

Abstract
The xeroderma pigmentosum group A protein (XPA) plays a central role in nucleotide excision repair (NER). To identify proteins that bind to XPA, we screened a HeLa cDNA library using the yeast two-hybrid system. Here we report a novel cytoplasmic GTP-binding protein, designated XPA binding protein 1 (XAB1). The deduced amino acid sequence of XAB1 consisted of 374 residues with a molecular weight of 41 kDa and an isoelectric point of 4.65. Sequence analysis revealed that XAB1 has four sequence motifs G1–G4 of the GTP-binding protein family in the N-terminal half. XAB1 also contains an acidic region in the C-terminal portion. Northern blot analysis showed that XAB1 mRNA is expressed ubiquitously, and immunofluorescence analysis revealed that XAB1 is localized mainly in the cytoplasm. Consistent with the GTP-binding motif, purified recombinant XAB1 protein has intrinsic GTPase activity. Using the yeast two-hybrid system, we elucidated that XAB1 binds to the N-terminal region of XPA. The deletion of five amino acids, residues 30–34 of XPA, required for nuclear localization of XPA abolished the interaction with XAB1. These results suggest that XAB1 is a novel cytoplasmic GTPase involved in nuclear localization of XPA.
INTRODUCTION
Nucleotide excision repair (NER) is a versatile DNA repair system correcting a broad spectrum of DNA damage, including ultraviolet (UV)-induced cyclobutane pyrimidine dimers and (6–4) photoproducts as well as chemical-carcinogen-induced lesion (1). There are two subpathways in NER (2). One is transcription-coupled repair (TCR), which efficiently removes the damage in the transcribed strand of transcriptionally active genes. The other is global genome repair (GGR), which occurs throughout the genome including the non-transcribed strand of active genes.
Xeroderma pigmentosum (XP) is an autosomal recessive human disease characterized by hypersensitivity to sunlight and a high incidence of skin cancer on sun-exposed skin (1,3). Cells from XP patients are hypersensitive to killing by UV irradiation. XP is classified into seven complementation groups (XP-A to XP-G) and a variant form (1). Except the variant form, the primary defect of XP resides in early steps of NER (3). Both TCR and GGR of NER are defective in groups A, B, D, F and G but not group C, in which only GGR is impaired (4–6). Cockayne syndrome (CS), another NER-defective hereditary disorder, shows photosensitivity, cachectic dwarfism and severe mental retardation but no predisposition to UV-induced skin cancer. In CS cells, the TCR subpathway is deficient, but GGR is proficient (7,8). CS is classified into two groups (CS-A and CS-B) (1,3). To date, all XP and CS genes have been cloned. In addition, the ERCC1 protein, encoded by a human gene that can correct the repair deficiency in a UV-sensitive, NER-defective, rodent mutant cell line, is also involved in NER. The core reaction of NER in humans has been reconstituted in vitro with purified XP and other proteins, and an outline of the GGR mechanism has been obtained (9). On the other hand, the molecular mechanism of TCR is not resolved for humans, although genetic and cell biological evidence indicates that CSA and CSB play key roles in TCR.
XPA, consisting of 273 amino acid residues, binds preferentially to UV- or chemical-carcinogen-damaged DNA (10,11). Several distinct functional regions have been identified in XPA. The N-terminal region (residues 4–29) is responsible for binding to the 34 kDa subunit of replication protein A (RPA34) (12,13) and the basic amino acid region (residues 30–42) is important for the localization of XPA to nucleus (14). The region containing the glutamic acid-cluster (E-cluster) (residues 78–84) is important for the interaction with ERCC1 (15,16). The C-terminal region (residues 226–273) is shown to bind to TFIIH (17,18). The central region (residues 98–219) is identified as the minimal polypeptide for preferential binding to damaged DNA (19). In addition, this polypeptide contains the region (residues 98–187) necessary for binding to the 70 kDa subunit of RPA (RPA70) (12,13).
