


Abstract
RegR is the response regulator of the RegSR two-component regulatory system in Bradyrhizobium japonicum. The only target known so far is the fixR-nifA operon, encoding the redox-responsive transcription factor NifA, which activates many genes required for symbiotic nitrogen fixation in soybean nodules. In previous in vivo studies, we identified a 32 bp upstream activating sequence located around position –68, which is essential for RegR-dependent expression of the fixR-nifA operon. Here, we used an in vitro binding-site selection assay (SELEX) to more precisely define the DNA-binding specificity of RegR. The selected sequences comprised an imperfect inverted repeat (GCGGC-N5-GTCGC) which is highly similar to an imperfect inverted repeat in the fixR UAS (GCGAC-N5-GACGC). In a parallel approach, band-shift experiments were performed with oligonucleotides comprising defined point or deletion mutations in the fixR UAS. This led to the identification of 11 critical nucleotides within a 17 bp minimal RegR binding site centered at position –64 upstream of the fixR-nifA transcription start site. Notably, all 11 critical nucleotides were located either within the half sites of the inverted repeat (four nucleotides in each half site) or in the 5 bp spacer that separates the half sites (three nucleotides). Based on these results, we defined a DNA motif comprising those nucleotides that are critical for RegR binding (RegR box; 5′-GNGAGCAGTTNNGNCGC-3′). A comparison of the RegR box with functional binding sites of the RegR-like regulator RegA of Rhodobacter capsulatus revealed considerable similarities. Thus, the RegR box may assist in the identification of new RegR target genes not only in B.japonicum but also in other α-proteobacteria possessing RegR-like response regulators.
INTRODUCTION
Regulation of nitrogen fixation in diazotrophic bacteria occurs at different levels including control of gene expression in response to various environmental signals. In most nitrogen-fixing proteobacteria, transcriptional control of genes required for nitrogen fixation is exerted by the NifA protein which activates transcription from –24/–12-type promoters that are associated with many nif and fix genes and recognized by the σ54 RNA polymerase (reviewed in 1,2). In symbiotic rhizobia such as Bradyrhizobium japonicum, the root nodule endosymbiont of soybean, NifA, is active only under microaerobic or anaerobic conditions. An increase of oxygen concentration is presumably sensed via a metal cofactor that is co-ordinated by essential cysteine residues in the NifA protein (3,4).
NifA of B.japonicum is encoded in the bicistronic fixR-nifA operon which is under the control of two disparately regulated, overlapping promoters (fixRp1, fixRp2; 5–7). The fixRp1 promoter belongs to the –24/–12 class, and it is autoactivated only under low-oxygen conditions via NifA which binds to imperfect binding sites loacted ∼100 nt upstream of the transcription start site (8). The fixRp2 promoter is active under both high and low oxygen conditions, and its activation is dependent on an upstream activating sequence (UAS) located around position –68. A protein which binds to the fixR UAS (RegR) was purified and the corresponding gene (regR) cloned (9). Null mutations in regR abolished aerobic fixR-nifA expression and also drastically reduced anaerobic expression. Moreover, those mutants showed only residual nitrogen fixation activity. An additional gene (regS) was found to be located upstream of regR, and we have recently shown that the products of regS and regR (RegS, RegR) are functional partners of a classical two-component regulatory system (10). A purified His-tagged derivative of a soluble, cytoplasmic portion of the sensor kinase RegS (RegSC) autophosphorylated in vitro and transferred the phosphoryl group to the His-tagged response regulator RegR. DNA-binding activity of RegR was demonstrated in gel retardation experiments using an oligonucleotide that spans the fixR-nifA UAS. Phosphorylation of RegR enhanced its DNA-binding activity substantially. Based on these data we proposed that RegR activates transcription from fixRp2 under both aerobic and anaerobic conditions in concert with a yet to be characterized RNA polymerase holoenzyme. Notably, the signal sensed and transduced by the RegSR system is also not known yet.
