

ABSTRACT
The past 10 years have been revolutionary for clostridial genetics. The rise of next-generation sequencing led to the availability of annotated whole-genome sequences of the important pathogenic clostridia: Clostridium perfringens, Clostridioides (Clostridium) difficile, and Clostridium botulinum, but also Paeniclostridium (Clostridium) sordellii and Clostridium tetani. These sequences were a prerequisite for the development of functional, sophisticated genetic tools for the pathogenic clostridia. A breakthrough came in the early 2000s with the development of TargeTron-based technologies specific for the clostridia, such as ClosTron, an insertional gene inactivation tool. The following years saw a plethora of new technologies being developed, mostly for C. difficile, but also for other members of the genus, including C. perfringens. A range of tools is now available, allowing researchers to precisely delete genes, change single nucleotides in the genome, complement deletions, integrate novel DNA into genomes, or overexpress genes. There are tools for forward genetics, including an inducible transposon mutagenesis system for C. difficile. As the latest addition to the tool kit, clustered regularly interspaced short palindromic repeat (CRISPR)-Cas9 technologies have also been adopted for the construction of single and multiple gene deletions in C. difficile. This article summarizes the key genetic technologies available to manipulate, study, and understand the pathogenic clostridia.
INTRODUCTION
The Clostridiaceae family (and within it the Clostridium genus) consists of an extremely diverse group of primarily Gram-positive bacteria that have traditionally been grouped together based on their anaerobic growth requirements and their ability to produce heat-resistant endospores. Historically, the members of the genus were very dissimilar, such that the genus lacked phylogenetic coherence, with over 200 species, at least 35 of which cause disease in humans and animals (1). Two recent studies have proposed a phylogenetic reorganization of the clostridia and subsequently changed the name of Clostridium difficile to Peptoclostridium difficile (2) and Clostridioides difficile (3, 4). However, the earlier name (P. difficile) did not comply with the Internal Journal of Systematic and Evolutionary Microbiology Bacterial Code and was rejected (3). Also recently, Clostridium sordellii has been reclassified as Paeniclostridium sordellii (5). For simplicity, in this article pathogenic members of the genera Clostridium, Clostridioides, and Paeniclostridium will all be referred to as pathogenic clostridia.
The most common feature of the pathogenic clostridia is that the cell and tissue damage that they cause primarily results from the production of potent extracellular toxins (6, 7). Although it is somewhat artificial in that it crosses family boundaries, it is useful to divide the pathogenic clostridia into three major groups based on their resultant disease pathology. These groups consist of the neurotoxic clostridia, which produce toxins that affect the nervous system, the enterotoxic clostridia, which produce toxins that affect the gastrointestinal tract, and the histotoxic clostridia, whose necrotic pathology results from the production of one or more toxins that affect the structural and functional integrity of host cells located at or near the site of infection. This article focuses on the genetic manipulation of the pathogenic clostridia and is divided by approaches and techniques rather than individual species.
The genetics and, in particular, the genetic manipulation of the clostridia have been revolutionized in the past decade. In large part, this has been made possible by advances in sequencing technology. The genomes of representatives of all of the pathogenic clostridia have now been completely sequenced, assembled, and annotated. The first Clostridium perfringens genome was published in 2002 (8), followed by Clostridium tetani in 2003 (9), C. difficile in 2006 (10), and Clostridium botulinum in 2007 (11). Multiple genome sequences are now available for these pathogens (12–19). The availability of these genome sequences has been important in the development of genetic manipulation technologies because precise sequences are required to design homology arms for homologous recombination, complementation, and use of newer technologies such as clustered regularly interspaced short palindromic repeats (CRISPR).
Genetic manipulation can be broken down into two main categories: forward and reverse genetics. Forward genetics involves random screens to identify genes that are responsible for a particular phenotype and can be employed without knowing the target gene. These methods encompass technologies such as transposon mutagenesis and transposon-directed insertion site sequencing, which are tools of random mutagenesis. Reverse genetics involves the inactivation of a particular gene to study the resulting phenotype and requires the target gene to be identified first. The technologies involved in this approach include insertional mutagenesis, allelic exchange to construct deletions and point mutations, and the insertion of genes or regions of DNA to disrupt genes.
