


Abstract
To explore the ability of triplex-forming oligodeoxyribonucleotides (TFOs) to inhibit genes responsible for dominant genetic disorders, we used two TFOs to block expression of the human rhodopsin gene, which encodes a G protein-coupled receptor involved in the blinding disorder autosomal dominant retinitis pigmentosa. Psoralen-modified TFOs and UVA irradiation were used to form photoadducts at two target sites in a plasmid expressing a rhodopsin–EGFP fusion, which was then transfected into HT1080 cells. Each TFO reduced rhodopsin–GFP expression by 70–80%, whereas treatment with both reduced expression by 90%. Expression levels of control genes on either the same plasmid or one co-transfected were not affected by the treatment. Mutations at one TFO target eliminated its effect on transcription, without diminishing inhibition by the other TFO. Northern blots indicated that TFO-directed psoralen photoadducts blocked progression of RNA polymerase, resulting in truncated transcripts. Inhibition of gene expression was not relieved over a 72 h period, suggesting that TFO-induced psoralen lesions are not repaired on this time scale. Irradiation of cells after transfection with plasmid and psoralen–TFOs produced photoadducts inside the cells and also inhibited expression of rhodopsin–EGFP. We conclude that directing DNA damage with psoralen–TFOs is an efficient and specific means for blocking transcription from the human rhodopsin gene.
INTRODUCTION
Gene-based therapies for autosomal dominant disorders require development of means to overcome or prevent the action of deleterious gene products. Triplex technology represents a promising tool for manipulating the expression and sequence of genes inside cells (1). Triplex-forming oligodeoxyribonucleotides (TFOs) can bind with high affinity and specificity to purine-rich sequences of double-stranded DNA. TFO binding has been shown to inhibit expression from both plasmid (2–6) and chromosomal (5,7–17) target genes. TFOs can block gene expression by binding to transcription factor binding sites in the promoter (8,9,18,19), by inhibiting transcription initiation (20) or by blocking progression of RNA polymerase (10) through the coding region of a gene. When covalently linked to a DNA damaging agent (e.g. psoralen), TFOs can cause site-specific damage in mammalian cells and induce localized mutations on both plasmid (21–23) and chromosomal (24,25) targets, as well as stimulate homologous recombination (26,27) on plasmid targets. The use of TFOs capable of creating site-specific DNA damage to block transcription, cause mutations or stimulate recombination is a promising approach to inactivate or correct mutant genes.
An important example of a disorder caused by deleterious protein expression is autosomal dominant retinitis pigmentosa (ADRP), a genetically heterogeneous disorder that affects the photoreceptor cells of the retina in ∼1.5 million people world wide (28–30). ADRP is clinically characterized by photoreceptor cell death and progressive retinal degeneration that ultimately leads to blindness (31). Although several different genes can cause ADRP (30), mutations in the rhodopsin gene are responsible for more cases than any other single gene.
Rhodopsin is the G protein-coupled receptor of the visual signal transduction cascade (32) and more than 100 rhodopsin mutations are associated with ADRP (33). Experiments with a rhodopsin heterozygous null mutation in mice (34,35) and the existence of a rhodopsin null mutation in haploid carriers without retinitis pigmentosa (36) indicate that ADRP is not caused by haploinsufficiency, but rather by the harmful expression of mutant rhodopsin. To treat dominantly inherited disorders such as ADRP successfully, it is necessary to develop treatments that inactivate, repair or replace the mutant allele.
We are testing the rhodopsin gene as a target for triplex-based approaches to gene therapy for ADRP. We have previously identified and characterized the triplex binding sites within the human rhodopsin gene (37) and have shown that psoralen-conjugated TFOs can form DNA photoadducts with high efficiency at multiple binding sites in the rhodopsin gene (38). In this paper we describe the use of fluorescence microscopy, fluorescence-activated cell sorting (FACS) analysis, spectrofluorometry, northern blotting and Southern blotting to investigate the ability of psoralen–TFOs to inhibit the expression of a rhodopsin–green fluorescent protein (GFP) fusion construct in human fibroblast cells. Two psoralen–TFOs were targeted to triplex binding sites located ∼2.0 kb apart in the rhodopsin gene. We found that these psoralen–TFOs are each capable of significantly reducing rhodopsin–GFP expression and that using both TFOs together can inhibit transcription to a greater extent than either TFO individually, in a way that is UVA-dependent and target-site specific. We also studied the ability of TFOs to inhibit rhodopsin–GFP expression over time and found that the level of inhibition remains nearly constant over a 72 h period, suggesting that the critical TFO-mediated psoralen photoadducts are not repaired in these human cells.
MATERIALS AND METHODS
Oligodeoxyribonucleotides
Unmodified oligodeoxyribonucleotides were purchased from Integrated DNA Technologies (Coralville, IA) and Oligos Etc. (Wilsonville, OR) and purified as previously described (38,39). Psoralen-modified TFOs were purchased from Oligos Etc. and used without further purification. For all TFOs the psoralen derivative 4′-(hydroxymethyl)-4,5′,8-trimethylpsoralen was joined to the TFO via a six carbon (C-6) linker. The 3′-ends of TFOs were protected by a 3′-propanolamine linker or by 3′-biotin (psoralen–TFO2–biotin). The control TFO is specific for a target site in the Chinese hamster adenine phosphoribosyltransferase gene (35); it has no binding site in any of the plasmids used here.