So far, the DNA target required for RegR binding was rather poorly defined by the endpoints of two deletion derivatives of the fixR UAS and a point mutation at position –68 (6). Accordingly, we have used in our previous gel retardation experiments a 32 bp oligonucleotide spanning the DNA region between positions –52 and –83 relative to the transcription start site of fixRp2 (9,10). In this study we have applied a variant of the SELEX strategy originally developed by Tuerk and Gold (11) in order to learn more about the DNA determinants required for RegR binding. The results from this approach combined with those from binding assays with a series of specifically mutated oligonucleotides enabled us to define a RegR box including 11 critical nucleotides that are part of an imperfect inverted repeat and the intervening spacer.
MATERIALS AND METHODS
Oligonucleotides and gel retardation assays
The double-stranded oligonucleotides FRPwt, FRP1–FRP3, UBP-36M1, UBP-36M2 and P1–P36, which correspond to the fixR UAS and deletion or point mutations thereof, were prepared as previously described (10; Table 1). Note that all oligonucleotides have single-strand overhangs consisting of 4 nt at the 5′-end of both strands. The sequence of the overhang in the top strand of all oligonucleotides is GATC and also in the bottom strand of oligonucleotides UBP-36M1 and UBP-36M2. All other oligonucleotides carry an AGTC overhang at the 5′-end of the bottom strand. All oligonucleotides were purchased from Microsynth (Balgach, Switzerland).
Table 1. Effect of mutations in the fixR UAS on RegR-binding activitya.

aOligonucleotides were incubated with 3 µM RegR and separated on a 6% non-denaturating polyacrylamide gel. The RegR-binding activity of individual oligonucleotides was determined as described in Materials and Methods, and it is indicated as percentage of the binding activity of the oligonucleotide FRPwt (A) which corresponds to the wild-type fixR UAS.
bOligonucleotides are listed in the following groups: (A), wild-type fixR UAS serving as a reference in all band shift assays; (B), deletion derivatives used for determination of the minimal RegR-binding site; (C), oligonucleotides used for determination of positions critical for RegR binding; (D), oligonucleotides carrying an improved inverted repeat. Deletions (Δ) and exchanges are indicated in the mutant oligonucleotides while unaltered positions are symbolized by a dash; (E), RegR box.
cNumbering of positions in (A) refers to the transcription start site P2 of the fixR-nifA operon (8).
dSingle nucleotide exchanges which resulted in >48% reduction of RegR-binding activity are indicated in red.
The randomized oligonucleotide pool ran-UAS2 was generated by PCR amplification of the 100 bp template oligonucleotide 5′-GGG GGA TCC GTT ACG ATA CGT AAA CGT ACA TAT G(N)32 CTG CAG AGT CGT TGG TCA ATC GCA CGA ATT CCC C-3′ with primer 5 (forward primer; 5′-GGG GGA TCC GTT ACG ATA CGT AAA CGT A-3′) and primer 6 (reverse primer; 5′-GGG GAA TTC GTG CGA TGA CCA ACG ACT-3′). Underlined nucleotides represent restriction enzyme recognition sites that are not present in the B.japonicum UAS. They were introduced for cloning purposes. The 100 bp oligonucleotide wt-UAS2 comprising 32 nt between position –83 and –52 of the fixR-UAS flanked by 34 nt on both sides was generated by PCR amplification of the template oligonucleotide 5′-CCC GCT AAG CTT TCG ATG TCA AAA CTA TCA TAT GC–83A TTC CGC GTG CGC GAC ATT AGG ACG CAA AAC–52 CTG CAG GCA TGC ATA TCC GAA TGC GTC TAG AGG G-3′ with primer 3 (5′-CCC GCT AAG CTT TCG ATG TCA AAA CTA T-3′) and primer 4 (5′-CCC TCT AGA CGC ATT CGG ATA TGC ATG C-3′).
