

Abstract
Intracerebral hemorrhage (ICH) is a prevalent hemorrhagic cerebrovascular emergency. Alleviating neurological damage in the early stages of ICH is critical for enhancing patient prognosis and survival rate. A novel form of cell death called ferroptosis is intimately linked to hemorrhage-induced brain tissue injury. Although studies have demonstrated the significant preventive impact of bovine serum albumin-stabilized selenium nanoparticles (BSA-SeNPs) against disorders connected to the neurological system, the neuroprotective effect on the hemorrhage stroke and the mechanism remain unknown. Therefore, based on the favorable biocompatibility of BSA-SeNPs, h-ICH (hippocampus-intracerebral hemorrhage) model was constructed to perform BSA-SeNPs therapy. As expected, these BSA-SeNPs could effectively improve the cognitive deficits and ameliorate the damage of hippocampal neuron. Furthermore, BSA-SeNPs reverse the morphology of mitochondria and enhanced the mitochondrial function, evidenced by mitochondrial respiration function (OCR) and mitochondrial membrane potential (MMP). Mechanistically, BSA-SeNPs could efficiently activate the Nrf2 to enhance the expression of antioxidant GPX4 at mRNA and protein levels, and further inhibit lipid peroxidation production in erastin-induced ferroptotic damages. Taken together, this study not only sheds light on the clinical application of BSA-SeNPs, but also provides its newly theoretical support for the strategy of the intervention and treatment of neurological impairment following ICH.
Keywords: BSA-selenium nanoparticles, Intracerebral hemorrhage, Ferroptosis, Cognitive function, Nrf2-GPX4 axis
Graphical abstract
Schematic diagram of the neuroprotective effect and mechanism of BSA-SeNPs in hemorrhagic stroke by inhibiting ferroptosis.
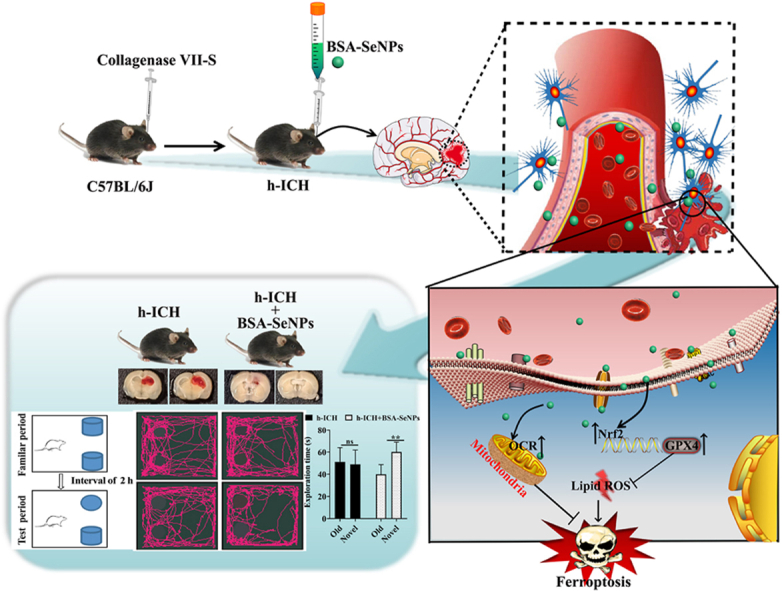
1. Introduction
Intracerebral hemorrhage (ICH) is a common hemorrhagic stroke subtype caused by the rupture of the vascular system. Along with the aging of population, the prevalence of ICH is growing rapidly, and it has become one of the serious diseases affecting human health. Moreover, ICH has high disability rate and fatality rate [1], about 90 % of the ICH survivors were left with varying degrees of cognitive dysfunction, which imposes a huge mental and social burden on patients [2]. At present, surgery is the main treatment for patients with cerebral hemorrhage [3]. Combined with symptomatic therapy can alleviate the primary injury after intracerebral hemorrhage to a certain extent, but it is ineffective to halt or restore the secondary brain injury (SBI) caused by cerebral hemorrhage [4]. More importantly, SBI after intracerebral hemorrhage is an important cause of neurological dysfunction [5]. Therefore, reducing secondary brain damage and relieving neurological dysfunction following cerebral hemorrhage have significant therapeutic implications for enhancing the prognosis and survival rate of patients with ICH.
There is a recent proposal linking the mechanisms of neurological impairment following ICH to ferroptosis, a type of regulated oxidative stress cell death that is iron-dependent [6]. It has been reported that excessive iron was released from broken blood vessels after hemorrhagic stroke [7]. The excessive iron activates oxidative stress, induces the accumulation of lipid per-oxidation, and eventually lead to secondary brain damage after hemorrhagic stroke, which induced neuronal death and neurological impairment [8]. Additionally, ferroptosis is the root cause of secondary brain damage [9]. Iron accumulation and lipid peroxidation are typical manifestations of ferroptosis. More importantly, a large number of studies have shown that DFO (iron chelator) or inhibiting lipid peroxides can mitigate brain damage and improve neurological function [10]. Furthermore, it has been reported that iron chelators conduct phase II clinical studies and increase the likelihood of a favorable clinical result for individuals with ICH [11]. Based on the above analysis, it is of great significance to find an effective strategy to improve neurological function in hemorrhagic stroke from the perspective of ferroptosis.
Selenium (Se), as an essential trace element for human health, can incorporated into selenoprotein and perform vital physiological functions such as scavenging free radicals and preventing oxidation [12]. Studies have shown that sodium selenite (an inorganic form of selenium) can inhibit ferroptosis by promoting the expression of GPX4 in the brain to exert neuroprotective effects [13]. However, sodium selenite has great toxicity and is not easily absorbed for human body, which is greatly limited in clinical application. It has been shown that using nanotechnology to facilitate antioxidant therapy is a viable treatment strategy for ICH caused by oxidative damage [14,15]. BSA-SeNPs,synthesized from sodium selenite as a raw material, are considered a new selenium substance compared to organic or inorganic selenium. Selenium nanoparticles (BSA-SeNPs) are highly sought-after materials because of its many advantages, such as its strong antioxidant, optimum biocompatibility, and ecologically friendly processes, enable its potential utilization as nanomedicine or nano-delivery in a therapeutic application [16]. Currently, it is unclear whether and how selenium nanoparticles mediate ferroptosis, to improve the repair of second brain damage following ICH. Therefore, we used BSA-SeNPs to fine-tune the antioxidant potential of hippocampal neuron in this work.
Nuclear factor erythroid-2-related factor 2 (Nrf2) is a key transcription regulator of cellular antioxidant response. It has been reported that the accumulation of Nrf2 in nucleus mitigated the brain damage after ICH [17]. Knock out of Nrf2 gene results in more pronounced hematoma volumes, neurological deficits, and increased brain edema after ICH [18]. The neuroprotective effect of Nrf2 is responsible for regulating the expression of genes that counteract oxidative stress, preventing lipid peroxidation, and further inhibiting ferroptosis [19]. Glutathione peroxidase 4 (GPX4), an extremely vital role involved in ferroptosis, is an established Nrf2 transcriptional target. After ICH, the treatment of GSH can alleviate neurological impairment [20] and the anti-oxidative function of GSH is catalyzed by GPX4 in ferroptosis [21]. Similarly, the increases of GPX4 (a kind of selenoprotein) by delivery of selenium can prevent ferroptosis and improve neural functional recovery following ICH [13,22]. Therefore, Nrf2-GPX4 axis may be a potential therapeutic target in the treatment of ICH.
