an EGR-1 (early growth response gene) promoter at the BglII restriction site of plasmid pGL2-basic (Promega). Both the TS:N25 selected and the TS:N25AS sequences were synthesized on an Applied Biosystems Model 391 DNA synthesizer and cloned into the HindIII restriction site of p644 to generate heterologous luciferase reporter constructs, p644/TS:N25 and p644/TS:N25AS.
U6 expression plasmid pGEMmU6 was a kind gift from Dr Leaf Huang (25). The TS:N25 DNA sequence was cloned into pGEMmU6 between the NsiI and XhoI restriction sites to yield pGEMmU6/TS:N25.
Plasmid pcDNA3.1(+):TS/N25 was constructed by inserting the TS:N25 sequence into pcDNA3.1(+):TS between the HindIII and KpnI restriction sites. The sequences and orientations of the cloned inserts were confirmed.
In vitro mRNA transcription
A luciferase cDNA sequence corresponding to nt 446–675 (luc/446–675) was PCR amplified using the pGEM-luc plasmid (Promega) as DNA template. The sequences of the primers were (underlined nucleotides represent the T7 promoter/primer sequence): luc (sense), d(GGA CCT CAG TTT GAC GCC AAC); luc (antisense), d(TAA TAC GAC TCA CTA TAG GGTTG CCA GGC TTA GGT CTA AAC).
A 32P-radiolabeled antisense luciferase RNA probe was synthesized by in vitro transcription using luc/446–675 as template and T7 RNA polymerase. The 32P-radiolabeled, β-actin antisense RNA probe was synthesized in vitro using the human pTR1-β-actin template (Ambion, Austin, TX) and T7 RNA polymerase. Full-length TS mRNA transcript (TS1–1524) and TS-3′UTR RNA were synthesized in vitro with SP6 RNA polymerase and the reagents included in the Ambion in vitro transcription kit (Ambion 13349). TS-30, TS:N25, TS:N25Δ, TSN25m1, TS:N25m2, TS:N25m3 and TS:N25m4 were chemically synthesized by the Keck Core Facility at Yale University. Once synthesized, all RNAs were resolved on a 15% polyacrylamide/8 M urea gel and subsequently gel purified. The concentration of RNA was then determined by UV absorbance at 260 nm.
Transient transfection and luciferase assay
To determine the effect of TS RNA sequences on the in vivo expression of the heterologous luciferase construct, transient transfection experiments were performed as described (26). Cells were harvested using reagents from the Promega dual luciferase kit (Promega E1910) and luciferase activity was measured using a luminometer.
Western immunoblot analysis
Cells were harvested and processed as previously described (6,11). Protein concentrations were determined by the Bio-Rad protein assay and an equivalent amount of cellular extract normalized to transfection efficiency was resolved by 10% SDS–PAGE as previously described (6,11). Proteins were probed with mouse monoclonal anti-His-tag antibody (1:300 dilution), followed by incubation with a horseradish peroxidase-conjugated secondary antibody (Bio-Rad Laboratories, Richmond, CA). Proteins were visualized with a chemiluminescence detection system using the Super Signal substrate (Pierce, Rockford, IL).
RNase protection assay
Total cellular RNA was extracted from cells by the method of Sacchi and Chomczynski (27). Total cellular RNA (10 µg) was incubated with 40 µg yeast tRNA and with at least a 4-fold molar excess of either radiolabeled luciferase antisense or β-actin antisense RNA probe (specific activity 2 × 108 c.p.m./µg, 5 × 105 c.p.m./reaction). Samples were processed according to the Ambion protocol (Ambion 1412) as described (9).
Immunoprecipitation and RT–PCR analysis
Whole cell extracts were prepared and TS ribonucleoprotein (RNP) complexes were immunoprecipitated with anti-TS 106 monoclonal antibody as described (6,11,28). The RNA isolated from cellular TS RNP complexes was subjected to reverse transcription and the resulting single-strand cDNA was then used as template for PCR amplification. The sequences of the primers were: EGR-1 (sense), d(GAG TCG CGA GAG ATC CAG); luc (antisense), d(CAA CAG TAC CGG AAT GCC); TS434–454 (sense), d(CCC GAG ACT TTT TGG ACA GC); TS634–614 (antisense), d(CGC ACA TGA TGA TTC TTC TGT CGT CA).
For the PCR reaction, samples were incubated at 95°C for 1 min, 60°C for 1 min and 72°C for 1.5 min for 30 cycles. At the end of the reaction, samples were incubated for an additional 10 min at 72°C. PCR products were resolved by electrophoresis on a 1.5% agarose gel stained with ethidium bromide. A series of control experiments were performed to determine the range of linear amplification for each gene.
