


Abstract
Two new photoreactive dATP analogs, N6-[4-azidobenzoyl–(2-aminoethyl)]-2′-deoxyadenosine-5′-triphosphate (AB-dATP) and N6-[4-[3-(trifluoromethyl)-diazirin-3-yl]benzoyl-(2-aminoethyl)]-2′-deoxyadenosine-5′-triphosphate (DB-dATP), were synthesized from 2′-deoxyadenosine-5′-monophosphate in a six step procedure. Synthesis starts with aminoethylation of dAMP and continues with rearrangement of N1-(2-aminoethyl)-2′-deoxyadenosine-5′-monophosphate to N6-(2-aminoethyl)-2′-deoxyadenosine-5′-monophosphate (N6-dAMP). Next, N6-dAMP is converted into the triphosphate form by first protecting the N-6 primary amino group before coupling the pyrophosphate. After pyrophosphorylation, the material is deprotected to yield N6-(2-aminoethyl)-2′-deoxyadenosine-5′-triphosphate (N6-dATP). The N-6 amino group is subsequently used to attach either a phenylazide or phenyldiazirine and the photoreactive nucleotide is then enzymatically incorporated into DNA. N6-dATP and its photoreactive analogs AB-dATP and DB-dATP were successfully incorporated into DNA using the exonuclease-free Klenow fragment of DNA polymerase I in a primer extension reaction. UV irradiation of the primer extension reaction with AB-dATP or DB-dATP showed specific photocrosslinking of DNA polymerase I to DNA.
INTRODUCTION
DNA photoaffinity labeling has been successfully used in probing the structural and functional features of the eukaryotic and prokaryotic transcription complexes, DNA replication complexes, chromatin structure and chromatin remodeling complexes (1–12). One approach to modifying DNA in a site-specific manner has been the enzymatic incorporation of a photoreactive nucleotide analog using a site-specific oligonucleotide annealed to target DNA (13). This method facilitates the incorporation of a radiolabel immediately adjacent to the photoreactive nucleotide. These nucleotide analogs are designed such that the photoreactive group is in the major groove of DNA and can therefore be used to differentiate protein contacts in the major versus the minor groove of DNA (14). Not only can the protein subunits be identified that interact at specific sites of DNA, but by mapping the site on the protein crosslinked to DNA the region of the protein in close proximity to those specific sites on DNA can be determined for more detailed structural information (14). Varying the length of the tether separating the photoreactive group from the nucleotide base has provided a more 3-dimensional perspective of these complexes and distinguished between proteins directly contacting DNA and proteins that don’t directly contact DNA, but are recruited through protein–protein interactions (3).
In these studies analogs of dTTP and dCTP have been used to study protein–DNA interactions (3,6–8,15,16). The lack of suitable purine photoreactive analogs has restricted the selection of positions in DNA for photocrosslinking. The photoreactive purine analog 8-azido-2′-deoxyadenosine carries a photoreactive group directly on the purine base and can be used for studying very close contacts in the major groove of DNA (17). We have chosen to utilize the N-6 amino group as the position to attach a phenylazido or phenyldiazirine by a short tether to adenine. Previously the N-6 amino group had been successfully used to attach a biotin group to dATP without interfering with its enzymatic incorporation into DNA and is routinely used to make biotinylated DNA (18). Two other positions that could have been utilized for modification of adenine are the C-8 and the C-2 positions. Recently arylazido analogs were coupled to the C-2 carbon of deoxyadenosine (2-[(p-azidophenacyl)thio]-2′-deoxyadenosine-5′-triphosphate) and used for photoinactivation of DNA polymerase I (4). This dATP analog is not suitable for incorporation into DNA because the orientation of the photosensitive group interferes with base pairing. The C-8 modified ATP analogs 8-azido-dATP, 8-azido-ATP and 8-[(4-azidophenacyl)thio]adenosine 5′-triphosphate have been synthesized and incorporated into DNA and RNA (17,19). There is some difficulty in extensively incorporating these C-8 substituted analogs, which might be caused by being primarily in the syn conformation and not the preferred anti conformation (20,21). The incorporation of 8-azido-ATP by Escherichia coli RNA polymerase inhibits further elongation of the RNA transcript (22) and 8-[(4-azidophenacyl)thio]adenosine 5′-triphosphate is not able to fully substitute for ATP, as is evident by not being able to incorporate two of these analogs sequentially in RNA (19).
We successfully synthesized two photoreactive dATP analogs that have a phenylazide or a phenyldiazirine tethered to the C-6 of adenine that can be enzymatically incorporated into DNA. We also demonstrate that these nucleotide analogs can be incorporated into DNA and used for probing protein–DNA interactions.