In the present work, we proposed using BSA-SeNPs-based nanotechnology to prevent ferroptosis in h-ICH model. We discovered that BSA-SeNPs have encouraging biocompatibility both in vitro and in vivo. Importantly, we observed that BSA-SeNPs could alleviate the cognitive impairments in the ICH mice model, stabilize the hemorrhagic region, and prevent the hippocampus cell damage. Furthermore, we revealed that BSA-SeNPs could upregulate the expressions of Nrf2 and GPX4. Attributed to such regulations, BSA-SeNPs could inhibit ferroptosis and lessen mitochondrial damage, including mitochondria respiration,mitochondrial membrane potential. In conclusion, we provide a novel approach for improving cognitive deficits following ICH by using nanotechnology, but not gene modification. Our innovative resolution will lay a theoretical foundation for the development of clinical drugs for neurological dysfunction after ICH.
2. Materials and methods
2.1. Materials
Hemin (Cat#51280) ,BSA (Bovine Serum Albumin,Cat# V900933), Sodium selenite (Cat #214485) were both purchased from Sigma. Beta Actin Ab (Cat #AF7018) and GPX4 Antibody (Cat#DF6701) were purchased from Affinity Bioscience; Nrf2 Ab (16396-1-AP) was purchased from protein tech. BODIPY C11581/591 (Cat#D3861, Invitrogen), Etramethylrhodamine (TMRM, Cat#T668, Invitrogen), and MitoTracker® Mitochondrion-Selective Probes (Cat#M22426, Invitrogen) were obtained from Thermo Fisher Scientific. Tubulin-Tracker Red (Cat# C1050) was obtained from beyotime. Malondialdehyde (MDA) detection kit (Cat# A003-4-1) was obtained from Nanjing Jiancheng Bioengineering Institute. Mouse 4-HNE (4-Hydroxynonenal) ELISA Kit (Cat# EM1583) was purchased from FineTest.
2.2. Preparation of BSA-SeNPs and labeled with FITC (FITC-BSA-SeNPs)
Na2SeO3 (25 mM, 2.5 mL) was dissolved in glutathione (GSH, 25 mM in 10 mL). 0.4 g of bovine serum albumin (BSA) was then added. After adjusting the solution pH to 7.2, NaOH was added dropwise, and the mixture was agitated for another 2 h. Then the aforementioned reaction solution was centrifuged for 30 min at 12000 rpm. The precipitate was washed two times with sterilized deionized water to obtain BSA-SeNPs. In order to prepare FITC-BSA-SeNPs, fluorescein isothiocyanate (FITC) solution and BSA-SeNPs suspension (1:50) were mixed overnight at room temperature. After centrifugation at 9000g for 30 min, FITC-BSA-SeNPs were produced following two washes with 70 % ethanol solution.
2.3. Characterization of the BSA-SeNPs
The size distribution and zeta potential of prepared BSA-SeNPs were characterized by dynamic light scattering (DLS, Malvern, Zetasizer Nano ZS), and the morphology of BSA-SeNPs was observed by transmission electron microscope (TEM, JEOL 2100F).
2.4. PBMC (peripheral blood mononuclear cells) isolation, in-vitro Vδ2 cell culture, and toxicity assessment in vitro
The whole blood of healthy participants was used to isolate PBMCs using a standard density gradient centrifugation technique based on Ficoll-Paque. Following the normal culture technique for 10 d prior to further experiments, isolated PBMCs were suspended in media (10 mL 10 % RPMI-1640) contained with 10 ng/mL IL-2 and 50 μM zoledronate. The cells were then seeded into 48-well plates. Following labeling for Vδ2 and CD3 molecules using fluorescent antibodies, Vδ2 cells were identified via flow cytometry. Unless otherwise indicated, Vδ2 cells that were employed in our tests ranged in maturity from 10 to 15 d, and their purity exceeded 90 % in total CD3+ cells. Following their respective treatments with BSA-SeNPs (6 or 12 h), the function of CD8 and Vδ2 cells were detected used flow cytometry.
2.5. In vivo toxicity assessment
Following collagenase injection, mice in various groups were given nanoparticles (40 μg/L or 80 μg/L) and were euthanized 1 and 7 d after the injury. For the serum biochemistry test, the blood of every mouse was separated. For hematoxylin and eosin (H&E) staining, the primary organs including the heart, liver, spleen, lungs, and kidneys were taken informally at 7 d after after the injury.
2.6. Quantitative real-time RT-PCR
From cell samples, total RNA was extracted using an earlier procedure [23]. The PrimeScript™ RT reagent Kit (Cat#: RR047A, Takara) was then used to reverse-transcribe the RNA into cDNA. Sangon Biotech provided the mRNA primers. GPX4 and Nrf2 mRNA expression levels (Table 1) were detected with a Real-Time System (Bio-Rad). Both GPX4 and Nrf2 genes were compared to β-actin.
Table 1.
RT-PCR primers.
| Genes | Genes Name | Sequence (5’→3′) |
|---|---|---|
| Nrf2 | Nrf2- F | GTTGCCACCGCCAGGACTAC |
| Nrf2- R | GTGCTCAGAAACCTCCTTCCAAAAC | |
| GPX4 | GPX4-F | ATAAGAACGGCTGCGTGGTGAAG |
| GPX4-R | TAGAGATAGCACGGCAGGTCCTTC |
2.7. Cell culture of HT22 cells and cell model of ICH
Mouse hippocampal neuron HT22 were cultured at 37 °C with 5 % CO2 in Dulbecco's modified Eagle's medium (DMEM), 10 % FBS, and puromycin (4 mg/mL; Sigma). Treatment with hemin (5, 25, 125 μM; Sigma) provoked cell death in mouse hippocampus neuron HT22. Cells were subjected to 50 μM hemin co-treatment with BSA-SeNPs (0.25–2 μM) or DFO (40 μΜ; Selleck) for 6 h in order to perform neurological protective studies.
2.8. Lipid ROS, cellular ROS, and mitochondrial membrane potential (MMP) identified using flow cytometry
The cells were cultured in 6-well plates, and when roughly 60 % of the cells had merged, the corresponding therapy was administered to the various groups. After cell harvesting, the cells were treated for half an hour with either tetramethylrhodamine (TMRM, staining MMP), DCFDA dye (cellular ROS), or 1 μM BODIPYTM581/591C11 (a fluorescent dye specific for lipid reactive oxygen species). After that, PBS was used twice to rinse the cells. The fluorescence intensity values were determined by flow cytometry. The amount of lipid ROS, intracellular ROS, or MMP level in the cells is represented by the mean values of each group's fluorescence intensity.
2.9. Seahorse analysis for phenotyping the mitochondrial respiration and glycolysis
Mitochondrial respiration and glycolysis are two crucial metabolic characteristics of live cells that may be provided by the Seahorse XF technology. The oxygen consumption ratio (OCR) and the extracellular and acidification rate (ECAR) both demonstrate how well cells can metabolize glucose using the Seahorse XF Cell Mito Stress Kit and the Glycolysis Stress Test Kit. The OCR and ECAR parameters were assessed by the Seahorse XFe 96 Extracellular Flux Analyzer (Agilent Technologies, Inc.). First, Seahorse XF 96 cell culture micro-plates were seeded with 2 × 104 hippocampal neuron HT22 per well and left incubating overnight. Oligomycin (1.5 μM), FCCP (0.5 μM), and antenone/antimycin A (0.5 μM) were successively injected to create the OCR curve, which was used to measure OCR. In order to produce the ECAR curve, glucose (10 mM), oligomycin (1.0 μM), and 2-DG (50 mM) were administered. The Seahorse XF 96 Wave software was then used to evaluate all of the data.
2.10. mRNA-Seq analysis of HT22 cells
HT22 cells that were either co-treated with BSA-SeNPs or not were collected, rinsed with cold PBS, and then 1 mL of trizol was used to extract total RNA in order to undertake transcriptome sequencing. We measured the amount and purity of RNA using a NanoDrop 2000 spectrophotometer. After the libraries were built, RNA integrity was checked. Then, RNA sequencing and analysis were carried out by the OE Biotech Co., Ltd. (Shanghai, China). Next, we used R language to perform a heatmap analysis of the differential genes, with a significance threshold of P < 0.05.