RESULTS
The RNA selection method used in this study is outlined in Figure 1. To effectively monitor the progress of the selection process, two different approaches were taken. The first employed a sequencing analysis of RNA sequences isolated from each round, while the second approach used the RNA competition gel shift assay to determine the relative binding affinity of RNAs isolated from each round. The transcribed RNA pool was initially subjected to four rounds of selection and amplification. As seen, there was no change in the pattern of the sequence derived from the second and fourth rounds (Fig. 2). Additional rounds of selection were subsequently performed and a detectable bias was observed in the RNA sequence only after the ninth and tenth rounds of selection. Subsequent rounds of selection failed to produce a change in the sequence pattern.
Figure 2.

RNA selection in vitro of TS-bound RNAs. Sequencing and labeling was performed as described in Materials and Methods. RNA transcripts were derived from amplified products after 2 (panel 1), 4 (panel 2), 9 (panel 3) and 10 (panel 4) rounds of selection.
As a complementary approach, the relative binding affinity of the pooled RNAs selected from each round for TS protein was determined. No improvement in binding of the RNA isolated from the second and fourth rounds of selection was observed when compared to the control degenerate oligoribonucleotide pool. In contrast, the binding affinity of the selected RNA was significantly enhanced after the ninth (IC50 0.15 nM) and tenth (IC50 0.10 nM) rounds of selection, with no subsequent increase in binding after the tenth round. The binding of the pooled RNAs to human TS protein from these two rounds of selection was 10- to 13-fold greater than the wild-type, full-length TS mRNA and 15- to 20-fold greater than the TS-30 RNA sequence.
These results suggest that the in vitro selection process identifies high affinity RNA ligands. The RNA selected from the tenth round was reverse transcribed and the resulting cDNA cloned into the pGEM4Z in vitro transcription plasmid. Twenty individual clones were selected and each clone that was analyzed expressed the identical sequence (TS:N25), 5′-CUCUUUACCGUGUCGGUUUCGUUGA-3′. This sequence was then subjected to Zuker RNA fold analysis (29) and the predicted stable stem–loop secondary structure is shown in Figure 3.
Figure 3.

Proposed secondary structure of selected TS:N25 RNA. Secondary structure analysis was performed according to the RNA fold program of Zuker and Stiegler (29).
An RNA mobility shift assay was then used to determine the relative binding affinity of the selected TS:N25 RNA sequence for human TS protein. As shown in Table 1, the TS:N25 RNA bound human recombinant TS protein with an affinity nearly 20-fold greater (IC50 0.08 nM) when compared to either the full-length TS mRNA (IC50 1.5 nM) or the 30 nt wild-type TS RNA sequence (IC50 2.0 nM). In contrast, a TS RNA corresponding to a sequence contained within the 3′-UTR was unable to compete for binding, even with concentrations of up to 100-fold molar excess.
Table 1. Relative binding affinity of mutant and deletion TS:N25 RNA sequences.
| Name | Sequence | IC50 (nM) |
|---|---|---|
| TS mRNA | TS1–1524 | 1.5 ± 0.3 |
| TS-30 | TS80–109 | 2.0 ± 0.2 |
| TS-3′UTR | TS1040–1240 | >100 |
| TS:N25 | CUC UUU ACC GUG UCG GUU UCG UUG A | 0.08 ± 0.01 |
| TS:N25Δ | CUC UUU ACC GCG GUU UCG UUG A | 90 ± 11 |
| TS:N25m1 | CUC UUU ACC GUA UCG GUU UCG UUG A | 0.20 ± 0.03 |
| TS:N25m2 | CUC UUU ACC GUC UCG GUU UCG UUG A | 0.30 ± 0.04 |
| TS:N25m3 | CUC UUU ACC GAG UCG GUU UCG UUG A | 0.15 ± 0.03 |
| TS:N25m4 | CUC UUU ACC GAA ACG GUU UCG UUG A | 0.56 ± 0.02 |
Binding of human TS protein to various RNA sequences was determined as described in Materials and Methods. The bold nucleotides represent point mutations. Each value represents the mean ± SE of between three and five experiments.