In addition to RPA34 and ERCC1, we isolated cDNAs for five unknown proteins which bound XPA in yeast two-hybrid screening of the HeLa cDNA library. Here we report the analysis of the cDNA for one of these unidentified XPA binding proteins, designated XPA binding protein 1 (XAB1).
MATERIALS AND METHODS
Two-hybrid screening
We generated a HeLa cDNA library in the pGAD GH vector (Clontech) for two-hybrid screening using a cDNA Synthesis Kit (Stratagene) according to the protocol supplied by the manufacturer. Screening was performed as described previously (20).
Cloning and rapid amplification of 5′ cDNA ends (5′-RACE) of XAB1
To obtain full-length cDNA for XAB1, we screened a HeLa cDNA library in λZAPII (provided by Dr H. Nojima, Osaka University) with a 541 bp EcoRI–PstI fragment of pGAD GH-17, one of the positive clones in the two-hybrid screening (20), as a probe. Plaque hybridization was carried out at 65°C in Church’s phosphate buffer (21) and washing was performed at 65°C in 2× SSC/0.1% SDS followed by 0.2× SSC/0.1% SDS.
5′-RACE was performed with 5′-AmpliFINDER RACE Kit (Clontech) using P1 primer (5′-GAGGATGCAAGGGCTTCAGTGATAA-3′) and P2 primer (5′-TCCCAGAAGCTGACCAGGTGAATAC-3′). Amplified DNA fragments were cloned and sequenced.
Northern blot analysis
Human Multiple Tissue northern blots were purchased from Clontech. Hybridization was carried out as described above. The signals were analyzed with a FUJIFILM BAS 2500 Bio-image Analyzer.
Preparation of recombinant XAB1 proteins
To prepare glutathione S-transferase (GST)-XAB1 fused protein, a 1169 bp EcoRI–XbaI fragment of pGAD GH-17, containing the entire coding region of the XAB1 cDNA, was ligated to pGEX-5X-3 (Pharmacia Biotech). Preparation of GST-XAB1 protein was performed as described previously (12), except that GST-XAB1 was induced at 37°C for 3 h and the salt concentration of the buffer was 150 mM.
For expression of hexahistidine-tagged XAB1 (His-XAB1), the EcoRI–XbaI fragment described above was ligated to pFastBac HTc (Life Technologies). Recombinant baculovirus was isolated as instructed by the protocol of the Bac-to-Bac Baculovirus Expression System (Life Technologies). Sf9 cells were infected with the recombinant baculovirus (1 plaque-forming unit/cell). After 48 h of infection, the cells were harvested, washed once with phosphate-buffered saline (PBS) and stored at –80°C. Cells were resuspended in ice-cold buffer A (20 mM Tris–HCl pH 8.0, 100 mM NaCl), frozen and thawed twice, then lyzed by sonication on ice. Lysates were centrifuged at 35 000 g for 30 min, and the cleared lysates were incubated with TALON Metal Affinity Resin (Clontech) for 1 h at 4°C. The resin was washed three times with 20 bed volumes of buffer A containing 10 mM imidazole. Bound proteins were eluted with buffer A containing 50 mM imidazole. The eluates were dialyzed against buffer B (50 mM Tris–HCl pH 8.0, 5 mM MgCl2, 100 mM KCl, 10% glycerol, 0.1 mM DTT) and applied to a Mini Q PC3.2/3 column (Pharmacia Biotech) equilibrated with buffer B. Proteins retained under these conditions were eluted with a linear gradient of 100–1000 mM KCl at a flow rate of 0.25 ml/min.
Analysis of subcellular localization of XAB1
Anti-XAB1 polyclonal antibody was raised against the GST-XAB1 protein as an antigen. Anti-XAB1 IgG was purified with protein G–Sepharose, followed by XAB1 affinity chromatography. For indirect immunofluorescence microscopy, HeLa cells were cultured on a glass slide overnight, fixed with 3.5% formaldehyde in PBS at room temperature for 20 min, and permeabilized with 0.2% Triton X-100 in PBS at room temperature for 20 min. The fixed cells were incubated with anti-XAB1 IgG (10 µg/ml in PBS) at room temperature for 1 h and washed three times with PBS. They were then incubated with FITC-conjugated anti-rabbit IgG (Cappel; 100-fold dilution with PBS) at room temperature for 1 h, washed three times with PBS and examined with a fluorescence microscope.