2.11. Mitochondria morphology observed under the transmission electronic microscopy
Hippocampal neuron HT22 cells were fixed with 2.5 % glutaraldehyde overnight. After washing the cells 5 times with PBS, fix the cells again with 1 % osmic acid for 2 h and subsequently washed with PBS for 3 times. The cells were dehydrated by 50 %, 70 %, 90 % acetone respectively for 20 min and dehydrated by 100 % acetone 20 min for 3 times. After that, the cells were soaked with pure acetone and resin (2:1) for 3–4 h at room temperature. The embedment polymerization was performed at 60 °C for 24 h. The ultrathin sections (50–70 nm, Leica, Germany) were collected and counterstained with uranyl acetate and lead citrate. Finally, all micrographs were taken by transmission electron microscope (H-7500, Hitachi, Japan).
2.12. Imaging by confocal microscopy
After treatment with selenium nanoparticles and hemin, the culture medium was discarded and the neurons were blocked with 5%BSA in PBS solution for 10 min at room temperature to eliminate non-specific staining. Then, the related fluorescent dyes (Mitotracker red) were used for staining. The changes of mitochondria in the prepared cell samples were observed under laser confocal.
2.13. Collagenase-induced h-ICH (hippocampus-intracerebral hemorrhage) mouse model
Male mice (8 weeks) were anesthetized with 1 % pentobarbital sodium (50 mg/kg). Under aseptic condition, mice were skin-prepared, erythromycin eye ointment was applied to the eyes, and iodine was used to disinfect the head position. Then, mice were fixed using a brain stereotaxic frame. The stereotaxic coordinates as follows: behind: 2.5 mm,left: 1.7 mm (x = −1.7 mm, y = −2.5 mm). Using a Hamilton syringe, 500 nL of collagenase (0.045 U/μL) was infused into the left hippocampus at a flow rate of 5 nL/s. After the injection, the needle was retained for 5 min and the needle was withdrawn slowly. Subsequently, the skin of mice was sutured and wiped with iodine. During the procedure, the mice were placed on a thermostatic heating pad. After modeling for 2 h, nanoparticles with different concentrations were administered immediately. The mice in the sham-operated group were injected with 500 nL PBS at the same coordinates.
2.14. Immunohistochemical staining
Nissl Staining is a method used to stain cells in neural tissue. HE (hematoxylin-eosin) staining is a frequently used staining method in histology to visualize cells and structures in tissues. The tissue from the brain was first permeated with 0.9 % normal saline, then fixed for 2 d with 4 % paraformaldehyde, and then dehydrated for 2 d using a solution of 30 % sucrose made with 0.01 M PBS. We used a cryostat microtome to serially slice the brain into 5 m-thick slices. We then stained the coronal sections with 0.1 % cresyl violet (Nissl) or hematoxylin and eosin (H&E) before mounting them with neutral glue. The samples were examined using image processing software (Budapest, Hungary) and the size of the hematoma was then measured using software.
2.15. Hemorrhage volume analysis
1 d or 7 d after h-ICH,the brain tissue of the mice was removed after anesthetized. The 1-mm brain slices were produced, and were fixed for 1 min in a 4 % paraformaldehyde solution (PFA). The images were analyzed using digital imaging and Image J software to determine the volume of hemorrhage.
2.16. Novel objective recognition test
There are three objects (A, B and C) in the environment, in which A and B are exactly the same, and C is completely different from A and B. Mice were first acclimated in the rearing cage for 5–7 d after purchase. 3 d before the test, animals were placed in the test environment, and the experimental mice were stroked for 1 min daily to allow the mice to acclimate to the tester and the test environment. At the beginning of the training period, objects A and B were placed on the left and right ends of one side wall, and the mice were put into the field with their backs facing the two objects, and moved freely for 10 min (familiarized phase). After that, the contact time between the mouse and the object was recorded using a stopwatch. Each mouse was given a 60-min interval before entering the test period. After that, the previous object B was changed to object C, and the mouse was still turned back to the two objects and observed for 2–5 min while being recorded with a video device. In this process, the exposure time of mice to object C was mainly observed. At the end of all experiments, all mice were returned to their original cages.
2.17. The content of 4-HNE detection
Mouse 4-HNE (4-Hydroxynonenal) ELISA Kit was used to measure 4-HNE content in compliance with the manufacturer's instructions (Fine test, Cat#EM1583). A microplate reader was used to quickly read and calculate the OD450 values at 450 nm. The standard curve, which was created beforehand, was used to compute the 4-HNE concentration.
2.18. MDA measurement
The MDA detection kit was used to measure MDA content in accordance with the manufacturer's protocol (Nanjing Jiancheng Bioengineering Institute). In short, a microplate reader (Thermo Fisher Scientific) was used to identify MDA content of the cells. The computational formula based on the manufacturer was used to compute the MDA concentration.
2.19. Western blotting
Western blotting was used to identify the expressions of Nrf2 and GPX4 proteins. From HT-22 cells, total protein was isolated. The BCA technique was used to measure the protein concentrations. A total of 30 μg protein was electro-blotted on polyvinylidene difluoride (PVDF Membrane (Millipore)) membranes after being separated by 10 % or 12.5 % SDS-PAGE at 80V electrophoresis. The membranes were then incubated for 2 h at room temperature in 10 % non-fat dry milk (Beyotime, Shanghai, China). Following that, the Rb anti- Nrf2 (1:1000) and Rb anti-GPX4 (1:1000) were respectively incubated under 4 °C for overnight. The PVDF membrane was then immersed in the appropriate secondary antibody (1:2000, Cat:AF7018, Affinity Biosciences, USA) and incubated for 2 h at room temperature following three TBST rinsing. An imaging system (Evolution-Capt Edge, Vilber, France) and an immunoblotting chemical luminescence kit (Thermo Fisher Scientific, Walsam, MA, USA) were used to display the optical density. The imageJ was used to analyze images.
2.20. Statistical analysis
Utilizing GraphPad Prism 8, all graphs and statistical analyses of all data (represented as mean S.E.M.) were completed. In triplicate, multiple individual experiments were performed. The two-tailed t-test was used to compare the two groups. The one-way analysis of variance (ANOVA) was used to compare more than two groups.
3. Results
3.1. Characterization and biocompatibility of BSA-SeNPs in vitro and in vivo
The generated BSA-SeNPs exhibit evenly dispersed spherical nanoparticles with a particle size of about 40 nm (The polydispersity index (PDI) of BSA-SeNPs is 0.0319) and the average zeta potential of BSA-SeNPs is around 10 mV (a positive potential charge), as illustrated in Fig. 1A and B, which were identified via transmission electron microscopy and dynamic light scattering detection. This is a very advantageous precondition for its biological uses, such as cellular activity and endocytosis [24] Furthermore, hippocampal neuronal cells were not rendered toxic after its treatment with BSA-SeNPs at varying concentrations (1–32 μM) for 6 h, but Na2SeO3 treatment at concentrations of 8–32 μM clearly caused cytotoxicity when compared to untreated cells (Fig. 1C), indicating that BSA-SeNPs have better biocompatibility in hippocampal neuronal cells. In order to demonstrate the biocompatibility of BSA-SeNPs in vitro, we separated immune cells such as CD8 (the primary effector cells of human acquired immunity) and Vδ2 cells (a crucial part of human T cells [25]) from peripheral blood. We then used flow cytometry to identify the apoptosis and function of CD8 and Vδ2 cells, respectively, after BSA-SeNPs treatment. As shown in Fig. 1D and E, BSA-SeNPs treatment (6 or 12 h) had no effect on the apoptosis of Vδ2 and CD8 cells. While Vδ2 and CD8 cells dramatically increased their rate of apoptosis when treated with sodium selenite (Na2SeO3, 12 h). Additionally, the functional markers TNF-a, IFN-γ, and PD-1 of Vδ2 and CD8 cells were unaffected by BSA-SeNPs treatment (Figure F–K).
Fig. 1.