The selected RNA sequence is predicted to form a stable stem–loop structure with a short 3 nt UGU loop aspect. Previous studies have documented the importance of such secondary structures in RNA recognition for several target mRNAs, including ferritin mRNA, transferrin mRNA, 5-aminolevulinate synthase mRNA and Drosophila Hsp70 mRNA (30–37). To investigate the potential relationship between the secondary structure of the selected RNA and binding to TS protein, a sequence with a deletion of the putative UGU loop (TS:N25Δ) was synthesized. Various mutant RNAs with point mutations in the loop were also synthesized. Their sequences are shown in Table 1. The binding affinity of these deletion and mutant RNA sequences to TS protein was investigated by RNA gel shift competition experiments. Two RNAs with either an A (TS:N25m1) or a C mutation (TS:N25m2) at the G nucleotide in the loop sequence showed lower binding affinity for TS, by 2.5- and 3.75-fold, respectively, when compared to the parent sequence (Table 1). A UGU→AGU mutation (TS:N25m3) resulted in only a 1.9-fold decrease in binding affinity. In contrast, mutation of all three nucleotides in the UGU loop sequence to AAA (TS:N25m4) decreased binding by 7-fold compared to the selected sequence. When the UGU loop sequence was completely deleted the relative binding affinity to TS was significantly decreased (IC50 90 nM) by >1000-fold. Zuker fold analysis predicts that the TS:N25m1, TS:N25m2 and TS:N25m3 mutant RNAs can still form a stem–loop motif. In contrast, the TS:N25m4 RNA is unable to adopt such an ordered structure. Taken together, these results suggest that the UGU loop sequence plays a critical role as a determinant of RNA recognition. Further studies, including chemical and enzymatic footprinting, are now in progress to elucidate the specific nucleotides that are in direct contact with TS protein.
We next investigated the potential biological relevance in vivo of the TS:N25 selected RNA sequence. For these studies the cDNA sequence corresponding to TS:N25 RNA was cloned into the p644 luciferase reporter plasmid which had been placed under control of an EGR-1 promoter element. The selected RNA sequence and its antisense sequence were inserted immediately upstream of the luciferase gene to yield p644/TS:N25 and p644/TS:N25AS (Fig. 4A). A significantly increased level of luciferase expression (3.8-fold) was observed in H630 cells transfected with p644/TS:N25 when compared to cells transfected with the parent p644 plasmid (Fig. 4B). In contrast, when the antisense sequence was placed immediately upstream of the luciferase gene, no change in luciferase activity was observed (Fig. 4B).
Figure 4.

(A) Structure of heterologous luciferase plasmids. Details of the constructions are presented in Materials and Methods. The solid lines represent sequences of the pGL2-basic plasmid. The solid box indicates the EGR-1 promoter and the hatched box represents the protein coding region of the luciferase gene. The open box represents the RNA sequences cloned into the HindIII restriction site of the p644 recombinant plasmid. (B) Effect of TS RNA sequences on luciferase expression in vivo. Luciferase heterologous constructs were transiently expressed in H630 cells. Luciferase activity was assayed 48 h post-transfection using the dual luciferase reporter system as described in Materials and Methods. The activity in cells transfected with the parent p644 plasmid was defined as 100%. Luciferase expression in cells transfected with p644/TS:N25 was significantly increased compared to cells transfected with p644 (paired Student’s t-test, *P < 0.01). Each value represents the mean ± SE of between three and five experiments.
As further evidence for the specificity of the selected sequence, the effect of an unrelated TS RNA sequence on luciferase expression was investigated. Previous experiments from our laboratory showed that human TS protein did not interact with the 3′-UTR of TS mRNA (5). Given these earlier findings in vitro, the sequence corresponding to nt 1040–1240 was cloned onto the 5′-end of the luciferase gene. The p644/TS1040–1240 heterologous construct was transiently expressed in H630 cells. As shown in Figure 4B, no alterations in luciferase were observed upon transfection of this construct when compared with the parent p644 plasmid. This finding provides further support for the specificity of the effect of the selected sequence. These transfection experiments suggest that the selected sequence TS:N25 expresses in vivo biological activity. However, in contrast to previously described TS response sequences, which function as repressor elements, our findings suggest that this sequence acts as an enhancer element by allowing induced expression of luciferase.
We next investigated the effect of the TS:N25 selected sequence on expression of human His-tagged TS in vivo. For these studies the cDNA corresponding to TS:N25 was cloned into pcDNA3.1(+):TS at the HindIII and KpnI restriction sites. After transient transfection of pcDNA3.1(+):TS and pcDNA3.1(+):TS/N25 into human colon cancer H630 cells the levels of human His-tagged TS protein were determined by western immunoblot analysis. As shown in Figure 5, expression of human His-tagged TS protein was increased by 2.3-fold in cells transfected with pcDNA3.1(+):TS/N25 when compared to those transfected with the pcDNA31.(+):TS plasmid. This finding suggests that the TS:N25 selected sequence is able to enhance the expression of TS in vivo.
Figure 5.
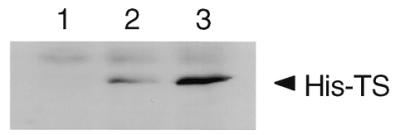
Effect of TS:N25 on the expression of human His-tagged TS in vivo. Human colon cancer H630 cells were transiently transfected in the absence (lane 1) or presence of pcDNA3.1(+):TS (lane 2) or pcDNA3.1(+):TS/N25 (lane 3). Luciferase activity was determined and an equivalent amount of cellular protein based on luciferase activity was resolved on a 10% SDS–polyacrylamide gel. Western immunoblot analysis was performed as described in Materials and Methods.