Plasmids
Recombinant PCR was used to eliminate the stop codon in exon 5 of the human rhodopsin gene and to fuse to it the coding region of Enhanced Green Fluorescent Protein (EGFP-N1; Clontech, Palo Alto, CA) via a linker encoding the peptide APVAT. The sequence of the region created by recombinant PCR was verified by DNA sequencing. The resulting rhodopsin–GFP fusion construct was cloned behind the CMV promoter as a SacI–HindIII fragment into pEGFP-N1 and named pSRG (Fig. 1). To create pSRGY, the CMV promoter and the coding region of yellow fluorescent protein (YFP, from pEYFP-N1; Clontech) were inserted in the same orientation, downstream of the rhodopsin–GFP fusion in pSRG.
Figure 1.

Plasmids and TFOs used in this study. (A) A genomic copy of the rhodopsin gene is fused to GFP cDNA and driven by the CMV promoter. Exons are shown as numbered boxes while introns and plasmid sequences are shown as lines. Exons and introns within the rhodopsin gene are drawn roughly to scale; other regions are not. The locations of the TFO2 and TFO9 binding sites are shown. (B) Psoralen–TFOs are aligned with their cognate binding sites and flanking regions. The mRNA identical strand in each binding site is marked with an asterisk at the 5′-end. (C) The sequence of the mutated TFO2 binding site in pSRG-M2 is shown with psoralen–TFO2. Nucleotide changes are underlined. The mRNA identical strand is marked with an asterisk at the 5′-end.
The binding site for psoralen–TFO2 was mutated using recombinant PCR. The mutated PCR fragment was cut with SacI and XmnI to give a 1.1 kb fragment. This fragment was used to replace the original 1.1 kb SacI–XmnI fragment from pSRG to yield pSRG-M2. The mutation in the TFO2 binding site was confirmed by nucleotide sequencing.
D. M. Spencer kindly provided the plasmid used in the alkaline phosphatase assays, pSRα-SEAP (40).
Targeted photoadduct formation
Plasmid substrates (3 µg pSRG, pSRG-M2 or pSRGY at ∼10–8 M final concentration and 1 µg pSRα-SEAP, as noted) were incubated with a 100-fold molar excess of psoralen–TFOs (10–6 M) at 37°C in triplex binding buffer (10 mM Tris–HCl, pH 7.6, 10 mM MgCl2 and 10% sucrose) for 18 h. Following incubation, the samples were either covered with a glass plate to eliminate short wavelength UV and irradiated with UVA (0.12 J/cm2/min) for 60 min (38) or they were directly transfected into HT1080 cells as described below.
In the latter case, 1 h after transfection the cells were washed extensively with phosphate-buffered saline (PBS) and UVA irradiated under a glass plate for 25 min at 0.2 J/cm2/min. After 36 h culture cells were harvested and analyzed by FACS as described below. To detect photoadducts directly, cells were incubated with pSRG or pSRG-M2 and biotinylated psoralen–TFO2. One hour after transfection the cells were washed extensively with PBS, irradiated as above and the DNA was then harvested and subjected to Southern blotting as described below.
Cell culture and transfections
The human fibrosarcoma cell line HT1080 was used for all experiments. Cells were grown in 6-well plates in MCDB302 medium (Sigma-Aldrich, St Louis, MO). HT1080 cells were transfected with 3 µg (0.15 nM final concentration) TFO-treated rhodopsin–GFP expression vector (pSRG, pSRG-M2 or pSRGY) and 1 µg (0.14 nM final concentration) pSRα-SEAP, when noted, using Fugene-6 transfection reagent (Roche, Indianapolis, IN) as recommended by the manufacturer. Final total TFO (reacted and unreacted) concentration was 15 nM. Fluorescent cells were visualized on an Olympus IX70 microscope (Olympus, Melville, NY) equipped with a Spot Camera (Diagnostic Instrument, Sterling Heights, MI) and images prepared using Adobe PhotoShop v.4.0.1.
FACS analysis
HT1080 cells were harvested in PBS and analyzed with a Coulter EPICS XL-MCL (Beckman-Coulter, Miami, FL) FACS using 488 nm excitation and standard EGFP optics. Fluorescence signals were recorded from two independent samples of 104 live cells for each condition. Mean GFP fluorescence intensities of samples were normalized to and plotted against the control (no TFO or UV treatment).
Alkaline phosphatase assay
After the indicated times and following heat treatment to inactivate endogenous activity as described (41), cellular supernatants were assayed for secreted alkaline phosphatase (SEAP) activity using the fluorogenic substrate 4-methylumbelliferyl phosphate (Sigma) and a Fluorescan II microplate fluorescence reader. SEAP activity was normalized to the untreated control.