Characterization of BSA-SeNPs and biocompatibility of BSA-SeNPs in vitro. (A–B) The distribution of zeta potential, particle size, and TEM images of BSA-SeNPs. (C) Hippocampal neuronal cells were treated with BSA-SeNPs and Na2SeO3 at concentrations of 1–32 μM, respectively, for 6 h. The living cells were detected by CCK-8. (D–E) Vδ2 and CD8 cells were double-stained with Annexin V and Propidium iodide (PI) after treatment with BSA-SeNPs (0.5 μM) or Na2SeO3 (0.5 μM) for 6 h or 12 h. (F–K) The expressions of TNF-a, IFN-γ, and PD-1 were detected by flow cytometry treatment with BSA-SeNPs (0.5 μM) for 6 h n = 3, ****P < 0.0001, ***P < 0.001, **P < 0.01, *P < 0.05, ns: no significance.
To further observe the biocompatibility of BSA-SeNPs in vivo, we isolated important tissues from mouse to perform HE staining. As shown in Fig. 2A, compared with the control group, there were no obvious organic changes in important tissues such as heart, liver, spleen, lung and kidney. At the same time, the outcomes of 6 biochemical analysis in the blood (Fig. 2B–G) showed that selenium nanoparticles did not affect the content of ALT (glutamic-pyruvic transaminase), AST (glutamic oxalacetic transaminase), TG (triglyceride), CHO (cholesterol), BUN (Blood urea nitrogen) and CK (creatine kinase) in the blood of mice.
Fig. 2.

Biosafety of BSA-SeNPs was tested in vivo. (A) HE staining of heart, liver, spleen, lung and kidney (n = 4,x100). (B–G) Results of 6 blood biochemical tests (n = 4); ns, no significance.
3.2. BSA-SeNPs reduced hemorrhagic area and hippocampal neuron damage in h-ICH mice during the acute phase
To further confirm the neuroprotective effect of BSA-SeNPs after hemorrhagic stroke in vivo, especially during the acute phase (hemorrhage that begins within 72 h [26]), we established a hippocampal intracerebral hemorrhage mouse model (h-ICH mouse), and then injected BSA-SeNPs into the hippocampus and isolated brain tissue for detection (Fig. 3A). The h-ICH model mice with saline injection showed large-area hemorrhage, as shown in Fig. 3B and C, however following injection of BSA-SeNPs in the hippocampus, hemorrhagic area was significantly decreased in h-ICH mice in a dose-dependent manner. For example, the saline group had a substantial hemorrhagic area of 4.39 %, however following injection of BSA-SeNPs (40 μg/L and 80 μg/L), the hemorrhagic area was reduced to 2.29 and 1.94 %, respectively. Furthermore, pathological analysis of brain tissue also revealed the therapeutic effects of BSA-SeNPs as a neuroprotective agent. First, hematoxylin and eosin (H&E) staining in brain slices of h-ICH animals revealed severe necrosis in the hippocampus-intracerebral hemorrhage area, whereas BSA-SeNPs administration significantly reduced the area of necrosis (Fig. 3D). As a result, we further used Nissl staining to investigate the damage to neurons in the hemorrhagic area of h-ICH mice. As shown in Fig. 3E, in the hemorrhagic area of h-ICH mice, the number of intact neurons was much lower than in the control group, and they also displayed uneven morphology and disorganized arrangement. Furthermore, after receiving BSA-SeNPs in h-ICH mice, the Nissl positive neurons in the brain tissue had no effect in DG field of the hippocampus (Fig. 3F), whereas the Nissl positive neurons in the CA1 (Fig. 3G) and CA3 (Fig. 3H) regions of the hippocampus had a significant effect. This suggested that BSA-SeNPs treatment alleviated neuron damage in the CA1 and CA3 regions of the hippocampus during the acute phase.
Fig. 3.

Situ injection of selenium nanoparticles in hippocampus reduces the area of hematoma and the damage of hippocampus neuron in h-ICH mice. (A) Illustrations of the experimental grouping and the treatment procedures. (B–C) Statistical data and representative images of brain slices after treatment with BSA-SeNPs (40 μg/L and 80 μg/L) in h-ICH mouse mode (n = 4). (D) Representative images of HE staining in the hippocampus (n = 4, scale bar: 200 μm, 20 μm). (E–H) Images of Nissl staining (n = 3, scale bar: 20 μm) of treated hippocampus DG (F), CA1 (G), and CA3 (H) fields. *P < 0.05, **P < 0.01, vs Control; #P < 0.05, ##P < 0.01, vs h-ICH; ns, no significance.
3.3. BSA-SeNPs attenuated the cognitive deficits and damage of hippocampal neuron in subacute period of h-ICH mice
To further explore the enhancement of brain protection of BSA-SeNPs, we also constructed C57 mice h-ICH model to examine the effects of BSA-SeNPs on the mice during the subacute period of hemorrhage (hemorrhage that begins within 3 d–21 d [26]). Mice were conducted a Novel Objective recognition test at seventh day after injection of BSA-SeNPs into the hippocampus (Fig. 4 A). Further, hemorrhage area, HE, and Nissl staining of the hippocampus in mice were evaluated (Fig. 4 A). Behavioral assessments were examined and shown in Fig. 4 B-D. Neither control nor h-ICH mice differed in exploration time for old or new objects, however injection of BSA-SeNPs in the hippocampus (Fig. 4 B and C) significantly increased the exploration time for novel objects in the h-ICH mice. Meanwhile, the discrimination index in the h-ICH mice group was even brought to a negative value; however, after undergoing BSA-SeNPs treatment during the test period, the discrimination index was dramatically recovered (Fig. 4D). Furthermore, the hemorrhage area of h-ICH mice was significantly restored by BSA-SeNPs treatment (Fig. 4E).
Fig. 4.

BSA-SeNPs shield the hippocampus from neuronal injury and cognitive decline in the subacute phase of h-ICH. (A) Illustrations of the experimental grouping and the treatment procedures. (B–D) Schematic diagram of the testing procedure (B), the exploration time (C), and the discrimination index (D) in the object recognition task (n = 4). (E) Hemorrhagic volume (F) Representative images of HE staining in the hippocampus (n = 4, scale bar: 200 μm, 20 μm). (G–J) Images of Nissl staining (n = 4) of treated hippocampus (DG, CA1, and CA3 fields, scale bar: 20 μm), DG: dentate gyrus. *P < 0.05, vs Control; #P < 0.05, vs h-ICH; ns, no significance.
We further clarified the therapeutic benefits of BSA-SeNPs as a neuroprotective substance by pathological investigation in the hippocampus. First, the H&E staining results in the hippocampus of ICH mice showed a trend toward a decrease in the number of intact neurons, along with irregular morphology and disorganized arrangement in the brain hemorrhage area. BSA-SeNPs, however, significantly decreased the area of hemorrhaging and enhanced the quantity of undamaged neurons (Fig. 4 F). Therefore, we also examined the damage to neurons in the hemorrhage area of ICH mice using Nissl staining. As shown in Fig. 4 G-J, compared with the control group, Nissl positive neurons were decreased in dentate gyrus (DG) region of the hippocampus. However, at seventh day after treatment with BSA-SeNPs, Nissl positive neurons showed a significant dose-dependent increase. In addition, at seventh day after BSA-SeNPs treatment, there was an increasing trend in Nissl positive neurons both in the CA1 and CA3 areas of the hippocampus.