MATERIALS AND METHODS
Synthesis of N1-(2-aminoethyl)-2′-deoxyadenosine-5′-monophosphate (N1-dAMP) (2)
First it was important to synthesize a dAMP analog with a primary amine that is tethered to the N-6 amino group by a two carbon linker. Synthesis of N1-dAMP (2) and its rearrangement to N6-(2-aminoethyl)-2′-deoxyadenosine-5′-monophosphate (N6-dAMP) (3) were carried out essentially as described (23) with a few modifications as follows. Deoxyadenosine 5′-monophosphate (1.9 g) (Sigma) was dissolved in 34.4 ml of sterile deionized water and the pH was adjusted to 4.0 with 0.1 M HClO4. An azaridine solution containing 1.13 ml of azaridine (ChemService) and 20 ml of sterile deionized water was adjusted to pH 4 before addition to the dAMP solution. The reaction was incubated at 25°C for 65 h and the pH of the reaction mixture maintained at 4.0 with 0.1 M HClO4. The reaction was stopped by addition of 600 ml of ice-cold acetone and the precipitate collected by centrifugation (4°C, 10 min, 4000 g) and washed twice with 100 ml of ice-cold acetone. The dried precipitate was dissolved in 50 ml of 5 mM formic acid and loaded onto a Dowex 1X2-200 column (1.7 × 20 cm). The product was eluted by washing with 5 mM formic acid and was present in the flow-through. The flow-through was passed through the same column for a second time to enhance removal of unreacted dAMP. After a second passage through the column, the product (2) was vacuum concentrated with a yield of 1.032 g (50.7%). Product purity was verified by cellulose thin layer chromatography (TLC) developed with 0.1 M K3PO4, pH 6.8, (NH4)2SO4, n-propanol (50 ml/30.5 g/1 ml) and showed only one spot with an Rf of 0.96.
Rearrangement of N1-dAMP (2)
One gram of N1-dAMP (2) was dissolved in 50 ml of sterile deionized water and the pH was adjusted to 11.0 by addition of 5 M NaOH. The solution was heated at 60°C for 5 h and the reaction stopped by addition of 2 M HClO4 to adjust the pH to 4.0. The rearrangement product was precipitated by the addition of 600 ml of ice-cold acetone. The precipitate was collected by centrifugation (4°C, 10 min, 4000 g) and washed twice with 100 ml of ice-cold acetone and air dried. The product (3) was dissolved in 25 ml of 0.1 M triethylammonium bicarbonate (TEAB) pH 8.0 and purified by DEAE–Sephadex A25 (1.5 × 20 cm) chromatography using a 1 liter linear gradient of 0.1–0.3 M TEAB. Peaks were collected and analyzed by cellulose TLC and UV spectroscopy. N6-dAMP (3)-containing fractions eluted at 0.2 M TEAB. Appropriate fractions were evaporated under vacuum with centrifugation and TEAB was removed by addition and evaporation of 100% ethanol. Yield of N6-dAMP (3) was 252 mg (0.68 mmol, 25.0%). N6-dAMP has a λmax at 266 nm (pH 8.0) and gives one spot on cellulose TLC with a Rf of 0.52 using the same TLC conditions as before. The presence of a primary amino group was verified by positive color development after spraying the spot with ninhydrin (24). 1H NMR (D2O) δ p.p.m.: 8.6 (s, H8), 8.4 (s, H2), 6.5 (t, H1′), 5.2–4.8 (m, H3′, H4′, 2H5′), 3.9–3.3 (m, 2Ha, 2Hb), 2.8–2.6 (m, 2H2′).
Protection of the primary amino group of N6-dAMP (3) by trifluoroacetylation
After obtaining N6-dAMP, the next step was to prepare the triphosphate form of the nucleotide. The phosphorylation of N6-dAMP required prior protection of the primary amino group. N6-dAMP (3) (0.7 mmol) was dissolved in 23.5 ml of ice-cold sterile deionized water (final concentration 30 mM). S-ethyl trifluorothioacetate (20 ml) (Aldrich) was added and the pH adjusted to 10.0 with 5 M LiOH. The reaction was stirred for 1 h at room temperature and the pH was occasionally re-adjusted to 10.0. After 1 h the pH was allowed to go to 8.0 and the water phase was transferred and diluted with 50 ml of 0.1 M TEAB pH 8.0. The solution was loaded onto a DEAE–Sephadex A25 column (1.5 × 20 cm) and eluted with a 1 liter gradient of 0.1–0.5 TEAB. The fractions containing N6-[trifluoroacetyl-(2-aminoethyl)]-2′-deoxyadenosine-5′-monophosphate (4) eluted at 0.15 M TEAB and were evaporated under vacuum centrifugation and TEAB removed by co-evaporation with 100% ethanol. The Rf value for the product on cellulose TLC was 0.20 and UV quantification showed a recovery of 207 mg (0.44 mmol, 63.7%).
Phosphorylation of N6-[trifluoroacetyl-(2-aminoethyl)]-2′-deoxyadenosine-5′-monophosphate (4)
Protected N6-dAMP (4) was rendered anhydrous by co-evaporation with anhydrous pyridine (2 × 10 ml) (Aldrich) and subsequently with anhydrous DMF (2 × 10 ml) (Aldrich). The compound was dissolved in 3 ml of DMF and 320 mg CDI (Aldrich) was added. The reaction mixture was stirred at room temperature in a desiccator for 30 min. Excess CDI was decomposed by addition of 130 µl of anhydrous CH3OH and incubation for an additional 30 min. Anhydrous tributylammonium pyrophosphate (Sigma), 2.3 ml of a 1 M solution in DMF, was added and the reaction was stirred overnight at room temperature under anhydrous conditions. The precipitated imidazolium pyrophosphate was removed by centrifugation and washed twice with 2 ml of DMF. Supernatant and washes were pooled and concentrated by co-evaporation with CH3OH under vacuum centrifugation. The concentrated sample was dissolved in 20 ml of 0.1 M TEAB pH 8.0 and loaded onto a DEAE–Sephadex A25 column (1.5 × 20 cm). The column was washed with 100 ml of 0.1 M TEAB and eluted with a 1 liter gradient of 0.1–1 M TEAB. N6-[trifluoroacetyl-(2-aminoethyl)]-2′-deoxyadenosine-5′-triphosphate (5) eluted at 0.45 M TEAB. The peak fractions were analyzed on PEI–cellulose developed with 1 M LiCl. The Rf value found for N6-[trifluoroacetyl-(2-aminoethyl)]-2′-deoxyadenosine-5′-triphosphate (0.55) was between the Rf values for ATP (0.41) and ADP (0.61). Fractions containing the triphosphate form were collected and evaporated under vacuum centrifugation and TEAB was removed by co-evaporation with 100% ethanol. The estimated recovery of 177 mg (0.281 mmol, 63.8%) of the triphosphate form was determined by spectroscopy based on the extinction coefficient for N6-dAMP.
