

Abstract
Recent advancements in spatial imaging technologies have revolutionized the acquisition of high-resolution multichannel images, gene expressions, and spatial locations at the single-cell level. Our study introduces xSiGra, an interpretable graph-based AI model, designed to elucidate interpretable features of identified spatial cell types, by harnessing multimodal features from spatial imaging technologies. By constructing a spatial cellular graph with immunohistology images and gene expression as node attributes, xSiGra employs hybrid graph transformer models to delineate spatial cell types. Additionally, xSiGra integrates a novel variant of gradient-weighted class activation mapping component to uncover interpretable features, including pivotal genes and cells for various cell types, thereby facilitating deeper biological insights from spatial data. Through rigorous benchmarking against existing methods, xSiGra demonstrates superior performance across diverse spatial imaging datasets. Application of xSiGra on a lung tumor slice unveils the importance score of cells, illustrating that cellular activity is not solely determined by itself but also impacted by neighboring cells. Moreover, leveraging the identified interpretable genes, xSiGra reveals endothelial cell subset interacting with tumor cells, indicating its heterogeneous underlying mechanisms within complex cellular interactions.
Keywords: explainable AI, spatial cell recognition, hybrid graph transformer, interpretable features
Introduction
Recent advances in spatial transcriptomic techniques have enabled commercially available platforms to measure mRNA expression in a tissue at molecular level spatial resolution, allowing biologists to gain novel insights about diseases [1]. Spatial locations of expressions are critical in understanding of cell–cell interactions and cell functioning in tissue microenvironment [2, 3]. Molecular imaging-based in situ hybridization approaches such as NanoString CosMx Spatial Molecular Imaging (SMI), an automated microscope imaging system, allows spatial in situ detection on formalin-fixed paraffin-embedded samples. CosMx SMI is capable of detecting both RNAs and proteins on the same tissue slide, allowing three-dimensional subcellular resolution image analysis with an accuracy of ~50 nm in the XY plane and high throughput (up to 1 million cells per sample) [4]. MERSCOPE [5], another commercial platform, utilizes the MERFISH [6] (multiplex error-robust fluorescence in situ hybridization) platform that captures hundreds to thousands of RNAs at the same time. Other noticeable commercial platforms that provide single-cell or subcellular spatial resolution include the 10x Genomics’ Xenium system and BGI’s SpaTial Enhanced REsolution Omics-Sequencing (Stereo-seq) in situ sequencing system using the DNA Nanoball technology.
The emerging spatial imaging technologies require tailored computational methods for data analysis, clustering, and enhancement. Some deep learning (DL)-based methods have been proposed for spatial transcriptomics data [7]. For example, one method [8] designed based on Graph Convolutional Neural Network (GCN), takes both gene expression data and spatial information of single cells as input, to cluster spatial transcriptomics data. The SiGra [9] method enhances the transcriptomics data and identifies spatial domains from the spatial multimodality data. SpaGCN [10] provides gene expression, spatial information, and histology information to the GCN network to identify spatial domains and spatially variable genes. stLearn [11] is developed for spatial transcriptomics to identify cell types, reconstruct cell trajectories, and detect microenvironment. BayesSpace [12] uses Bayesian approach and spatial information to enhance gene expression signals. STAGATE [13] combines gene expression and cell locations to train a graph autoencoder for low-dimensional embedding and spatial domain detection. conST [14] integrates gene expression, histology features extracted using MAE [15] model, and spatial information using GCN and contrastive learning to learn embeddings, which aid in several downstream tasks like trajectory analysis and spatial domain identification. ConGI [16] utilizes contrastive learning to jointly learn common representations across gene expression and histology image features for spatial domain identification. Other methods such as Seurat [17] and Scanpy [18] also provide functionalities for analysis of spatial transcriptomics data and domain identification. Clustering methods such as Louvain [19] perform domain recognition using expression data but do not utilize spatial information.
Although the above DL-based methods prove to identify spatial cells or domains with high accuracy, their intrinsic black-box nature inhibits explainability, making it unclear which genes and cells are utilized by these methods to achieve accurate spatial identities [20]. Such explainability issue is common when applying advanced deep learning approaches [21]. Explaining the model decisions can aid to find any limitations and validate model functioning using known knowledge [22]. Several works are done to make the graph neural network models interpretable. For example, Saliency [23] assigns the square of gradients as importance scores for the input features. Guided backpropagation [24] uses only positive gradients for backpropagation while setting negative gradients to 0. Those positive gradients at the input layer are used as the importance score of input features. InputXGradient [25] assigns importance score for input features as gradients multiplied by input. Deconvolution [26, 27] computes the gradient of output class with respect to the input selectively propagating only the non-negative gradients and utilizes that as the importance score for input features.
In this study, we have proposed a novel method, i.e. xSiGra, to identify interpretable features contributing to the identification of spatial cell types. xSiGra takes advantage of the multimodality spatial data including multichannel histology images, spatial information, and gene expression data. xSiGra not only demonstrates its superior performance than existing state-of-the-art methods in the identification of spatial cell types, but also provides explainable information about the importance of cells and genes for the identification. The explainable capability of xSiGra also shows better performance than existing several graph-explainable algorithms, which facilitates biological insights from spatial imaging data.
Results
Overview of the xSiGra model
The primary function of the xSiGra model is to elucidate the significance of each gene for biological annotations, such as cell identities, within high-resolution spatial data. As depicted in the overall diagram (Fig. 1), cells of the same type exhibit diverse gene expression patterns and interact with their environment differently across various spatial locations and microenvironments. We illustrate this conceptually through two types of endothelial–stromal interactions: firstly, via growth factors secreted by stromal cells and receptors expressed on endothelial cell surfaces, and secondly, through the expression of specific structural proteins in the extracellular matrix (ECM) by stromal cells and the endothelial integrins responsible for recognizing and binding to these proteins.
Figure 1.

Overview of xSiGra model. (a) xSiGra uses spatial transcriptomics data to gain insights about the cellular microenvironment. Spatial transcriptomics profiles are represented by graph structure with each cell in spatial graph having associated gene expression and multichannel images. xSiGra generates enhanced gene expressions and identifies spatial cell types, with related interpretable features for biological insights. (b) xSiGra performs image augmented learning to enhance transcriptomics data (SiGra+) and recognize spatial cell types. It also provides interpretable features that can be used for biological interpretation.
The xSiGra model is composed of a SiGra+ module and an explainable module. The explainable module uses a novel graph gradient-weighted class activation mapping (graph Grad-CAM) algorithm to understand the contribution of each gene for different cell types within the spatial slides. The SiGra+ module learns the latent representation of multimodal spatial transcriptomics data, which is used by the explainable module to explain which spatial gene expression patterns are used by SiGra+ for spatial cell type identification. The SiGra+ module also further enhances the original SiGra [9] model by using a VGG feature extractor [28] to improve histochemistry image feature extraction, and by introducing a Kullback–Leibler (KL) divergence loss [29] to better balance the contributions from the transcriptomics and the imaging data.
xSiGra accurately identifies spatial cell types in various datasets
We have compared our method with nine state-of-the-art methods, i.e. SiGra [9], SpaGCN [10], stLearn [11], BayesSpace [12], STAGATE [13], conST [14], ConGI [16], Seurat [17], and Scanpy [18]. Here, we use 8 NanoString CosMx lung tissue slices (see Data Availability) for performance comparisons. Adjusted Rand index (ARI) score and normalized mutual information (NMI) are used for evaluation of the detected spatial cell types, including tumor, fibroblasts, lymphocyte, myeloid, mast, neutrophil, endothelial, and epithelial cells. Figure 2a shows the ARI results of each of the methods across eight lung cancer NanoString CosMx SMI tissues. xSiGra achieved a median ARI of 0.565, better than SiGra (ARI = 0.505), stLearn (ARI = 0.360), Seurat (ARI = 0.315), ConGI (ARI = 0.305), and BayesSpace (ARI = 0.265), as well as STAGATE (ARI = 0.245), Scanpy (ARI = 0.235), SpaGCN (ARI = 0.20), and conST (ARI = 0.07). Specifically for Lung-13 tissue, which consists of 20 field of views (FOVs) and 77 643 cells, Fig. 2b presents the spatial cell types identified by different methods. The cell types identified by xSiGra match well with the ground truth with an overall ARI of 0.64, higher than SiGra (ARI = 0.45), stLearn (ARI = 0.60), Seurat (ARI = 0.35), Scanpy (ARI = 0.32), STAGATE (ARI = 0.29), BayesSpace (ARI = 0.28), ConGI (ARI = 0.26), SpaGCN (ARI = 0.26), and conST (ARI = 0.07). Figure 2c shows the spatial cell types at the FOV level for better clarity. The cells identified by xSiGra show consistency with the ground truth, identifying the continuous tumor region along with infiltrated immune cells. On the contrary, in FOV1, BayesSpace mislabels some lymphocytes and fibroblast as mast and endothelial cells while stLearn identifies some tumor cells and fibroblast incorrectly as mast and epithelial cells. In FOV5, BayesSpace misrecognizes some tumor cells as neutrophils; fibroblasts are mislabeled as mast cells. stLearn mixes fibroblasts with neutrophil cells as well as tumor cells with mast cells.