3.4. BSA-SeNPs ameliorated ferroptosis in hemin/erastin-exposed hippocampal neuronal cells
Hippocampal neuronal cells were co-treated with hemin and BSA-SeNPs for 6 h, BSA-SeNPs overtly abrogated hemin-induced cytotoxicity (Fig. 5A and B). To confirm the effects of BSA-SeNPs on anti-ferroptosis in hippocampal neuronal cells, we examined the classical indicators (lipid ros, 4-HNE, MDA) of ferroptosis. Data demonstrated that BSA-SeNPs (0.5 μM) significantly inhibited hemin-induced lipid peroxidation (Fig. 5B). More importantly, cytometry showed that BSA-SeNPs and Fer-1 (ferroptosis Inhibitor) significantly decreased ferroptosis inducer erastin-induced lipid peroxidation in hippocampal neuronal cells (Fig. 5C). Besides, erastin-increased 4-HNE, MDA contents were dramatically reduced by the co-treatments of BSA-SeNPs and Fer-1 (Fig. 5D and E). Collectively, BSA-SeNPs efficiently shielded hippocampus neuronal cells against ferroptic damage caused by hemin/erastin.
Fig. 5.

BSA-SeNPs inhibited hemin/erastin-induced ferroptotic damages in vitro. Hippocampal neuronal cells were co-treated with hemin or erastin and BSA-SeNPs for 6 h. (A–B) The relative living cell viability was imaged and detected by CCK-8 and after co-treatment with hemin and BSA-SeNPs (0.5 μM) or Na2SeO3 (0.5 μM) for 6 h. (C–D) The content of lipid ros was examined by flow cytometry. (E–F) The content of 4-HNE, MDA were respectively detected by cellular mouse 4-HNE (4-Hydroxynonenal) elisa kit and MDA elisa kit. ****P < 0.0001, **P < 0.01, *P < 0.05.
3.5. BSA-SeNPs mitigate hemin-induced mitochondrial damage in hippocampal neurons
Mitochondrial shrinkage is a typical morphological feature of ferroptosis [27] so we examined whether BSA-SeNPs alter mitochondria morphology in hippocampal neurons. Using transmission electron microscopy, we observed that compares with hemin treatment alone, the mtiochondrial morphology of the hippocampal neurons co-treated with hemin and BSA-SeNPs was stretched (Fig. 6 A). Then, we used confocal imaging to detect the co-localization of BSA-SeNPs (Green) and mitochondrion (Red) in hippocampal neurons and further ascertain the effect of BSA-SeNPs on the mitochondria (Fig. 6 B). Meanwhile, we also observed that BSA-SeNPs inhibited hemin-increased lipid ROS (Green), which mainly occurred in the mitochondria of hippocampal neurons (Supplementary Fig. 2). In addition, we explored the effects of BSA-SeNPs on the two major energy production pathways, mitochondrial respiration (OCR) and glycolysis (ECAR), in hippocampal neurons. As shown in Fig. 7 A-F, after co-treated with BSA-SeNPs and hemin, basal respiration (Fig. 7C), ATP-production (Fig. 7 D), H+ proton leak (Fig. 7 E), maximal respiration (Fig. 7 F), were all elevated compared with hemin treatment. Co-treated with BSA-SeNPs and hemin did not have effect on glycolysis (ECAR) in hippocampal neurons (Supplementary Fig. 1). Also, we detected the mitochondrion biosynthesis and mitochondrial membrane potential (MMP, TMRM staining) after co-treated with BSA-SeNPs and hemin by flow cytometry. We found that compared with hemin treatment, mean fluorescence intensity of TMRM was weaken with BSA-SeNPs treatment after hemin exposure (BSA-SeNPs + Hemin group, Fig. 7G–I) and mean fluorescence intensity of mitotracker red was made no difference (Supplementary Fig. 4A). These data indicated that BSA-SeNPs only enhanced mitochondrial function and did not facilitate mitochondrial biosynthesis of hippocampal neurons.
Fig. 6.

Effects of BSA-SeNPs on mitochondrial morphology. (A) Transmission electron microscopy (TEM) images of hippocampal neuronal cells treated with Hemin (6 h), BSA-SeNPs + Hemin (6 h), BSA-SeNPs (6 h). Shrunken mitochondria were marked as single white arrowheads (upper scale bar: 2 μm, bottom scale bar: 500 nm). (B) Internalization of BSA-SeNPs into mitochondria of hippocampal neurons was imaged by confocal microscopy (scale bar: 16 μm). The nucleus of hippocampal neuron was stained with Hoechst (blue); mitochondria were stained with Mitotracker Red (red).
Fig. 7.

Effects of BSA-SeNPs on mitochondrial function and respiration in hippocampal neurons. (A) The profile of the Agilent Seahorse XF Cell Mito Stress Test [28] illustrates the essential elements of mitochondrial function. (B–F) Key parameters of mitochondrial function: basal respiration (C), ATP-production (D), H+ proton leak (E), maximal respiration (F) of Mitochondria respiration in hippocampal neurons before and after BSA-SeNPs treatment were detected by Seahorse XF96 Mito Stress Kit. (G–I) Mitochondrial membrane potential (TMRM staining) of hippocampal neurons before and after BSA-SeNPs treatment was analyzed by confocal microscopy (G, scale bar: 16 μm) and flow cytometry (H–I). ***P < 0. 001, **P < 0.01, *P < 0.05, ns: no significance.
The main mechanism of ferroptosis is to catalyze the high expression of unsaturated fatty acids on the cell membrane, resulting in lipid peroxidation, which induce cell death [29]. In view of this, we evaluated whether BSA-SeNPs could interact with cytoskeletal structures (phospholipid bilayer). We observed the co-localization of FITC-BSA-SeNPs (Green) and tubulin (Red, a cytoskeletal protein) in hippocampal neurons by confocal imaging (Supplementary Fig. 2). Then, with or without treatment of BSA-SeNPs after hemin exposure, we detected the expressions of tubulin in hippocampal neurons by using flow cytometry and found that treatment of BSA-SeNPs did not have effect on the expression of tubulin (Supplementary Fig. 4B).
3.6. BSA-SeNPs modulates ferroptosis via up-regulating Nrf2-GPX4 axis
To further explore the mechanism of BSA-SeNPs modulates ferroptosis in hippocampal neurons,we performed mRNA sequence on hippocampal neuronal cells after BSA-SeNPs treatment (Fig. 8A–D). As the result showed that there are 2234 differentially expressed genes (DEGs) in Hemin vs Control (Fig. 8 A),37 DEGs in BSA-SeNPs_Hemin vs Hemin (Fig. 8 B), and only 15 DEGs in BSA-SeNPs vs Control (Fig. 8C). Then, we performed Gene Set Enrichment Analysis (GSEA) based on the gene sets and we found that the ‘glutathione metabolic process’ was enriched in BSA-SeNPs_Hemin phenotype (Fig. 8 D). The glutathione metabolic process was closely associated with ferroptosis [30]. To our note, GPX4 palys a key role in the glutathione metabolic process and also act as a “star factor” in ferroptosis [31]. Moreover, Nrf2 is considered as a master regulator of the antioxidant response and inhibition of Nrf2, or several of its downstream target genes, is linked to increased cell death [19]. It is worth noting that GPX4, as a kind of selenoproteins, is also a direct downstream target gene of Nrf2. In addition, pharmacological selenium (Se) augments GPX4 to protect neurons [13]. Emerging evidence have been reported that Nrf2-GPX4 axis was closely related with ferroptosis [32,33] Therefore, we speculated that Nrf2-GPX4 axis may be invovled in BSA-SeNPs modulating ferroptosis.
Fig. 8.

BSA-SeNPs reduced GPX4-mediated lipid peroxidation by activating Nrf2. (A–C) Differences in gene expression level cluster analysis. Red represented relatively high expression protein-coding genes, blue represented protein-coding genes expressed in relatively low. Hemin vs Control (2234);BSA-SeNPs_Hemin vs Hemin (37);BSA-SeNPs vs Control (15). (D) GSEA plots of the gene sets upon the BSA-SeNPs_Hemin and Hemin group. (E, F) The expressions of GPX4 and Nrf2 mRNA in hippocampal neuronal cells were assessed by q-PCR. (G–I) The protein expressions of GPX4 and Nrf2 were assessed by Western blot analysis after treatment with THBQ ((5 μM, 24h). (J–K) The level of lipid ROS was assessed by flow cytometry after co-treated with BSA-SeNPs (0.5 μM, 6h), THBQ, PKU ((10 μM, 24h) respectively in erastin (8 μM, 6h)-exposed hippocampal neurons. (L–M) The protein expressions of Nrf2 (M) and GPX4 (N) were assessed by Western blot analysis after treatment with mL385 (5 μM, 24 h). (O) Lipid ROS level was assessed by flow cytometry after co-treated with mL385 and BSA-SeNPs in erastin-exposed hippocampal neurons. *P < 0.05, **P < 0.01, ****P < 0.0001, ns: no significance.