Deprotection of N6-[trifluoroacetyl-(2-aminoethyl)]-2′-deoxyadenosine-5′-triphosphate (5)
The protected triphosphate (5) (0.281 mmol) was dissolved in 4 ml of sterile deionized water and the pH adjusted to 11.5 by addition of 5 M NaOH. The solution was stirred at room temperature for 2.5 h and the pH was occasionally re-adjusted to 11.5. After incubation, the pH was brought to 8.0 with 1 M HCl and the solution diluted in 40 ml of 0.1 M TEAB pH 8.0 before loading onto a DEAE–Sephadex A25 column (1.5 × 20 cm). The column was washed with 100 ml of 0.1 M TEAB and eluted with a 1 liter gradient of 0.1–0.75 M TEAB pH 8. A single peak eluted at 0.45 M TEAB and was analyzed by cellulose TLC (Rf = 0.67). The presence of the primary amino group was confirmed by positive color development with ninhydrin. Yield of N6-(2-aminoethyl)-2′-deoxyadenosine-5′-triphosphate (N6-dATP) (6) was estimated to be 106 mg (0.195 mmol, 69.4%) by UV spectroscopy.
Incorporation of N6-dATP (6) into double-stranded (ds)DNA by the Klenow fragment of DNA polymerase
The reactions for incorporation of N6-dATP (6) into DNA contained 2 pmol p2B2D single-stranded (ss)DNA (15) and 3 pmol of oligonucleotide d(pGAAGAAAGAGTATACTA) in 18 µl of buffer A. The mixture was heated for 3 min at 90°C and then for 30 min at 37°C, before addition of 3.3 pmol of [α-32P]dCTP (sp. act. 3000 Ci/mmol, final concentration 0.1 µM) and 18 µl buffer A supplemented with 0.2 mg/ml BSA. The mixture was divided into 4 µl aliquots. One microliter of an appropriate nucleotide dilution plus 0.5 U of the Klenow fragment were added to each aliquot. The incorporation mixture was incubated at 37°C for 5 min, 2 µl from each sample removed and combined with 10 µl of 95% formamide + dye and analyzed by 10% urea–PAGE.
Coupling of N6-dATP (6) to photoreactive groups: 4-azidobenzoate and 4-[3-(trifluoromethyl)-diazirin-3-yl]benzoate
Two different photoreactive groups were separately attached to N6-dATP and both procedures were carried out under indirect lighting with a 40 W incandescent lamp. The reaction for coupling with 4-azidobenzoate contained 480 µl of 100 mM 4-azidobenzoate N-hydroxysuccinimide (Molecular Probes) in DMF, 240 µl of 38.9 mM N6-dATP (9.3 µmol) in 100 mM sodium borate pH 8.5 and was incubated at room temperature for 8 h. N6-[4-azidobenzoyl-(2-aminoethyl)]-2′-deoxyadenosine-5′-triphosphate (AB-dATP) (7) was purified on a DEAE–Sephadex A25 column essentially as described previously (13). The reaction mixture was diluted with 3 ml of E-pure sterile water and loaded onto a DEAE–Sephadex A25 column (0.5 × 8 cm). The column was washed with a 100 ml gradient of 0.1–1.5 M TEAB pH 8. The UV absorbance of small portions of each fractions was measured at 266 nm; the AB-dATP (7) eluted at 0.5–0.6 M TEAB. The appropriate fractions were collected and evaporated under vacuum centrifugation as before. Concentrated fractions were analyzed by TLC on PEI–cellulose developed with 1 M LiCl and on cellulose developed with 0.1 M K3PO4 pH 6.8, (NH4)2SO4, n-propanol (50 ml/30.5 g/1 ml). The Rf value for AB-dATP (7) on PEI–cellulose was 0.19 and on cellulose was 0.04. The extinction coefficient for AB-dATP was found to be 22 000 l/mol cm at 266 nm in a solution containing 10 mM Tris–HCl pH 8.0 and 1 mM EDTA. The yield of AB-dATP was 5.2 µmol (56%).
