


Abstract
Members of the RecQ family of DNA helicases are involved in processes linked to DNA replication, DNA recombination and gene silencing. RecQ homologues of various animals have been described recently. Here, for the first time for plants, we characterised cDNAs of all in all six different RecQ-like proteins that are expressed to different extents in Arabidopsis thaliana. Surprisingly, three of these proteins are small in size [AtRecQl1, AtRecQl2, AtRecQl3—606, 705 and 713 amino acids (aa), respectively], whereas the two bigger proteins result from a duplication event during plant evolution [AtRecQl4A and AtRecQl4B—1150 and 1182 aa, respectively]. Another homologue (AtRecQsim, 858 aa) most probably arose by insertion of an unrelated sequence within its helicase domain. The presence of these homologues demonstrates the conservation of RecQ family functions in higher eukaryotes. We also detected a small gene (AtWRNexo) encoding 285 aa which, being devoid of any RecQ-like helicase domain, reveals a striking homology to the exonuclease domain of human Werner protein, a prominent RecQ helicase of larger size. By means of the two-hybrid assay we were able to detect an interaction between AtWRNexo and AtRecQl2, indicating that activities that reside in a single protein chain in mammals might in plants be complemented in trans.
INTRODUCTION
The RecQ family of DNA helicases has recently attracted wide attention as members of this family are involved in biological processes such as DNA replication and recombination in pro- and eukaryotes (1–3). Mutations of three different members of the human RecQ family result in severe genetic disorders: Bloom syndrome (4), Werner syndrome (5) and Rothmund–Thomson syndrome (6). Very recently a connection between the action of RecQ homologues and gene expression was demonstrated by the involvement of QDE3 of Neurospora crassa in post-transcriptional gene silencing (7).
The RecQ protein family is composed of different members that vary in size and in the presence or absence of specific functional domains. Besides RecQ of Escherichia coli, in the last couple of years more than 20 different members have been described for most ‘model’ organisms as Saccharomyces cerevisiae, Schizosaccharomyces pombe, N.crassa, Drosophila melanogaster, Caenorhabditis elegans, Xenopus laevis, Mus musculus and Homo sapiens but not for plants. Whereas in the genome of E.coli and yeast only one RecQ homologue is present, five different homologues have been found so far in the human genome. The investigated members of the RecQ family show a remarkable sequence conservation within all seven helicase domains (8). Beyond these domains sequence comparisons revealed considerable variation in length and sequence motives ranging from very small members like RecQ itself or RecQL (9) to extremely large members like FFA from X.laevis (10) and QDE3 from N.crassa (7). In general there are three different groups of proteins belonging to the RecQ family: first, the small RecQ-like proteins of 400–750 amino acids (aa); secondly, the longer RecQ-like homologues such as BLM or QDE3 of 1000–1900 aa; and thirdly, large proteins harbouring an additional exonuclease domain like WRN and FFA.
RecQ itself has been shown to be a repressor of illegitimate recombination in E.coli (11). All described phenotypes resulting from mutations of genes belonging to the RecQ helicase family reveal enhanced genomic instabilities, in particular chromosomal aberrations. Such phenomena might result from the improper resolution of unusual DNA structures that occur during replication and recombination processes in the cells (3). In line with this suggestion a number of reports describe the capability of RecQ family members to interact with DNA substrates such as G4-DNA (12–14) and synthetic Holliday junctions (15). It has been demonstrated that several RecQ homologues interact with topoisomerases (16–20) and that this interaction might be required for the disruption of joint molecules in control of DNA recombination (21).
In this work we describe the molecular characterisation of five true RecQ-like proteins from Arabidopsis thaliana. Surprisingly, three of these predicted proteins are small in size (AtRecQl1, AtRecQl2, AtRecQl3—606, 705 and 713 aa, respectively), whereas the larger two proteins result from a recent duplication event (AtRecQl4A and AtRecQl4B—1150 and 1182 aa, respectively). We were also able to identify two more cDNAs (AtRecQsim and AtWRNexo) with similarities to RecQ homologues in the Arabidopsis genome. A protein (AtRecQsim, 858 aa) which carries within its helicase domain a putative insertion was characterised, as well as a small protein of 285 aa which reveals a striking homology to the exonuclease domain of hWRN protein. By two-hybrid analysis we were able to demonstrate that AtWRNexo interacts with AtRecQl2, indicating that two activities that reside in a single polypeptide chain within the hWRN in animals might be complemented in plants in trans.