Then, we performed q-PCR and western blotting to dertemine the expressions of Nrf2 and GPX4 after cotreatment with BSA-SeNPs. As shown in Fig. 8 E-F, Nrf2 and GPX4 mRNA were decreased in hippocampal neurons after treatment with ferroptosis activitor erastin. While, co-treatment with BSA-SeNPs significantly increased the mRNA expressions of Nrf2 and GPX4. Consistent with the mRNA results, the expressions of GPX4 and Nrf2 proteins were significantly increased after BSA-SeNPs treatment (Fig. 8G–H). To confirm the regulatory role of Nrf2 on GPX4 in hippocampal neurons, we further used western blotting to detect changes in GPX4 expression after activating Nrf2 expression by THBQ treatment. As shown in Fig. 8 I, overexpression of Nrf2 led to an increase of GPX4 expression. Then, to further assess whether Nrf2-GPX4 axis was invovled in ferroptosis, activation of Nrf2 by THBQ treatment or activation of GPX4 by PKU both attenuated erastin-elevated lipid ros (Fig. 8J–K). Moreover, to evaluate whether Nrf2-GPX4 axis-mediated the protective effects of BSA-SeNPs against ferroptosis, we used ML385 (the Nrf2 specific inhibitor, also inhibited its targeted proteins) to pretreated hippocampal neurons for 18 h and then con-treated with or without BSA-SeNPs for 6 h (Fig. 8L–O). The results showed that treatment with ML385 (5 μM, 24 h) significantly inhibited the expressions of Nrf2 protein (Fig. 8 M) and GPX4 protein (Fig. 8 M). Then, pretreated with ML385 abrogated the inhibited effect of BSA-SeNPs on lipid ROS level (Fig. 8 O). Altogether, our data demonstrated that BSA-SeNPs alleviated lipid peroxidation by activating the Nrf2-GPX4 axis.
4. Discussion
ICH is the most prevalent subtype of hemorrhagic stroke. Currently, most treatment approaches available are limited to alleviating the primary injury of ICH. Therefore, it is vital to elucidate the pathogenesis of secondary brain injury (SBI) in order to create medications that can gradually improve neurological impairment of ICH. Previous studies indicates that ferrotosis is the cause of secondary brain injury [34,35], which is consistent with our findings in ICH model in vitro or in vivo. BSA-SeNPs protected the hippocampus and improved the cognitive function of h-ICH mouse model. Furthermore, it was shown that BSA-SeNPs inhibited cellular lipid peroxidation, preserving the functionality of neural cells in a manner similar to that of the ferroptosis antagonist Fer-1. The mechanism involved is that BSA-SeNPs activated Nrf2 and its downstream protein GPX4 expression, hence lowering the level of lipid peroxidation in neural cells and further preventing ferroptosis.
Recent researches revealed that a series of nanoparticles, such as ceria nanoparticles (CeNP) [15] and curcumin nanoparticles [14], have anti-oxidative stress properties that may protect neurons in ICH mouse model. Among these, BSA-SeNPs attracted our attention as it has good biocompatibility in vivo and in vitro. After injection of BSA-SeNPs, there was no discernible change in the blood parameters for the liver (AST and ALT), heart (CK), TG, CHO, BUN, or BUN. In addition, treatment of BSA-SeNPs had no effect on the apoptosis and function of CD8 and Vδ2 cells isolated from human peripheral blood. Further research showed that situ injection of BSA-SeNPs significantly decreased the hippocampal damage (CA1 and CA3 regions of the hippocampus) and hematoma area during the acute phase of hemorrhagic stroke. Maybe, it is too early to observe any effect in the hippocampal DG region 24 h after BSA-SeNPs injection. Furthermore, BSA-SeNPs alleviated behavioral impairment, hippocampal damage (DG and CA1 of the hippocampus), and hematoma area in h-ICH mice during the subacute phase. These results indicated that BSA-SeNPs protected hippocampal neurons from damage, hence mitigating ICH pathogenesis.
The concealed mechanism of ferroptosis has been progressively disclosed since this form of cell death induced by erastin (a small molecule) was discovered in 2012 [27]. Ferroptosis is characterized by membrane lipid peroxidation and the accompanying accumulation of oxidative products [22]. Thus, one strategy to inhibit ferroptosis in mammalian cells may be to limit lipid peroxidation. In our study, BSA-SeNPs effectively reduced neurotoxicity and lipid peroxide production induced by hemin treatment (to mimic hemorrhagic stroke in vitro [36]). Subsequent research revealed that BSA-SeNPs and Fer-1 (ferroptosis inhibitor) eliminated the production of lipid peroxide and oxidative products (4-HNE and MDA) induced by ferroptosis inducer erastin. These data indicate that BSA-SeNPs alleviates ferroptosis-related lipid peroxidation. Mitochondria, an organelle bound by double membrane, play a key role in ferroptosis [37]. Ferroptotic cells generally have smaller mitochondria with increased membrane density under electron microscopy [27]. Furthermore, mitochondrial dysfunction was involved in the progression of neurodegenerative diseases [38]. After treated with hemin, the morphology of mitochondrial was shrunken in hippocampal neurons, but it was dramatically restored by BSA-SeNPs treatment which mainly co-located with mitochondria. Increasing datas indicate that cellular metabolism is necessary for ferroptosis, most likely because different stages of cellular metabolism are the primary source of lipid reactive oxygen species (ROS) [39,40]. Mitochondria played a central role in oxidative metabolism [41]. Given this, we thoroughly investigated the effects of BSA-SeNPs on the two main cellular metabolic routes, glycolysis (ECAR) and mitochondrial respiration (OCR). Following co-treatment with BSA-SeNPs, ATP generation, maximal respiration, H+ proton leak, and baseline respiration were all increased. However, BSA-SeNPs showed no effect on hippocampal neurons' glycolysis (ECAR). These results indicated that BSA-SeNPs mainly enhance mitochondrial respiratory function to promote cell metabolism. To further determine the effect of BSA-SeNPs on mitochondrial, we observed the change of mitochondrial membrane potential (MMP) and the MMP was diminished following BSA-SeNPs treatment. Emerging evidences demonstrated that steady-state redox balance regulating by antioxidants is directly linked to the development of the nervous system disease [42,43]. Nrf2 is recognized as a principal regulator of the antioxidant response pathway, since a large number of its downstream target genes are engaged in redox-imbalance prevention or correction in the cell [44]. Additionally, it has been demonstrated that Nrf2 is essential for modulating other important metabolic pathways, such as lipid metabolism [45]. Dysregulation of the Nrf2 pathway has been linked to the onset or progression of numerous diseases [45]. After ICH, Nrf2 is crucial in minimizing brain damage [17]. More importantly, Nrf2 pathway has been identified as target to exert their neuroprotective effects by antioxidants [46], such as phenolic acid derivative ferulic acid (FA) [47]. Here, Nrf2 activation has a significant protective effect on hippocampus damage. The expression of Nrf2 mRNA and protein was both declined in erastin-treated hippocampal neurons, which was impressively restored by BSA-SeNPs treatment. In addition,there is growing evidence of a strong link between Nrf2 and GPX4 [48,49]. Glutathione peroxidase 4 (GPX4) is a crucial anti-ferroptotic suppressor of lipid peroxides and is also a known Nrf2 transcriptional target [49]. In this study, we found that erastin induced significant inhibitions of GPX4 in hippocampal neurons, which was effectively recovered after BSA-SeNPs treatment. We confirmed that Nrf2 activation by THBQ up-regulated GPX4 expression, suppressing the production of toxic lipid ros. BSA-SeNPs effectively averted ferroptosis caused by erastin. In summary, our findings provide more evidence that BSA-SeNPs, through stimulating Nrf2/GPX4-mediated lipid peroxidation, are a significant therapeutic target for ICH.