The reaction for coupling of 4-[3-(trifluoromethyl)-diazirin-3-yl]benzoate contained 248 µl of 50 mM 4-[3-(trifluoromethyl)-diazirin-3-yl]benzoate N-hydroxysuccinimide in DMSO, 124 µl of 12.5 mM N6-dATP (1.5 µmol) in 100 mM sodium borate pH 8.5 and was incubated at room temperature for 12 h. The reaction mixture was diluted with 6 ml of E-pure sterile water and loaded onto DEAE–Sephadex A25, purified and analyzed essentially as for AB-dATP. The fractions containing N6-[4-[3-(trifluoromethyl)-diazirin-3-yl] benzoyl-(2-aminoethyl)]-2′-deoxyadenosine-5′-triphosphate (DB-dATP) (8) eluted at 1.2 M TEAB. The Rf value for DB-dATP on PEI–cellulose was 0.12 and on cellulose was 0.07. The extinction coefficient of DB-dATP was found to be 7500 l/mmol cm at 266 nm in a solution containing 10 mM Tris–HCl pH 8.0 and 1 mM EDTA. The yield of DB-dATP was 1.4 µmol (87%).
Incorporation of AB-dATP (7) and DB-dATP (8) into dsDNA by the Klenow fragment of DNA polymerase I and photocrosslinking of the Klenow fragment
The optimal concentration for incorporation was determined by primer extension assay with an immobilized template. Biotinylated ssDNA was immobilized on streptavidin-coated Dynabeads M 280 as described by Sengupta et al. (1). Two picomoles of immobilized ssDNA were washed three times with buffer A (50 mM Tris–HCl pH 8, 50 mM KCl, 7 mM MgCl2, 1 mM β-mercaptoethanol, 0.05% Tween 20). A sample containing 2 pmol of washed immobilized DNA and 8 pmol of complementary oligonucleotide d(pCCATCCAAGTACTAACCAGGCCC) in 20 µl of buffer A supplemented with 0.2 mg/ml BSA was heated for 3 min at 70°C and incubated for 30 min at 37°C with brief vortexing every 2 min. Beads were washed three times with buffer A and resuspended in 25 µl of buffer A containing 7 pmol of [α-32P]dGTP (sp. act. 6000 Ci/mmol, final concentration 0.27 µM). The labeling reaction was initiated by addition of 0.25 U of exonuclease-free Klenow fragment of DNA polymerase I and incubated for 5 min at 37°C with brief vortexing every 1–2 min. One microliter of sample was removed after incubation and added to 9 µl of 95% formamide with 0.1% xylene cyanol and bromophenol blue for analysis on a 10% polyacrylamide gel containing 8.3 M urea. The remaining beads were washed five times with 50 µl of buffer A until all unincorporated radioactivity was removed, resuspended in 48 µl of buffer A and divided into 4 µl aliquots. To each aliquot 6 µl of appropriate nucleotide dilution in buffer A along with 0.25 U of Klenow fragment were added. After 5 min incubation, 3 µl of the sample was combined with 7 µl of 95% formamide with dye for PAGE analysis. The remaining sample was added to 1 µl of chase mix (dATP, dTTP, dGTP and dCTP, 2 mM in buffer A) and incubated for 5 min at 37°C. Four microliters of each sample were combined with 6 µl of 95% formamide + dye and analyzed by 10% urea–PAGE.
Crosslinking experiments were done with 10 µg p2B2D ssDNA (15) combined with 5.6 pmol of oligonucleotide d(pGAAGAAAGAGTATACTA) in 34 µl of buffer A. The mixture was heated for 3 min at 90°C and then for 30 min at 37°C, before combining with 6.3 pmol of [α-32P]dCTP (sp. act. 3000 Ci/mmol, final concentration 0.1 µM) and diluting in 34 µl of buffer A supplemented with 0.2 mg/ml BSA. The mixture was divided into 8 µl aliquots. Two microliters of an appropriate nucleotide dilution plus 0.5 U of the Klenow fragment were added to each aliquot. The incorporation mixture was incubated at 37°C for 5 min, 2 µl from each sample was removed and combined with 10 µl of 95% formamide + dye. The remaining sample was irradiated as described (9). Two microliters of each irradiated sample were combined with 10 µl of 95% formamide + dye and analyzed on a 10% polyacrylamide gel containing 8.3 M urea. Some of the crosslinked sample was digested with DNase I (Gibco BRL) and S1 nuclease (Gibco BRL) for SDS–PAGE. Two microliters of the irradiated reactions with AB-dATP (7) or DB-dATP (8) were diluted with 8 µl of buffer A and crosslinked DNA was digested with 12 U of DNase I for 10 min at room temperature. DNase I was denatured upon addition of 10% SDS (final concentration 0.5%) with heating at 90°C for 3 min. The pH was adjusted to 4.5 with acetic acid and ZnSO4 was added to 1 mM final concentration. Upon digestion with 10 U of S1 nuclease at 37°C for 10 min, the pH of the sample was brought to ∼7 with 0.5 µl of 1 M Tris. Samples were denatured with 5× SDS–PAGE loading buffer (70 mM Tris–HCl pH 6.8, 5% β-mercaptoethanol, 2% SDS; 25) and upon heating for 3 min at 90°C were analyzed by 8% SDS–PAGE.
RESULTS
Two photoreactive deoxyadenosine nucleotides were synthesized by tethering either a phenylazide or a phenyldiazirine to the C-6 carbon of adenine. Previously, selective modification of the C-6 position has been done by preparation of the 6-chloropurine-2′-deoxyriboside (26) followed by nucleophilic displacement of the chloro group. Instead of trying to prepare the nucleotide version of 6-chloropurine, it was easier to use another approach that begins with dAMP. This second approach first modifies adenine by alkylation of the N1 nitrogen of adenosine and subsequent rearrangement to the N6 position to produce N6-(2-aminoethyl)adenosine. The aminoethylation of adenine was originally developed and optimized for modification of NAD+ (27) and AMP (23). The primary amino group of the 2-aminoethyladenosine can be coupled to photoreactive moieties using N-hydroxysuccinimidyl esters of 4-azidobenzoic acid and 4-diazirinylbenzoic acid. We found that this procedure could be utilized with dAMP, although with a lower efficiency than previously reported for AMP and requiring further purification.