His-tagged RegR protein was purified as reported by Emmerich et al. (10). Labeling of the oligonucleotides and gel retardation assays were performed as described previously (9). Because we demonstrated previously that purified RegR protein is at least partially phosphorylated (10) no extra in vitro phosphorylation step was applied. Intensities of radioactive signals in band shift gels were quantified with a phosphorimager and the program IMAGEQUANT (Molecular Dynamics). RegR-binding activities were calculated as the ratio between the intensity of radioactive signals of the retarded DNA and the combined intensities of radioactive signals of retarded plus free DNA. Unspecific background noise was subtracted from all signal intensities. Relative RegR-binding activities were calculated as the percentage of binding activity of specific oligonucleotides relative to that of the wild-type fixR UAS or the DIVS sequence (for dominant in vitro selected sequence; see Results). Wild-type fixR UAS or the DIVS sequence were present on each band shift gel to enable the calculation of relative RegR-binding activities.
In vitro binding site selection assay
Approximately 100 pg of the 5′-end-labeled double-stranded oligonucleotides ran-UAS2 and wt-UAS2 (∼50 000 c.p.m.) were incubated in separate reactions with RegR (1.5–3 µM in the initial four selection cycles and 1.5 µM in later cycles) and subjected to a gel retardation assay. The gel was dried under vacuum on a Whatman 3MM filter and exposed to a phosphorimager screen. The gel region harboring putative ran-UAS2–RegR complexes was localized by comparison with the electrophoretic mobility of the wt-UAS2–RegR complex which was loaded on the adjacent control lane. An appropriate gel slice was excised, rehydrated by adding 250–500 µl of 2 mM ethylenediaminetetraacetic acid and 50–100 µl 3 M Na-acetate, and the DNA was extracted by the freeze–squeeze method (12) without the filtration step described in the original protocol. The eluted DNA was amplified by PCR with primers 5 and 6, and the PCR products were purified by preparative 3% agarose gel electrophoresis. DNA fragments of an apparent length of 90–140 bp were isolated and subjected to a new cycle of labeling and gel retardation. Surprisingly, up to cycle six, the major products obtained by amplification of ran-UAS2 had an apparent length of ∼140 bp whereas those obtained with the control wt-UAS2 had the expected size of 100 bp (see also Results). After the sixth cycle, two distinct DNA populations of ∼100 and ∼140 bp in size were observed with ran-UAS2, and both were processed separately in subsequent cycles. The 100 bp DNA population became dominant in the amplification products of cycles 10 and 11. Individual species of the 100 bp amplification products were cloned by digesting appropriate samples with EcoRI and BamHI, ligation with EcoRI–BamHI-digested pUC19 vector DNA and subsequent transformation into Escherichia coli DH5α using standard cloning procedures (13). Plasmid DNA was isolated from individual white colonies grown on Luria–Bertani medium plates (14) supplemented with isopropyl thio-β-d-galactoside (IPTG; 200 µg·ml–1), 5-bromo-4-chloro-3-indoxyl-β-d-galactoside (X-gal; 20 µg·ml–1) and ampicillin (200 µg·ml–1). Inserts were sequenced with the chain-termination method (15) using an ABI PRISM 310 DNA sequencer (Applied Biosystems, Foster City, CA) and M13 and M13reverse standard primers.