TS translational regulation is mediated directly by alterations in translational efficiency of TS mRNA (4,8). To begin to identify the specific molecular event(s) regulating the changes in luciferase activity, an RNase protection assay was employed to determine expression of luciferase mRNA in transfected H630 cells. Luciferase mRNA levels were identical in cells transfected with p644 (Fig. 6, lane 2), p644/TS1040–1240 (Fig. 6, lane 3) and p644/TS:N25 (Fig. 6, lane 4). As a complementary approach, an RT–PCR analysis was used to determine luciferase-specific mRNA levels in cells transfected with the p644, p644/TS:N25 and p644/TS1040–1240 plasmids. Using this method, expression of luciferase mRNA was identical in cells transfected with each of these reporter constructs (data not shown). Taken together, these results suggest that the selected TS:N25 RNA exerts its effects at the translational level.
Figure 6.
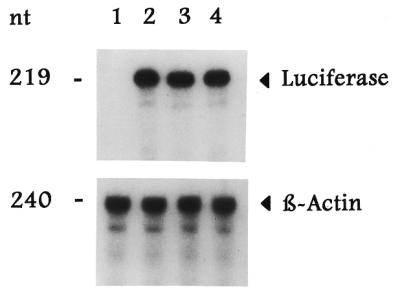
Effect of the selected RNA sequence on luciferase mRNA expression. Human colon cancer H630 cells were transiently transfected in the absence (lane 1) or presence of p644 (lane 2), p644/TS:N25 (lane 3) or p44/TS1040–1240 (lane 4). Total cellular RNA was isolated and then subjected to RNase protection analysis as described in Materials and Methods. The arrowheads indicate the positions of the luciferase- and β-actin-associated bands.
An immunoprecipitation/RT–PCR method was developed in our laboratory to isolate RNP complexes composed of TS protein and TS mRNA from human colon cancer H630 cells (6,11). In the present report we have employed a similar strategy to isolate RNP complexes from H630 cells transfected with the heterologous reporter plasmid, p644/TS:N25. TS RNP complexes were immunoprecipitated with an anti-TS monoclonal antibody and the specific RNA fraction bound to TS was isolated and subjected to RT–PCR amplification. Using primers corresponding to sequences of the EGR-1 promoter and the luciferase gene as outlined in Figure 7B, an 85 bp DNA product was amplified from cells transfected with p644/TS:N25 (Fig. 7A, lane 1), but not from cells transfected with p644 (Fig. 7A, lane 3) or p644/TS1040–1240 (Fig. 7A, lane 4). This band migrated at the same position as the DNA product obtained from a control PCR reaction using the p644/TS:N25 plasmid as DNA template (Fig. 7A, lane 5). Furthermore, when the immunoprecipitation reaction was performed in the absence of antibody (Fig. 7A, lane 2) or with a control antibody such as anti-α-tubulin monoclonal antibody (data not shown), no amplified DNA product was observed. Control experiments were performed in which RNAs isolated from TS RNP complexes were directly PCR amplified in the absence of the RT step, and the 85 bp DNA product was not amplified (Fig. 7A, lane 6). Taken together, the results of the immunoprecipitation/RT–PCR analysis confirm a direct interaction between TS protein and the selected RNA sequence TS:N25 in intact human colon cancer H630 cells.
Figure 7.

Isolation of TS RNP complexes in human colon cancer cells transfected with luciferase heterologous constructs. (A) Ethidium bromide stain. TS RNP complexes were isolated by the immunoprecipitation/RT–PCR method described in Materials and Methods. H630 cells were transiently transfected with p644/TS:N25 (lanes 1, 2 and 6), p644 (lane 3) or p644/TS1040–1240 (lane 4). Whole cell extracts were immunoprecipitated with an anti-TS monoclonal antibody or with no antibody (lane 2). Lane 5 represents a control PCR reaction in which the p644/TS:N25 DNA template was subjected to PCR amplification using the same primer set. Lane 6 represents isolated nucleic acid that was treated with RNase A prior to reverse transcription. Lane M represents the size marker. (B) p644/TS:N25 sequence. The 85 bp fragment predicted to be RT–PCR amplified is shown in relation to its position on the full-length p644 plasmid. The positions of the EGR-1 promoter and the luciferase gene are indicated.