For immunoblot analysis, cytosol and nuclear extracts from HeLa cells (2 × 106) were fractionated as described previously (22).
Two-hybrid binding assay
The entire coding sequence of XAB1 was inserted into pGBT9 vector and expressed as a fusion protein to the DNA binding domain of Gal4. Both wild-type and mutant XPA cDNAs (14) were subcloned into pGAD GH vector and expressed as fusion proteins with the activation domain of Gal4. For measurement of protein interaction, plasmids were cotransformed using lithium acetate into a yeast strain SFY526 (MATa, ura3-52, his3-200, ade2-101, lys2-801, trp1-901, leu2-3, 112, canr, gal4-542, gal80–538, URA3::GAL1UAS-GAL1TATA-lacZ) (23). After 5 days of growth on SD lacking leucine and tryptophan plates, β-galactosidase activity was measured in a filter assay using X-gal and in a liquid assay using chlorophenol red-β-d-galactopyranoside as a substrate.
Assay for GTPase and ATPase activity of His-XAB1
GTPase activity was measured with purified His-XAB1 (0.1 mg/ml) in buffer B containing 13 nM [α-32P]GTP (3000 Ci/mmol) and 0.2 mM unlabeled GTP at 37°C. The reaction was stopped at various time points by adding an equal volume of a solution containing 2 mM EDTA and 0.5% SDS. Aliquots of the samples were spotted onto a polyethylenimine-cellulose thin-layer chromatography plate (Merck) and resolved with a solution containing 1.6 M LiCl or 1 M acetic acid and 1 M LiCl. The signals were analyzed with a FUJIFILM BAS 2500 Bio-image Analyzer. ATPase assay was performed as above, except that 13 nM [α-32P]ATP (3000 Ci/mmol) and 0.2 mM unlabeled ATP were used.
RESULTS
Two-hybrid screening
We carried out yeast two-hybrid screening to identify proteins that bind to XPA. Yeast strain HF7c was sequentially transformed with the pGBT9 XPA and pGAD GH HeLa cDNA library (8.7 × 107) that we constructed. Transformants (1.5 × 107) were subjected to dual selection (HIS+, LacZ+) and 281 independent clones were isolated. These clones were classified into seven groups by dot blot hybridization. Two of them encoded known proteins that bind to XPA; 145 clones were ERCC1 and 45 clones were RPA34. The other five groups were found to be unknown. Two of the five unknown groups, group 1 and group 2, had been detected in a previous screening (20). Out of 281 clones, 20 and 54 belonged to group 1 and group 2, respectively. In this study, we analyzed the cDNA belonging to the group 1 clones.
Isolation of cDNA for XAB1
Clone GH-17 had the longest insert, 2.5 kb, in group 1 and an open reading frame of 1125 bp. Sequence analysis of the other cDNAs from this group revealed that the 3′-untranslated region of the GH-17 insert was rearranged. To isolate full-length cDNAs, a HeLa cDNA library in λZAPII was screened with the insert of GH-17 as a probe. Eight positive clones with inserts of 1.2, 1.5, 1.6, 1.7, 1.75 and 1.8 kb were obtained by the screening of 1.5 × 106 plaques. All of them lacked the 5′ portion of the open reading frame of GH-17 and had the 3′-untranslated sequence containing the poly(A) tail. To obtain the missing 5′-end of the cDNAs, 5′-RACE was performed using poly(A)+ RNA from human placenta and WI38 cells. Four different clones from each RNA template were obtained. They had the same nucleotide sequence of GH-17 in the open reading frame region. The 5′-untranslated sequence of the longest clone was identical to that of GH-17. The nucleotide sequence around the first ATG codon of GH-17 agreed with the consensus sequence for eukaryotic initiation codon described by Kozak (24), and immunoblot analysis revealed that the in vitro translation product from GH-17 was the same size as the protein in HeLa cell lysate (data not shown). Thus, GH-17 encodes the entire coding region and the protein coding region starts from the first ATG of the open reading frame.