RESULTS
In vitro selection of RegR binding sites
From our previous work it was known that purified RegR binds to a 32 bp oligonucleotide corresponding to positions –52 to –83 of the fixR-nifA promoter region (9,10). In order to more precisely define the RegR binding site, we synthesized a population of oligonucleotides comprising 32 randomized nucleotide positions flanked by defined sequences of 34 bp on both sides (ran-UAS2), and used purified RegR protein to select high-affinity DNA targets as described in Materials and Methods. As a control, we used the oligonucleotide wt-UAS2 corresponding to the wild-type UAS flanked by similar border sequences (see Materials and Methods). After 11 cycles of selection, a distinct protein–DNA complex was detected which had the same electrophoretic mobility as the RegR–wt-UAS2 complex (Fig. 1). Portions of the retarded DNA from cycles 9, 10 and 11 were amplified, cloned and characterized by DNA sequencing (Fig. 2). Out of a total of 40 clones that were sequenced after the tenth and the eleventh cycles, 30 clones had an identical nucleotide sequence at those 32 positions which were randomized in the original ran-UAS2 pool [hereafter referred to as DIVS (dominant in vitro selected) sequence]. Eight additional clones contained a similar sequence with deviations at one or two positions. Two clones contained a totally different sequence that did not bind RegR in subsequent experiments (not shown in Fig. 2). Aligning the DIVS sequence with the fixR UAS revealed a total of 16 identical nucleotides most of which are located in the 5′ portion of the DIVS sequence. Interestingly, related imperfect inverted repeats consisting of 5 nt per half site and 5 intervening nt are present in the region of maximal similarity between the fixR UAS and the DIVS sequence [GCGAC-N5-GACGC (fixR UAS); GCGGC-N5-GTCGC (DIVS); see Fig. 2]. The inverted repeat of the DIVS sequence includes at its 5′-end a guanosine residue which was not part of the randomized sequence portion but corresponds to the last position of the defined 5′ sequence of ran-UAS2.
Figure 1.
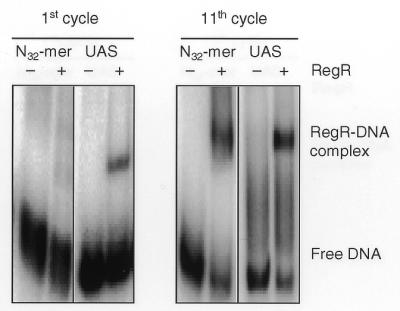
Representative band shift experiments of the first and the eleventh cycles of the SELEX procedure used for the enrichment of RegR binding sites. Approximately 100 pg (50 000 c.p.m.) of end-labeled oligonucleotides ran-UAS2 (N32mer) and wt-UAS2 (UAS) were incubated with buffer (–) or with 3 µM (first cycle) or 1.5 µM RegR protein (eleventh cycle). After separation on a 6% non-denaturating polyacrylamide gel, the gel was dried and bands were visualized by phosphorimager analysis.
Figure 2.

Comparison of the fixR UAS with individual oligonucleotides selected after 10 (top) and 11 (bottom) cycles of the SELEX procedure. The alignment was created with the GCG ‘PILEUP’ program of the software package (version 10.0) of the UWGCG (Genetics Computer Group of the University of Wisconsin, Madison, WI). Individual oligonucleotides are specified at the left margin with a code and, in parentheses, with the number of their appearance in a total of 40 clones analyzed after 10 or 11 rounds of selection. Two clones showed a completely different sequence, and thus were not included in the figure. 10-DIVS and 11-DIVS refer to identical sequences that were found most frequently after round 10 and 11. Capital letters denote nucleotides located within the stretch of the 32 originally randomized nucleotides, and lowercase letters mark unique flanking sequences that were not covered by the primers used for amplification. Nucleotides indicated in red are common to all sequences shown while green nucleotides in the selected sequences specify positions that deviate from the DIVS sequence. Horizontal arrows mark an imperfect inverted repeat present in the fixR UAS and in the DIVS sequence. The numbering of the fixR UAS refers to the transcription start site P2 of the fixR-nifA operon (8). The numbers indicated at the top refer to position numbers of the 100 bp ran-UAS2 oligonucleotides.