EARLY ADAPTATIONS OF MUTAGENESIS TECHNOLOGIES
Genetic manipulation methods have now been established for most pathogenic clostridia but are most refined for C. difficile and C. perfringens. The clostridia are not naturally competent, but transformation via electroporation has been described for several of the pathogenic species. Electroporation into C. botulinum was demonstrated by Zhou et al. in 1983 (20), and C. tetani was transformed with the Enterococcus faecalis shuttle vector pAT19 in 1998 (21). So far, however, there have been no further reports of successful transformation (via electroporation) into either of these two species. Whether DNA transfer via electroporation is successful partly depends on the restriction and modification status of the organism and the transformation frequency that can be obtained. Electroporation is not currently feasible for C. difficile, despite numerous attempts in many laboratories. It has been successful for C. perfringens strain 13 (22, 23), but many other strains of C. perfringens are not amenable to DNA transformation.
The first defined mutants in a pathogenic Clostridium species were constructed in C. perfringens by allelic exchange utilizing double crossover events (22, 24). These studies made use of the fact that derivatives of the gas gangrene strain 13 have an apparent lack of active restriction and modification systems, thereby making them very amenable to genetic analysis (8, 25). There are several well-characterized shuttle plasmids that can reliably and reproducibly be introduced into C. perfringens cells by electroporation-mediated transformation (23) or by conjugation from Escherichia coli (26). The most widely used C. perfringens-E. coli shuttle vectors for complementation and other genetic studies are pJIR750 and pJIR751 (27), which are derivatives of the shuttle vector pJIR418 (28). These plasmids all contain the origin of replication lacZ′ gene and multiple cloning sites of the E. coli plasmid pUC18 and the origin of replication and rep gene of the C. perfringens bacteriocin plasmid pIP404. Detection of recombinants can be achieved by X-Gal screening in E. coli and transformants selected on chloramphenicol or erythromycin in either E. coli or C. perfringens, respectively. Subsequently, derivatives of pJIR750 and pJIR751 were constructed that carried the oriT site from plasmid RP4, designated pJIR1456 and pJIR1457, respectively (29). The construction of these vectors facilitated RP4-mediated conjugative transfer of these plasmids from E. coli to C. perfringens, allowing strains that were not amenable to DNA transformation to be genetically manipulated.
The first genetic manipulation of C. difficile was successfully performed using the oriT-containing C. perfringens vectors, allowing regulatory genes to be introduced in trans into this bacterium (30). The first mutants generated in C. difficile were derived by homologous recombination from single crossover events. In 2006 O’Connor et al. (31) constructed a chromosomal mutant in CD3255 (virR) using pJIR1456 (26). This replicon is relatively unstable in C. difficile, and hence it can act as a pseudo-suicide plasmid in this bacterium. While this development represented an important step forward in the genetic manipulation of C. difficile, the mutants were sometimes unstable when selective pressure was removed, which could lead to the generation of revertants (31, 32).
A prerequisite for the mutagenesis of C. difficile was the use of an erythromycin resistance marker, and hence an erythromycin-sensitive recipient strain was required. C. difficile 630 has become the primary strain for laboratory manipulation, because it is amenable to DNA transfer (via conjugation, as discussed below), and it was the first C. difficile strain to have its genome sequenced (10). However, C. difficile 630 is erythromycin resistant. After a number of passages two erythromycin-sensitive strains were eventually obtained, namely 630E or JIR8094 (31) and 630Δerm (33). Recently, it was discovered through whole-genome sequencing that both of these erythromycin-sensitive derivatives obtained additional mutations during their passaging, which differed between the two derivatives (34–36). JIR8094 showed a larger number of seemingly important changes compared to the parental strain. In light of these differences, which are described in more detail by Collery et al. (34), 630Δerm is now the strain of choice in studies to elucidate pathways of virulence, regulation, and metabolism in C. difficile.