The reconstituted cDNA was 1829 nucleotides in length (DDBJ/EMBL/GenBank accession no. AB044661) and contained an open reading frame of 1125 bp, encoding a polypeptide of 374 amino acid residues with a predicted molecular weight of 41 kDa and a calculated isoelectric point of 4.65. We designated this protein as XAB1. The N-terminal region of the deduced amino acid sequence contained several structural motifs that were conserved within GTP-binding proteins: the phosphate/magnesium binding motif G-1 (-G26MAGSGKT), G-2 (-G69IRDT), G-3 (-D122TPG) and the putative guanine ring recognition motif G-4 (-N189KTD) (25–27). But XAB1 did not have the guanine base recognition site with consensus sequence G-5 [(T/G)(C/S)A] (26,27). The C-terminal region of XAB1 contained a cluster of acidic amino acid residues.
In order to identify homologous proteins, the predicted amino acid sequence of XAB1 was used to search the National Center for Biotechnology Information (NCBI) database using the BLAST command. The sequence of one unknown partial cDNA, Homo sapiens mRNA for a putative ATP (GTP)-binding protein (CAA09376), was a perfect match. And the most highly related protein was the Caenorhabditis elegans hypothetical protein (P46577) (52% identity, 68% similarity), followed by the Saccharomyces cerevisiae hypothetical protein Yjr072cp (6322532) (48% identity, 62% similarity), Drosophila melanogaster hypothetical protein (CAB51688) (47% identity, 64% similarity) and Schizosaccharomyces pombe hypothetical protein (CAA17930) (47% identity, 64% similarity). The alignment of these sequences is shown in Figure 1. The NCBI database showed that the gene encoding the putative ATP (GTP)-binding protein (CAA09376) maps to 2p23.3.
Figure 1.

Alignment of the amino acid sequences of XAB1 and related proteins. XAB1 is aligned with hypothetical proteins of C.elegans (accession no. P46577), S.cerevisiae (accession no. 6322532), D.melanogaster (accession no. CAB51688) and S.pombe (accession no. CAA17930). Amino acids are shown in the single-letter code. Identical residues are marked by boxes. The sequence motifs G1–G4 that are conserved in GTP-binding proteins are underlined. The basic region is double underlined.
Expression of XAB1 mRNA
To determine the size of the mRNA and tissue-specific expression of XAB1, northern blot analysis of poly(A)+ RNA from different human tissues was performed (Fig. 2). A major transcript with an approximate size of 2.1 kb, which is consistent with the size of the cDNA clone, was identified. This gene was expressed ubiquitously in adult organs, with the highest level in testis. Two minor transcripts of 2.9 and 1.4 kb were also detected. The 1.4 kb transcript was expressed exclusively in testis.
Figure 2.

Northern blot analysis of XAB1 mRNA in different tissues. Human multiple tissue northern blots (Clontech) were probed with a 0.54 kb fragment of XAB1 cDNA. Each lane contained 2 µg of poly(A)+ RNA from each tissue: spleen (lane 1), thymus (lane 2), prostate (lane 3), testis (lane 4), ovary (lane 5), small intestine (lane 6), colon (lane 7), leukocyte (lane 8), heart (lane 9), brain (lane 10), placenta (lane 11), lung (lane 12), liver (lane 13), skeletal muscle (lane 14), kidney (lane 15) and pancreas (lane 16). The β-actin cDNA was used to probe the same blot as a control. The mobility of RNA size standards is shown on the left of the blots.
Subcellular localization of XAB1
To determine the subcellular localization of XAB1, immunofluorescence analysis was performed using anti-XAB1 antibody. As shown in Figure 3A, intense signal was detected predominantly in cytoplasm. Western blot analysis of fractionated cellular proteins also showed that XAB1 is predominantly expressed in cytosol (Fig. 3B).
Figure 3.