MATERIALS AND METHODS
Accession numbers
The cDNA sequences for all the genes characterised in this study have been deposited in the EMBL GenBank under the accession numbers AJ404470–AJ404476.
Computer analysis
BLAST searches were performed within the Stanford genomic database resources of A.thaliana using the Bloom and Werner syndrome protein sequences of H.sapiens and TBLASTN (22). Sequence analysis was done with DNASTAR (Lasergene) and multiple alignments on the internet using CLUSTALW (Version 1.7). Pair wise comparisons were done with the MEGALIGN program (Lipman–Pearson method) of the DNASTAR package (Lasergene) using a Ktuple of 2 and a gap penalty of 4.
RNA and DNA isolation
Arabidopsis thaliana ecotype columbia RNA was isolated using peqGOLD TriFast (peqlab, Erlangen) following the instructions of the manufacturer. The following tissues were used: mature rosette leaves, flower shoots, complete flowers or 2–4-week-old sterile grown seedlings. Genomic DNA of A.thaliana ecotype columbia was isolated from 2–4-week-old sterile grown seedlings with the Plant DNA Midi Kit (peqlab, Erlangen).
Southern analysis
Genomic DNA (6 µg) of A.thaliana ecotype columbia was digested with the respective restriction enzymes overnight. After electrophoresis in 0.9% agarose the gel was denatured, neutralised, and transferred overnight onto a Hybond-NX nylon membrane (Amersham, Braunschweig) by capillary transfer using 20× SSC as transfer buffer. After blotting, the membrane was cross-linked with 120 mJ/cm2 in a Stratalinker 1800 (Stratagene, Amsterdam). Hybridisation of the Southern blots and signal detection was done with the Digoxigenin chemiluminescent labelling and detection system from Roche Molecular Biochemicals (Mannheim). The Dig DNA labelling Kit was used for labelling the probe, Dig Easy Hyb as hybridisation buffer and CPD Star as chemiluminescent substrate. The different probes used to detect the described genes were in all cases homologous to non-conserved parts of the genes to avoid cross hybridisation.
Reverse transcription (RT)–PCR and RACE
RT–PCR as well as 5′- and 3′-RACE experiments were done according to a modified SMART-protocol from Clontech (Heidelberg) (23) as described (24). Sequence analysis of the RT–PCR products was done as described (24).
Two-hybrid assay
To test possible protein–protein interactions between the proteins of interest, we used the MATCHMAKER Two-Hybrid System 2 from Clontech (Heidelberg) with the GAL4AD-vector pGAD-GH and the GAL4BD-vector pAS2-1. The X-gal staining was performed by colony-lift filter assay according to the Clontech protocol. The clones were also tested for self-activation by growing on HIS minus medium with 25 mM 3-AT and by the same X-gal staining procedure. After the staining assay we rechecked for the presence of the inserted genes in all tested yeast clones.
RESULTS
Identification of RecQ homologues in Arabidopsis and cDNA analysis
The rapidly growing sequence database of A.thaliana provides a powerful tool for identifying putative homologous proteins by database searches with sequence motives of genes of known function from different organisms. Such a database search using TBLASTN with the conserved helicase motives of the Bloom and the Werner syndrome protein sequences of H.sapiens resulted in six significant hits with genomic Arabidopsis sequences. Starting from the conserved domain, partial cDNAs were amplified by RT–PCR followed by RACE analysis to establish the complete cDNA sequences as described (24). The exon–intron composition of the genes was determined by comparison of the determined cDNA sequences with the genomic BAC clones listed in Table 1. Figure 1 shows schematically the intron–exon structure of all six identified RecQ-like genes of Arabidopsis. A summary of the relevant data is presented in Table 1. The genes contain a relatively high number of introns compared to the average of A.thaliana, which is around five per gene (25). Interestingly, two of the introns (Fig. 1, marked by black boxes) are present in nearly conserved positions in all six RecQ-like genes. One intron is located shortly after or in the DEAH box and the second shortly upstream of the FGMG box. All other intron positions vary considerably between the different RecQ homologues.