Fluorometer assay
Plasmid substrate (3 µg pSRG, pSRGY or pEYFP-N1) was incubated with 1 µM psoralen–TFO at 37°C overnight, UVA irradiated and subsequently transfected into HT1080 cells. Eighteen hours after transfection, cells were washed and harvested by trypsinization. For each treatment, 2 × 105 cells in 1 ml PBS were analyzed in an ISS PC1 photon counting spectrofluorometer (ISS, Champaign, IL). Cells were excited at 460 nm and the emission spectrum was recorded from 490 to 550 nm. Fluorescence curves of pSRGY represent the sum of fluorescence from GFP and YFP. To compare GFP expression relative to YFP, curves were first normalized to their values at 530 nm (emission maximum of YFP) and then the background fluorescence at 505 nm (emission maximum of GFP) of pEYFP-N1 (YFP, alone) was subtracted. ‘Normalized green fluorescence’ represents the remaining fluorescence at 505 nm normalized to the emission value for the untreated pSRGY plasmid on a scale of 0 (pEYFP value) to 100 (untreated pSRGY value).
Northern blot analysis
Plasmid substrate (5 µg pSRG or pSRG-M2) was incubated with 5 µM psoralen–TFO at 37°C overnight and UVA irradiated. Cells were transfected and total RNA was isolated using a High Pure RNA Isolation kit (Roche, Indianopolis, IN) 24 h after transfection. Approximately 20 µg total RNA from each sample were separated by formaldehyde–agarose gel electrophoresis and blotted onto nylon using a NorthernMax-Plus Kit (Ambion, Austin, TX). The blot was probed with a 550 bp fragment corresponding to the first exon of rhodopsin (42) and the GAPDH probe supplied with the NorthernMax-Plus Kit. Final washing stringency was 0.1× SSC, 0.1% SDS at 62 (rhodopsin) or 52°C (GAPDH).
Southern blot analysis
Plasmid DNA from cells treated with biotinylated psoralen–TFO2 and post-transfection UVA irradiation was isolated using a QIAprep Spin Miniprep Kit (Qiagen, Valencia, CA) and digested with SmaI. The restriction fragments were resolved on a 1% agarose gel and transferred to nylon membranes by capillary transfer. The membranes were blocked and probed with alkaline phosphatase-conjugated streptavidin using an Ambion Brightstar Biodetect kit according to the manufacturer’s instructions and the resulting chemiluminescence was recorded on X-ray film. A standard Southern, using a radiolabeled probe hybridizing to the 2.2 kb fragment containing the TFO2 binding site, served as a control for loading and transfer efficiency.
RESULTS
EGFP fluorescence as a reporter for transcription from the rhodopsin gene
Using an expression construct that contains all introns and exons of the human rhodopsin gene as well as cDNA encoding EGFP, we have been able to use fluorescence as a sensitive and quantitative monitor of the effects on transcription of intron-directed TFOs. Simultaneous expression assays for control genes on the same plasmid or on a co-transfected plasmid, as well as use of target site mutants and control TFOs, have allowed us to verify the specificity of rhodopsin gene suppression by psoralen–TFOs.
Psoralen–TFOs form photoadducts with high efficiency
The psoralen–TFOs used in our experiments (Fig. 1A) have previously been characterized as binding with high affinity (37) to their target sequences (Fig. 1A) and forming photoadducts with high efficiency at synthetic target sites (38). In accordance, using alkaline and neutral agarose gel electrophoresis we found that psoralen–TFO2 generated 88% photoadducts and 70% photocrosslinks, while psoralen–TFO9 generated 70% photoadducts and 55% photocrosslinks (data not shown) when incubated with a 3.2 kb pSRG plasmid fragment containing both target sites. The two TFOs together generated 85% photocrosslinks (data not shown).
Inhibition of rhodopsin gene expression by TFOs
In the absence of TFO treatment, pSRG was efficiently transfected and exhibited robust expression as observed by fluorescence microscopy (Fig. 2A). UVA treatment alone (Fig. 2B), treatment with psoralen–TFO2 or psoralen–TFO9 in the absence of UVA (Fig. 2C and E) and UVA treatment in the presence of a control psoralen–TFO (Fig. 2G) had no detectable effect on expression. In contrast, UVA irradiation combined with either psoralen–TFO2 (Fig. 2D) or psoralen–TFO9 (Fig. 2F) dramatically decreased expression levels. Psoralen–TFO2 was somewhat more effective than psoralen–TFO9, but the greatest reduction in expression was observed when both rhodopsin-specific TFOs were used together along with UVA irradiation (Fig. 2H).
Figure 2.

Fluorescence microscopy of HT1080 cells transfected with pSRG treated as indicated: (A) no treatment; (B) UVA alone; (C) psoralen–TFO2, no UVA; (D) psoralen–TFO2 and UVA; (E) psoralen–TFO9, no UVA; (F) psoralen–TFO9 and UVA; (G) control psoralen–TFO and UVA; (H) psoralen–TFO2, psoralen–TFO9 and UVA. Images were acquired 18 h post-transfection using EGFP optics.