Figure 2.

Performance evaluation on lung cancer slices. (a) Box plot of adjusted Rand index for 8 NanoString SMI datasets is shown for each of the methods, i.e. conST, SpaGCN, Scanpy, STAGATE, BayesSpace, ConGI, Seurat, stLearn, SiGra, and xSiGra. The middle line represents the median and limits are first and third quartiles. (b) Spatial cell types of ground truth and those identified by stLearn and BayesSpace are shown. (c) Spatial cell types for 2 FOVs of ground truth and those identified by stLearn and BayesSpace are shown. (d) UMAP visualizations for raw and enhanced gene expressions are shown for non-tumor cells. (e) Violin plot of raw and enhanced gene expressions of marker genes in different cell types.
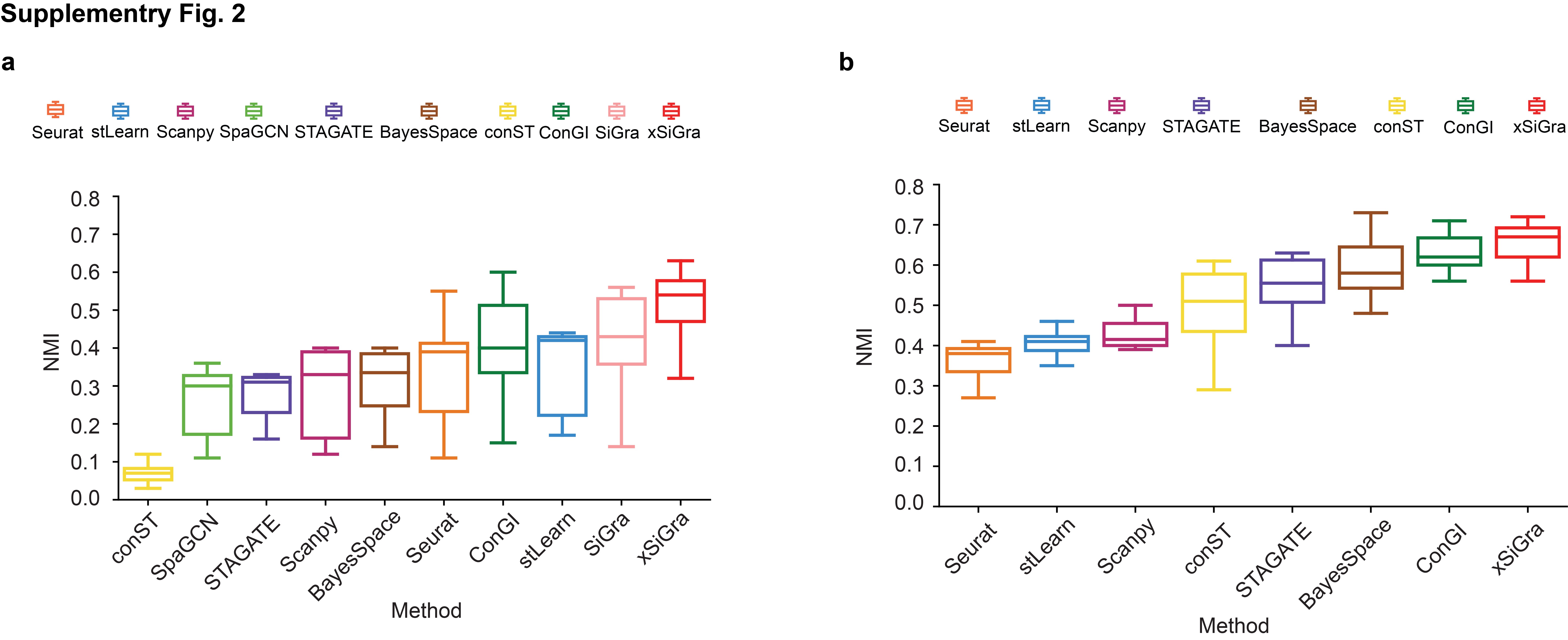
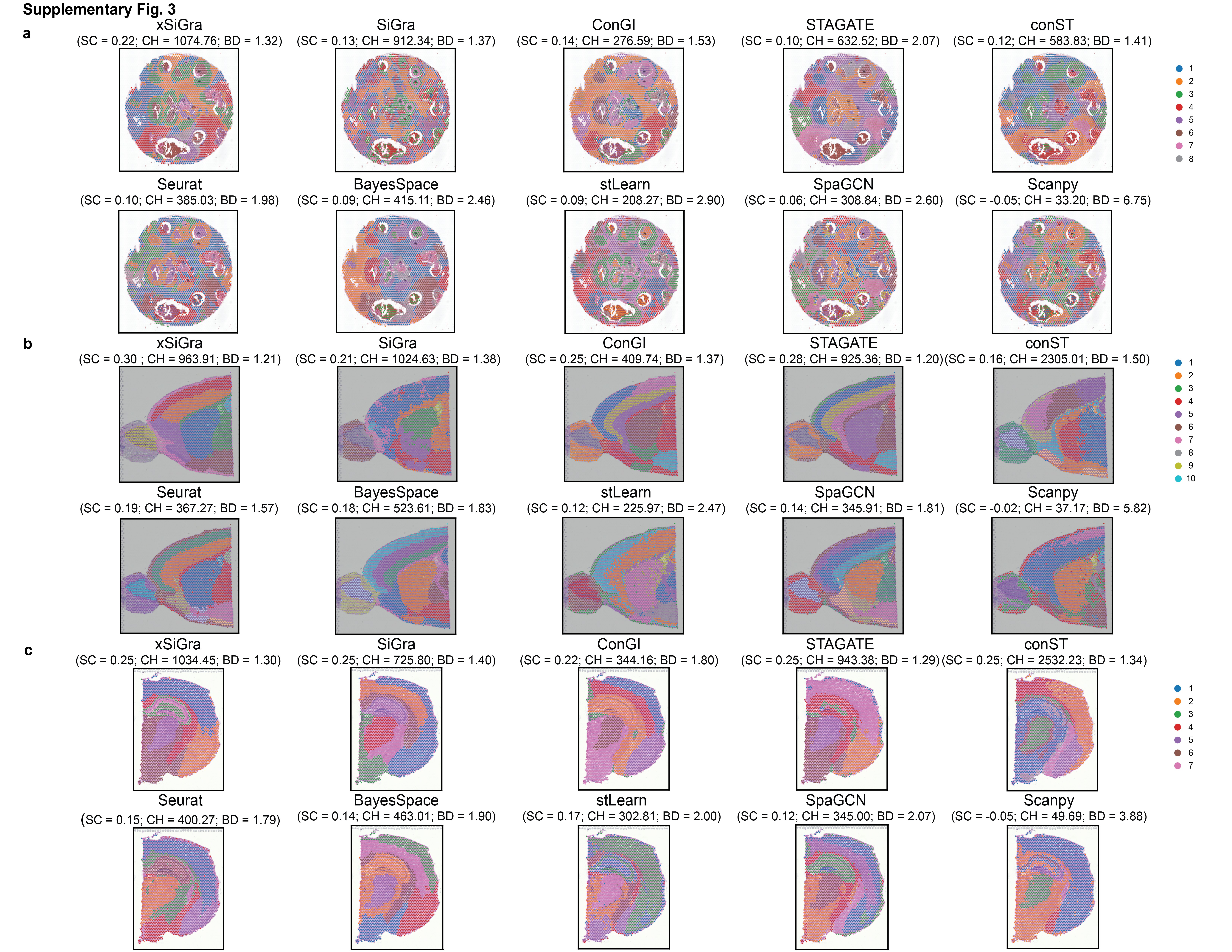
In addition to single-cell spatial data, xSiGra also proves to perform better in spot-based spatial data such as 10x Visium spatial slices. As shown in Supplementary Fig. 1a, xSiGra achieves a higher ARI score compared to the state-of-the-art methods across 12 Visium datasets (see Data Availability). Supplementary Fig. 1b–d present the spatial domains accurately identified by xSiGra. This demonstrates that in comparison with the existing methods, our method can more accurately identify the spatial domains in spot-based spatial data and distinguish the different domains in the complex microenvironment. Moreover, xSiGra also enhances gene expression data for downstream analysis. From the Uniform Manifold Approximation and Projection (UMAP) analysis on enhanced data and raw data, enhanced data present clearer separation of different cell types than raw data (Fig. 2d). The enhanced gene expression data are further supported by the cell type markers whose expressions are highly expressed in their corresponding cell types (Fig. 2e). Supplementary Fig. 2a shows the NMI results of each of the methods across eight lung cancer NanoString CosMx SMI tissues. xSiGra achieved a median ARI of 0.54, better than SiGra (ARI = 0.43), stLearn (ARI = 0.42), ConGI (ARI = 0.4), Seurat (ARI = 0.39), and BayesSpace (ARI = 0.335), as well as STAGATE (ARI = 0.31), Scanpy (ARI = 0.33), SpaGCN (ARI = 0.3), and conST (ARI = 0.07). In Supplementary Fig. 2b, xSiGra achieves a higher NMI score compared to the state-of-the-art methods across 12 DLPFC Visium datasets.