THE ROADMAP TO CLOSTRIDIAL GENETICS
Overcoming Restriction and Modification Systems
As mentioned above, complete genome sequences are now available for all of the pathogenic clostridia, which has opened up the field for the development of more sophisticated genetic tools. Technologies such as Illumina sequencing are now quick and affordable; however, obtaining accurate reads in repetitive regions is difficult with these technologies because of the short reads that they produce. This problem can be overcome by the use of long-read technologies such as MinION and, in particular, PacBio. When these latter methods are used in combination with Illumina sequencing, they can provide an accurately assembled genome, which is crucial to locate genes and identify up- and downstream defined genetic elements and, importantly, potential restriction-modification systems (37).
Restriction-modification systems have held back much of the genetic development of the clostridia, because they can protect the recipient bacterial cell from foreign DNA, making the transfer of foreign DNA for genetic manipulation difficult or impossible (38). A common method used to overcome restriction barriers is in vitro or in vivo methylation of the DNA to be introduced, so that the recipient recognizes it as its own. To this end, it becomes important again to have knowledge of the genome sequence, which can reveal all the methylases and restriction-modification systems present. For many years pAN2, which contains a bacteriophage methyltransferase gene, has been used to methylate DNA before transformation into Clostridium acetobutylicum (39). Recently, similar systems have been developed for Clostridium pasteurianum (40), Clostridium saccharobutylicum, (41, 42), and Clostridium cellulovorans (43).
In all fields of bacteriology, researchers work mostly on a few select laboratory strains, which are for one reason or another easy to manipulate. For example, C. difficile 630 has no active restriction-modification systems, so it is easier to conjugate with, and its genome was the first to be sequenced, so most molecular studies have been carried out with this strain and its derivatives. Similarly, C. perfringens strain 13 is the most widely used strain of that species, with transformation protocols having been successfully developed at an early stage. Finally, ATCC 9714 is the laboratory strain of choice for P. sordellii studies. While these strains are great tools to unravel the molecular basis of pathogenicity and metabolism, it has become evident that they often have phenotypic limitations. For this reason, clinical isolates need to be considered and used for research studies, particularly those related to virulence. Different ribotypes of C. difficile and different toxinotypes of C. perfringens, for example, display very different phenotypes, causing different diseases or disease severity (44, 45). Therefore, it is important to understand and develop ways to overcome the barrier to DNA transfer into these organisms, and the use of the genetics roadmap suggested in reference 37 can help to facilitate such an analysis. In this workflow, the genome sequence of a strain is determined first, which gives insight into its restriction-modification system. This information in turn helps to overcome the barriers to DNA transfer, as well as providing the genetic blueprint of the strain, making genetic manipulations such as gene deletion, substitutions or additions (46), and further investigation possible.
Plasmid Cloning Vectors
Conjugation as a method of DNA transfer has been very successful in the pathogenic clostridia. C. perfringens strains that cannot be transformed by electroporation have successfully been genetically manipulated using conjugation from E. coli, using plasmids pJIR1456 or pJIR1457, as discussed earlier (22, 29, 30). Conjugative manipulation was first established for C. botulinum using pJIR1457 (47) with the RP4-carrying E. coli strain S17-1 as the donor strain. Conjugation is also used to introduce DNA into P. sordellii and has been used successfully to generate numerous mutants in this clostridial species (48–52).