Table 1. Compilation of molecular properties of the isolated cDNAs.
| Gene | Accession number | cDNA/ORF (nt) | Protein (aa) | Exons (no.) | Kozak optimal |
|---|---|---|---|---|---|
| cDNA/ | (–3) (+4) | ||||
| genomic BACs | A/G–ATGG | ||||
| AtRecQl1 | AJ404470 | 2060/1821 | 606 | 10 | A–ATGA |
| AC012393 | |||||
| AtRecQl2 | AJ404471 | ∼2316/2118 | 705 | 20 | T–ATGG |
| AC007654 | |||||
| AtRecQl3 | AJ404472 | 2415/2142 | 713 | 19 | G–ATGA |
| AL031135 | |||||
| AtRecQl4A | AJ404473 | 3916/3549 | 1182 | 26 | C–ATGA |
| AC007259 | |||||
| AtRecQl4B | AJ404474 | 3877/3453 | 1150 | 26 | T–ATGG |
| AC018908 | |||||
| AtRecQsim | AJ404475 | 2988/2577 | 858 | 16 | T–ATGG |
| AC007478 | |||||
| AtWRNexo | AJ404476 | 1146/858 | 285 | 6 | G–ATGT |
| AL035528 |
Figure 1.

Schematic genomic structure of six RecQ-like homologues of A.thaliana. Introns are shown as white boxes within the grey shaded sequence. Introns with a conserved position throughout the six genes are shown as black boxes and are numbered above. The letters below the schematic sequence structure denote the most conserved amino acids of helicase domains II and V (see also Fig. 3).
Peculiarities of the different RecQ homologues
Except for AtRecQl4A, all genes analysed possess semi-optimal Kozak sequences around their respective translational start codons. The putative initiation site of AtRecQl4A, however, deviates in both the –3 and +4 position from the general rule indicating an unfavourable environment for initiation of translation. However, as no alternative ATG with a better consensus sequence could be detected we nevertheless assume that the identified ATG is indeed the start codon.
In the case of AtRecQl2, 5′-RACE could not be applied to determine the exact length of the expressed mRNA. This was due to the fact that a sequence motif of three cytosines is present in the mRNA before the putative start codon. Because the RACE-specific primer contains a motif of three guanines, cDNA synthesis was initiated at this position. Using a primer that bound to the putative mRNA sequences before the CCC motif indicated that the mRNA was indeed slightly longer. However, using another primer further upstream of the binding site that included another methionine in frame with the putative translation start site, gave a band of only ∼10% intensity of the PCR product in comparison to the primers further downstream, indicating at least that the majority of mRNAs did not include this methionine. Nevertheless, we cannot exclude that in a subpopulation of mRNAs this methionine might be used as translational start site and longer mRNAs of AtRecQl2 may exist. As we were not able to detect larger mRNAs in the respective northern blot (data not shown) and by the use of other primers complementary to sequences further upstream, we can at least exclude the possibility that such a AtRecQl2 subform exists in large quantities in Arabidopsis.
AtRecQl3 harbours two very small exons (numbers 3 and 11) of only 8 and 4 nt, respectively (Fig. 1). Such mini-exons are currently under investigation as to their unusual splicing mechanism, which involves specific sequences in the preceding introns (26). AtRecQl4A and 4B both possess an intron in their 5′-UTR.
Sequence analysis of the AtRecQsim gene revealed two special features. First, it has an unusual structure within its seven conserved helicase domains (Fig. 2). Although AtRecQsim possesses all seven domains and the homology is in good agreement with the other AtRecQ-like sequences, between domains III and IV a unique sequence of ∼160 aa is present (Fig. 2) which shows no similarity to any other RecQ homologue or any other gene. As the putative insertion disrupts the highly conserved distance within the helicase domains we assume that the protein might not be a functional RecQ-like helicase any more and decided to classify the protein as RecQ-similar and not as RecQ-like. Secondly, the gene possesses an intron in the 3′-UTR located 19 nt downstream of the stop codon (Fig. 1).
Figure 2.