We also compared the performance of xSiGra on spot-based data, which did not provide ground truth information using clustering metrics Silhouette coefficient (SC), Caliniski–Harabasz index (CH), and Davies–Bouldin index (BD). For SC and CH, larger values indicate better clustering performance while for BD smaller values indicate better performance.
Supplementary Fig. 3 provides the comparison results on spatial domain detection of tissue slices from human breast cancer, mouse brain anterior, and mouse brain coronal sections, respectively. For example, in the human breast cancer tissue (Supplementary Fig. 3a), each method was evaluated based on their SC, CH, and BD. Notably, xSiGra exhibited the highest SC and CH value (SC: 0.22, CH: 1074.76), and the lowest BD value of 1.32, compared with SiGra (SC = 0.13; CH = 912.34; BD = 1.37), ConGI (SC = 0.14; CH = 276.59; BD = 1.53), STAGATE (SC = 0.10; CH = 632.52; BD = 2.07), conST (SC = 0.12; CH = 583.83; BD = 1.41), Seurat (SC = 0.10; CH = 385.03; BD = 1.98), BayesSpace (SC = 0.09; CH = 415.11; BD = 2.46), stLearn (SC = 0.09; CH = 208.27; BD = 2.90), SpaGCN (SC = 0.06; CH = 308.84; BD = 2.60), and Scanpy (SC = −0.05; CH = 33.20; BD = 6.75). This confirms the superior performance of xSiGra than other methods in identifying spatial domains. In addition, for the mouse brain anterior tissue (Supplementary Fig. 3b) and mouse brain coronal section (Supplementary Fig. 3c), xSiGra still achieves the best performance in identifying spatial domains. These findings underscore the outperformance of xSiGra in delineating spatial domains across diverse tissue types, providing valuable insights into cellular organization within complex microenvironment.
xSiGra presents better interpretability for explainable features
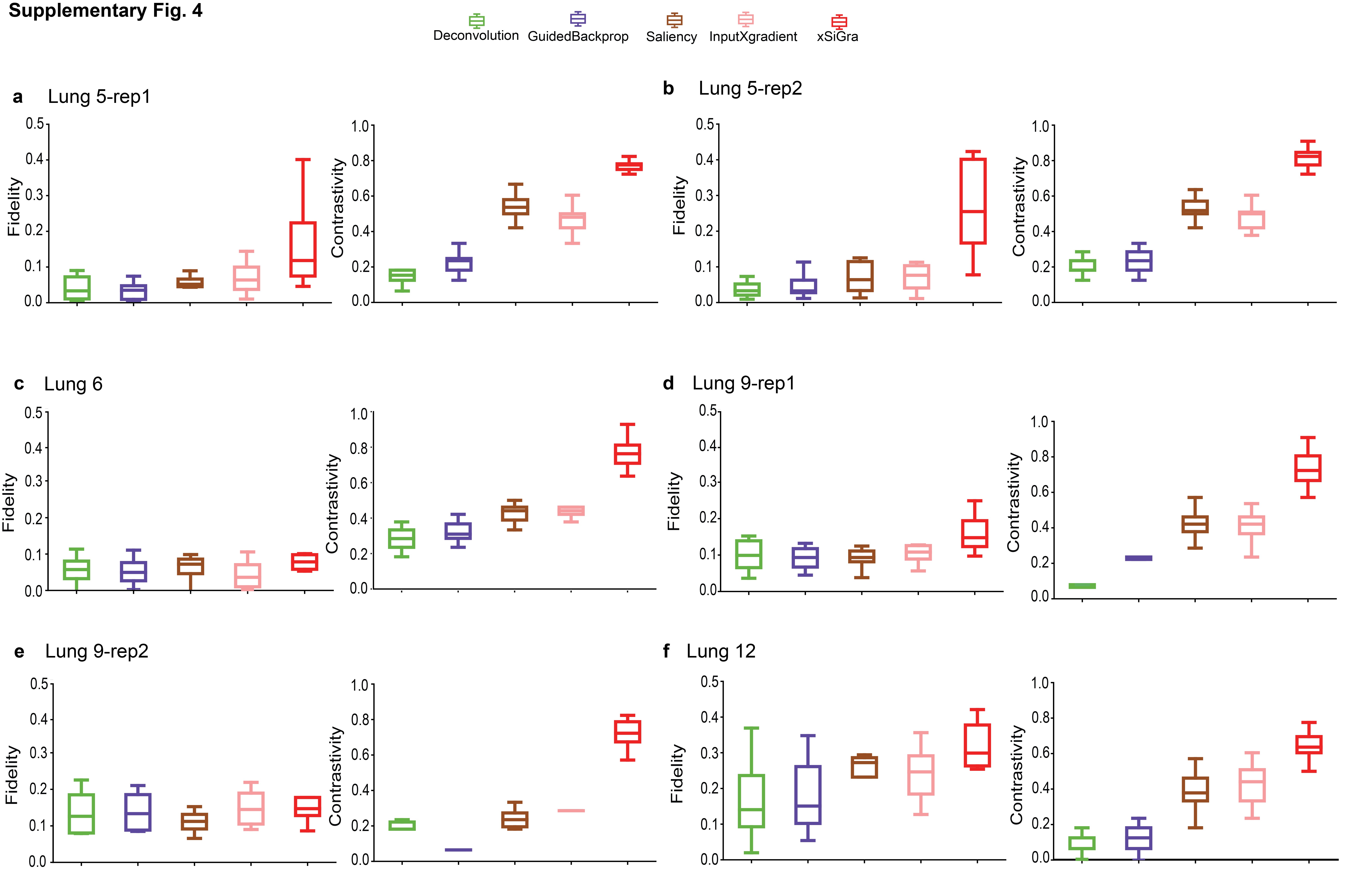
Our method uses a novel graph Grad-CAM to identify the important cells and genes for spatial cell types. The model interpretability can be evaluated with two key metrics: fidelity and contrastivity [30]. Fidelity measures how much the detection accuracy is reduced when the important genes are excluded. Contrastivity measures the differences in important genes identified across different spatial cell types. Based on these two metrics, we have compared xSiGra with four existing explainable methods, including Saliency [23], InputXGradient [25], GuidedBackprop [24], and Deconvolution [26] on each of the lung tissue slices (see Data Availability). Specifically, Fig. 3a shows the fidelity and contrastivity scores of each of the methods on the Lung-13 tissue slice, where xSiGra achieved a higher median fidelity score of 0.16, which is the best compared to Saliency (median: 0.10), InputXGradient (median: 0.09), GuidedBackprop (median: 0.05), and Deconvolution (median: 0.04). xSiGra also obtained better contrastivity score (median: 0.77) than the other methods, especially Deconvolution (median: 0.18). Figure 3b shows that the fidelity and contrastivity scores of different methods on this Lung-5 rep3 tissue slice. Across all eight lung cancer tissue slices (Fig. 3 and Supplementary Fig. 4), xSiGra presents superior performance in both fidelity score (median = 0.16) and contrastivity score (median = 0.76), compared to the other methods (Saliency: 0.09, 0.45; InputXGradient: 0.08, 0.46; GuidedBackprop: 0.06, 0.16; and Deconvolution: 0.05, 0.23). Collectively, xSiGra demonstrates superior performance of interpretability than other explainable methods.
Figure 3.

Evaluation results of model explainability. (a) Box plot of fidelity and contrastivity of deconvolution, GuidedBackprop, Saliency, InputXGradient, and xSiGra methods on Lung-13 tissue slice. (b) Box plot of fidelity and contrastivity of those methods on Lung 5-rep3 tissue slice.
xSiGra identifies important genes and cells for spatial cell types
Given the strong interpretability of xSiGra, it is able to reveal the important genes and cells contributing to the identified spatial cell types. To visualize the cell-level contributions, as shown in Fig. 4a, we use gradient colors representing cells ranging from low to high contribution for each of the major cell population (tumor, fibroblast, myeloid, and lymphocytes). It is observed that, for a specific cell type, not only the cells within this cell type, but also some neighboring cells contribute to the identification of this specific cell type. To further confirm this observation, we utilize four distinct colors to differentiate cells of the particular cell type that exhibit importance and those not belonging to the specific cell type but still demonstrate importance (Fig. 4b).
Figure 4.

Spatial figure to visualize cell-level contribution in identifying spatial cell types. (a) Visualization of the contribution of cells in identifying spatial cell types. (b) Contribution of cells in identifying the specific cell type. (c) Violin plot to visualize the contribution of cells to each cell type.