In early studies, C. perfringens-based vectors were used for the genetic manipulation of C. difficile (30). For example, plasmid pJIR1457 was used to study the functional capacity of the toxin regulator TcdR (then called TxeR) in C. difficile employing RP4-mediated conjugative transfer of a tcdR-carrying recombinant plasmid from an E. coli donor (30). Note that the difficulty of introducing DNA into some strains using the RP4-based system from E. coli resulted in the construction of a novel plasmid transfer system that exploited the conjugation apparatus encoded by the broad-host range transposon Tn916 together with the cognate oriT site encoded by this element (53). This system, which uses a Tn916-carrying C. perfringens or Bacillus subtilis strain as a donor and oriTTn916-carrying shuttle plasmids, facilitated the genetic manipulation of previously recalcitrant C. difficile, C. perfringens, P. sordellii, and Clostridium septicum strains (54). More recently, heat shock has also been used to increase conjugation efficiency in C. difficile, and the choice of media was also shown to affect conjugation frequency (55).
The construction of specific C. difficile shuttle vectors was realized by Purdy et al. in 2002 (56). The plasmids derived in this work can replicate autonomously in C. difficile and contain either replicon CD6, which was amplified from a C. difficile plasmid isolated during the same study, the replicon from pCB102, or that from pIP404; the latter two plasmids were derived from Clostridium beijerinckii (57) and C. perfringens (28), respectively. A much higher frequency of transfer was achieved using the native replicon pCD6, and plasmids carrying this replicon were shown to be maintained more stably in C. difficile. The plasmid constructed in this study, containing the pCD6 replicon, a Gram-negative replication site, possibly including oriT for transfer and an erythromycin gene for antibiotic selection, was originally called pMTL9301 and was later renamed pMTL960 (56). pMTL960 has been used successfully in numerous studies, including those involving an examination of the function of the C. difficile cell wall protein V, the lipoprotein CD0873 (an adhesin), and the cysteine proteases Cwp84 and Cwp13 (58–60).
In 2009, Heap et al. published a modular vector system that has been useful in a number of clostridial species (61). This system includes four main modules: a Gram-negative replicon, a Gram-positive replicon, an antibiotic resistance marker, and an insertion module. All of these modules can easily be swapped and replaced by others, making this system easy to modify depending on the purpose and organism for which it will be used (Fig. 1, Table 1). For example, four different antibiotic resistance markers can be used, and two Gram-negative replicons are included. The use of specific restriction sites that were employed to build the plasmid series allows the straightforward incorporation of new modules incorporating, for example, new Gram-positive replicons. The development of this vector series has helped to standardize the genetic work in many clostridial species. All these vectors can be used as shuttle vectors and can be transferred from E. coli via RP4-mediated conjugation, or directly in some species by electroporation, and have successfully been used in many C. difficile studies investigating virulence regulation, biofilm formation, colonization, and sporulation (62–64). These plasmids have also been used in other pathogenic clostridial species, including an investigation into the role of two-component systems in neurotoxin regulation of C. botulinum (65). If used without the Gram-positive replicon, they act as suicide vectors in the clostridia and have been effectively used in this capacity in studies involving the nonpathogenic species C. beijerinckii and Clostridium autoethanogenum (66, 67).
FIGURE 1.
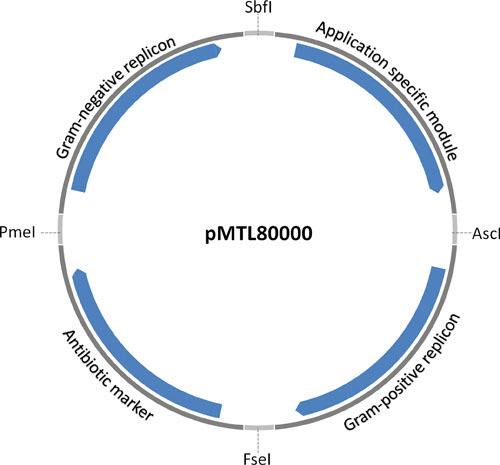
Illustration of the pMTL80000 modular vector series. The figure highlights the four modules separated by the unique restriction sites: SbfI, AscI, FseI, and PmeI. The modules consist of a Gram-positive replicon module, a selectable marker, a Gram-negative replicon unit with optional transfer (tra) genes, and an application-specific module.
TABLE 1.