Schematic structure of RecQ-like proteins from different organisms. The deduced proteins were aligned according to the helicase domains (shown as filled boxes). Stretches of acidic or basic amino acids are represented as light grey or dark grey boxes, respectively. The exonuclease domain of the human WRN protein is indicated. The size of the predicted proteins is given on the right.
Comparison of the different AtRecQ homologues with RecQ-like proteins of other organisms
All isolated RecQ-like genes from A.thaliana fitted very well in an alignment of the seven conserved helicase domains (Fig. 3) (8). The identity at amino acid level within this area is between 31% (different AtRecQls to HsRecQl4) and 53% (AtRecQl4A to BLM). Interestingly, the two longer RecQ homologues AtRQL4A and 4B show the highest amino acid identity with the human BLM protein (53%) and the yeast SGS1 (46%) when the aligned region is restricted to the most conserved region within the seven helicase domains (Fig. 3). In general there is also a reasonable number of highly conserved amino acids outside of the helicase domains. In particular, the amino acid sequence LLYVTPE between domains Ia and II and the three amino acids tyrosine (Y) histidine (H) and alanine (A) between domains IV and V are nearly 100% conserved (Fig. 3), a phenomenon that has already been described (8) and also applies for A.thaliana.
Figure 3.

Multiple alignment of the seven highly conserved helicase domains of RecQ-like proteins from A.thaliana and various other organisms. The multiple alignment was done with CLUSTALW (version 1.7; www.ibc.wustl.edu ). Gaps are represented by dashes, amino acids conserved in at least eight out of the ten sequences are grey shaded and the consensus amino acid is given in bold below the sequences. The helicase domains are numbered above the sequences. Amino acids considered as conserved in a given position are: (D,E); (K,R); (I,L and V). The organisms included in the alignment are, from top to bottom: E.coli; A.thaliana; N.crassa; H.sapiens; M.musculus and S.cerevisiae.
In Figure 2 the protein structure of different members of the RecQ family is shown schematically. All together the different members exhibit a great variation in size and organisation around the seven conserved helicase domains. Ranging from 606 to 1955 aa, the proteins can classified into two groups by size (Fig. 2, upper and lower parts). Except for the RecQ protein of E.coli, all other members of the family depicted in Figure 2 possess reasonable stretches of acidic amino acids (indicated as grey boxes). These acidic areas are not conserved in their relative position between family members. In the case of AtRecQsim there is an additional stretch of basic amino acids at its C-terminus (Fig. 2).
AtRecQl4A and B originate from a duplication event during plant evolution
Sequence comparison of AtRecQl4A and 4B revealed that both genes were closely related and apparently arose by a recent sequence duplication. Further analysis of sequence data from the Arabidopsis genome revealed that the duplication on chromosome I is not limited to the area of AtRecQl4A and B. The whole duplication consists of a 33–35 kb region in which eight predicted genes are located. Five of these genes, including AtRecQl4A and B, are highly conserved (70% protein identity) between both loci. Interestingly, both RecQ-like genes are ~70% identical at nucleic acid and protein level and all 25 intron positions are identical. Even more striking is the fact that all introns have the same phase in both genes. Thus, intron positions and phases are much more conserved than the nucleotide sequence.
All AtRecQ-like genes are present once in the Arabidopsis genome
To test whether the isolated cDNA originate from single copy genes, Southern blots were performed. For all Southern blots 6 µg of genomic DNA from A.thaliana ecotype columbia was restricted with ClaI, EcoRI or EcoRV and hybridised with labelled single-stranded DNA (500–1000 nt) of the genes. The observed banding pattern was, in all cases except for AtRecQL4A, in accordance with the assumption of single copy genes and according to theoretical calculations using the sequence information of the respective BAC clones. The duplicated genes AtRecQl4A and B showed cross hybridisation (Fig. 4), which demonstrates that 75% identity at nucleotide level is sufficient to give a hybridisation signal under the conditions used. However, the observed banding pattern reveals that the two genes are also present as single copies in the A.thaliana genome.
Figure 4.

Southern analysis of the A.thaliana members of the RecQ family. The indicated fragment sizes are calculated from the sequence information of the corresponding genomic BAC clones. All genes show a banding pattern that is in accordance with a presumed single copy status in the Arabidopsis genome.