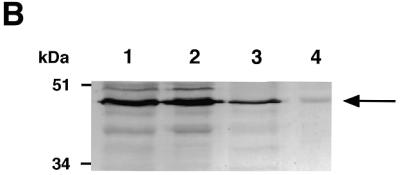
Subcellular localization of XAB1. (A) Immunofluorescence analysis with anti-XAB1 antibody in HeLa cells. (B) Immunoblot analysis of fractionated cellular proteins with anti-XAB1 antibody. Lane 1, whole cell extracts; lane 2, cytosol; lane 3, nuclear extracts; lane 4, nuclear precipitates.
Interaction of XPA deletion mutants with XAB1
Several functional domains have been identified in XPA (12–19). To delineate the binding region of XPA for XAB1, yeast two-hybrid assay was performed with a reverse combination in screening, full-length XAB1-bait and XPA deletion mutant-prey. The ability of each mutant to interact with XAB1 was determined as β-galactosidase activity (Fig. 4 and Table 1). The truncated XPA with deletion of the C-terminal region between amino acid residues 98 and 273 (CΔ176) and deletion of the region between residues 61 and 92 (Δ61–92) were still able to interact with XAB1. Deletion of the N-terminal end of XPA between residues 1 and 52 (NΔ52) and deletion of the region between residues 37 and 58 (Δ37–58) abolished the activation of the reporter gene. Furthermore, the truncated XPA with deletion of basic amino acid residues between 30 and 34 (Arg-Lys-Arg-Gln-Arg; Δ30–34) lost the ability to interact with XAB1. These results indicate that XAB1 interacts with the N-terminal region of XPA and that the five basic amino acid residues (residues 30–34) of XPA are important for the binding to XAB1.
Figure 4.

Localization of XAB1 binding region in XPA. A plasmid encoding Gal4 activation domain/XPA deletion mutant was cotransformed with pGBT9-XAB1 or control plasmid and β-galactosidase activity was measured. The structural and functional domains of XPA are illustrated at the top.
Table 1. Protein–protein interaction between XAB1 and XPA was measured by quantitative liquid β-galactosidase assay of a yeast two-hybrid system.
| DNA binding domain | Activation domain | β-Galactosidase activity (Miller units) |
|---|---|---|
| pGBT9-XAB1 | pGADGH-XPA | 5.1 |
| pGADGH-NΔ29 | 0.3 | |
| pGADGH-NΔ52 | 0.2 | |
| pGADGH-NΔ137 | 0.4 | |
| pGADGH-CΔ130 | 118.7 | |
| pGADGH-CΔ176 | 60.7 | |
| pGADGH-Δ30–34 | 0.3 | |
| pGADGH-Δ30–42 | 0.3 | |
| pGADGH-Δ37–58 | 0.3 | |
| pGADGH-Δ61–92 | 24.7 | |
| pGADGH-Δ98–128 | 14.2 | |
| pGBT9 | pGADGH-XPA | 0.1 |
| pGADGH-NΔ29 | 0.3 | |
| pGADGH-NΔ52 | 0.2 | |
| pGADGH-NΔ137 | 0.4 | |
| pGADGH-CΔ130 | 0.3 | |
| pGADGH-CΔ176 | 0.4 | |
| pGADGH-Δ30–34 | 0.4 | |
| pGADGH-Δ30–42 | 0.4 | |
| pGADGH-Δ37–58 | 0.4 | |
| pGADGH-Δ61–92 | 0.3 | |
| pGADGH-Δ98–128 | 0.4 |
Values are the averages of three to 10 independent transformants.
GTPase activity of recombinant His-XAB1
XAB1 has the motifs that were conserved in GTP-binding proteins, raising the possibility that XAB1 has intrinsic GTPase activity. To test the possibility, we measured GTPase activity using His-XAB1 produced by the baculovirus system. The protein was purified by metal affinity resin and Mini Q column chromatography (Fig. 5). His-XAB1 was eluted from the Mini Q column at 200–300 mM KCl. GTPase activity was detected in the fractions containing His-XAB1 (fractions 13 and 14), and the elution profile of GTPase activity was correlated with that of His-XAB1. We also tested whether XAB1 has ATPase activity. However, we could not detect the ATPase activity (Fig. 5D).