Specifically, in the case of tumor cells, the majority of these cells, along with a few non-tumor cells, exhibit significant contributions to the identification of this cell type. However, there are also a few tumor cells that do not contribute to its identification. Similarly, for fibroblasts, most of these cells contribute to their identification along with a few non-fibroblast cells. Comparable patterns are observed for myeloid and lymphoid cells as well. Notably, for each spatial cell type, the cells that contribute to it despite not belonging to that specific cell type are primarily neighboring cells. Figure 4c provides quantitative measurements of the contributions of eight different cell types for identifying a particular spatial cell type. In tumor identification, tumor cells make the major contribution, while fibroblasts, along with a few myeloid and lymphocyte cells, contribute significantly to the identification of the fibroblast cell type. Myeloid cells, as well as some lymphocytes and fibroblasts, contribute to the identification of the myeloid cell type. Similarly, lymphocyte cells, along with some myeloid and fibroblast cells, show contributions to the identification of the lymphocyte cell type. Overall, xSiGra offers valuable insights into spatial cell type identification, providing specific information about how cells and their neighboring cells are involved in deciphering spatial cellular heterogeneity.
Explainable xSiGra uncovers endothelial subset involved in ECM-related interactions
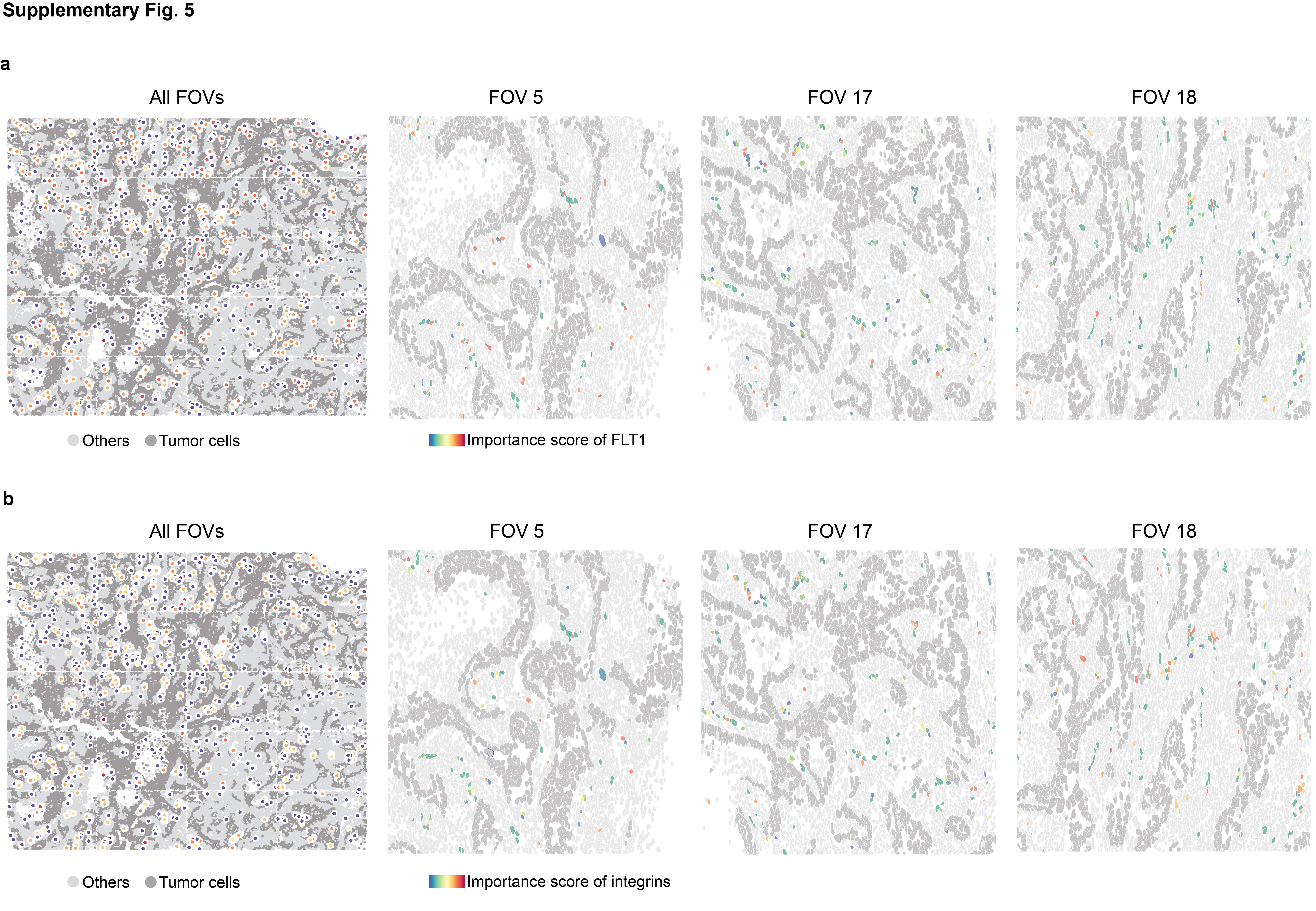
With the interpretable features and their importance scores, we delve deeper into the analysis of cell–cell interactions (CCIs) involving known ligand–receptor (L-R) pairs between adjacent cells (Materials and Methods). The CCI analysis reveals significant L-R pairs along with their associated cell types, providing valuable biological insights (Fig. 5a). Figure 5b illustrates the quantification of L-R interactions within CCIs across different cell types, highlighting a strong interaction between endothelial cells and tumor cells, particularly driven by ECM-related interactions. Through interrogation of the ECM-related interactions, notable receptors such as FLT1 and integrins (ITGA2, ITGA3, ITGA6, ITGAV) in endothelial cells were identified. Specifically, in FOV 14, spatial visualization of FLT1 and integrins’ importance scores in endothelial cells is shown (Fig. 5c), with further visualization across the whole tissue slice and individual FOVs provided in Supplementary Fig. 5. These observations underscore the diverse importance of FLT1 or integrins among endothelial cells, suggesting their distinct roles in ECM-related interactions. Subsequently, we conduct a comparative analysis between endothelial cells with higher and lower importance scores through differential expressed gene analysis, revealing overexpressed genes and enriched pathways in endothelial cells with high FLT1 importance scores (Fig. 5d), as well as the enriched pathways in those with high integrins importance scores (Fig. 5e). The top enriched ECM-related pathways emphasize that endothelial cells with high importance scores play a pivotal role in their interaction with tumor cells, indicating a heterogeneous underlying mechanism within this cell type in the complex cellular communications.
Figure 5.

Downstream analysis using the identified important cells and genes. (a) Circular plot of cell–cell interactions. (b) Heatmap of cell–cell interactions with the number of involved L-R interactions from both raw data and importance scores. (c) Spatial visualization of blood vessel endothelial cells in FOV 14. The zoom-in panels show the importance scores of FLT1 and integrins. (d) Gene enrichment analysis of endothelial cells with high importance scores of FLT1. (e) Gene enrichment analysis of endothelial cells with high importance scores of ECM receptors ITGA2, ITGA3, ITGA6, and ITGAV.
Discussion
Recent advances in spatial transcriptomics technologies have enabled subcellular RNA profiling in tissue slice [1, 31]. Platforms like NanoString CosMx SMI [5] and Vizgen MERSCOPE [4] have enabled the spatial profiling of thousands of RNA targets. However, such spatial imaging data faces challenges of missing values and data noise, which can negatively affect downstream analysis such as spatial domain detection [31, 32]. Several deep learning models have been proposed to improve noisy transcriptomics data and perform data analysis [7], but most of them are black-box approaches that lack transparency and interpretability [21, 33]. To address this challenge, we have proposed xSiGra, which not only accurately identifies spatial cell types and enhances gene expression profiles, but also offers quantitative insights about which cells and genes are important for the identification of spatial cell types, thus making it an interpretable model.
Morphology features have been shown to be linked with gene expression data and combining information from both can lead to better cell prediction performance [34, 35]. xSiGra is engineered to use the potential of multimodal data such as multichannel cell images and their microenvironment. It includes two major modules, i.e. SiGra+ and graph Grad-CAM. The SiGra+ module is evolved from our recently published SiGra model with an improvement by using a VGG28 feature extractor to better leverage the histochemistry images, which are as part of the spatial imaging data, and by introducing a KL divergence loss item to better balance the contributions from transcriptomics and imaging. xSiGra learns the latent space representation, which is used to identify the spatial cell types by Leiden clustering [36]. To make the model explainable, we use a variant of gradient-weighted class activation map (Grad-CAM) [30, 37]. To integrate Grad-CAM with SiGra+, we introduce two linear layers with intermediate nonlinear transformation. We train these layers to predict the cell type membership probability through minimizing the cross-entropy loss, using the latent representation as input and vendor-provided cell types as ground truth. With the new layers replacing Leiden clustering, xSiGra is able to provide the cell and gene importance scores for the spatial cell types. The derived quantitative measures of gene and cell importance for each cell type facilitates downstream analysis like cell–cell interactions and gene enrichment analysis. Importantly, xSiGra is one of the first attempts to quantitatively measure the gene and cell importance to spatial cell types, making it a transparent and trustworthy solution for users.
xSiGra’s strength lies in its comprehensive integration of multimodal data including gene expression, spatial information, and histology features leading to better performance than existing methods. While most of the existing methods utilize black-box models providing little insights into their decision-making processes, xSiGra’s unique strength lies in providing interpretable features that aid researchers in gaining deeper biological insights and understanding the underlying mechanisms driving model’s predictions. In addition to the advantages, xSiGra holds considerable promise for continuous improvement in the future. It can be adapted and expanded in several ways. Due to its hybrid architecture, it can easily incorporate emerging modalities in omics data including novel image modalities. It can be further improved by integrating with 3D images and spatial information to use richer information for data analysis. xSiGra relies on pretrained VGG models with ImageNet weights for feature extraction. While effective, these models may not be optimized for histology images, potentially limiting performance. Future improvements could involve using models pretrained specifically on histology data to enhance feature extraction accuracy. While the current focus of xSiGra is on providing explanations for spatial imaging data, it can be easily extended to provide interpretability for other spatial omics data such as spatial proteomics data. The adaptability of xSiGra for ongoing evolution in spatial technologies is anticipated to empower its utility in the field.