Isolation of a short WRN-like gene (AtWRNexo)
It has been claimed that a WRN-like gene is present in A.thaliana genome on chromosome 4 (27). We isolated the corresponding cDNA (Table 1) and confirmed the 5′- and 3′-ends by RACE (Fig. 5A). The gene consists of six exons and encodes a small protein of 285 aa. The sequence analysis demonstrated that the gene contains no RecQ-like helicase domains and that the homology is restricted to the N-terminal part, the exonuclease domain of the hWRN protein. A sequence comparison is given in Figure 5B. As demonstrated by Southern blotting performed under highly stringent conditions, the gene occurs as one copy in the Arabidopsis genome (data not shown).
Figure 5.

The small WRNexo gene from A.thaliana. (A) Schematic intron/exon structure of the gene. The five introns are represented as white boxes in the grey shaded sequence. (B) Alignment of AtWRNexo with the N-terminus of the WRN protein from mouse and the X.laevis FFA protein. Amino acids conserved in all three positions are given as letters below the sequence. Highly conserved amino acid stretches are represented as grey shaded boxes. Amino acids considered as conserved in a given position are: (D,E); (K,R); (I,L and V).
The two-hybrid assay revealed that AtWRNexo and AtRecQl2 can interact
As we were surprised to find that AtWRNexo, in contrast to hWRN, did not carry RecQ-like sequence motives, we were tempted to speculate that AtWRNexo might interact in trans with one or the other of the smaller RecQ homologues that we detected in A.thaliana. Therefore, we performed two-hybrid assays of AtWRNexo cloned in the GAL4AD-vector pGAD-GH and with the RecQ-like proteins 1, 2 and 3 of A.thaliana in the GAL4BD-vector pAS2-1. As shown in Figure 6 we were able to detect a strong interaction between AtWRNexo and AtRecQl2 but not between AtWRNexo and AtRecQl1 or between AtWRNexo and AtRecQl3.
Figure 6.
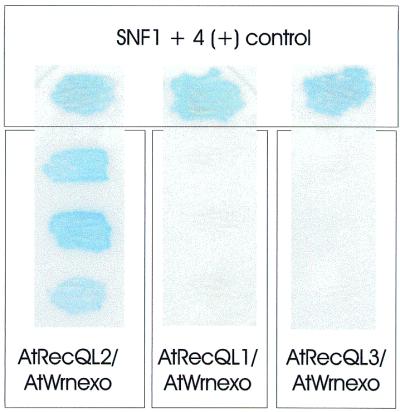
Two-hybrid analysis to detect interactions of AtWRNexo with AtRecQl1 to 3. AtWRNexo and AtRecQl2 showed a strong interaction after 4 h X-gal staining (left). The panels on the right with AtRecQl1 or 3 and AtWRNexo were X-gal stained for 12 h without resulting in a positive signal. As a control the SNF1 + 4 interaction is given in the upper part of the figure.
Expression analysis
Northern blots of mRNA from different tissues (leaves, flowers and stems) of A.thaliana revealed a low but detectable expression of the AtRecQ-like genes in all tissues tested. No organ-specific expression of any gene could be detected; the strongest signals were obtained in flower and shoot tissue. In all cases only one prominent band could be detected that was in accordance with the size calculated from the respective cDNA analysis (data not shown). For direct comparisons of expression levels comparative RT–PCRs were performed (Fig. 7A and B). Using mRNA isolated from rosette leaves, shoots, flowers and 3-week-old seedlings, AtRecQl1 to 4B and AtWRNexo specific primers, we were able to demonstrate different degrees of expression. The expression of the RecQ-like genes 1, 2, 4A and 4B was higher in shoots and flowers than in leaves and seedlings, indicating an expression preference for dividing tissues. In contrast, the mRNA level of AtWRNexo and the AtRecQl3 did not differ much between all examined tissues (Fig. 7A). To characterise the expression levels of the individual genes we performed comparative RT–PCR with mRNA from flowers in a dilution series ranging from 1 to 100. We were able to detect the following degrees of expression in flowers: for AtRecQl1, 2 and 4B ∼100 times weaker than for AtWRNexo and for AtRecQl3 and 4A about 10 times weaker expression than for AtWRNexo (Fig. 7B). This is also in accordance with the fact that expressed sequence tags (ESTs) with high homology to WRNexo can be found by computer search in several plant species whereas we were not able to identify any EST of the RecQ-like genes. The expression of the AtRecQsim gene was in all tissues much lower than the other AtRecQ-like genes (data not shown).