Gene suppression as a function of time
The qualitative effects observed by fluorescence microscopy (Fig. 2) were confirmed quantitatively by FACS analysis (Fig. 3A). Results from cells transfected with variously treated plasmids and harvested at 12 h post-transfection intervals revealed that expression of rhodopsin–EGFP peaked between 24 and 48 h and then decreased (Fig. 3A). The untreated plasmid, the UVA-treated plasmid and the plasmid treated with the control psoralen–TFO and UVA all gave comparable levels of expression. As observed at 18 h (Fig. 2), expression levels were dramatically lower in cells transfected with plasmids incubated with psoralen–TFO2, psoralen–TFO9 or both TFOs and UVA irradiated. At each time point expression of rhodopsin–GFP was reduced by ∼80% for psoralen–TFO2, 70% for psoralen–TFO9 and 90% for the combination of both psoralen–TFOs.
Figure 3.

Rhodopsin–GFP fluorescence and alkaline phosphatase activity over time. (A) The mean fluorescence intensity of cells transfected with various plasmid samples was determined by FACS analysis and plotted (arbitrary units) as a function of time after transfection. (B) Alkaline phosphatase activity was determined from an aliquot of the cell medium from each sample. Activity, monitored by the fluorescence intensity of the dephosphorylated substrate and described by an arbitrary number, is plotted as a function of time after transfection. Data points represent the average of two parallel experiments with the range of values indicated by error bars, where they exceed the size of the symbol.
To demonstrate that the psoralen–TFO effects on rhodopsin–GFP expression were not due to differences in transfection efficiency or to general suppression of expression, 1 µg of pSRα-SEAP, which expresses a secreted alkaline phosphatase, was mixed with pSRG prior to any treatment of the samples shown in Figure 3A. Immediately before FACS analysis (Fig. 3A) an aliquot of medium was assayed for alkaline phosphatase activity. Phosphatase activities exhibited little variation among treated samples for each time point (Fig. 3B), demonstrating that the effects of psoralen–TFOs and UVA are specific for the rhodopsin–EGFP construct.
Effect of photoadducts on plasmid stability
In order to determine whether photoadducted TFOs were blocking transcription from an otherwise intact plasmid or, alternatively, simply triggering preferential destruction of the modified plasmid, we assessed the effects of the same set of treatments on plasmid pSRGY. This construct carries genes for the rhodopsin–EGFP fusion and for YFP, each driven by independent CMV promoters. If psoralen–TFO-mediated photoadducts inhibit rhodopsin expression without plasmid destruction, YFP expression should be unaffected; if photoadducts lead to plasmid destruction, both genes should be affected in parallel. Expression levels of rhodopsin–EGFP and YFP were assessed at 18 h after transfection by scanning spectrofluorometry, in order to resolve the closely overlapping emissions of EGFP and YFP (Fig. 4A). Again, control treatments did not significantly affect expression of rhodopsin–EGFP relative to YFP, while treatment with UVA plus psoralen–TFO2 or psoralen–TFO9 gave effective suppression of rhodopsin–EGFP, and combining the two psoralen–TFOs reduced rhodopsin–GFP expression by ∼90%. Interestingly, the absolute levels of YFP expression were somewhat higher in the samples with TFO/UVA suppression of rhodopsin–EGFP expression (data not shown), suggesting some competition between the two at the level of transcription or translation and leading to the unequivocal conclusion that the treatments which block rhodopsin–EGFP expression do not lead to plasmid destruction.
Figure 4.

Expression of pEYFP (yellow) and pSRGY (green and yellow). (A) Emission spectra of cells transfected with pSRGY treated as indicated or with untreated pEYFP, normalized to emission at 530 nm. (B) Normalized green fluorescence at 505 nm, calculated from the emission spectra as described in the text.
Suppression requires a triplex binding site
To test the dependence of TFO2 effects on target site sequence, we mutated the binding site for psoralen–TFO2 in pSRG to create plasmid pSRG-M2 (Fig. 1A and C). Plasmids pSRG and pSRG-M2 were treated in parallel and the results quantified by FACS analysis as in Figure 3 (Fig. 5A and B). The fluorescence of cells transfected with pSRG-M2 was 98% of those cells transfected with pSRG, indicating that the mutated psoralen–TFO2 binding site did not affect transcription or splicing of rhodopsin–GFP. Comparison of Figure 5A and B shows that psoralen–TFO9 had comparable effects on rhodopsin–GFP expression in both plasmids, whereas psoralen–TFO2 had a negligible effect on pSRG-M2, either alone or in combination with psoralen–TFO9. These results confirm that the target site for TFO2 is essential for gene suppression and support the conclusion that the effects of psoralen–TFO2 and psoralen–TFO9 are mediated through triplex formation.
Figure 5.

Loss of TFO2 effects by mutation of its target site. (A) Fluorescence was analyzed as in Figure 3 after pSRG was treated with psoralen–TFOs and UVA irradiation and transfected into HT1080 cells. (B) As (A) except that plasmid pSRG-M2, containing the TFO2 target site mutation, was used. The mean fluorescence intensity of pSRG-M2 was within 5% of the mean fluorescence intensity of pSRG. (C) Northern blot analysis of rhodopsin–GFP mRNA levels was performed on ∼20 µg total RNA using a rhodopsin-specific probe. Predicted sizes of various transcripts are indicated by numbers. The arrows represent the migration positions of the 2.37 (top arrow) and 1.35 kb (bottom arrow) molecular weight markers and the 18S rRNA (1.9 kb, middle arrow). Exons are represented by boxes and introns represented by lines. The shaded box represents the coding region of EGFP.