The synthesis of AB-dATP (7) and DB-dATP (8) was achieved in six steps (Fig. 1). In the first step dAMP was modified by azaridine on N1 as described by Sicsic et al. (23). N1-dAMP (2) was purified by Dowex anion exchange chromatography. At acidic pH the positive charge of the quaternary amine formed on the purine ring compensates for the negative charge of the phosphate and aminoethylated dAMP does not bind to the anion exchange slurry, whereas dAMP does bind under low salt conditions. The purified product was recovered with an overall 50.7% conversion and yields a single spot on cellulose TLC.
Figure 1.

Synthesis of N6-[4-azidobenzoyl-(2-aminoethyl)]-2′-deoxyadenosine-5′-triphosphate. (1) 2′-deoxyadenosine-5′-monophosphate; (2) N1-(2-aminoethyl)-2′-deoxyadenosine-5′-monophosphate; (3) N6-(2-aminoethyl)-2′-deoxyadenosine-5′-monophosphate; (4) N6-[trifluoroacetyl-(2-aminoethyl)]-2′-deoxyadenosine-5′-monophosphate; (5) N6-[trifluoroacetyl-(2-aminoethyl)]-2′-deoxyadenosine-5′-triphosphate; (6) N6-(2-aminoethyl)-2′-deoxyadenosine-5′-triphosphate; (7) N6-[4-azidobenzoyl-(2-aminoethyl)]-2′-deoxyadenosine-5′-triphosphate; (8) N6-[4-[3-(trifluoromethyl)-diazirin-3-yl]benzoyl-(2-aminoethyl)]-2′-deoxyadenosine-5′-triphosphate.
Conversion of N1-dAMP to N6-dAMP (3) is carried out by the Dimroth rearrangement (28). Contrary to the results of Sicsic et al. with AMP (23), the rearrangement of dAMP gave less of the desired product (25 versus 98%) even after complete disappearance of the starting material (N1-dAMP). A control experiment with N1-aminoethylated AMP showed complete conversion of AMP and lack of co-product. Lower recovery and the presence of an undesired co-product made it necessary to further purify the material by DEAE–Sephadex A25 anion exchange chromatography. Fractions containing N6-modified dAMP eluted close to 0.2 M TEAB. The purified N6-dAMP had a Rf value of 0.67 on cellulose TLC and a λmax of 266 nm, corresponding to parameters published for the AMP derivative. The 1H NMR spectrum is compatible with the described spectrum for aminoethylated AMP with extra peaks for two C2′ protons. The presence of the primary amino group was verified by the ninhydrin test. The rearrangement co-product eluted in an earlier part of the gradient, had a higher Rf value on cellulose TLC (0.75) and a different λmax (262 nm). The 1H NMR spectrum of the co-product showed non-stoichiometric aminoethylation and two extra peaks with smaller shifts of 1.2 and 1.4 p.p.m.
Purified N6-dAMP (3) is converted to the triphosphate form by application of the Hoard–Ott method (29). The primary amino group is first protected by treatment with S-ethyl trifluorothioacetate under alkaline conditions (18). The protected N6-[trifluoroacetyl-(2-aminoethyl)]-2′-deoxyadenosine-5′-monophosphate (4) was purified by DEAE–Sephadex A25 chromatography with a yield of 63% and eluted at 0.15 M TEAB. The protected monophosphate nucleotide was rendered anhydrous and treated with CDI under anhydrous conditions. Excess CDI was decomposed by treatment with anhydrous CH3OH and the intermediate imidazolidate was reacted with inorganic pyrophosphate to form the protected N6-dATP (5). The final product was purified by DEAE–Sephadex A25 chromatography and eluted at 0.55 M TEAB. The product was analyzed by PEI–cellulose TLC developed with 1 M LiCl. The Rf value for the protected triphosphate form was between the Rf values of ADP and ATP.
Deprotection of the amino group was achieved by alkaline treatment with a 69.4% yield. The deprotected N6-dATP (6) was purified by DEAE–Sephadex A25 chromatography and eluted at 0.45 M TEAB and showed a single spot on cellulose TLC developed with 0.1 M K3PO4 pH 6.8, (NH4)2SO4, n-propanol. The presence of the primary amino group was shown by the ninhydrin test. N6-dATP was successfully incorporated into dsDNA by DNA polymerase I (Fig. 2).
Figure 2.
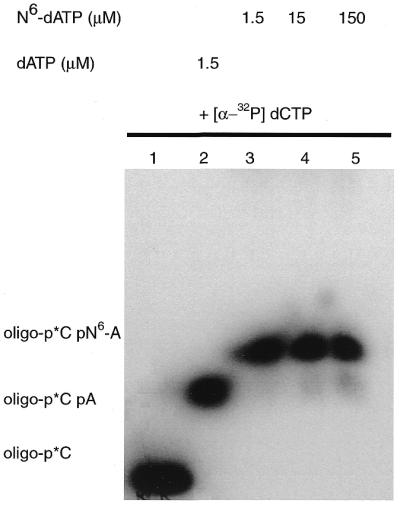
Incorporation of N6-dATP. DNA primer extension reactions were performed as described in Materials and Methods using the oligonucleotide d(pGAAGAAAGAGTATACTA) annealed to M13 ssDNA construct p2B2D. Nucleotides [α-32P]dCMP and N6-dAMP were incorporated in the primer extension reaction and products analyzed on a 10% polyacrylamide gel containing 8.3 M urea. The concentration of N6-dATP in the reaction was varied from 1.5 to 150 µM.