Module choices for pMTL80000 plasmids
| Gram+ replicon | Marker | Gram– replicon | Application-specific |
|---|---|---|---|
| 0. Spacer* | 0. Spacer* | ||
| 1. catP | 1. P15a | 1. MCS | |
| 2. pBP1 | 2. ermB | 2. p15a + tra | 2. Pthl + MCS |
| 3. pCB102 | 3. aad6 | 3. Pfdx + MCS | |
| 4. pCD6 | 4. tetA | 4. ColE1 | 4. catP reporter |
| 5. pIM13 | 5. ColE1 + tra |
Abbreviations: MCS, multiple cloning site; Pthl, thialase promoter; Pfdx, ferrodoxin promoter
Spacer: a module may consist of a short spacer in place of a functional component
Inducible Vector Systems
In many genetic studies it is essential to be able to control or regulate the expression of cloned genes. The usual method is to place the cloned gene behind an inducible or repressible promoter so that expression of the gene of interest is induced by the addition of a low-molecular-weight effector molecule to the culture medium. However, only a limited number of such systems are available for use in the pathogenic clostridia.
In C. perfringens, an inducible expression system was made available by the development of the vector pKRAH1 (68). This shuttle plasmid encodes the C. perfringens BgaR transcriptional regulator and the inducible promoter, PbgaL, which is activated by BgaR in the presence of lactose. The addition of lactose to the culture medium therefore leads to the expression of genes cloned downstream of PbgaL. This system initially was validated by cloning the gusA reporter gene behind PbgaL and measuring β-glucuronidase (β-GusA) activity in C. perfringens SM101 cells with and without lactose induction. Substituting isopropyl β-d-1-thiogalactopyranoside (IPTG) for lactose did not result in increased activity, suggesting that IPTG is either not an inducer or cannot be transported into the cell (68). Subsequently, pKRAH1 was constructed and used to express a yfp-pilB construct, which allowed the resultant fusion protein to be localized (68). pKRAH1 has been used in other studies of the biogenesis of type IV fimbriae in C. perfringens (69), and the BgaR/PbgaL system was also used to study the role of the endonuclease RNaseY (70) and carbonic anhydrase (71) in C. perfringens and for the analysis of acetogenesis in Clostridium ljungdahlii (72).
Other workers developed a xylose-inducible gene system for use in C. perfringens (73). The vectors, pXCH (confers chloramphenicol resistance) and pXEH (confers erythromycin resistance), encode the XylO transcriptional repressor and the xylB promoter from C. difficile. Genes are cloned downstream of PxylB, and XylO repression is subsequently relieved by the addition of xylose. The vectors were validated in C. perfringens strain 13 by using them to regulate the expression of chloramphenicol acetyltransferase and α-toxin reporter constructs. The xylose-inducible system has been used by other workers in combination with the lactose system (69).
Classic IPTG-inducible systems are not effective in C. difficile, but in 2011 Fagan et al. developed an anhydrous tetracycline-inducible system for this bacterium (74). The system is based on pRMC2 from Staphylococcus aureus (75), combining the tetracycline-inducible promoter system with a codon-optimized gusA gene from pCBR023 on plasmid pRPF185, including the fdx terminator from C. pasteurianum. The plasmid backbone is taken from the E. coli-C. difficile shuttle vector pMTL960. When GusA activity was monitored as a measure for leakiness of the system, no GusA activity was measured in the absence of anhydrous tetracycline (the inducer), providing supporting evidence that the system is tightly regulated (74). This system has been used successfully in other studies involving C. difficile to determine the functional role of TcdE (76) and to investigate the spore differentiation pathway, including the creation of an inducible spo0A gene most recently (77, 78). Additionally, it has been used to study spore coat proteins of C. difficile (79) and has been employed in other clostridial species, such as P. sordellii, in which it was used to examine a new conjugation system (51). A second inducible system has also been described and employed in C. difficile. Purcell et al. created a nisin-inducible system in which the cpr promoter, responsible for the transcription of an ABC transporter, which confers resistance against nisin, is cloned upstream of the target gene. This system was used to study the role of cyclic di-GMP in motility and aggregation of C. difficile (80).