Photoadduct formation results in truncated transcripts
To determine whether the reduction in rhodopsin–GFP fluorescence was due to a reduction in rhodopsin mRNA levels, northern blot analysis was performed on RNA isolated from cells transfected with psoralen–TFO-treated and untreated plasmids. The primary transcript of rhodopsin–GFP is ∼2.6 kb when fully spliced (Fig. 5C, lane 2). Plasmid pSRG treated with psoralen–TFO2 produced truncated transcripts, which migrated at a size consistent with blockage of RNA polymerase at the TFO2 binding site. No truncated transcripts were formed with psoralen–TFO2-treated pSRG-M2, consistent with the absence of a TFO2 binding site. Plasmids treated with psoralen–TFO9 produced two major transcripts (Fig. 5C, lanes 4, 8 and 9). The size of the shorter transcript was consistent with a truncated transcript whose first intron had been removed by splicing. The longer transcript was the appropriate size for an unspliced transcript truncated at the TFO9 binding site. These results clearly indicate that TFO-directed psoralen photoadducts block progression of RNA polymerase, which results in truncated rhodopsin transcripts and a decrease in overall rhodopsin expression.
Post-transfection photoadduct formation
After determining that crosslinking of psoralen–TFOs to their targets results in a highly efficient and specific block of transcription under conditions where we could ensure efficient formation of crosslinks, we investigated whether triplex-dependent crosslinking could occur in living cells. Cells were transfected with plasmid pSRG and psoralen–TFO2, psoralen–TFO9 or control TFO, washed to remove any DNA left outside the cells and then subjected to UVA irradiation. As shown in Figure 6A, transcription of rhodopsin–GFP was reduced under conditions for specific crosslinking; 30% inhibition was observed with psoralen–TFO9, 45% with psoralen–TFO2 and nearly 50% with both specific TFOs, but not under control conditions (psoralen–control TFO or no irradiation). These results imply that triplex-induced crosslinking can occur inside the cells. Further verification that stable photoadducts could be formed in the cells was obtained with biotinylated psoralen–TFO2. Again, the cells were irradiated after transfection and, after removal of external plasmid and TFO, the plasmid was isolated and restriction fragments were analyzed by Southern blotting and detected with streptavidin–alkaline phosphatase (Fig. 6B). A strong signal was observed at the correct position for the restriction fragment containing the triplex target site, but not at other positions and not when the plasmid used was pSRG-M2, which lacks the target site. Thus, two different experimental approaches demonstrate that triplex-directed crosslinks can be formed at the target site in the rhodopsin gene inside living cells.
Figure 6.
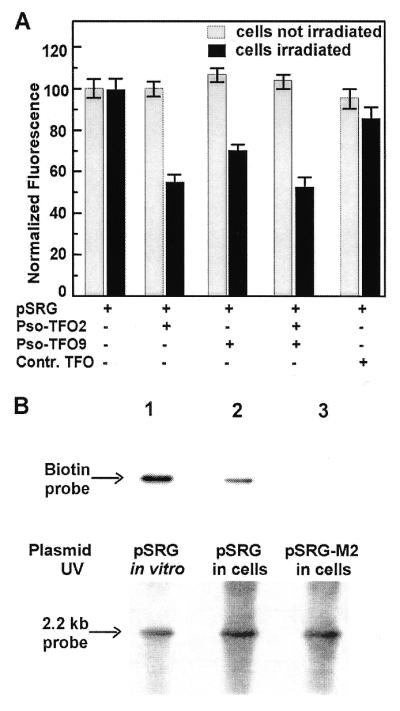
Formation of TFO-targeted crosslinks within living cells. (A) FACS results from control cells and cells treated with pSRG and psoralen–TFO2 and/or psoralen–TFO9 prior to UVA irradiation of the washed cells. Grey bars represent samples that did not receive any UVA irradiation after transfection, whereas black bars show samples that were irradiated 1 h after transfection. Three independent experiments were carried out and the results plotted are the means, normalized to the control (transfection with plasmid but no TFO), for each condition, with error bars indicating the standard error. (B) Southern blot of plasmid DNA extracted from cells after treatment with pSRG or pSRG-M2 and psoralen–TFO2–biotin prior to UVA irradiation of the washed cells (lanes 2 and 3) and from cells transfected with the TFO already crosslinked to pSRG (lane 1). (Top) The result of probing with alkaline phosphatase-conjugated streptavidin followed by recording of chemiluminescence for detection of the biotin tag. (Bottom) The result of probing with a 32P-labeled probe specific for the plasmid fragment containing the triplex site, as a control for equal loading and transfer, and to verify that the photoadduct corresponds to the intended fragment. Arrows show the migration position corresponding to 2.2 kb.