Figure 7.

Comparative RT–PCR of AtRecQl1 to 4B and AtWRNexo using mRNA from different tissues (A) and a quantification of expression in flowers (B). (A) Each cDNA was amplified with 38 PCR cycles starting with reverse transcription of 10 ng mRNA from Arabidopsis rosette leaves, shoots, flowers or seedlings. (B) Each cDNA was amplified with 35 PCR cycles starting with reverse transcription of 10 ng mRNA from Arabidopsis flowers or a 1:10 or 1:100 dilution of this RT, respectively. (A and B) One-fifth of the PCR was subjected to gel electrophoresis and EtBr staining.
DISCUSSION
In the present communication we report on the isolation of six cDNA sequences from Arabidopsis with similarities to the RecQ gene of E.coli. As sequencing of the Arabidopsis genome is still in progress, more RecQ homologues might be found in due course. Nevertheless, the presence of six different genes in the Arabidopsis genome all with RecQ helicase-specific motives is surprising, as in all other organisms fewer homologues have been identified so far. In the human genome five RecQ-like genes were identified, two smaller (HRecQL, HRecQ4 of 649 and 410 aa, respectively) and three larger genes (BLM, WRN and HRecQ5 of 1417, 1432 and 1208 aa, respectively) (2). Therefore we were surprised to detect three smaller homologues of 606, 705 and 713 aa in Arabidopsis. We wondered whether one or the other might be transported to the chloroplast. To address this question we used the ChloroP-Server (http://www.cbs.dtu.dk/services/ChloroP/ ) to find putative transit peptide sequences. Within all RecQ-homologues isolated from Arabidopsis no indication of the presence of a transit peptide was found (data not shown). As on the other side a sequence motif that was homologous to the exonuclease domain of the Werner protein was detected in the Arabidopsis genome (27) and our analysis revealed the presence of a respective cDNA devoid of any helicase domain, it was tempting to speculate that the AtWRNexo protein might interact in trans with one or more of the short RecQ homologues. Indeed, via two-hybrid analysis we were able to detect such an interaction with AtRecQl2 but not AtRecQl1 and 3. Although further biochemical studies like co-immunoprecipitation are required to sustain the biological significance of our finding, the two-hybrid interaction of AtWRNexo with AtRecQl2 indicates that both functions, present in a single polypeptide chain in mammals, might be supplied in plants in trans. We also tried to apply the two-hybrid analysis for the other RecQ homologues. Unfortunately the cDNAs proved to be unstable, both in yeast (AtRecQl4A and 4B) or during cloning in E.coli (AtRecQsim). Therefore, for the moment further conclusions about an interaction of these proteins with the AtWRNexo protein have to await alternative interaction studies with purified proteins.
We were able to detect, via EST search, homologues of AtWRNexo in Medicago sativa, Oryza sativa, Lotus japonicus and glycine max (data not shown) indicating that this gene is common at least to higher plants. The only significant homologies that we were able to detect with proteins outside the plant kingdom were to the exonuclease domains of the WRN proteins from mouse, human and X.laevis. Therefore it is tempting to speculate that during eukaryotic genome evolution two unlinked motives were fused to a single gene. Although such an event must have been extremely rare, the selective advantage of carrying both activities in a single peptide chain seems to have conserved the fusion in animal evolution.
As the two longer RecQ homologues of Arabidopsis (RecQl4A and RecQl4B of 1150 and 1182 aa, respectively) arose by a recent gene duplication event during plant evolution, only one ‘kind’ of long RecQ homologue could be identified. Notably RecQl4A and RecQl4B are most similar to the human BLM protein.
The molecular structure of the AtRecQsim gene indicates that the gene most probably descends from a RecQ-like gene into which an unrelated sequence was inserted. Interestingly, this sequence contains a high number of acidic amino acids. Notably, acidic domains are found in many other RecQ homologues like AtRecQl1, AtRecQl4A and 4B, QDE3, BLM, WRN, SGS1 outside of the helicase domain (Fig. 2). It will therefore be especially interesting to determine the biochemical activities of AtRecQsim.