The primary amino group of N6-dATP was coupled to the photoreactive groups 4-azidobenzoate and 4-[3-(trifluoromethyl)-diazirin-3-yl]benzoate. The photoreactive analogs AB-dATP (7) and DB-dATP (8) were purified by anion exchange chromatography with DEAE–Sephadex A25 and analyzed by PEI–cellulose TLC as described. AB-dATP and DB-dATP eluted at 0.5–0.6 and 1.2 M TEAB, respectively. Evidence for the presence of the photosensitive group was seen by the product turning brown upon UV irradiation when spotted on a TLC plate. Synthesis and recovery are summarized in Table 1 and Figure 1.
Table 1. Recovery in individual steps of synthesis, shown as molar percent.
| Recovery (%) | ||
|---|---|---|
| Step | Overall | |
| 2′-deoxyadenosine-5′-monophosphate | 100 | 100 |
| N1-(2-aminoethyl)-2′-deoxyadenosine-5′-monophosphate | 50.9 | 50.9 |
| N6-(2-aminoethyl)-2′-deoxyadenosine-5′-monophosphate | 25.0 | 12.7 |
| N6-[trifluoroacetyl-(2-aminoethyl)]-2′-deoxyadenosine-5′-monophosphate | 63.7 | 8.1 |
| N6-[trifluoroacetyl-(2-aminoethyl)]-2′-deoxyadenosine-5′-triphosphate | 63.8 | 5.2 |
| N6-(2-aminoethyl)-2′-deoxyadenosine-5′-triphosphate | 69.4 | 3.6 |
| N6-[4-azidobenzoyl-(2-aminoethyl)]-2′-deoxyadenosine-5′-triphosphate | 56.2 | 2.0 |
| N6-[4-[3-(trifluoromethyl)-diazirin-3-yl]benzoyl-(2-aminoethyl)]-2′-deoxyadenosine-5′-triphosphate | 86.0 | 3.1 |
The incorporation of N6-dATP, AB-dATP and DB-dATP analogs into dsDNA was achieved by primer extension with the Klenow fragment of DNA polymerase I. The optimal concentration for incorporation of AB-dATP and DB-dATP was determined in an incorporation assay using immobilized ssDNA as template. After labeling of DNA primer with [α-32P]dGTP, unincorporated nucleotides were removed by washing the immobilized DNA and additional synthesis carried out by adding different amounts of the photoreactive analog (Fig. 3A). Both AB-dATP and DB-dATP were efficiently incorporated into dsDNA. A concentration of AB-dATP ≥ 0.03 µM is sufficient for complete incorporation, as is evident from the shift in electrophoretic mobility without any significant amount of longer, read-through products (Fig. 3B, lanes 3, 5, 7, 9, 11 and 13). DB-dATP can also be efficiently incorporated, as is evident from the complete shift of electrophoretic mobility at concentrations ≥0.09 µM (Fig. 3C, lanes 3, 5, 7, 9, 11 and 13). After addition of all four nucleotides all bands (Fig. 3B and C lanes 3′, 5′, 7′, 9′, 11′ and 13′) are chased to full length, demonstrating that incorporation of the modified nucleotide does not interfere with continued chain synthesis.
Figure 3.



Incorporation of AB-dATP and DB-dATP by DNA polymerase I is efficient and does not preclude further primer extension. (A) Scheme of incorporation. The primer extension reaction was performed on immobilized DNA template pXP10/XEH annealed with complementary oligonucleotide d(pCCATCCAAGTACTAACCAGGCCC). In the first step [α-32P]dGMP was incorporated using the exonuclease-free Klenow fragment of DNA polymerase I and the immobilized DNA was extensively washed to remove unincorporated nucleotide. Primer extension was continued with varying amounts of modified dATP and later all four nucleotides were added to chase the product to full length. (B) Samples were loaded onto a 10% polyacrylamide gel containing 8.3 M urea and the dried gel analyzed by autoradiography. Lane 1 is the sample after incorporation of [α-32P]dGTP and lanes 3, 5, 7, 9, 11 and 13 are samples containing the [α-32P]dGMP-labeled primer incubated with the Klenow fragment and varying concentrations of AB-dATP. In lanes 2, 4, 6, 8, 10 and 12 dATP was added in place of AB-dATP. Lanes 2′–13′ are samples from lanes 2–13 in which all four dNTP were added. (C) Lane 1 is the sample after incorporation of [α-32P]dGTP and lanes 3, 5, 7, 9, 11 and 13 are samples with varying concentration of DB-dATP. In lanes 2, 4, 6, 8, 10 and 12 dATP was added in place of DB-dATP and in lanes 2′–13′ the samples with DB-dATP were subsequently chased by addition of all four dNTPs.