DISCUSSION
Potent and persistent suppression of rhodopsin transcription by triplex photoadducts
To be useful, any scheme for inactivating the rhodopsin gene must be able to block the very robust expression from the rhodopsin promoter in rod cells and do so without causing transcriptional inhibition or modification of other genes and maintain the transcription block for a long time, if not permanently. Our results indicate that UVA-induced psoralen–TFO photoadducts are an extremely potent means for blocking transcription directed by a very strong promoter. The 90% suppression seen when both TFOs were used, along with our finding that crosslinks constitute the majority of the photoadducts, in accord with previous studies (38), suggests that a psoralen–TFO crosslink represents an insurmountable block to transcription. The presence of apparently stable truncated transcripts in transfected cells suggests that RNA polymerase was stalled, as occurs in vitro at psoralen–TFO crosslinks but not at psoralen–TFO monoadducts (43). Moreover, our results demonstrating undiminished YFP expression from the same plasmid make it clear that the effects are due to site-specific transcription inhibition rather than to plasmid degradation. Our results also demonstrate the specificity inherent in the triplex-based approach. Genes lacking the rhodopsin target sequences were unaffected and a psoralen–TFO not directed against any rhodopsin sequences was without effect. Also of interest for further applications is the persistent nature of the transcription block (Fig. 2). There does not seem to be an efficient mechanism for repair of this type of lesion in HT1080 cells, as reported previously for HeLa cells (6,44). The ability of terminally differentiated neurons such as photoreceptors to repair this kind of DNA damage is untested, but these results are encouraging.
Triplex-induced photoadduct formation in cells
Although several reports have documented transcription inhibition by TFOs in cells (9,10,12,16,45,46), there has been some concern that the intracellular environment may have adverse effects on triplex stability (e.g. high potassium ion concentrations or competition with transcription complexes or histones). Our results (Fig. 6A) clearly show that our TFOs can induce specific inhibition of rhodopsin–EGFP synthesis even when the crosslinking occurs inside the cell. Although, not surprisingly, the effect is not as great as observed when the crosslinking is carried out in vitro in advance of transfection, the inhibition is robust enough to suggest that with further optimization it should be possible to achieve triplex-directed transcription blocks with high efficiency. Detection of biotin-tagged psoralen–TFO2 covalently attached to the plasmid fragment bearing its triplex target site (Fig. 6B) provides direct evidence of crosslinking in cells after transfection.
Implications for triplex targeting strategies
While our focus has been on approaches to inactivating the rhodopsin gene, our results suggest useful directions for triplex-based gene inactivation in general. Previous studies (3,7–11,47) have demonstrated gene suppression of other genes by unmodified TFOs, however, our results clearly indicate that UVA irradiation is required for psoralen activation and inactivation of rhodopsin–EGFP expression. As discussed above, in the absence of effective repair mechanisms, psoralen–TFO crosslinks impose a permanent block on transcription.
Because the reagents used were designed to optimize crosslinking, rather than monoadduct formation (38), these experiments do not address the ability of monoadducts to exert similar effects on transcription; such an effect would be of interest because it would expand the range of useful target sites. Previous reports (21,43,48–50) suggest that in addition to their differences in ability to block transcription, repair mechanisms for psoralen monoadducts and psoralen crosslinks differ. Our preliminary results suggest that cell lines differ considerably in their ability to overcome transcription block by psoralen photoadducts (Z.Intody, unpublished observations).
Another important conclusion from our results is the enhanced efficacy achieved by simultaneous targeting of two different triplex sites within a single gene. All our experiments clearly indicate that treating a plasmid with two psoralen–TFOs can reduce expression to a lower level than either psoralen–TFO separately (Figs 1 and 5). The simplest explanation is that each psoralen–TFO upon irradiation produces stable crosslinks with a finite probability so that treatment with more than one simply increases the probability that a given copy of a gene will have at least one TFO crosslink. Previously we found that not only does the rhodopsin gene contain multiple triplex sites, but that eukaryotic genes in general are likely to have several such sites for directing TFO-mediated damage (37).
Quantifying triplex-mediated gene suppression by FACS
Our results also demonstrate the utility of FACS analysis as a method of quantifying triplex effects on gene expression. By targeting psoralen–TFOs to the gene for a rhodopsin–GFP fusion protein we were able to visually evaluate changes in expression by fluorescence microscopy and to evaluate these changes quantitatively using FACS analysis. FACS analysis is advantageous because of rapid and reproducible data collection. We have quantified fluorescence in 70 cell samples in ∼60 min. The technique allows us to examine not only the aggregate expression level, but also the distribution of expression levels and take note of cells expressing the gene at abnormally high or abnormally low levels.
Prospects for therapeutic applications
We have developed a technology that seems to be capable of imposing a potent and specific transcription block on the rhodopsin gene and possibly any gene of interest. Our current results are limited to a gene located on an extrachromosomal plasmid, with initial triplex formation under optimal conditions. However, they define a well-characterized, facile system for optimizing various aspects of TFO treatment, including TFO modifications, delivery methods, triplex stability in cells and time and duration of irradiation. Although several studies suggest that chromosomal sites can indeed be be targeted by TFOs (5,7,9–16,51,52), it is clear that the efficiency of TFO-mediated inhibition of chromosomal transcription will need to be improved if it is to be therapeutically useful. Even if we achieve our eventual goal of selective inactivation of the rhodopsin gene in rod photoreceptors, considerably more work will be necessary to determine whether this powerful technology can be developed into safe and effective therapies.