Next we determined the binding activity of RegR for the in vitro selected oligonucleotides listed in Figure 2. RegR bound equally well to the dominant DIVS sequence and the wt-UAS2 oligonucleotide comprising the wild-type fixR UAS (data not shown). Oligonucleotides 10-15 and 10-20 both carrying two alterations at positions 41 plus 72 and 30 plus 35, respectively, showed a slight reduction (15–20%) in binding activity relative to the DIVS sequence (Fig. 3, left). Binding of oligonucleotide 11–10 which differs only at position 36 from the DIVS sequence was greatly reduced (∼5% residual binding activity; Fig. 3, right; see also Discussion). RegR binding to the five remaining oligonucleotides 10-1, 10-5, 10-8, 11-7, 11-8 altered at positions 60, 30, 58, 59 and 30 plus 65, respectively, did not differ from binding to the DIVS sequence (data not shown).
Figure 3.

RegR-binding activity of three in vitro selected sequences that deviate from the DIVS sequence at one or two positions (see Fig. 2). Approximately 100 pg (50 000 c.p.m.) of end-labeled oligonucleotides 10-15, 10-20, 11-10, and DIVS were incubated with buffer (–) or with 3 µM RegR protein (+). After separation on 6% non-denaturating polyacrylamide gels, the gels were dried and bands were visualized by phosphorimager analysis. Upon quantification of the signal intensities, relative binding activities were calculated as described in Materials and Methods.
To evaluate whether the DIVS sequence was already present after round 9, a portion of the retarded band of cycle 9 was cloned and 18 clones were analyzed by DNA sequencing. Five clones contained the DIVS sequence whereas the remaining 13 clones had other sequences. A compilation of all 18 sequences revealed that 31 of 44 nucleotide positions were biased in favor of the DIVS sequence (data not shown).
Determination of the minimal binding site of RegR
To identify the minimal binding site for RegR, we performed gel retardation experiments using a series of oligonucleotides carrying progressive deletions at one or both ends and determined their relative RegR-binding activity (Table 1B). A 17 bp oligonucleotide (P20) still exhibited 70% relative binding activity as compared to the 32 bp reference oligonucleotide FRPwt which corresponds to the wild-type fixR UAS. Deletion of one additional nucleotide at each end (P24) abolished RegR binding almost completely. On the basis of these results we concluded that the minimal RegR-binding in the fixR UAS comprises 17 nt from A–56 to C–72.
Identification of nucleotides critical for RegR binding
To determine those nucleotides that are critical for binding of RegR to the minimal 17 bp element, we first performed gel retardation experiments with oligonucleotides comprising multiple exchanges at several (3–12) positions (FRP1, FRP2, FRP3, P5, P11 and P12; Table 1C). All of these oligonucleotides were severely impaired in RegR binding. Next, we used oligonucleotides carrying nucleotide exchanges. Oligonucleotides P7, P9, P13, P14, P15, P17 representing transversions G–71→T, G–69→T, A–66→C, T–65→G, T–64→G, G–61→T, respectively, showed strongly reduced relative binding activities between 0 and 29%. Relative binding activities between 32 and 51% were observed with oligonucleotides UBP-36M1, P28, P31, P10, P32 (A–68→C, C–67→A, C–59→A, G–58→A, C–57→A). Exchanges at positions –73 (P6), –72 (P26), –70 (P27), –63 (P29), –60 (P30) and –56 (P33) had little effects on RegR binding. The transversions A–68→C and A–66→C resulted in a significant decrease of RegR-binding activity. In the DIVS sequence, guanosines are present at the positions corresponding to A–68 and A–66 in the fixR UAS. We therefore determined the binding activity of RegR to oligonucleotides P34, P35 and P36 carrying the transitions A–68→G, A–66→G and A–68→G plus A–66→G, respectively. RegR exhibited strong binding activity to all three oligonucleotides, which is in perfect accordance with the binding of RegR to the DIVS sequence. Based on these results, we defined a motif comprising the nucleotides critical for RegR binding as ‘RegR box’ (5′-GNGAGCAGTTNNGNCGC-3′; Table 1E). The RegR box comprises 15 nt of which 11 are critical for RegR binding and eight form an imperfect inverted repeat.