The Klenow fragment of DNA polymerase I is stalled on the primed DNA template after incorporation of [α-32P]dCMP and AB-dATP or DB-dATP, because of a lack of the other two nucleotides. The effectiveness of the modified DNA template for protein–DNA photoaffinity labeling was tested by crosslinking the stalled DNA polymerase to modified DNA by UV irradiation. The electrophoretic mobility of the labeled DNA primer is greatly reduced once crosslinked to DNA polymerase and can be detected by loading the sample onto a denaturing polyacrylamide gel (Fig. 4B and D, lanes 5, 7, 9, 11 and 13). Although some of the DNA photoproducts after crosslinking are labeled DNA crosslinked to DNA polymerase, there are also some that react with the solvent and cause the labeled primer to not migrate as a single band. The large shift of labeled DNA is specific for incorporation of AB-dATP or DB-dATP. Irradiated samples containing dATP did not show a significant amount of material with a large shift in electrophoretic mobility or diffuse bands in the lower part of the gel, typical of those when AB-dATP or DB-dATP are incorporated and irradiated (compare lanes 2 and 3 to 4 and 5 in Fig. 4B and D).
Figure 4.





Specific photocrosslinking of DNA polymerase I by DNA templates with AB-dATP or DB-dATP incorporated near the 3′-side of the replicated DNA strand. (A) Scheme of DNA polymerase I photocrosslinking. DNA primer extension reactions were performed as described in Materials and Methods using the oligonucleotide d(pGAAGAAAGAGTATACTA) annealed to M13 ssDNA construct p2B2D. The radiolabeled nucleotide [α-32P]dCTP and varying amounts of modified dATP are incorporated in a one step primer extension reaction. The stalled DNA polymerase complex was crosslinked by UV irradiation and analyzed on a 10% polyacrylamide gel containing 8.3 M urea (B and D) or after extensive digestion of DNA analyzed by 8% SDS–PAGE (C and E). Samples in (B) and (C) contained AB-dATP and those in (D) and (E) contained DB-dATP.
The size of the protein crosslinked to the DNA primer was determined by degrading most of the DNA primer with DNase I and S1 nuclease to leave a small piece of radiolabeled DNA crosslinked to DNA polymerase. The sample was then analyzed by SDS–PAGE, the gel dried and the radiolabeled protein detected by autoradiography or phosphorimaging (see Fig. 4C and E). The observed electrophoretic mobility of the photoaffinity labeled protein corresponds to that of the Klenow fragment (68 kDa). The signal decrease in lanes 4 and 5 (Fig. 4D and E) corresponds to incomplete incorporation of photoreactive dATP at 60 and 20 nM concentrations consistent with this nucleotide being responsible for the crosslinking of DNA polymerase.
The efficiency of crosslinking for both photoreactive analogs was determined by phosphorimager analysis of the denaturing gels. The amount of material migrating near the same position as before irradiation was compared to the amount of the material with a much slower electrophoretic mobility, presumably caused by crosslinking DNA to the Klenow fragment of DNA polymerase. The efficiency of crosslinking ranges from ~15% for AB-dATP (Fig. 4B, lanes 5, 7, 9 and 11) and ~9% for DB-dATP (Fig. 4D, lanes 5, 7, 9 and 11).
DISCUSSION
In this work we present the synthesis of AB-dATP and DB-dATP, two new photoreactive dATP analogs containing the photoreactive moiety tethered by a two carbon spacer to the N6 amino group. The N6 position was chosen for attachment of the photoreactive group so as not to interfere with normal base pairing in dsDNA. The synthesis of our analog was achieved in a six step procedure, starting with azaridine aminoethylation on N1 and continuing with Dimroth rearrangement (28). Protection of the generated primary amino group was achieved by trifluoroacetylation and followed by phosphorylation according to the Hoard–Ott method (29). The deprotected dATP analog was coupled to 4-azidobenzoate or 4-[3-(trifluoromethyl)-diazirin-3-yl]benzoate photoreactive moieties. The structure of the modified analog was verified by 1H NMR, TLC and UV spectroscopy at each step of the chemical synthesis. In contrast to aminoethylation of AMP as done by Sicsic et al. (23), the most problematical step in our procedure seems to be the Dimroth rearrangement, which shows the presence of co-product and a low conversion that necessitates the insertion of a purification step following rearrangement. The synthesis summarized in Figure 1 and Table 1 shows good conversion at the other steps, with an overall recovery of 2.0% for AB-dATP and 3.1% for DB-dATP. Incorporation of these new photoreactive analogs into dsDNA was achieved with DNA polymerase I. In our procedure concentrations >90 nM were sufficient for complete shift of the labeled DNA and did not show any significant misincorporation (Fig. 3B and C). The addition of all four nucleotides after incorporation of photoreactive AB-dATP or DB-dATP resulted in complete shift of the labeled DNA to the full length of DNA, indicating that incorporation of modified dAMP does not preclude further extension of the DNA (Fig. 3B and C).
DNA polymerase I was shown to be crosslinked to a primed DNA template in an efficient manner requiring the incorporation of either AB-dATP or DB-dATP (see Fig. 4B–E). The loss of signal with low concentrations of photoreactive dATP analogs (Fig. 4C and E) corresponds to incomplete incorporation of the modified dATP analog. The overall efficiency of crosslinking was reasonably high for photoaffinity labeling, ranging from 9 to 15%, with slightly lower values for DB-dATP.