Acknowledgments
ACKNOWLEDGEMENTS
We wish to thank Dr David Spencer for the gift of plasmid pSRα-SEAP and Dr Jeremy Nathans for the human rhodopsin cDNA. We also thank Dr Karen Vasquez and Dr David Spencer for helpful comments on the manuscript. We also wish to thank Fung Chan for help in the design and construction of plasmid pSRG. We would also like to thank the Flow Cytometry Core Facility at Baylor College of Medicine for their valuable assistance. This work was funded by a grant from the National Institutes of Health to J.H.W. (EY11731). B.D.P. was supported by a training grant from the National Institutes of Health (T32 EY07102).
REFERENCES
- 1.Vasquez K.M. and Wilson,J.H. (1998) Trends Biochem. Sci., 23, 4–9. [DOI] [PubMed] [Google Scholar]
- 2.Ing N.H., Beekman,J.M., Kessler,D.J., Murphy,M., Jayaraman,K., Zendegui,J.G., Hogan,M.E., O’Malley,B.W. and Tsai,M.J. (1993) Nucleic Acids Res., 21, 2789–2796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kovacs A., Kandala,J.C., Weber,K.T. and Guntaka,R.V. (1996) J. Biol. Chem., 271, 1805–1812. [DOI] [PubMed] [Google Scholar]
- 4.Svinarchuk F., Debin,A., Bertrand,J.R. and Malvy,C. (1996) Nucleic Acids Res., 24, 295–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kochetkova M. and Shannon,M.F. (1996) J. Biol. Chem., 271, 14438–14444. [DOI] [PubMed] [Google Scholar]
- 6.Musso M., Wang,J.C. and Van Dyke,M.W. (1996) Nucleic Acids Res., 24, 4924–4932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Orson F.M., Thomas,D.W., McShan,W.M., Kessler,D.J. and Hogan,M.E. (1991) Nucleic Acids Res., 19, 3435–3441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Postel E.H., Flint,S.J., Kessler,D.J. and Hogan,M.E. (1991) Proc. Natl Acad. Sci. USA, 88, 8227–8231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McShan W.M., Rossen,R.D., Laughter,A.H., Trial,J., Kessler,D.J., Zendegui,J.G., Hogan,M.E. and Orson,F.M. (1992) J. Biol. Chem., 267, 5712–5721. [PubMed] [Google Scholar]
- 10.Scaggiante B., Morassutti,C., Tolazzi,G., Michelutti,A., Baccarani,M. and Quadrifoglio,F. (1994) FEBS Lett., 352, 380–384. [DOI] [PubMed] [Google Scholar]
- 11.Okada T., Yamaguchi,K. and Yamashita,Y. (1994) Growth Factors, 11, 259–270. [DOI] [PubMed] [Google Scholar]
- 12.Thomas T.J., Faaland,C.A., Gallo,M.A. and Thomas,T. (1995) Nucleic Acids Res., 23, 3594–3599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tu G.C., Cao,Q.N. and Israel,Y. (1995) J. Biol. Chem., 270, 28402–28407. [DOI] [PubMed] [Google Scholar]
- 14.Porumb H., Gousset,H., Letellier,R., Salle,V., Briane,D., Vassy,J., Amor-Gueret,M., Israel,L. and Taillandier,E. (1996) Cancer Res., 56, 515–522. [PubMed] [Google Scholar]
- 15.Rininsland F., Johnson,T.R., Chernicky,C.L., Schulze,E., Burfeind,P. and Ilan,J. (1997) Proc. Natl Acad. Sci. USA, 94, 5854–5859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Aggarwal B.B., Schwarz,L., Hogan,M.E. and Rando,R.F. (1996) Cancer Res., 56, 5156–5164. [PubMed] [Google Scholar]
- 17.Oh D.H. and Hanawalt,P.C. (1999) Nucleic Acids Res., 27, 4734–4742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Grigoriev M., Praseuth,D., Guieysse,A.L., Robin,P., Thuong,N.T., Helene,C. and Harel-Bellan,A. (1993) Proc. Natl Acad. Sci. USA, 90, 3501–3505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Giovannangeli C., Diviacco,S., Labrousse,V., Gryaznov,S., Charneau,P. and Helene,C. (1997) Proc. Natl Acad. Sci. USA, 94, 79–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Duval-Valentin G., Thuong,N.T. and Helene,C. (1992) Proc. Natl Acad. Sci. USA, 89, 504–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sandor Z. and Bredberg,A. (1994) Nucleic Acids Res., 22, 2051–2056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang G., Levy,D.D., Seidman,M.M. and Glazer,P.M. (1995) Mol. Cell. Biol., 15, 1759–1768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang G., Seidman,M.M. and Glazer,P.M. (1996) Science, 271, 802–805. [DOI] [PubMed] [Google Scholar]
- 24.Majumdar A., Khorlin,A., Dyatkina,N., Lin,F.L., Powell,J., Liu,J., Fei,Z., Khripine,Y., Watanabe,K.A., George,J., Glazer,P.M. and Seidman,M.M. (1998) Nature Genet., 20, 212–214. [DOI] [PubMed] [Google Scholar]
- 25.Vasquez K.M., Wang,G., Havre,P.A. and Glazer,P.M. (1999) Nucleic Acids Res., 27, 1176–1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Faruqi A.F., Seidman,M.M., Segal,D.J., Carroll,D. and Glazer,P.M. (1996) Mol. Cell. Biol., 16, 6820–6828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sandor Z. and Bredberg,A. (1995) Biochim. Biophys. Acta, 1263, 235–240. [DOI] [PubMed] [Google Scholar]
- 28.Shastry B.S. (1994) Am. J. Med. Genet., 52, 467–474. [DOI] [PubMed] [Google Scholar]
- 29.Berson E.L. (1996) Proc. Natl Acad. Sci. USA, 93, 4526–4528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dryja T.P. and Li,T. (1995) Hum. Mol. Genet., 4, 1739–1743. [DOI] [PubMed] [Google Scholar]
- 31.Heckenlively J.R. (1988) Retinitis Pigmentosa. J.B. Lippincott, Philadelphia, PA.