We next investigated whether induced expression of TS:N25 RNA could compete away some of the TS mRNA bound to TS protein in vivo. For these studies we overexpressed TS:N25 RNA in human colon cancer H630 cells using the U6 expression plasmid. U6 is a small, stable RNA that is constitutively and highly expressed in cells (up to 500 000 molecules U6 RNA/cell) as an abundant small nuclear ribonucleoprotein where it plays a central role in splicing and spliceosome assembly. It is transcribed by RNA polymerase III. The U6 promoter has been engineered to express short RNA sequences at high intracellular levels. H630 cells were transfected with either pGEMmU6 or pGEMmU6/TS:N25 and the TS RNP complexes were then isolated to quantitate the amount of TS mRNA bound to TS protein. As shown in Figure 8, the amount of TS mRNA bound to TS protein was significantly decreased (3.8-fold) in cells transfected with pGEMmU6 containing the TS:N25 sequence when compared with the pGEMmU6 plasmid alone.
Figure 8.

Isolation of TS RNP complexes in human colon cancer cells transfected with U6 heterologous plasmids. Human colon cancer H630 cells were transiently transfected in the absence (lane 1) or presence of pGEMmU6 (lane 2) or pGEMmU6/TS:N25 (lane 3). Whole cell extracts were immunoprecipitated with an anti-TS monoclonal antibody and TS RNP complexes were isolated as described in Materials and Methods. PCR products were resolved by electrophoresis on a 1.0% agarose gel stained with ethidium bromide.
Additional experiments were performed to show that the biological activity of the selected TS:N25 sequence was dependent upon TS. For these studies we investigated luciferase expression from the TS-responsive p644/TS:N25 heterologous construct in human colon cancer HCT-C18 cells. This cell line was selected because HCT-C18 cells express a functionally inactive TS protein. Recently we established an HCT-C:His-TS(+) cell line by stable transfection of HCT-C18 cells with a human His-tagged TS cDNA. Biochemical analysis using the TS catalytic assay confirmed that parent HCT-C18 cells express essentially no TS enzyme activity while the TS-transfected HCT-C:TS(+) cells express significant levels of TS enzyme activity (8.03 pmol/min/mg protein). p644 and p644/TS:N25 were each transiently transfected into HCT-C18 and HCT-C:His-TS(+) cells. As seen in Figure 9, luciferase activity in TS-deficient HCT-C18 cells was similar following transfection with either reporter construct. However, luciferase activity was significantly increased by nearly 4-fold in HCT-C:His-TS(+) cells transfected with p644/TS:N25 compared with cells transfected with the parent p644 plasmid. This series of experiments provide further supportive evidence that a functionally active TS is required for the TS:N25 sequence to display its complete translational regulatory effects.
Figure 9.

Effect of TS:N25 RNA on luciferase expression in HCT-C18 and HCT-C:His-TS+ cells. Human colon cancer HCT-C18 (open square) and HCT-C:His-TS+ (hatched square) cells were transiently transfected with p644, p644/TS:N25 or p644/TS:N25(AS). Luciferase activity was measured 48 h post-transfection using the dual luciferase assay system as described in Materials and Methods. The activity in cells transfected with the p644 plasmid was taken as 100%. Transfection with p644/TS:N25 resulted in a significant increase in luciferase activity (P < 0.01), as indicated by the asterisk, when compared to transfection with p644. Statistical analysis was performed using the paired Student’s t-test. Each value represents the mean ± SE of between three and five experiments.
DISCUSSION
We employed an in vitro RNA selection method to identify an RNA sequence that binds with high affinity to human TS protein. Of note, this selected RNA sequence binds with nearly 20-fold higher affinity to TS than native TS mRNA cis-acting sequences previously identified. A secondary structure analysis predicts that this sequence adopts a stable stem–loop structure with a 3 base loop (Fig. 3). Subsequent mutational and deletional analysis reveals that the UGU loop is a critical element for recognition by TS protein. Transfection experiments confirmed that this element is sufficient to confer the property of translational regulation onto a heterologous luciferase reporter gene. Moreover, its biological activity in vivo is observed only in human colon cancer cells expressing a functional TS protein. These findings, taken together, provide critical support for the role of this selected RNA sequence as a specific binding site for TS in vitro and in vivo.
Previous work from this laboratory focused on the cis-acting sequences located within the 5′-upstream and protein coding regions of TS mRNA. Careful sequence analysis has failed to identify a consensus nucleotide sequence between these two elements and the selected RNA sequence. One potential limitation of such an analysis is that the well-characterized RNA binding proteins appear to recognize a combination of sequence and secondary structure. Thus, identification of a consensus RNA binding site by sequence analysis alone may be insufficient.