We have shown the synthesis of new photoreactive dATP analogs, AB-dATP and DB-dATP, which are readily incorporated into dsDNA by DNA polymerase I. These two photoreactive dATP analogs help overcome the restriction of modifying potential DNA sites, because of the availability primarily of pyrimidine nucleotide analogs for site-specific DNA–protein photocrosslinking.
Acknowledgments
ACKNOWLEDGEMENT
This work was supported by Public Health Service grant GM48413 from the National Institute of General Medical Sciences.
REFERENCES
- 1.Sengupta S.M., Persinger,J., Bartholomew,B. and Peterson,C.L. (1999) Methods, 19, 434–446. [DOI] [PubMed] [Google Scholar]
- 2.Pruss D., Bartholomew,B., Persinger,J., Hayes,J., Arents,G., Moudrianakis,E.N. and Wolffe,A.P. (1996) Science, 274, 614–617. [DOI] [PubMed] [Google Scholar]
- 3.Persinger J. and Bartholomew,B. (1996) J. Biol. Chem., 271, 33039–33046. [DOI] [PubMed] [Google Scholar]
- 4.Moore B.M., Jalluri,R.K. and Doughty,M.B. (1996) Biochemistry, 35, 11642–11651. [DOI] [PubMed] [Google Scholar]
- 5.Lagrange T., Kim,T.K., Orphanides,G., Ebright,Y.W., Ebright,R.H. and Reinberg,D. (1996) Proc. Natl Acad. Sci. USA, 93, 10620–10625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Braun B.R., Bartholomew,B., Kassavetis,G.A. and Geiduschek,E.P. (1992) J. Mol. Biol., 228, 1063–1077. [DOI] [PubMed] [Google Scholar]
- 7.Bartholomew B., Braun,B.R., Kassavetis,G.A. and Geiduschek,E.P. (1994) J. Biol. Chem., 269, 18090–18095. [PubMed] [Google Scholar]
- 8.Bartholomew B., Durkovich,D., Kassavetis,G.A. and Geiduschek,E.P. (1993) Mol. Cell. Biol., 13, 942–952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bartholomew B., Kassavetis,G.A. and Geiduschek,E.P. (1991) Mol. Cell. Biol., 11, 5181–5189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Naryshkin N., Revyakin,A., Kim,Y., Mekler,V. and Ebright,R.H. (2000) Cell, 101, 601–611. [DOI] [PubMed] [Google Scholar]
- 11.Kim T.K., Lagrange,T., Wang,Y.H., Griffith,J.D., Reinberg,D. and Ebright,R.H. (1997) Proc. Natl Acad. Sci. USA, 94, 12268–12273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Capson T.L., Benkovic,S.J. and Nossal,N.G. (1991) Cell, 65, 249–258. [DOI] [PubMed] [Google Scholar]
- 13.Bartholomew B., Tinker,R.L., Kassavetis,G.A. and Geiduschek,E.P. (1995) Methods Enzymol., 262, 476–494. [DOI] [PubMed] [Google Scholar]
- 14.Persinger J., Sengupta,S.M. and Bartholomew,B. (1999) Mol. Cell. Biol., 19, 5218–5234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bartholomew B., Kassavetis,G.A., Braun,B.R. and Geiduschek,E.P. (1990) EMBO J., 9, 2197–2205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lannutti B.J., Persinger,J. and Bartholomew,B. (1996) Biochemistry, 35, 9821–9831. [DOI] [PubMed] [Google Scholar]
- 17.Meffer R. and Dose,K. (1988) FEBS Lett., 239, 190–194. [DOI] [PubMed] [Google Scholar]
- 18.Gebeyehu G., Rao,P.Y., SooChan,P., Simms,D.A. and Klevan,L. (1987) Nucleic Acids Res., 15, 4513–4534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Costas C., Yuriev,E., Meyer,K.L., Guion,T.S. and Hanna,M.M. (2000) Nucleic Acids Res., 28, 1849–1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Woody A.Y., Vader,C.R., Woody,R.W. and Haley,B.E. (1984) Biochemistry, 23, 2843–2848. [DOI] [PubMed] [Google Scholar]
- 21.Kapuler A.M. and Spiegelman,S. (1970) Proc. Natl Acad. Sci. USA, 66, 539–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Browser A.C. and Hanna,M.M. (1991) J. Mol. Biol., 220, 227–239. [DOI] [PubMed] [Google Scholar]
- 23.Sicsic S., Leonil,J. and Le Goffic,F. (1989) Eur. J. Biochem., 179, 435–440. [DOI] [PubMed] [Google Scholar]
- 24.Ruhemann S. (1911) Trans. Chem. Soc., 97, 1438. [Google Scholar]
- 25.Laemmli U.K. (1970) Nature, 227, 680–685. [DOI] [PubMed] [Google Scholar]
- 26.Yoshikawa M., Kato,T. and Takenishi,T. (1967) Tetrahedron Lett., 50, 5065–5068. [DOI] [PubMed] [Google Scholar]
- 27.Schmidt H.L. and Grenner,G. (1976) Eur. J. Biochem., 67, 295–302. [DOI] [PubMed] [Google Scholar]
- 28.Dimroth O. (1909) Justus Liebigs Ann. Chem., 363, 183. [Google Scholar]
- 29.Hoard D.E. and Ott,D.G. (1965) J. Am. Chem. Soc., 87, 1785–1788. [DOI] [PubMed] [Google Scholar]