Figure 5.


Recombinant hexahistidine-tagged XAB1 hydrolyzes GTP but not ATP. (A) Affinity purified His-XAB1 (In) was loaded onto a Mini Q column. The bound proteins were eluted with a linear gradient of KCl and fractions were analyzed by SDS–polyacrylamide gel electrophoresis and Coomassie blue staining. (B) Samples eluted from the Mini Q column were assayed for GTPase activity. Mini Q-purified His-XAB1 (0.1 mg/ml) was assayed for GTPase (C) or ATPase (D) activity. Incubations were carried out for 0, 15 and 30 min. Thin layer chromatography was performed with a solution containing 1.6 M LiCl (B) or 1 M acetic acid and 1 M LiCl (C and D). The positions of GTP, GDP and ATP are indicated.
DISCUSSION
We had searched for proteins that bind to XPA by yeast two-hybrid screening and had shown that the RPA34 bound to XPA (20). In the present study, we screened approximately 12 times the number of HeLa cDNA clones (1.5 × 107) screened previously using XPA as bait. Among the positive clones, there were ERCC1, RPA34 and five unknown cDNAs. We designated these unknown proteins XABs. XAB1 and XAB2 were detected in the previous and the present screening. Recently, we have shown that XAB2 is a novel tetratricopeptide repeat protein involved in transcription-coupled DNA repair and transcription (28). From a periodic database search, XAB3 and XAB5 were identified as a putative metallopeptidase PRSM1 (29) and a Golgi reassembly stacking protein GRASP65 (30), respectively. XAB4 has a region homologous to XAB5. Here we showed that XAB1, consisting of 374 amino acid residues with a predicted molecular weight of 41 kDa, is a novel cytoplasmic protein having GTPase activity.
GTP-binding proteins constitute a large superfamily which comprises at least three subfamilies: large heterotrimeric signal-transducing G-proteins, low molecular weight G-proteins and GTPases involved in protein synthesis (26). The α-subunits of heterotrimeric G-proteins have a built-in GTPase-activating protein domain that facilitates GTP hydrolysis and accounts for their high intrinsic GTPase activity (26). The p21ras and ras-related low molecular weight GTPases exhibit high affinity for GTP and GDP, have low intrinsic GTPase and require GTPase-activating protein for efficient hydrolysis of GTP and release of GDP (26). Recently, a new group of large molecular GTP-binding proteins (e.g. dynamin) has been identified (31). In the GTP binding motif, these proteins harbor only the four characteristic motifs for GTPase, G1–G4 (31). In the dynamin family, MxA protein is well characterized in biochemical studies. MxA exhibits low affinity for GTP, lower affinity for GDP and a high Km for the GTP hydrolysis reaction (32,33).
In size, XAB1 resembles the α-subunits of the heterotrimeric G-proteins. On the other hand, in terms of its GTP-binding motif, XAB1 resembles the dynamin family. We tried to detect intrinsic GTP-binding activity of recombinant XAB1 protein by nitrocellulose filter binding assay, but we could not detect any GTP or GDP-binding activity (data not shown). However, we have detected GTPase activity of recombinant XAB1 protein in solution (Fig. 5). Filter binding assay is less suitable for measuring low affinity interaction between protein and nucleotide than binding experiments in solution (33). Our data suggest that XAB1 would exhibit low affinity for GTP and GDP, and high GTPase activity like MxA protein. XAB1 resembles the dynamin family with respect to GTPase activity, but differs from them in molecular weight.
A group of hypothetical proteins showed high degrees of sequence similarity with XAB1 (Fig. 1). At the amino acid level, all the proteins share ∼47–52% sequence identity. It is likely that these proteins constitute a novel subfamily of GTP-binding proteins since they all contain the same G1–G4 motifs and possess a similar molecular size (355–385 amino acid residues), which is smaller than that of the dynamin family.