The RNA Zuker fold program predicts that the selected RNA sequence adopts a stable stem–loop structure. Moreover, the UGU loop is required for RNA binding, as point mutations within the loop sequence or complete deletion of this 3 nt sequence results in significantly altered affinity for TS protein. These findings suggest that non-Watson–Crick G-U base pairings may maintain the loop in a stable structure that allows for optimal interaction with TS protein. Although this stem–loop structure differs from that predicted for the cis-acting elements located within either the 5′-upstream or protein coding region, further studies are in progress to elucidate more precisely the specific structural requirements for TS protein binding. In addition, it is conceivable that the uridine nucleotides in the UGU loop form covalent Michael adducts with cysteine sulfhydryl groups on TS protein. The formation of a covalent bond between the target mRNA and a cysteine moiety has been observed for other well-characterized RNA binding proteins, such as R17 bacteriophage coat protein, yeast tRNA synthase and iron regulatory factor (38–40).
One interesting aspect of this work is that the selected RNA functions in vivo as a positive enhancer element. Our transfection experiments demonstrate that the selected sequence enhances not only expression of luciferase in vivo but that of TS as well. Although sequence analysis failed to detect a homologous sequence within human TS mRNA, a preliminary secondary structure analysis identified a sequence contained within nt 620–635 that adopts a similar stem–loop structure to the TS:N25 RNA. Further studies are underway to determine whether this native sequence may, indeed, function as an enhancer element for TS mRNA translation. The mechanism(s) underlying the positive effect of the selected sequence on luciferase and TS mRNA translation remains unclear. However, several possiblities exist to explain this process. It is conceivable that the binding of TS protein to the selected sequence enhances subsequent binding of the critical translational intitiation factors near or at the cap site. Another possibility is that formation of the RNP complex between TS:N25 RNA and TS protein facilitates, in some manner, the process of ribosomal scanning. Finally, this RNP complex may alter the conformation of the luciferase mRNA in such a manner as to promote its subsequent translation.
The positive effect of TS:N25 RNA on cellular gene expression is in marked contrast to the repressor effect of the previously identified elements in the 5′-upstream and protein coding regions (4,11). Although most of the well-characterized cis-acting translational regulatory sequences function as repressor elements, a growing list of RNA sequences act as enhancer elements (41–44). Furthermore, there are examples, including ferritin mRNA and hepatitis C mRNA, in which both negative and positive regulatory sequences reside on the same mRNA (45–47).
Several other issues are raised by the current study, including why the wild-type sequences were not identified in the selection process. It is conceivable that the native, cognate TS RNA sequences may have been present in the earlier rounds of selection but were overtaken by the selected sequence after successive rounds of selection. An alternative possibility is that insufficient clones were selected and analyzed and thus, for purely statistical reasons, native TS RNA sequences were not identified through this in vitro iterative approach.
Another issue concerns the potential biological relevance of the selected sequence. Clearly, the 25 nt RNA selected from the degenerate RNA library binds TS protein with high affinity and specificity. The question is whether there are other native cellular RNAs with a similar structural motif. As already noted, a preliminary secondary structure analysis suggests that a similar stem–loop structure may reside within nt 620–635 of TS mRNA. Furthermore, a search of matching eukaryotic sequences identified a number of mRNA sequences with homology to the selected RNA sequence, including those corresponding to apoptosis-associated tyrosine kinase (ATK), eIF2B δ-subunit, ATPase II and the family of keratins, including 1, 3 and 5, (Table 2). Interestingly, a Zuker fold analysis of each of these sequences predicts the formation of a stem–loop structure nearly identical to that observed with the 25 nt sequence selected by in vitro selection. A preliminary series of RNA binding experiments were performed to determine whether human TS protein can interact with these cellular RNAs. As shown in Table 3, the RNAs corresponding to eIF2B δ-subunit and ATK bind to TS with a similar affinity to TS:N25 RNA. This preliminary analysis suggests that TS may, indeed, recognize various cellular mRNAs with this structural motif in vivo.
Table 2. Sequences obtained from a GenBank profile search that most closely match the in vitro selected RNA sequence.
| mRNA | Nucleotides | Sequence homology (%) |
|
|---|---|---|---|
| Full sequence | Core sequence | ||
| Keratin 1 | 232–256 | 56 | 100 |
| Keratin 3 | 305–315 | 56 | 100 |
| Keratin 5 | 349–373 | 56 | 100 |
| eIF2B | 823–847 | 65 | 91 |
| ATPase II | 26–50 | 48 | 91 |
| ATK | 4553–4577 | 60 | 91 |
Core sequence refers to the 11 nt stem–loop motif of TS:N25.
Table 3. Relative binding affinity of cellular mRNAs with TS protein.
| RNA | IC50 (nM) |
|---|---|
| TS:N25 | 0.08 |
| eIF2B | 0.10 |
| ATK | 0.12 |
| TS:N25AS | >10 |
The binding affinity of ATK and eIF2B RNA for human recombinant TS was determined by the RNA gel shift assay as described in Materials and Methods. Each value represents the mean of between three and five experiments and the SE for each experiment was <10%.