Biologically, xSiGra offers a robust tool for comprehensive insights into tissue architecture and cellular interactions. This is particularly valuable in understanding the spatial heterogeneity of tissues, which can reveal crucial information about disease mechanisms, developmental processes, and cellular microenvironments. The interpretability provided by xSiGra further enhances the biological relevance of the findings. The potential real-world implications of xSiGra are profound. In clinical settings, the model can aid in the diagnosis and prognosis of diseases by providing detailed spatial maps of gene expression and histological features. For instance, xSiGra could help identify tumor subregions with distinct molecular characteristics, leading to more targeted and effective treatment strategies. Additionally, xSiGra can be used to evaluate the spatial effects of drug candidates on tissues, enhancing the precision and efficacy of therapeutic interventions. Overall, xSiGra serves as a pivotal tool in advancing both basic research and clinical applications.
Materials and Methods
Data processing
Single-cell spatial transcriptomics datasets including 8 lung cancer tissue slices of NanoString CosMx SMI, 12 brain tissue slices of 10x Visium DLPFC, human breast cancer, mouse brain anterior, and mouse brain coronal tissue slices are used. Gene expressions are first normalized by multiplying by 10 000 and then log-transformed. (1) For the NanoString datasets, each whole tissue sample includes a certain number of FOVs. For each cell within a FOV, a 120-pixel  120-pixel image is cropped with the cell at the center. For NanoString SMI data, a spatial graph is constructed using spatial locations of cells. Cells at a distance <80 pixels (14.4 μm) are considered as neighbors in the graph. Consider the graph as
120-pixel image is cropped with the cell at the center. For NanoString SMI data, a spatial graph is constructed using spatial locations of cells. Cells at a distance <80 pixels (14.4 μm) are considered as neighbors in the graph. Consider the graph as 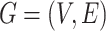 where each
where each 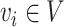 is a cell node (
is a cell node (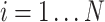 and
and 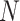 is the total number of cells) and each
is the total number of cells) and each 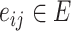 is the distance between cell
is the distance between cell  and
and 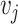 . Each node has gene expression
. Each node has gene expression 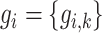 (
(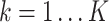 and
and 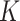 are total genes) and image features
are total genes) and image features 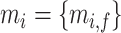 where
where 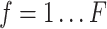 . (2) In 10x Visium DLPFC data, for each spot, 50-pixel
. (2) In 10x Visium DLPFC data, for each spot, 50-pixel 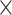 50-pixel image is cropped with the spot as the center. To construct the spatial graph for 10x Visium data, spots at a distance <150 pixels (116 μm) are considered as neighbors in the graph. (3) In 10x Visium human breast cancer data, for each spot, 132-pixel
50-pixel image is cropped with the spot as the center. To construct the spatial graph for 10x Visium data, spots at a distance <150 pixels (116 μm) are considered as neighbors in the graph. (3) In 10x Visium human breast cancer data, for each spot, 132-pixel 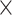 132-pixel image is cropped with spot as the center. For the spatial graph, spots at a distance <400 pixels (116 μm) are considered as neighbors. (4) For mouse brain anterior and coronal tissue sections, for each spot, 31-pixel
132-pixel image is cropped with spot as the center. For the spatial graph, spots at a distance <400 pixels (116 μm) are considered as neighbors. (4) For mouse brain anterior and coronal tissue sections, for each spot, 31-pixel 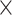 31-pixel image is cropped with spot as the center and spots at a distance <188 pixels (116 μm) are considered as neighbors. Similar to NanoString datasets, for 10x Visium datasets the graph
31-pixel image is cropped with spot as the center and spots at a distance <188 pixels (116 μm) are considered as neighbors. Similar to NanoString datasets, for 10x Visium datasets the graph 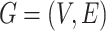 has node
has node 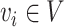 representing a cell (
representing a cell (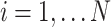 and
and 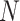 is the total number of cells) and
is the total number of cells) and 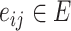 representing the edge between the neighboring cell
representing the edge between the neighboring cell 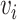 and
and 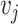 . Each node has gene expression
. Each node has gene expression 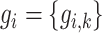 (
(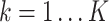 and
and 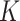 are total genes) and image features
are total genes) and image features 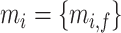 where
where 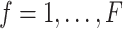 .
.
The xSiGra model
The xSiGra model consists of the (1) SiGra+ to reconstruct gene expressions and (2) graph gradient-weighted class activation mapping (graph Grad-CAM) model to identify interpretable features.
(1) SiGra+
SiGra+ is a hybrid graph encoder-decoder framework, consisting of three modules: (i) the image module, (ii) the gene module, and (iii) the hybrid module. Specifically, (i) in the image module, for each node 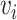 in the graph, features from the images
in the graph, features from the images 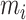 are first extracted using a pretrained VGG-16 network. The extracted image features are then passed through graph transformer layers [38, 39] to obtain the latent space
are first extracted using a pretrained VGG-16 network. The extracted image features are then passed through graph transformer layers [38, 39] to obtain the latent space 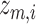 . This latent space representation further goes through additional graph transformer layers to reconstruct the gene expressions of node
. This latent space representation further goes through additional graph transformer layers to reconstruct the gene expressions of node 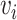 . Here,
. Here, 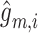 represents the reconstructed gene expression from
represents the reconstructed gene expression from 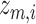 . (ii) In the gene module, for each node
. (ii) In the gene module, for each node 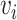 in the graph, gene expression features are passed through graph transformer layers to obtain the latent space
in the graph, gene expression features are passed through graph transformer layers to obtain the latent space 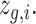 This latent space representation goes through graph transformer layers
This latent space representation goes through graph transformer layers 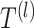 (
(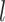 denotes the
denotes the 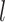 -th layer) to reconstruct the gene expression counts for the node
-th layer) to reconstruct the gene expression counts for the node 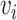 . Here,
. Here, 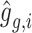 represents the reconstructed gene expression from
represents the reconstructed gene expression from 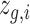 . (iii) In the hybrid module, the two latent space features
. (iii) In the hybrid module, the two latent space features 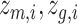 are concatenated and provided as input to graph transformer layer, which projects it to new latent space
are concatenated and provided as input to graph transformer layer, which projects it to new latent space 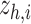 . The hybrid embedding
. The hybrid embedding 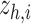 is further passed through graph transformer layers to reconstruct the gene expressions of the node
is further passed through graph transformer layers to reconstruct the gene expressions of the node 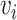 . Here,
. Here, 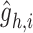 represents the reconstructed gene expression from hybrid embeddings.
represents the reconstructed gene expression from hybrid embeddings.
With the input graph 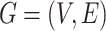 , the multimodal features (image and gene expression) of neighboring nodes also contribute to the reconstructed gene expressions of node
, the multimodal features (image and gene expression) of neighboring nodes also contribute to the reconstructed gene expressions of node 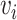 .
.
The hybrid graph encoder-decoder is trained to learn gene expressions with the loss function (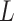 ) as
) as
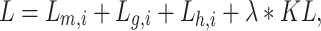 |
where 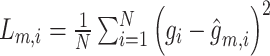 ,
, 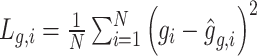 ,
, 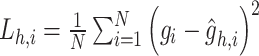 . Meanwhile,
. Meanwhile,
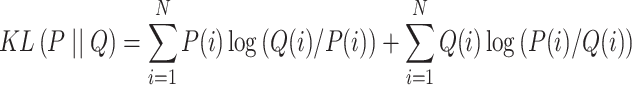 |
where 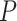 and
and 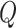 are reconstructed gene expressions pairs
are reconstructed gene expressions pairs 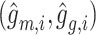 ,
, 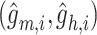 , and
, and 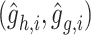 . Here,
. Here, 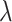 is chosen as 0.001 using hyperparameter tuning.
is chosen as 0.001 using hyperparameter tuning. 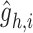 is used as the final reconstructed gene expression and
is used as the final reconstructed gene expression and 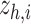 is used for clustering to identify spatial cell types using the Leiden algorithm [36].
is used for clustering to identify spatial cell types using the Leiden algorithm [36].