Improvement and/or extension of the imperfect inverted repeat enhances RegR-binding activity
Two mutations (G–75→T in UBP-36M2 and G–62→T in P16) strongly increased RegR binding. The G–62→T transversion in P16 results in the addition of a sixth pair of complementary nucleotides at the proximal borders of the imperfect inverted repeat. Similarly, the G–75→T mutation in UBP-36M2 extends the complementarity of the half sites at their distal ends (positions –75/–74 and –54/–53). The additional complementary positions, however, are separated by two non-complementary positions (positions –73/–72 and –56/–55) from the core elements of the imperfect inverted repeat. These observations prompted us to study in more detail the effects of improved complementarity between the half sites of the imperfect inverted repeat on RegR binding with the help of the oligonucleotides listed in section D of Table 1. RegR-binding activity was progressively increased by step-wise improvement and extension of the complementarity of the imperfect inverted repeat in the minimal RegR-binding site (compare oligonuclotides P20, P21, P22 and P23 in Table 1). Maximal enhancement of RegR-binding (∼3-fold) was observed when the optimized inverted repeat consisting of 6 nt per half site was flanked by the genuine sequences present in the fixR UAS (oligonucleotide P19).
DISCUSSION
In the present study, we have defined the DNA determinants required for binding of the response regulator RegR from B.japonicum by applying two different in vitro approaches: (i) a SELEX strategy by which a RegR-binding site was selected from a pool of randomized oligonucleotides, and (ii) directed mutagenesis of the fixR UAS, the only RegR-binding site known so far in B.japonicum.
The SELEX strategy yielded a dominant oligonucleotide species (DIVS sequence) with RegR-binding abilities comparable to the fixR UAS and eight additional species of different RegR-binding activities, which deviated from the DIVS sequence at one or two positions. Surprisingly, oligonucleotide 11-10 showed only very weak RegR-binding activity although it apparently passed through 11 cycles of selection. It is possible, however, that the point mutation which distinguishes this oligonucleotide from the DIVS sequence was introduced during the PCR amplification after the final round of selection. Similarly, PCR errors are the likely cause of the nucleotide sequence variability detected at positions 30 and 72 which both belong to the region with a defined sequence in the original pool of oligonucleotides (ran-UAS2). After all, PCR errors may have stimulated the SELEX procedure by increasing the complexity of the oligonucleotide pool.
The functional role of the highly similar imperfect inverted repeats present in the fixR UAS and the DIVS sequence (GCGAGC-N5-GATCGC) was studied by analyzing the RegR-binding activity of a collection of oligonucleotides carrying progressive deletions at their borders or nucleotide exchanges. The minimal-length oligonucleotide still exhibiting RegR-binding activity (P20) perfectly coincided with the location of the inverted repeat. On the basis of our mutational analysis, we conclude that the half sites of the imperfect inverted repeat are necessary but not sufficient for RegR-binding. Critical nucleotides also include the spacer region between the two half sites as documented by the effects of exchanges at positions A–66, T–65 and T–64. The combined results from the SELEX approach and the directed mutagenesis led us to define the RegR box (5′-GNGAGCAGTTNNGNCGC-3′). Formally, this definition of the RegR box should not be regarded as invariant because certain individual positions were not exchanged by all three alternative nucleotides. The type of exchange may indeed be important as documented by our finding that A→G transitions at positions –68 and –66 did not affect RegR binding whereas A→C transversions at these positions strongly reduced it.