The presented data are a prerequisite for the elucidation of the biological function of RecQ homologues in plants. It will be interesting to investigate which of the AtRecQ homologues is able to at least partial complement the Sgs1 mutation in yeast (28). It will be possible to define interactions with topoisomerases (16–20) or other proteins involved in DNA repair and DNA replication (29–33). In vitro expression of the proteins will lead to further characterisation of their function (15,21). Insertion mutants or plants in which the expression of the respective gene is suppressed will provide us with new insight into the function of the respective genes in DNA repair and replication in vivo. It will be of specific interest to test, as has been demonstrated for QDE3 in N.crassa, whether RecQ homologues are involved in post-transcriptional gene silencing phenomena in plants (34).
Our study clearly exemplifies the evolution of RecQ homologues in higher eukaryotes. Whereas E.coli and lower eukaryotes like S.cerevisiae and S.pombe only contain one RecQ homologue in their genome, both in the animal and plant kingdoms multiple homologues are found. These homologues seem to have evolved first by duplication and then by consecutive shuffling or fusion with other protein domains. Comparing AtRecQl4A and AtRecQl4B we were able to detect such a recent duplication event. Analysing AtRecQsim we found strong indications for the insertion of an acidic domain into the conserved helicase domains of a RecQ homologue. The presence of independent genes for the WRN-exonuclease (AtWRNexo) and RecQ domains (AtRecQ2) in Arabidopsis that resides within a single protein in mammals (WRN) is a strong argument for a fusion event during animal evolution.
Acknowledgments
ACKNOWLEDGEMENTS
We would like to thank I. Schubert for critical comments on the manuscript, and Anja Ipsen and Andrea Kunze for help and discussions. The work was partly funded by grant A15 of the SFB 363 of the Deutsche Forschungsgemeinschaft.
DDBJ/EMBL/GenBank accession nos AJ404470–AJ404676
REFERENCES
- 1.Karow J.K., Wu,L. and Hickson,I.D. (2000) Curr. Opin. Genet. Dev., 10, 32–38. [DOI] [PubMed] [Google Scholar]
- 2.Shen J.C. and Loeb,L.A. (2000) Trends Genet., 16, 213–220. [DOI] [PubMed] [Google Scholar]
- 3.Chakraverty R.K. and Hickson,I.D. (1999) Bioessays, 21, 286–294. [DOI] [PubMed] [Google Scholar]
- 4.Ellis N.A., Groden,J., Ye,T.Z., Straughen,J., Lennon,D.J., Ciocci,S., Proytcheva,M. and German,J. (1995) Cell, 83, 655–666. [DOI] [PubMed] [Google Scholar]
- 5.Yu C.E., Oshima,J., Fu,Y.H., Wijsman,E.M., Hisama,F., Alisch,R., Matthews,S., Nakura,J., Miki,T., Ouais,S., Martin,G.M., Mulligan,J. and Schellenberg,G.D. (1996) Science, 272, 258–262. [DOI] [PubMed] [Google Scholar]
- 6.Kitao S., Shimamoto,A., Goto,M., Miller,R.W., Smithson,W.A., Lindor,N.M. and Furuichi,Y. (1999) Nature Genet., 22, 82–84. [DOI] [PubMed] [Google Scholar]
- 7.Cogoni C. and Macino,G. (1999) Science, 286, 2342–2344. [DOI] [PubMed] [Google Scholar]
- 8.Kitao S., Ohsugi,I., Ichikawa,K., Goto,M., Furuichi,Y. and Shimamoto,A. (1998) Genomics, 54, 443–452. [DOI] [PubMed] [Google Scholar]
- 9.Puranam K.L. and Blackshear,P.J. (1994) J. Biol. Chem., 269, 29838–29845. [PubMed] [Google Scholar]
- 10.Yan H., Chen,C.Y., Kobayashi,R. and Newport,J. (1998) Nature Genet., 19, 375–378. [DOI] [PubMed] [Google Scholar]