Taken together, these data demonstrated that BSA-SeNPs, which alleviates lipid peroxidation by directly activating the Nrf2-GPX4 axis, is powerful in inhibiting ferroptosis of hippocampal neurons and the pathology of h-ICH mice. Therefore, BSA-SeNPs may have the potential to be used as an ICH therapeutic candidate, as the anti-feroptosis effect delays the progression of neurodegenerative diseases.
Funding
The authors sincerely thank all participants involved in this study. The study was supported by GuangDong Basic and Applied Basic Research Foundation (Cat: 2022A1515140039; 2022A1515140056); Key Technologies R&D Program of Huizhou (2022CZ010428); Huizhou Outstanding Youth Science and Technology Talent project (2023EQ050027, 2023EQ050026); Medical Science Foundation of Guangdong Province (A2024602, B2021152).
CRediT authorship contribution statement
Xiao-Na Li: Writing – original draft, Visualization, Validation, Methodology, Conceptualization. Li Lin: Writing – review & editing, Funding acquisition, Data curation, Conceptualization. Xiao-Wei Li: Writing – original draft, Methodology, Conceptualization. Qian Zhu: Visualization, Funding acquisition. Zhen-Yan Xie: Validation. Yong-Zhen Hu: Software, Funding acquisition. Qing-Shan Long: Funding acquisition. Xiao-Bing Wei: Formal analysis. Yi-Qi Wen: Resources. Li-Yang Zhang: Software. Qi-Keng Zhang: Validation. Ying-Chao Jing: Resources. Xin-Hua Wei: Writing – review & editing, Supervision, Conceptualization. Xue-Song Li: Writing – review & editing, Supervision, Funding acquisition, Conceptualization.
Declaration of competing interest
All authors declare that there are no competing interests.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.redox.2024.103268.
Contributor Information
Xin-Hua Wei, Email: eyxinhuawei@scut.edu.cn.
Xue-Song Li, Email: lxs75cedar@163.com.
Appendix A. Supplementary data
The following is the Supplementary data to this article.
Data availability
Data will be made available on request.
References
- 1.Lim T.C., Mandeville E., Weng D., Wang L.S., Kurisawa M., Leite-Morris K., et al. Hydrogel-based therapy for brain repair after intracerebral hemorrhage. Translational stroke research. 2020;11:412–417. doi: 10.1007/s12975-019-00721-y. [DOI] [PubMed] [Google Scholar]
- 2.Qureshi AI, Mendelow AD, Hanley DF. Intracerebral haemorrhage. Lancet. 2009;373(9675):1632–1644. doi: 10.1016/S0140-6736(09)60371-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zille M, Farr TD, Keep RF, Römer C, Xi G, Boltze J. Novel targets, treatments, and advanced models for intracerebral haemorrhage. EBioMed. 2022 Feb;76 doi: 10.1016/j.ebiom.2022.103880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Loan JJ, Kirby C, Emelianova K, Dando OR, Poon MT, Pimenova L, Hardingham GE, McColl BW, Klijn CJ, Al-Shahi Salman R, Schreuder FH, Samarasekera N. Secondary injury and inflammation after intracerebral haemorrhage: a systematic review and meta-analysis of molecular markers in patient brain tissue. J. Neurol. Neurosurg. Psychiatr. 2022 Feb;93(2):126–132. doi: 10.1136/jnnp-2021-327098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Puy L, Parry-Jones AR, Sandset EC, Dowlatshahi D, Ziai W, Cordonnier C. Intracerebral haemorrhage. Nat. Rev. Dis. Primers. 2023 Mar 16;9(1):14. doi: 10.1038/s41572-023-00424-7. [DOI] [PubMed] [Google Scholar]
- 6.Xu Y., Li K., Zhao Y., Zhou L., Liu Y., Zhao J. Role of ferroptosis in stroke. Cell. Mol. Neurobiol. 2022 doi: 10.1007/s10571-022-01196-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Duan X., Wen Z., Shen H., Shen M., Chen G. Intracerebral hemorrhage, oxidative stress, and antioxidant therapy. Oxid. Med. Cell. Longev. 2016;2016 doi: 10.1155/2016/1203285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wan J., Ren H., Wang J. vol. 4. 2019. pp. 93–95. (Iron Toxicity, Lipid Peroxidation and Ferroptosis after Intracerebral Haemorrhage). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cao Y., Li Y., He C., Yan F., Li J.R., Xu H.Z., et al. Selective ferroptosis inhibitor Liproxstatin-1 Attenuates neurological deficits and Neuroinflammation after Subarachnoid hemorrhage. Neurosci. Bull. 2021;37:535–549. doi: 10.1007/s12264-020-00620-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zeng L., Tan L., Li H., Zhang Q., Li Y., Guo J. Deferoxamine therapy for intracerebral hemorrhage. A systematic review. 2018;13 doi: 10.1371/journal.pone.0193615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Selim M. Deferoxamine mesylate: a new hope for intracerebral hemorrhage: from bench to clinical trials. Stroke. 2009;40:S90–S91. doi: 10.1161/STROKEAHA.108.533125. [DOI] [PubMed] [Google Scholar]
- 12.Mojadadi A., Au A., Salah W., Witting P. vol. 13. 2021. (Role for Selenium in Metabolic Homeostasis and Human Reproduction). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Alim I., Caulfield J.T., Chen Y., Swarup V., Geschwind D.H., Ivanova E., et al. Selenium Drives a transcriptional Adaptive Program to Block ferroptosis and Treat stroke. Cell. 2019;177:1262–1279 e25. doi: 10.1016/j.cell.2019.03.032. [DOI] [PubMed] [Google Scholar]
- 14.Yang C., Han M., Li R., Zhou L., Zhang Y., Duan L., et al. Curcumin nanoparticles inhibiting ferroptosis for the enhanced treatment of intracerebral hemorrhage. International journal of nanomedicine. 2021;16:8049–8065. doi: 10.2147/IJN.S334965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zheng J., Lu Jn, Mei S., Wu H., Sun Z., Fang Y., et al. Ceria nanoparticles ameliorate white matter injury after intracerebral hemorrhage: microglia-astrocyte involvement in remyelination. J. Neuroinflammation. 2021;18 doi: 10.1186/s12974-021-02101-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mikhailova E.O. Selenium nanoparticles: Green Synthesis and Biomedical application. Molecules. 2023;28 doi: 10.3390/molecules28248125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang Y., Yu W., Liu Y., Chang W., Wang M., Zhang L. Regulation of nuclear factor erythroid-2-related factor 2 as a potential therapeutic target in intracerebral hemorrhage. Front. Mol. Neurosci. 2022;15 doi: 10.3389/fnmol.2022.995518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cheng Y., Liu M. iTRAQ-based Quantitative Proteomics indicated Nrf2/OPTN-mediated Mitophagy inhibits NLRP3 Inflammasome activation after intracerebral hemorrhage. 2021;2021 doi: 10.1155/2021/6630281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dodson M., Castro-Portuguez R., Zhang D.D. NRF2 plays a critical role in mitigating lipid peroxidation and ferroptosis. Redox Biol. 2019;23 doi: 10.1016/j.redox.2019.101107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Diao X., Zhou Z., Xiang W., Jiang Y., Tian N., Tang X., et al. Glutathione alleviates acute intracerebral hemorrhage injury via reversing mitochondrial dysfunction. Brain Res. 2020;1727 doi: 10.1016/j.brainres.2019.146514. [DOI] [PubMed] [Google Scholar]