In previous studies, the role of the fixR UAS had been studied in vivo by genomic footprinting (8) and mutagenesis (6). Remarkably, G–75 which was protected from in vivo methylation is not part of the minimal RegR-binding site and its mutation does not interfere with RegR binding in vitro. This could mean that RegR protects its target DNA to a larger extent than is required for binding. Conversely, no protection of guanosines at positions –71, –69, –62, –61 and –58 was observed even though exchanges at these positions affected RegR binding to a variable extent. It seems obvious, however, that contacts of RegR with critical nucleotides does not automatically prevent their modification with the small molecule dimethyl sulphate used for in vivo methylation. Alternatively, RegR could become re-positioned on the DNA in vivo by other transcription factors. An A–68→C mutation reduced fixR-nifA expression by >90% in aerobically grown cells. By contrast, oligonucleotide UBP-36M1, which carries the very same exchange still exhibited considerable RegR-binding activity (40%). This may indicate that RegR-mediated transcriptional activation in vivo requires strong DNA-binding. Alternatively, it is possible that the A–68→C mutation, though it still enables DNA binding by RegR, affects productive interaction of RegR with the RNA polymerase, e.g., by altering the topology of the protein–DNA complex.
Bradyrhizobium japonicum RegR is highly similar to RegA, the response regulator of the Rhodobacter capsulatus RegBA two-component system (80% similarity; 9; for reviews, see 16,17). Most notably, a C-terminal region of 23 amino acids comprising the putative helix–turn–helix motif is completely conserved in the two proteins suggesting that they bind to comparable DNA target sequences. In fact, we recently demonstrated that RegR binds to oligonucleotides comprising binding sites for the constitutively active RegA variant, RegA* (R.Emmerich, H.Hennecke and H.M.Fischer, in press). The RegA-binding sites in the promoter regions of the R.capsulatus puc-, puf- and senC-regA-hvrA-operons and of the Rhodobacter sphaeroides cycA gene had been characterized by DNase I footprinting (18–21). Inspection of the regions protected by RegA indeed revealed similarities to the RegR box (Fig. 4). All RegA-binding sites include an inverted repeat which differ from each other by the length of the half sites (3–8 bp) and the extent of the intervening spacer DNA (1–7 bp). The variations in the spacing between half sites may indicate that RegR and RegA can bind to one half site only or that they bind as monomers. Moreover, it should be noted that unlike all other examples listed in Figure 4 the promoter of R.capsulatus senC is negatively controlled by RegA, which may be reflected by a distinct architecture of the corresponding binding site. The half sites of the inverted repeats exhibit variable but substantial sequence similarity to the RegR box which is most conserved in the R.capsulatus puc and puf promoter regions. It seems well possible that optimal DNA-binding sites for RegR and RegA show subtle differences though both proteins share identical helix–turn–helix motifs.
Figure 4.

Comparison of the B.japonicum RegR box and known DNA-binding sites of R.capsulatus RegA*, a constitutively active variant of RegA (19). Black horizontal lines mark the DNA regions upstream of the R.capsulatus puf- (Rc puf), puc- (Rc puc), and senC-regA-hvrA-operons (Rc senC) and of the R.sphaeroides cycA gene (Rs cycA) which are protected from DNase I attack by RegA* (19–21). Position numbers are indicated relative to the transcription start site of the respective gene or operon. Nucleotides in red are identical in the RegR box and in the RegA*-binding sites. Horizontal arrows denote the half sites of (imperfect) inverted repeats centered at the positions marked by a plus sign. Gaps were introduced manually to align the center of the inverted repeats.
In conclusion, we believe that the RegR box defined in this work will assist in the identification of new binding sites for RegR in the genome of B.japonicum or even for RegR homologs in other α-proteobacteria such as ActR of Sinorhizobium meliloti (22), PrrA of R.sphaeroides (23) and RegA of R.capsulatus, Rhodovulum sulfidophilum and Roseobacter denitrificans (24,25) all of which possess identical helix–turn–helix DNA-binding domains.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Enrique Morett and Humberto Barrios for providing plasmids containing mutated RegR-binding sites, which were used in initial experiments. We also appreciate the valuable comments of an anonymous reviewer. This work was supported by the Federal Institute of Technology Zürich.