(2) Graph Grad-CAM model
xSiGra uses a novel post hoc graph Grad-CAM model to reveal the importance of genes and cells for biological functions of interest, such as the identified spatial cell types. Since the spatial cell types are identified through unsupervised clustering of the latent space 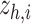 , it is challenging to directly interpret the results. Herein, we first train an auxiliary AI classifier component to map spatial cell types to the corresponding ground truth, then use graph Grad-CAM to explore the importance of genes and cells for the predicted probabilities of spatial cell types.
, it is challenging to directly interpret the results. Herein, we first train an auxiliary AI classifier component to map spatial cell types to the corresponding ground truth, then use graph Grad-CAM to explore the importance of genes and cells for the predicted probabilities of spatial cell types.
For the auxiliary classifier component, we choose a dense multilayer perceptron as the classifier, with intermediate nonlinear ReLU [40] activations as the hidden layers and a log softmax activation layer to predict probabilities of spatial cell types. The log softmax activation is used as it shows better stability and faster gradient optimization over softmax activation [41]. Briefly,
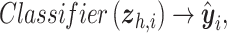 |
where, for cell 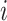 ,
, 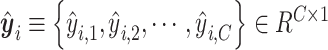 is a vector of the predicted probabilities of cell
is a vector of the predicted probabilities of cell 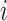 belonging to each of the
belonging to each of the 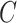 spatial cell types. For the NanoString CosMx data, the vendor-provided cell type annotations
spatial cell types. For the NanoString CosMx data, the vendor-provided cell type annotations 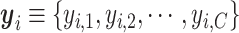 are used as ground truth for training the classifier, where
are used as ground truth for training the classifier, where  is the cell type label and for each cell there is only one non-zero label. The cross-entropy loss used for training is
is the cell type label and for each cell there is only one non-zero label. The cross-entropy loss used for training is
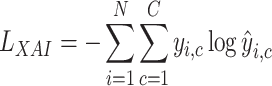 |
This classifier maps each cell from the latent space 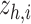 to its corresponding cell type and thus allows interpreting the gene and cell importance.
to its corresponding cell type and thus allows interpreting the gene and cell importance.
Then, we developed a novel graph Grad-CAM algorithm to explore the contributions of each gene in each cell to the prediction of each cell type with backpropagation. Consider the spatial graph with 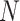 cells (nodes) and
cells (nodes) and 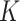 genes, the
genes, the 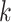 -th feature at
-th feature at 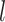 -th layer in the SiGra+ module for cell
-th layer in the SiGra+ module for cell  is denoted as
is denoted as 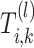 . For spatial cell type
. For spatial cell type 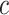 , its specific weights for gene
, its specific weights for gene 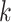 (or latent feature
(or latent feature 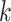 for
for 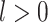 ) of cell
) of cell 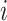 at the
at the 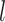 -th layer
-th layer 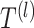 of the SiGra+ gene module is given by
of the SiGra+ gene module is given by
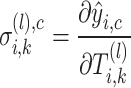 |
The feature importance at the 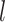 -th layer is determined through backward propagation:
-th layer is determined through backward propagation:
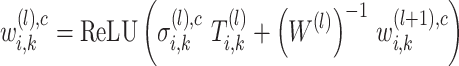 |
where 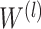 is the learned parameter of
is the learned parameter of 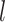 -th transformer layer
-th transformer layer 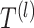 . The importance score of gene
. The importance score of gene 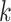 in cell
in cell 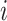 for spatial cell type
for spatial cell type 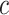 is determined as
is determined as
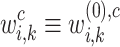 |
Similarly, the importance score of cell 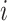 for spatial cell type
for spatial cell type 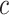 is determined as
is determined as
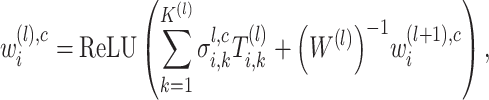 |
where 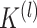 is the number of latent features at the
is the number of latent features at the 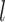 -th layer, and
-th layer, and 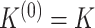 . For simplicity, in xSiGra, we only use the input layer, i.e.
. For simplicity, in xSiGra, we only use the input layer, i.e. 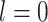 , to compute gene and cell importance. Specifically, the importance of gene
, to compute gene and cell importance. Specifically, the importance of gene 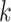 in cell
in cell 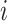 is computed as
is computed as
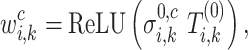 |
and the importance of cell 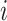 for determining cell type
for determining cell type 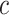 is
is
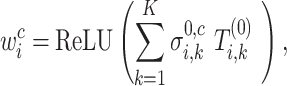 |
where 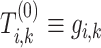 is gene expression count at layer
is gene expression count at layer 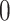 for cell
for cell 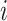 and gene
and gene 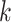 . Thus, the importance of genes and cells for each spatial cell type is determined. In this work, through fine-tuning, we determine the hyperparameters as two layers for the auxiliary classifier with the dimensions of each layer as 1024 and 8.
. Thus, the importance of genes and cells for each spatial cell type is determined. In this work, through fine-tuning, we determine the hyperparameters as two layers for the auxiliary classifier with the dimensions of each layer as 1024 and 8.
Performance benchmarking
(1) For benchmarking on the accuracy of identified spatial cell types or domains with ground truth, we compare our method with nine existing methods, including SiGra [9], SpaGCN [10], stLearn [11], BayesSpace [12], STAGATE [13], conST [14], ConGI [16], Seurat [17], and Scanpy [18]. The performance of different methods is evaluated using the ARI score and NMI. That is, suppose 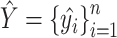 represent the spatial cell types or domains, and
represent the spatial cell types or domains, and 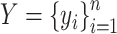 represent the ground truth, i.e.
represent the ground truth, i.e. 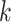 cell types or spatial domains from
cell types or spatial domains from 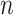 cells or spots, ARI is calculated as
cells or spots, ARI is calculated as
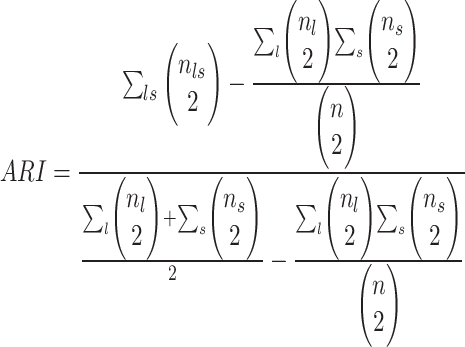 |
where 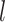 and
and 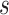 denote the
denote the 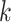 cell types or domains,
cell types or domains, 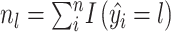 ,
, 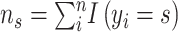 ,
, 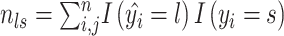 , and
, and 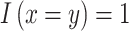 when
when 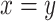 , else
, else 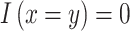 .
.
NMI is calculated as
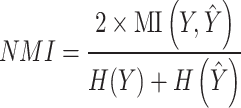 |
where 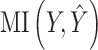 denotes the mutual information between
denotes the mutual information between 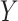 and
and 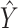 and
and 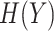 ,
, 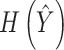 denotes their entropies.
denotes their entropies.
(2) For benchmarking on the accuracy of identified spatial domains without ground truth annotations, we compare our method with nine existing methods, including SiGra [9], SpaGCN [10], stLearn [11], BayesSpace [12], STAGATE [13], conST [14], ConGI [16], Seurat [17], and Scanpy [18]. The performance of different methods is evaluated with the clustering metrics such as SC, CH, and BD, which are used for benchmarking performance on datasets when ground truth information is not available.
SC measures clustering performance using pair-wise distance within and between clusters and a higher value of the score denotes well-defined clusters. SC is calculated as
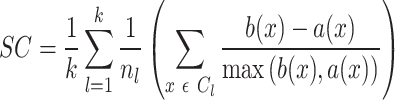 |
where 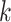 is the number of clusters,
is the number of clusters, 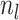 denotes number of samples in cluster
denotes number of samples in cluster 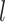 ,
, 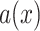 denotes average distance between sample
denotes average distance between sample 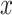 and all samples from same cluster,
and all samples from same cluster, 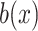 denotes average distance between sample
denotes average distance between sample 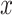 and all samples from nearest cluster, and
and all samples from nearest cluster, and 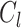 denotes cluster
denotes cluster 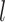 .
.
CH measures the closeness within clusters and a higher value of CH index refers to better grouped clusters. CH is calculated as follows:
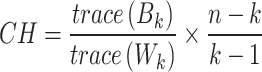 |
where 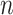 is number of samples in data,
is number of samples in data, 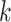 is the number of clusters,
is the number of clusters, 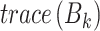 denotes between-cluster dispersion matrix, and
denotes between-cluster dispersion matrix, and 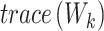 denotes within-cluster dispersion matrix.
denotes within-cluster dispersion matrix.
BD computes the similarities between clusters and a lower value of this index denotes better separation between different clusters. BD is calculated as
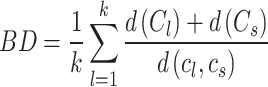 |
where 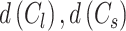 denote the average distance between each cell in cluster
denote the average distance between each cell in cluster 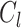 and its centroid
and its centroid 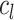 and the average distance between each cell in cluster
and the average distance between each cell in cluster 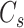 and its centroid
and its centroid 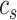 , respectively, where
, respectively, where 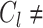
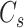 .
. 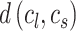 denote the distance between cluster centroids
denote the distance between cluster centroids 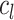 and
and 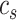 .