- 21.Bersuker K., Hendricks J.M., Li Z., Magtanong L., Ford B., Tang P.H., et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature. 2019;575:688–692. doi: 10.1038/s41586-019-1705-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ursini F., Maiorino M. Lipid peroxidation and ferroptosis: the role of GSH and GPx4. Free Radic. Biol. Med. 2020;152:175–185. doi: 10.1016/j.freeradbiomed.2020.02.027. [DOI] [PubMed] [Google Scholar]
- 23.Chen D., Si W., Shen J., Du C., Lou W., Bao C., et al. miR-27b-3p inhibits proliferation and potentially reverses multi-chemoresistance by targeting CBLB/GRB2 in breast cancer cells. Cell Death Dis. 2018;9:188. doi: 10.1038/s41419-017-0211-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zha S., Liu H., Li H., Li H., Wong K.L., All A.H. vol. 18. 2024. pp. 1820–1845. (Functionalized Nanomaterials Capable of Crossing the Blood-Brain Barrier). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hu Y., Hu Q., Li Y. γδ T cells: origin and fate. subsets, diseases and immunotherapy. 2023;8:434. doi: 10.1038/s41392-023-01653-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jiang C., Guo H., Zhang Z., Wang Y., Liu S., Lai J., et al. Molecular, pathological, clinical, and therapeutic Aspects of Perihematomal edema in different stages of intracerebral hemorrhage. Oxid. Med. Cell. Longev. 2022;2022:1–38. doi: 10.1155/2022/3948921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dixon S.J., Lemberg K.M., Lamprecht M.R., Skouta R., Zaitsev E.M., Gleason C.E., et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149:1060–1072. doi: 10.1016/j.cell.2012.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Divakaruni A.S., Paradyse A., Ferrick D.A., Murphy A.N., Jastroch M. Analysis and Interpretation of microplate-based oxygen consumption and pH data. 2014;547:309–354. doi: 10.1016/B978-0-12-801415-8.00016-3. [DOI] [PubMed] [Google Scholar]
- 29.Sun Y., Li Q., Guo H., He Q. Ferroptosis and iron metabolism after intracerebral hemorrhage. Cells. 2022;12:90–107. doi: 10.3390/cells12010090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gao M., Monian P., Quadri N., Ramasamy R., Jiang X. Glutaminolysis and Transferrin regulate ferroptosis. Molecular cell. 2015;59:298–308. doi: 10.1016/j.molcel.2015.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ursini F., Maiorino M. Lipid peroxidation and ferroptosis: the role of GSH and GPx4. Free radical biology & medicine. 2020;152:175–185. doi: 10.1016/j.freeradbiomed.2020.02.027. [DOI] [PubMed] [Google Scholar]
- 32.Shi Y.S., Chen J.C., Lin L., Cheng Y.Z., Zhao Y., Zhang Y., et al. Dendrobine rescues cognitive dysfunction in diabetic encephalopathy by inhibiting ferroptosis via activating Nrf2/GPX4 axis. Phytomedicine : international journal of phytotherapy and phytopharmacology. 2023;119 doi: 10.1016/j.phymed.2023.154993. [DOI] [PubMed] [Google Scholar]
- 33.Yang S., Wang L., Zeng Y., Wang Y., Pei T., Xie Z., et al. Salidroside alleviates cognitive impairment by inhibiting ferroptosis via activation of the Nrf2/GPX4 axis in SAMP8 mice. Phytomedicine : international journal of phytotherapy and phytopharmacology. 2023;114 doi: 10.1016/j.phymed.2023.154762. [DOI] [PubMed] [Google Scholar]
- 34.Wang Y., Wu S., Li Q., Sun H., Wang H. Pharmacological inhibition of ferroptosis as a therapeutic target for neurodegenerative diseases and strokes. 2023;10 doi: 10.1002/advs.202300325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Long H., Zhu W., Wei L., Zhao J. Iron homeostasis imbalance and ferroptosis in brain diseases. MedComm. 2023;4 doi: 10.1002/mco2.298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zille M., Karuppagounder S.S., Chen Y., Gough P.J., Bertin J., Finger J., et al. Neuronal death after hemorrhagic stroke in vitro and in vivo Shares features of ferroptosis and Necroptosis. Stroke. 2017;48:1033–1043. doi: 10.1161/STROKEAHA.116.015609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gan B. Mitochondrial regulation of ferroptosis. The Journal of cell biology. 2021;220 doi: 10.1083/jcb.202105043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Johnson J., Mercado-Ayon E., Mercado-Ayon Y., Dong Y.N., Halawani S., Ngaba L., et al. Mitochondrial dysfunction in the development and progression of neurodegenerative diseases. Arch. Biochem. Biophys. 2021;702 doi: 10.1016/j.abb.2020.108698. [DOI] [PubMed] [Google Scholar]
- 39.Liang D., Minikes A.M., Jiang X. Ferroptosis at the intersection of lipid metabolism and cellular signaling. Molecular cell. 2022;82:2215–2227. doi: 10.1016/j.molcel.2022.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li D., Li Y. The interaction between ferroptosis and lipid metabolism in cancer. Signal Transduct Target Ther. 2020;5:108. doi: 10.1038/s41392-020-00216-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lenaz G., Bovina C., D'Aurelio M., Fato R., Formiggini G., Genova M.L., et al. Role of mitochondria in oxidative stress and aging. Ann. N. Y. Acad. Sci. 2002;959:199–213. doi: 10.1111/j.1749-6632.2002.tb02094.x. [DOI] [PubMed] [Google Scholar]
- 42.Calabrese V., Colombrita C., Guagliano E., Sapienza M., Ravagna A., Cardile V., et al. Protective effect of Carnosine during Nitrosative stress in Astroglial cell cultures. Neurochem. Res. 2005;30:797–807. doi: 10.1007/s11064-005-6874-8. [DOI] [PubMed] [Google Scholar]
- 43.Areti A., Yerra V.G., Naidu V., Kumar A. Oxidative stress and nerve damage: role in chemotherapy induced peripheral neuropathy. Redox Biol. 2014;2:289–295. doi: 10.1016/j.redox.2014.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Murakami S., Kusano Y., Okazaki K., Akaike T., Motohashi H. 2023. NRF2 Signalling in Cytoprotection and Metabolism. [DOI] [PubMed] [Google Scholar]
- 45.Dodson M., de la Vega M.R., Cholanians A.B., Schmidlin C.J., Chapman E., Zhang D.D. Modulating NRF2 in disease: Timing is Everything. Annu. Rev. Pharmacol. Toxicol. 2019;59:555–575. doi: 10.1146/annurev-pharmtox-010818-021856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Calabrese V., Cornelius C., Dinkova-Kostova A.T., Calabrese E.J., Mattson M.P. Cellular stress responses, the hormesis paradigm, and vitagenes: novel targets for therapeutic intervention in neurodegenerative disorders. Antioxidants Redox Signal. 2010;13:1763–1811. doi: 10.1089/ars.2009.3074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Catino S., Paciello F., Miceli F., Rolesi R., Troiani D., Calabrese V., et al. Ferulic acid Regulates the Nrf2/Heme Oxygenase-1 system and counteracts Trimethyltin-induced neuronal damage in the human Neuroblastoma cell line SH-SY5Y. Front. Pharmacol. 2015;6:305. doi: 10.3389/fphar.2015.00305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang Q., Bin C., Xue Q., Gao Q., Huang A. GSTZ1 sensitizes hepatocellular carcinoma cells to sorafenib-induced ferroptosis via inhibition of NRF2/GPX4 axis. 2021;12:426. doi: 10.1038/s41419-021-03718-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shakya A., McKee N.W., Dodson M., Chapman E., Zhang D.D. Anti-ferroptotic effects of Nrf2: beyond the antioxidant response. Mol. Cell. 2023;46:165–175. doi: 10.14348/molcells.2023.0005. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data will be made available on request.