To identify the function of XAB1 in NER, we tested the effects of the addition of recombinant XAB1 protein or anti-XAB1 antibody on cell-free NER assay. But there were no significant effects of recombinant XAB1 and anti-XAB1 antibody (data not shown). XAB1 is expressed ubiquitously and its homologs are conserved in different species. The gene for the S.cerevisiae homolog is essential to the viability of yeast (data not shown). Consequently, XAB1 could have an essential function. We showed that XAB1 bound to the N-terminal region (residues 1–52) of XPA and the deletion of 30–34 amino acid residues of XPA abolished the ability to interact with XAB1 (Fig. 4). Previously, we have shown that the NLS of XPA is localized in a region of 30–42 amino acid residues (14). These results suggest that XAB1 is involved in the nuclear localization of XPA.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Dr Hiroshi Nojima for providing the λZAPII cDNA library, and Drs Yoshihiro Yoneda and Taro Tachibana for discussions. This work was supported by a Grant-in-Aid for Scientific Research from the Ministry of Education, Science, Sports and Culture of Japan and by CREST of Japan Science and Technology.
DDBJ/EMBL/GenBank accession no. AB044661
REFERENCES
- 1.Friedberg E.C., Walker,G.C. and Siede,W. (1995) DNA Repair and Mutagenesis. ASM Press, Washington, DC.
- 2.Hanawalt P.C. and Spivak,G. (1999) In Dizdaroglu,M. and Karakaya,A.E. (eds), Advances in DNA Damage and Repair. Kluwer Academic/Plenum Publishers, New York, pp. 169–179.
- 3.Bootsma D., Kreamer,K.H., Cleaver,J.E. and Hoeijmakers,J.H.J. (1998) In Vogelstein,B. and Kinzler,K.W. (eds), The Genetic Basis of Human Cancer. McGraw Hill, New York, pp. 245–274.
- 4.Evans M.K., Robbins,J.H., Ganges,M.B., Tarone,R.E., Nairn,R.S. and Bohr,V.A. (1993) J. Biol. Chem., 268, 4839–4847. [PubMed] [Google Scholar]
- 5.Venema J., van Hoffen,A., Natarajan,A.T., van Zeeland,A.A. and Mullenders,L.H. (1990) Nucleic Acids Res., 18, 443–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Venema J., van Hoffen,A., Karcagi,V., Natarajan,A.T., van Zeeland,A.A. and Mullenders,L.H. (1991) Mol. Cell. Biol., 11, 4128–4134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Venema J., Mullenders,L.H., Natarajan,A.T., van Zeeland,A.A. and Mayne,L.V. (1990) Proc. Natl Acad. Sci. USA, 87, 4707–4711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.van Hoffen A., Natarajan,A.T., Mayne,L.V., van Zeeland,A.A., Mullenders,L.H. and Venema,J. (1993) Nucleic Acids Res., 21, 5890–5895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.de Laat W.L., Jaspers,N.G. and Hoeijmakers,J.H.J. (1999) Genes Dev., 13, 768–785. [DOI] [PubMed] [Google Scholar]
- 10.Jones C.J. and Wood,R.D. (1993) Biochemistry, 32, 12096–12104. [DOI] [PubMed] [Google Scholar]
- 11.Asahina H., Kuraoka,I., Shirakawa,M., Morita,E.H., Miura,N., Miyamoto,I., Ohtsuka,E., Okada,Y. and Tanaka,K. (1994) Mutat. Res., 315, 229–237. [DOI] [PubMed] [Google Scholar]