.
(3) To evaluate the explainable power of xSiGra, we have compared it with the existing explainable methods, namely, Saliency [23], InputXGradient [25], GuidedBackprop [24], and Deconvolution [26]. The model explainability is measured with two metrics, i.e. fidelity score and contrastivity score. Specifically, (1) fidelity score is computed as the differences between the model’s C-index score (area under the receiver operating characteristic curve) when all genes are used and when the top 30 important genes are masked. The fidelity score is computed separately for different spatial cell types and the median value of fidelity scores is used for comparison. A higher fidelity score indicates better explainable capability. (2) Contrastivity score is computed as 1 − Jaccard similarity for each pair of different spatial cell types. Specifically, if 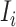 represents the list of top 30 important genes identified for cell type
represents the list of top 30 important genes identified for cell type 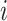 ,
, 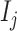 is the list of top 30 important genes identified for cell type
is the list of top 30 important genes identified for cell type 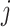 , then the contrastivity score for cell types
, then the contrastivity score for cell types 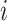 and
and 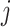 is computed as
is computed as 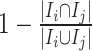 . The median value of contrastivity scores across all pairs of spatial cell types is used for performance evaluation.
. The median value of contrastivity scores across all pairs of spatial cell types is used for performance evaluation.
Enrichment analysis
Gene sets of the Reactome and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway database are downloaded from the MSigDB Collections [42, 43]. Functional enrichment based on the above respective databases is assessed by hypergeometric test, which is used to identify an a priori–defined gene set that shows statistically significant enrichment. The test is performed by the Scanpy package. We further correct the P-values by Benjamini–Hochberg and those with <.05 are considered as statistically significant.
Cell–cell interaction analysis
To conduct the cell–cell interaction analysis, we first built a spatial graph using cell location. Cells at a distance <80 pixels (14.4 μm) are considered as neighbors in the graph. Then, we use a known set of ligand–receptor pairs [44] to compute interaction scores for all cells in the spatial graph. For each neighbor cell pair 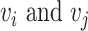 , we have
, we have 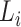 and
and 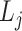 as the ligand-related expression or importance score in cell
as the ligand-related expression or importance score in cell 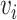 and cell
and cell 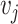 , while
, while 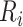 and
and 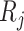 as the receptor-related expression or importance score in cell
as the receptor-related expression or importance score in cell 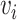 and cell
and cell 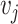 , respectively. The interaction scores are computed as score 1 =
, respectively. The interaction scores are computed as score 1 = 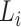 ×
× 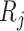 and score 2 =
and score 2 = 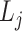 ×
× 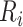 . Next, the interaction scores for each unique ligand gene–cell type and receptor gene–cell type neighbor pairs are aggregated using average. To select the statistically significant interactions, we compute z-scores and choose the interactions having False Discovery Rate (FDR) adjusted P-value <.05. The significant interactions identified using the analysis are further used to gain biological insights.
. Next, the interaction scores for each unique ligand gene–cell type and receptor gene–cell type neighbor pairs are aggregated using average. To select the statistically significant interactions, we compute z-scores and choose the interactions having False Discovery Rate (FDR) adjusted P-value <.05. The significant interactions identified using the analysis are further used to gain biological insights.
Key Points
xSiGra leverages histology images, gene expression data, and spatial cell locations to identify spatial cell types along with their interpretable features.
xSiGra incorporates explainable artificial intelligence to gain insights about the interpretable cells and genes. xSiGra is provided as an open-source tool for spatial imaging data processing.
The application of xSiGra on lung cancer tissue reveals that the activity of each cell type is not only influenced by itself but also by its neighbors. Furthermore, it highlights the pivotal role of an endothelial cell subset in ECM-related interactions with tumor cells.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Contributor Information
Aishwarya Budhkar, Luddy School of Informatics, Computing, and Engineering, Indiana University Bloomington, 107 S Indiana Ave, Bloomington, IN 47405, United States.
Ziyang Tang, Department of Computer and Information Technology, Purdue University, 610 Purdue Mall, West Lafayette, IN 47907, United States.
Xiang Liu, Department of Biostatistics and Health Data Science, Indiana University School of Medicine, 340 W 10th St, Indianapolis, IN 46202, United States.
Xuhong Zhang, Luddy School of Informatics, Computing, and Engineering, Indiana University Bloomington, 107 S Indiana Ave, Bloomington, IN 47405, United States.
Jing Su, Department of Biostatistics and Health Data Science, Indiana University School of Medicine, 340 W 10th St, Indianapolis, IN 46202, United States; Gerontology and Geriatric Medicine, Wake Forest School of Medicine, 475 Vine St, Winston-Salem, NC 27101, United States.
Qianqian Song, Department of Health Outcomes and Biomedical Informatics, College of Medicine, University of Florida, Gainesville, FL 32611, United States.
Funding
Q.S. is supported by the National Institute of General Medical Sciences of the National Institutes of Health (R35GM151089). J.S. was supported partially by the National Library of Medicine of the National Institutes of Health (R01LM013771). J.S. was also supported by the Indiana University Precision Health Initiative and the Indiana University Melvin and Bren Simon Comprehensive Cancer Center Support Grant from the National Cancer Institute (P30CA 082709).
Data availability
In this study, we have used the NanoString CosMx SMI non-small-cell lung cancer (NSCLC) formalin-fixed paraffin-embedded (FFPE) Dataset, 10x Visium datasets from human dorsolateral prefrontal cortex (DLPFC) [45], 10x Visium data from human breast cancer tissue, and mouse brain coronal and anterior tissue sections. NanoString dataset contains eight different samples from five NSCLC tissues, Lung 5-rep1, Lung 5-rep2, Lung 5-rep3, Lung6, Lung 9-rep1, Lung 9-rep2, Lung-12, and Lung-13. For each dataset, gene expression data and multichannel images are provided with five channels: MembraneStrain, PanCK, CD45, CD3, and DAPI. For each staining, a rich gray-scale image composed of a combination of multiple FOVs is also provided. Along with that, metadata about cells such as identified cell coordinates, cell area, width, and height is provided. Lung-13 sample consists of 20 field of views (FOVs) and 77 643 cells. The cells were grouped into eight cell types: tumor, fibroblasts, lymphocyte, mast, neutrophil, and endothelial and epithelial cells. 10x Visium DLPFC dataset consists of 12 samples, 151676, 151675, 151674, 151673, 151672, 151671, 151670, 151669, 151510, 151509, 151508, and 151507, with up to 6 cortical layers and white matter manual annotation. The hematoxylin and eosin (H&E) images of the tissue sections along with the transcriptomics data are provided. Mouse brain anterior and coronal sections and human breast cancer 10x Visium datasets consist of one sample each with transcriptomics data and H&E images of the tissue sections.
Conflict of interest: None declared.
Code availability
The xSiGra model is provided as an open-source python package in GitHub (https://github.com/QSong-github/xSiGra), with detailed manual and tutorials.