- 11.Hanada K., Ukita,T., Kohno,Y., Saito,K., Kato,J. and Ikeda,H. (1997) Proc. Natl Acad. Sci. USA, 94, 3860–3865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fry M. and Loeb,L.A. (1999) J. Biol. Chem., 274, 12797–12802. [DOI] [PubMed] [Google Scholar]
- 13.Sun H., Karow,J.K., Hickson,I.D. and Maizels,N. (1998) J. Biol. Chem., 273, 27587–27592. [DOI] [PubMed] [Google Scholar]
- 14.Sun H., Bennett,R.J. and Maizels,N. (1999) Nucleic Acids Res., 27, 1978–1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Karow J.K., Constantinou,A., Li,J.L., West,S.C. and Hickson,I.D. (2000) Proc. Natl Acad. Sci. USA, 97, 6504–6508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gangloff S., McDonald,J.P., Bendixen,C., Arthur,L. and Rothstein,R. (1994) Mol. Cell. Biol., 14, 8391–8398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Watt P.M., Louis,E.J., Borts,R.H. and Hickson,I.D. (1995) Cell, 81, 253–260. [DOI] [PubMed] [Google Scholar]
- 18.Wu L., Davies,S.L., North,P.S., Goulaouic,H., Riou,J.F., Turley,H., Gatter,K.C. and Hickson,I.D. (2000) J. Biol. Chem., 275, 9636–9644. [DOI] [PubMed] [Google Scholar]
- 19.Johnson F.B., Lombard,D.B., Neff,N.F., Mastrangelo,M.A., Dewolf,W., Ellis,N.A., Marciniak,R.A., Yin,Y., Jaenisch,R. and Guarente,L. (2000) Cancer Res., 60, 1162–1167. [PubMed] [Google Scholar]
- 20.Shimamoto A., Nishikawa,K., Kitao,S. and Furuichi,Y. (2000) Nucleic Acids Res., 28, 1647–1655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Harmon F.G., DiGate,R.J. and Kowalczykowski,S.C. (1999) Mol. Cell, 3, 611–620. [DOI] [PubMed] [Google Scholar]
- 22.Altschul S.F., Gish,W., Miller,W., Myers,E.W. and Lipman,D.J. (1990) J. Mol. Biol., 215, 403–410. [DOI] [PubMed] [Google Scholar]
- 23.Matz M., Shagin,D., Bogdanova,E., Britanova,O., Lukyanov,S., Diatchenko,L. and Chenchik,A. (1999) Nucleic Acids Res., 27, 1558–1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hartung F. and Puchta,H. (2000) Nucleic Acids Res., 28, 1548–1554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Deutsch M. and Long,M. (1999) Nucleic Acids Res., 27, 3219–3228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Simpson C.G., Hedley,P.E., Watters,J.A., Clark,G.P., McQuade,C., Machray,G.C. and Brown,J.W. (2000) RNA, 6, 422–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mayer K. et al. (1999) Nature, 402, 769–777. [DOI] [PubMed] [Google Scholar]
- 28.Yamagata K., Kato,J., Shimamoto,A., Goto,M., Furuichi,Y. and Ikeda,H. (1998) Proc. Natl Acad. Sci. USA, 95, 8733–8738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Brosh R.M. Jr, Orren,D.K., Nehlin,J.O., Ravn,P.H., Kenny,M.K., Machwe,A. and Bohr,V.A. (1999) J. Biol. Chem., 274, 18341–18350. [DOI] [PubMed] [Google Scholar]
- 30.Lebel M., Spillare,E.A., Harris,C.C. and Leder,P. (1999) J. Biol. Chem., 274, 37795–37799. [DOI] [PubMed] [Google Scholar]
- 31.Kamath-Loeb A.S., Johansson,E., Burgers,P.M. and Loeb,L.A. (2000) Proc. Natl Acad. Sci. USA, 97, 4603–4608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cooper M.P., Machwe,A., Orren,D.K., Brosh,R.M., Ramsden,D. and Bohr,V.A. (2000) Genes Dev., 14, 907–912. [PMC free article] [PubMed] [Google Scholar]
- 33.Wang Y., Cortez,D., Yazdi,P., Neff,N., Elledge,S.J. and Qin,J. (2000) Genes Dev., 14, 927–939. [PMC free article] [PubMed] [Google Scholar]
- 34.Cogoni C. and Macino,G. (1999) Curr. Opin. Microbiol., 2, 657–662. [DOI] [PubMed] [Google Scholar]