These studies enhance our understanding of the specific molecular elements controlling TS-directed translational regulation. In particular, they provide insights into the critical nucleotide elements required for protein recognition. Thus, the in vitro RNA selection approach represents a powerful method for characterizing the molecular basis underlying the TS mRNA–TS protein interaction. Moreover, our preliminary studies suggest that the selected RNA sequence can inhibit TS catalytic activity. Thus, such an approach may provide the rational basis for molecular drug design to identify novel small molecules that can be used to inhibit and/or sequester TS enzyme activity and function.
Acknowledgments
ACKNOWLEDGEMENTS
The authors thank Drs John Schmitz, Jingfang Ju and Ashwin Gollerkeri for their critical reading of the manuscript. This work was supported in part by grants from the National Science Foundation (MCB97-24071 to G.F.M.), the National Cancer Institute (CA44355 to F.M., and CA16359 and CA75712 to E.C.) and the Department of Veterans Affairs (VA Merit Review to E.C.).
REFERENCES
- 1.Friedkin M. and Kornberg,A. (1957) Chemical Basis of Heredity. Johns Hopkins University Press, Baltimore, MD, pp. 609–614.
- 2.Danenberg P.V. (1977) Biochim. Biophys. Acta, 473, 73–79. [DOI] [PubMed] [Google Scholar]
- 3.Carreras C. and Santi,D.V. (1995) Annu. Rev. Biochem., 64, 721–762. [DOI] [PubMed] [Google Scholar]
- 4.Chu E., Koeller,D.M., Casey,J.L., Drake,J.C., Chabner,B.A., Elwood,P.C., Zinn,S., Allegra,C.J. (1991) Proc. Natl Acad. Sci. USA, 88, 8977–8981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chu E., Voeller,D., Koeller,D.M., Drake,J.C., Takimoto,C.H., Maley,G.F., Maley,F., Allegra,C.J. (1993) Proc. Natl Acad. Sci. USA, 90, 517–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chu E., Voeller,D.M., Jones,K.L., Takechi,T., Maley,G.F., Maley,F., Segal,S. and Allegra,C.J. (1994) Mol. Cell. Biol., 14, 207–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Voeller D.M., Changchien,L.M., Maley,G.F., Maley,F., Takechi,T., Turner,R.E., Montfort,W.R., Allegra,C.J. and Chu,E. (1995) Nucleic Acids Res., 23, 869–875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chu E. and Allegra,C.J. (1996) Bioessays, 18, 191–198. [DOI] [PubMed] [Google Scholar]
- 9.Lin X., Voeller,D.M., Allegra,C.J., Maley,F. and Chu,E. (2000) Nucleic Acids Res., 28, 1381–1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ju J.F., Pedersen-Lane,J., Maley,F. and Chu,E. (1999) Proc. Natl Acad. Sci. USA, 96, 3769–3774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chu E., Takechi,T., Jones,K.L., Voeller,D.M., Copur,S.M., Maley,G.F., Maley,F., Segal,S. and Allegra,C.J. (1995) Mol. Cell. Biol., 15, 179–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Parsels L.A., Ju,J.F., Pedersen-Lane,J., Maley,F. and Chu E. (1999) Proc. Am. Assoc. Cancer Res., 40, 437. [Google Scholar]
- 13.Tuerk C. and Gold,L. (1990) Science, 249, 505–510. [DOI] [PubMed] [Google Scholar]
- 14.Szostak J.W. (1992) Trends Biochem. Sci., 17, 89–93. [DOI] [PubMed] [Google Scholar]
- 15.Szostak J.W. and Ellington,A.D. (1993) In Gesteland,R.F. and Atkins J.F. (eds), The RNA World. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, pp. 511–533.