References
- 1. Rao A, Barkley D, França GS. et al. Exploring tissue architecture using spatial transcriptomics. Nature 2021;596:211–20. 10.1038/s41586-021-03634-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Armingol E, Officer A, Harismendy O. et al. Deciphering cell–cell interactions and communication from gene expression. Nat Rev Genet 2021;22:71–88. 10.1038/s41576-020-00292-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Berglund E, Maaskola J, Schultz N. et al. Spatial maps of prostate cancer transcriptomes reveal an unexplored landscape of heterogeneity. Nat Commun 2018;9:2419. 10.1038/s41467-018-04724-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. He S, Bhatt R, Brown C. et al. High-plex imaging of RNA and proteins at subcellular resolution in fixed tissue by spatial molecular imaging. Nat Biotechnol 2022;40:1794–806. 10.1038/s41587-022-01483-z. [DOI] [PubMed] [Google Scholar]
- 5. Emanuel G, He J. Using MERSCOPE to generate a cell atlas of the mouse brain that includes lowly expressed genes. Microscopy Today 2021;29:16–9. 10.1017/S1551929521001346. [DOI] [Google Scholar]
- 6. Chen KH, Boettiger AN, Moffitt JR. et al. RNA imaging. Spatially resolved, highly multiplexed RNA profiling in single cells. Science 2015;348:aaa6090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Li Y, Stanojevic S, Garmire LX. Emerging artificial intelligence applications in spatial transcriptomics analysis. Comput Struct Biotechnol J 2022;20:2895–908. 10.1016/j.csbj.2022.05.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Li J, Chen S, Pan X. et al. Cell clustering for spatial transcriptomics data with graph neural networks. Nat Comput Sci 2022;2:399–408. 10.1038/s43588-022-00266-5. [DOI] [PubMed] [Google Scholar]
- 9. Tang Z, Li Z, Hou T. et al. SiGra: single-cell spatial elucidation through an image-augmented graph transformer. Nat Commun 2023;14:5618. 10.1038/s41467-023-41437-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hu J, Li X, Coleman K. et al. SpaGCN: integrating gene expression, spatial location and histology to identify spatial domains and spatially variable genes by graph convolutional network. Nat Methods 2021;18:1342–51. 10.1038/s41592-021-01255-8. [DOI] [PubMed] [Google Scholar]
- 11. Pham D, Tan X, Xu J. et al. stLearn: integrating spatial location, tissue morphology and gene expression to find cell types, cell-cell interactions and spatial trajectories within undissociated tissues. Cold Spring Harbor: Cold Spring Harbor Laboratory, 2020, 1–18. [Google Scholar]
- 12. Zhao E, Stone MR, Ren X. et al. Spatial transcriptomics at subspot resolution with BayesSpace. Nat Biotechnol 2021;39:1375–84. 10.1038/s41587-021-00935-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Dong K, Zhang S. Deciphering spatial domains from spatially resolved transcriptomics with an adaptive graph attention auto-encoder. Nat Commun 2022;13:1739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zong Y, Yu T, Wang X. et al. conST: an interpretable multi-modal contrastive learning framework for spatial transcriptomics. BioRxiv. 2022 Jan 17:2022-01. [Google Scholar]
- 15. He K, Chen X, Xie S. et al. Masked autoencoders are scalable vision learners. In Proceedings of the IEEE/CVF Conference on Computer Vision and Pattern Recognition, 2022, pp. 16000–9. [Google Scholar]
- 16. Zeng Y, Yin R, Luo M. et al. Identifying spatial domain by adapting transcriptomics with histology through contrastive learning. Brief Bioinform 2023;24:bbad048. [DOI] [PubMed] [Google Scholar]
- 17. Butler A, Hoffman P, Smibert P. et al. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat Biotechnol 2018;36:411–20. 10.1038/nbt.4096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wolf FA, Angerer P, Theis FJ. SCANPY: large-scale single-cell gene expression data analysis. Genome Biol 2018;19:15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. De Meo P, Ferrara E, Fiumara G. et al. Generalized louvain method for community detection in large networks. In 2011 11th International Conference on Intelligent Systems Design and Applications. IEEE, 2011 Nov 22, pp. 88–93. [Google Scholar]
- 20. Zhou Z, Hu M, Salcedo M. et al. Xai meets biology: A comprehensive review of explainable ai in bioinformatics applications. arXiv preprint arXiv:2312.06082. 2023 Dec 11. [Google Scholar]
- 21. Karim R, Islam T, Shajalal M. et al. Explainable AI for bioinformatics: methods, tools, and applications. Brief Bioinform 2022;24:bbad236. [DOI] [PubMed] [Google Scholar]
- 22. Tjoa E, Guan C. A survey on explainable artificial intelligence (XAI): toward medical XAI. IEEE Trans Neural Netw Learn Syst 2021;32:4793–813. 10.1109/TNNLS.2020.3027314. [DOI] [PubMed] [Google Scholar]
- 23. Simonyan K, Vedaldi A, Zisserman A. Deep inside convolutional networks: Visualising image classification models and saliency maps. arXiv preprint arXiv:1312.6034. 2013 Dec 20. [Google Scholar]
- 24. Springenberg JT, Dosovitskiy A, Brox T. et al. Striving for simplicity: The all convolutional net. arXiv preprint arXiv:1412.6806. 2014 Dec 21. [Google Scholar]
- 25. Shrikumar A, Greenside P, Shcherbina A. et al. Not just a black box: Learning important features through propagating activation differences. arXiv preprint arXiv:1605.01713. 2016 May 5. [Google Scholar]
- 26. Zeiler MD, Fergus R. in Computer Vision–ECCV 2014: 13th European Conference, Zurich, Switzerland, September 6–12, 2014, Proceedings, Part I 13 818–833. Springer, 2014. [Google Scholar]
- 27. Mahendran A, Vedaldi A. Salient deconvolutional networks. In Computer Vision–ECCV 2016: 14th European Conference. Amsterdam, The Netherlands: Springer International Publishing, October 11-14, 2016, Proceedings, Part VI 14 2016, pp. 120–35. [Google Scholar]
- 28. Liu S, Deng W. Very deep convolutional neural network based image classification using small training sample size. In 2015 3rd IAPR Asian conference on pattern recognition (ACPR). IEEE, 2015 Nov 3, pp. 730–734. [Google Scholar]
- 29. Carreira-Perpinan MA, Hinton G.. On contrastive divergence learning. In International workshop on artificial intelligence and statistics. PMLR, 2005 Jan 6, pp. 33–40. [Google Scholar]
- 30. Pope PE, Kolouri S, Rostami M. et al. Explainability methods for graph convolutional neural networks. In Proceedings of the IEEE/CVF Conference on Computer Vision and Pattern Recognition, 2019, 10772–81. [Google Scholar]
- 31. Moses L, Pachter L. Museum of spatial transcriptomics. Nat Methods 2022;19:534–46. 10.1038/s41592-022-01409-2. [DOI] [PubMed] [Google Scholar]
- 32. Liu J, Tran V, Vemuri VNP. et al. Concordance of MERFISH spatial transcriptomics with bulk and single-cell RNA sequencing. Life Sci Alliance 2023;6:e202201701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Yu Y, Kossinna P, Li Q. et al. Explainable autoencoder-based representation learning for gene expression data. Cold Spring Harbor: Cold Spring Harbor Laboratory, 2021. [Google Scholar]
- 34. Cutiongco MFA, Jensen BS, Reynolds PM. et al. Predicting gene expression using morphological cell responses to nanotopography. Nat Commun 2020;11:1384. 10.1038/s41467-020-15114-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Tan X, Su A, Tran M. et al. SpaCell: integrating tissue morphology and spatial gene expression to predict disease cells. Bioinformatics 2020;36:2293–4. 10.1093/bioinformatics/btz914. [DOI] [PubMed] [Google Scholar]
- 36. Traag VA, Waltman L, Van Eck NJ. From Louvain to Leiden: guaranteeing well-connected communities. Sci Rep 2019;9:5233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Selvaraju RR, Cogswell M, Das A. et al. Grad-cam: Visual explanations from deep networks via gradient-based localization. In Proceedings of the IEEE International Conference on Computer Vision, 2017, 618–26. [Google Scholar]
- 38. Shi Y, Huang Z, Feng S. et al. Masked label prediction: Unified message passing model for semi-supervised classification. arXiv preprint arXiv:2009.03509. 2020 Sep 8. [Google Scholar]
- 39. Vaswani A, Shazeer N, Parmar N. et al. Attention is all you need. Advances in Neural Information Processing Systems, Volume 30. 2017. [Google Scholar]
- 40. Agarap AF. Deep learning using rectified linear units (relu). arXiv preprint arXiv:1803.08375. 2018 Mar 22. [Google Scholar]
- 41. Mahima R, Maheswari M, Roshana S. et al. A comparative analysis of the most commonly used activation functions in deep neural network. In 2023 4th International Conference on Electronics and Sustainable Communication Systems (ICESC). IEEE, 2023 Jul 6, 1334–39. [Google Scholar]
- 42. Subramanian A, Tamayo P, Mootha VK. et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci 2005;102:15545–50. 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Liberzon A, Birger C, Thorvaldsdóttir H. et al. The molecular signatures database Hallmark gene set collection. Cell Syst 2015;1:417–25. 10.1016/j.cels.2015.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Tang Z, Zhang T, Yang B. et al. spaCI: deciphering spatial cellular communications through adaptive graph model. Brief Bioinform 2022;24:bbac563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Maynard KR, Collado-Torres L, Weber LM. et al. Transcriptome-scale spatial gene expression in the human dorsolateral prefrontal cortex. Nat Neurosci 2021;24:425–36. 10.1038/s41593-020-00787-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
In this study, we have used the NanoString CosMx SMI non-small-cell lung cancer (NSCLC) formalin-fixed paraffin-embedded (FFPE) Dataset, 10x Visium datasets from human dorsolateral prefrontal cortex (DLPFC) [45], 10x Visium data from human breast cancer tissue, and mouse brain coronal and anterior tissue sections. NanoString dataset contains eight different samples from five NSCLC tissues, Lung 5-rep1, Lung 5-rep2, Lung 5-rep3, Lung6, Lung 9-rep1, Lung 9-rep2, Lung-12, and Lung-13. For each dataset, gene expression data and multichannel images are provided with five channels: MembraneStrain, PanCK, CD45, CD3, and DAPI. For each staining, a rich gray-scale image composed of a combination of multiple FOVs is also provided. Along with that, metadata about cells such as identified cell coordinates, cell area, width, and height is provided. Lung-13 sample consists of 20 field of views (FOVs) and 77 643 cells. The cells were grouped into eight cell types: tumor, fibroblasts, lymphocyte, mast, neutrophil, and endothelial and epithelial cells. 10x Visium DLPFC dataset consists of 12 samples, 151676, 151675, 151674, 151673, 151672, 151671, 151670, 151669, 151510, 151509, 151508, and 151507, with up to 6 cortical layers and white matter manual annotation. The hematoxylin and eosin (H&E) images of the tissue sections along with the transcriptomics data are provided. Mouse brain anterior and coronal sections and human breast cancer 10x Visium datasets consist of one sample each with transcriptomics data and H&E images of the tissue sections.