- 12.Saijo M., Kuraoka,I., Masutani,C., Hanaoka,F. and Tanaka,K. (1996) Nucleic Acids Res., 24, 4719–4724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li L., Lu,X., Peterson,C.A. and Legerski,R.J. (1995) Mol. Cell. Biol., 15, 5396–5402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Miyamoto I., Miura,N., Niwa,H., Miyazaki,J. and Tanaka,K. (1992) J. Biol. Chem., 267, 12182–12187. [PubMed] [Google Scholar]
- 15.Nagai A., Saijo,M., Kuraoka,I., Matsuda,T., Kodo,N., Nakatsu,Y., Mimaki,T., Mino,M., Biggerstaff,M., Wood,R.D., Sijbers,A., Hoeijmakers,J.H.J. and Tanaka,K. (1995) Biochem. Biophys. Res. Commun., 211, 960–966. [DOI] [PubMed] [Google Scholar]
- 16.Li L., Peterson,C.A., Lu,X. and Legerski,R.J. (1995) Mol. Cell. Biol., 15, 1993–1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Park C.H., Mu,D., Reardon,J.T. and Sancar,A. (1995) J. Biol. Chem., 270, 4896–4902. [DOI] [PubMed] [Google Scholar]
- 18.Nocentini S., Coin,F., Saijo,M., Tanaka,K. and Egly,J.M. (1997) J. Biol. Chem., 272, 22991–22994. [DOI] [PubMed] [Google Scholar]
- 19.Kuraoka I., Morita,E.H., Saijo,M., Matsuda,T., Morikawa,K., Shirakawa,M. and Tanaka,K. (1996) Mutat. Res., 362, 87–95. [DOI] [PubMed] [Google Scholar]
- 20.Matsuda T., Saijo,M., Kuraoka,I., Kobayashi,T., Nakatsu,Y., Nagai,A., Enjoji,T., Masutani,C., Sugasawa,K., Hanaoka,F., Yasui,A. and Tanaka,K. (1995) J. Biol. Chem., 270, 4152–4157. [DOI] [PubMed] [Google Scholar]
- 21.Church G.M. and Gilbert,W. (1984) Proc. Natl Acad. Sci. USA, 81, 1991–1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Saijo M., Sakai,Y., Kishino,T., Niikawa,N., Matsuura,Y., Morino,K., Tamai,K. and Taya,Y. (1995) Genomics, 27, 511–519. [DOI] [PubMed] [Google Scholar]
- 23.Harper J.W., Adami,G.R., Wei,N., Keyomarsi,K. and Elledge,S.J. (1993) Cell, 75, 805–816. [DOI] [PubMed] [Google Scholar]
- 24.Kozak M. (1987) Nucleic Acids Res., 15, 8125–8148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dever T.E., Glynias,M.J. and Merrick,W.C. (1987) Proc. Natl Acad. Sci. USA, 84, 1814–1818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bourne H.R., Sanders,D.A. and McCormick,F. (1990) Nature, 348, 125–132. [DOI] [PubMed] [Google Scholar]
- 27.Sprang S.R. (1997) Annu. Rev. Biochem., 66, 639–678. [DOI] [PubMed] [Google Scholar]
- 28.Nakatsu Y., Asahina,H., Citterio,E., Rademarkers,S., Vermeulen,W., Kamiuchi,S., Yeo,J.P., Khaw,M.C., Saijo,M., Kodo,N., Matsuda,T., Hoeijmakers,J.H. and Tanaka,K. (2000) J. Biol. Chem., in press. [DOI] [PubMed] [Google Scholar]
- 29.Scott I.C., Halila,R., Jenkins,J.M., Mehan,S., Apostolou,S., Winqvist,R., Callen,D.F., Prockop,D.J., Peltonen,L. and Kadler,K.E. (1996) Gene, 174, 135–143. [DOI] [PubMed] [Google Scholar]
- 30.Barr F.A., Puype,M., Vandekerckhove,J. and Warren,G. (1997) Cell, 91, 253–262. [DOI] [PubMed] [Google Scholar]
- 31.van der Bliek A.M. (1999) Trends Cell Biol., 9, 96–102. [DOI] [PubMed] [Google Scholar]
- 32.Melen K., Ronni,T., Lotta,T. and Julkunen,I. (1994) J. Biol. Chem., 269, 2009–2015. [PubMed] [Google Scholar]
- 33.Richter M.F., Schwemmle,M., Herrmann,C., Wittinghofer,A. and Staeheli,P. (1995) J. Biol. Chem., 270, 13512–13517. [PubMed] [Google Scholar]