- 16.Henderson R.B., Menotti,E., Bonnard,C. and Kuhn,C.L. (1994) J. Biol. Chem., 269, 17481–17489. [PubMed] [Google Scholar]
- 17.Tsai D.E., Harper,D.S. and Keene,J.D. (1991) Nucleic Acids Res., 19, 4931–4938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bartel D.P., Zapp,M.L., Green,M.R. and Szostak,J.W. (1991) Cell, 67, 529–536. [DOI] [PubMed] [Google Scholar]
- 19.Park J.G., Oie,H.K., Sugarbaker,P.H., Henslee,J.G., Chen,T.R., Johnson,B.E. and Gazdar,A. (1987) Cancer Res., 47, 6710–6718. [PubMed] [Google Scholar]
- 20.Hoganson D.K., Williams,A. and Berger,S. (1999) Biochem. Pharmacol., 58, 1529–1537. [DOI] [PubMed] [Google Scholar]
- 21.Lu Y.Z., Aiello,P.D. and Matthews,R.G. (1984) Biochemistry, 18, 6870–6876. [Google Scholar]
- 22.Davisson V.J., Sirawaporn,W. and Santi,D.V. (1989) J. Biol. Chem., 264, 9145–9148. [PubMed] [Google Scholar]
- 23.Pedersen-Lane J., Maley,G.F., Chu,E. and Maley,F. (1997) Protein Expr. Purif., 10, 256–262. [DOI] [PubMed] [Google Scholar]
- 24.Liebold E. and Munro,H.N. (1988) Proc. Natl Acad. Sci. USA, 85, 2171–2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.He Y. and Huang,L. (1997) Cancer Res., 57, 3993–3999. [PubMed] [Google Scholar]
- 26.Felgner P.L., Gadek,T.R., Holm,M., Roman,R., Chan,H.W., Wenz,M., Northrop,J.P., Ringold,G.M. and Danielsen,M. (1987) Proc. Natl Acad. Sci. USA, 84, 7413–7417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chomczynski P. and Sacchi,N. (1987) Anal. Biochem., 162, 156–159. [DOI] [PubMed] [Google Scholar]
- 28.Steitz J.A. (1989) Methods Enzymol., 180, 468–481. [DOI] [PubMed] [Google Scholar]
- 29.Zuker M. and Stiegler,P. (1981) Nucleic Acids Res., 9, 133–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Person R.D., Bartel,D.P., Szostak,J.W., Horvath,S.J. and Feigon,J. (1994) Biochemistry, 33, 5357–5366. [DOI] [PubMed] [Google Scholar]
- 31.Valegard K., Murray,J.B., Stockley,P.G., Stonehouse,N.J. and Liljas,L. (1994) Nature, 371, 623–626. [DOI] [PubMed] [Google Scholar]
- 32.Kikinis Z., Eisenstein,K.S., Bettary,A.J. and Munro,H.N. (1995) Nucleic Acids Res., 23, 4190–4195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Borer P.N., Lin,Y., Wang,S., Roggenbuck,M.W., Gott,J.M., Uhlenbeck,O.C. and Pelczer,I. (1995). Biochemistry, 34, 6488–6503. [DOI] [PubMed] [Google Scholar]
- 34.Schlegl J., Gegout,V., Schlager,B., Hentze,M.W., Westhof,E., Ehresmann,C., Ehresmanmn,B. and Romby,P. (1997) RNA, 3, 1159–1172. [PMC free article] [PubMed] [Google Scholar]
- 35.Addess K.J., Basilio,J.P., Klausner,R.D., Rouault,T.A. and Pardi,A. (1997) J. Mol. Biol., 274, 72–83. [DOI] [PubMed] [Google Scholar]
- 36.Hess M.A. and Duncan,R.F. (1996) Nucleic Acids Res., 24, 2441–2449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zimmerman G.R., Jenison,R.D., Wick,C.L., Simorre,J.P. and Pardi,A. (1997) Nat. Struct. Biol., 4, 644–649. [DOI] [PubMed] [Google Scholar]
- 38.Romaniuk P.J. and Uhlenbeck,O.C. (1985) Biochemistry, 24, 4239–4244. [DOI] [PubMed] [Google Scholar]
- 39.Starzyk R.M., Koontz,S.W. and Schimmel,P. (1982) Nature, 298, 136–140. [DOI] [PubMed] [Google Scholar]
- 40.Klausner R.D., Rouault,T.A. and Harford,J.B. (1993) Cell, 72, 19–28. [DOI] [PubMed] [Google Scholar]
- 41.Gallie D.R., Sleat,D.E., Watts,J.W., Turner,P.C. and Wilson,T.M.A. (1988) Nucleic Acids Res., 16, 883–893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Falcone D. and Andrews,D.W. (1991) Mol. Cell. Biol., 11, 2656–2664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mizokami A. and Chang,C. (1994) J. Biol. Chem., 269, 25655–25659. [PubMed] [Google Scholar]
- 44.Rogers J.T., Leiter,L.M., McPhee,J., Cahill,C.M., Zhan,S.S., Potter,H. and Nilsson,L.N. (1999) J. Biol. Chem., 5, 6421–6431. [DOI] [PubMed] [Google Scholar]
- 45.Rogers J.T., Andriotakis,J.L., Lacroix,L., Durmowicz,G.P., Kasschau,K.D. and Bridges,K.R. (1994) Nucleic Acids Res., 22, 2768–2686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dix D.J., Lin,P.N., Kimata,Y. and Theil,E.C. (1992) Biochemistry, 31, 2818–2822. [DOI] [PubMed] [Google Scholar]
- 47.Yoo B.J., Spaete,R.R., Geballe,A.P., Selby,M., Houghton,M. and Han,J.H. (1992) Virology, 191, 889–899. [DOI] [PubMed] [Google Scholar]