- 32.Nathans J. (1992) Biochemistry, 31, 4923–4931. [DOI] [PubMed] [Google Scholar]
- 33.al-Jandal N., Farrar,G.J., Kiang,A.S., Humphries,M.M., Bannon,N., Findlay,J.B., Humphries,P. and Kenna,P.F. (1999) Hum. Mutat., 13, 75–81. [DOI] [PubMed] [Google Scholar]
- 34.Humphries M.M., Rancourt,D., Farrar,G.J., Kenna,P., Hazel,M., Bush,R.A., Sieving,P.A., Sheils,D.M., McNally,N., Creighton,P., Erven,A., Boros,A., Gulya,K., Capecchi,M.R. and Humphries,P. (1997) Nature Genet., 15, 216–219. [DOI] [PubMed] [Google Scholar]
- 35.Lem J., Krasnoperova,N.V., Calvert,P.D., Kosaras,B., Cameron,D.A., Nicolo,M., Makino,C.L. and Sidman,R.L. (1999) Proc. Natl Acad. Sci. USA, 96, 736–741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rosenfeld P.J., Cowley,G.S., McGee,T.L., Sandberg,M.A., Berson,E.L. and Dryja,T.P. (1992) Nature Genet., 1, 209–213. [DOI] [PubMed] [Google Scholar]
- 37.Perkins B.D., Wilson,J.H., Wensel,T.G. and Vasquez,K.M. (1998) Biochemistry, 37, 11315–11322. [DOI] [PubMed] [Google Scholar]
- 38.Perkins B.D., Wensel,T.G., Vasquez,K.M. and Wilson,J.H. (1999) Biochemistry, 38, 12850–12859. [DOI] [PubMed] [Google Scholar]
- 39.Vasquez K.M., Wensel,T.G., Hogan,M.E. and Wilson,J.H. (1995) Biochemistry, 34, 7243–7251. [DOI] [PubMed] [Google Scholar]
- 40.MacCorkle R.A., Freeman,K.W. and Spencer,D.M. (1998) Proc. Natl Acad. Sci. USA, 95, 3655–3660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Spencer D.M., Wandless,T.J., Schreiber,S.L. and Crabtree,G.R. (1993) Science, 262, 1019–1024. [DOI] [PubMed] [Google Scholar]
- 42.Sung C.H., Schneider,B.G., Agarwal,N., Papermaster,D.S. and Nathans,J. (1991) Proc. Natl Acad. Sci. USA, 88, 8840–8844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang Z. and Rana,T.M. (1997) Proc. Natl Acad. Sci. USA, 94, 6688–6693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Guieysse A., Praseuth,D., Giovannangeli,C., Asseline,U. and Helene,C. (2000) Nucleic Acids Res., 296, 373–383. [DOI] [PubMed] [Google Scholar]
- 45.Roy C. (1994) Eur. J. Biochem., 220, 493–503. [DOI] [PubMed] [Google Scholar]
- 46.Kochetkova M., Iversen,P.O., Lopez,A.F. and Shannon,M.F. (1997) J. Clin. Invest., 99, 3000–3008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Degols G., Clarenc,J.P., Lebleu,B. and Leonetti,J.P. (1994) J. Biol. Chem., 269, 16933–16937. [PubMed] [Google Scholar]
- 48.Calsou P., Sage,E., Moustacchi,E. and Salles,B. (1996) Biochemistry, 35, 14963–14969. [DOI] [PubMed] [Google Scholar]
- 49.Segal D.J., Faruqi,A.F., Glazer,P.M. and Carroll,D. (1997) Mol. Cell. Biol., 17, 6645–6652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wang G. and Glazer,P.M. (1995) J. Biol. Chem., 270, 22595–22601. [DOI] [PubMed] [Google Scholar]
- 51.Postel E.H. (1992) Ann. N. Y. Acad. Sci., 660, 57–63. [DOI] [PubMed] [Google Scholar]
- 52.Belousov E.S., Afonina,I.A., Kutyavin,I.V., Gall,A.A., Reed,M.W., Gamper,H.B., Wydro,R.M. and Meyer,R.B. (1998) Nucleic Acids Res., 26, 1324–1328. [DOI] [PMC free article] [PubMed] [Google Scholar]