REFERENCES
- 1.Dixon R. (1998) Arch. Microbiol., 169, 371–380. [DOI] [PubMed] [Google Scholar]
- 2.Fischer H.M. (1994) Microbiol. Rev., 58, 352–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fischer H.M., Fritsche,S., Herzog,B. and Hennecke,H. (1989) FEBS Lett., 255, 167–171. [DOI] [PubMed] [Google Scholar]
- 4.Fischer H.M. and Hennecke,H. (1987) Mol. Gen. Genet., 209, 621–626. [DOI] [PubMed] [Google Scholar]
- 5.Barrios H., Fischer,H.M., Hennecke,H. and Morett,E. (1995) J. Bacteriol., 177, 1760–1765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Thöny B., Anthamatten,D. and Hennecke,H. (1989) J. Bacteriol., 171, 4162–4169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Thöny B., Fischer,H.M., Anthamatten,D., Bruderer,T. and Hennecke,H. (1987) Nucleic Acids Res., 15, 8479–8499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Barrios H., Grande,R., Olvera,L. and Morett,E. (1998) Proc. Natl Acad. Sci. USA, 95, 1014–1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bauer E., Kaspar,T., Fischer,H.M. and Hennecke,H. (1998) J. Bacteriol., 180, 3853–3863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Emmerich R., Panglungtshang,K., Strehler,P., Hennecke,H. and Fischer,H.M. (1999) Eur. J. Biochem., 263, 455–463. [DOI] [PubMed] [Google Scholar]
- 11.Tuerk C. and Gold,L. (1990) Science, 249, 505–510. [DOI] [PubMed] [Google Scholar]
- 12.Beutel B.A. and Gold,L. (1992) J. Mol. Biol., 228, 803–812. [DOI] [PubMed] [Google Scholar]
- 13.Sambrook J., Fritsch,E.F. and Maniatis,T. (1989) Molecular Cloning: A Laboratory Manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 14.Miller J.H. (1972) Experiments in Molecular Genetics. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 15.Sanger F., Nicklen,S. and Coulson,A.R. (1977) Proc. Natl Acad. Sci. USA, 74, 5463–5467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sganga M.W. and Bauer,C.E. (1992) Cell, 68, 945–954. [DOI] [PubMed] [Google Scholar]
- 17.Bauer C.E., Elsen,S. and Bird,T.H. (1999) Annu. Rev. Microbiol., 53, 495–523. [DOI] [PubMed] [Google Scholar]
- 18.Mosley C.S., Suzuki,J.Y. and Bauer,C.E. (1994) J. Bacteriol., 176, 7566–7573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Du S., Bird,T.H. and Bauer,C.E. (1998) J. Biol. Chem., 273, 18509–18513. [DOI] [PubMed] [Google Scholar]
- 20.Du S., Kouadio,J.L.K. and Bauer,C.E. (1999) J. Bacteriol., 181, 4341. [Google Scholar]
- 21.Karls R.K., Wolf,J.R. and Donohue,T.J. (1999) Mol. Microbiol., 34, 822–835. [DOI] [PubMed] [Google Scholar]
- 22.Tiwari R.P., Reeve,W.G., Dilworth,M.J. and Glenn,A.R. (1996) Microbiology, 142, 1693–1704. [DOI] [PubMed] [Google Scholar]
- 23.Eraso J.M. and Kaplan,S. (1994) J. Bacteriol., 176, 32–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Inoue K., Kouadio,J.L.K., Mosley,C.S. and Bauer,C.E. (1995) Biochemistry, 34, 391–396. [DOI] [PubMed] [Google Scholar]
- 25.Masuda S., Matsumoto,Y., Nagashima,K.V.P., Shimada,K., Inoue,K., Bauer,C.E. and Matsuura,K. (1999) J. Bacteriol., 181, 4205–4215. [DOI] [PMC free article] [PubMed] [Google Scholar]