Some clostridial genes are toxic when overexpressed in E. coli and therefore are difficult to clone, making complementation studies in the original clostridial host problematic. This problem can be overcome by use of the vector pJIR3422 (81, 82). This vector exploits the phenotypic features of the clostridial Tn4451/3 site-specific recombinase TnpX, specifically, the specificity of binding of this protein to a transposon-derived promoter. TnpX represses expression from its own promoter, PattCI, which is usually located at the joint of the circular form of the Tn4451/3 elements (83). This promoter is present in pJIR3422, and expression from this promoter is repressed in the presence of a catalytically inactive derivative of TnpX, present in the E. coli strain used for cloning, but is not repressed in the clostridial host because of the absence of TnpX. The use of this system facilitated the cloning of the tcpG conjugation gene and the feoB ferrous uptake gene in E. coli and the complementation of mutants of these genes in C. perfringens, which could not be achieved without this system (81).
Reporter Assays
Historically, protein reporter systems for the analysis of protein localization have been lacking in most pathogenic clostridia, particularly in C. difficile. Widely used reporter proteins such as green fluorescent protein for fluorescence or luciferase for chemiluminescence require oxygen for correct reporter protein folding or full enzymatic activity and hence have been thought to be of limited value in anaerobic bacteria. In the past 5 years, however, a number of assay systems have been established (77, 84, 85), which have made the functional characterization of genes and their encoded products more feasible. Reporters can be split into two main categories: visualizable reporters and enzymatic reporters. The former are utilized to study phenotypic heterogeneity and localizing proteins, whereas the latter serve as read-outs on transcription.
Visualizable reporters
Several of these systems are based on fluorescence. Ransom et al. successfully developed a codon optimized cyan-fluorescent protein and mCherry (mCherryOpt) in C. difficile to perform a number of subsequent studies (84, 86). They created two vectors using mCherryOpt, one to study localization, which they used to examine septal location of two cell division proteins, MldA and ZapA, in fixed cells (86). The other vector allows cloning of a promoter upstream of the mCherryOpt gene to study gene expression and has been exemplified by studying the pdaV operon, which is required for lysozyme resistance. (84). The variants were successfully expressed in C. difficile but required exposure to oxygen for chromophore maturation (84). Recently, work by Ribis et al. used mCherry fusions to establish the importance of SpoIVa and SipL in spore coat assembly and to elucidate the mechanism of engulfment of the forespore by the mother cell in C. difficile (87, 88). Other recent examples of the use of mCherry in C. difficile include a study of flagellum and toxin phase variation, demonstrating the potential to study single-cell heterogeneity, and work demonstrating that toxin gene expression is bistable in C. difficile (89, 90).
Yellow fluorescent protein fusions have recently been used successfully in C. perfringens to examine localization and function of PilB, an ATPase responsible for pili assembly (69).
iLOV, a light oxygen voltage domain, has also been optimized for use in C. difficile. LOV domains are plant or bacterial blue light receptors and are part of flavin mononucleotide-based fluorescent proteins (FbFPs) (91, 92). These fluorescent reporters can be utilized with and without oxygen, and they are smaller (13 kDa) than green fluorescent protein (25 kDa), which makes them ideal fusion proteins with a reduced probability of disrupting native protein function. iLOV has been used as a fluorescent reporter in C. difficile (85), as a translational fusion to study the cell division protein FtsZ. This study also proved that iLOV fusions can be successfully transported out of the cell by constructing a fusion to FliC, the flagella subunit. Functional iLOV fusions in P. sordellii and the nonpathogenic C. acetobutylicum were also generated in this study. The brightness of FbFPs is lower than that of green fluorescent protein, and hence a few technical modifications have been suggested to improve their usefulness, such as lowering background noise and improving the quality of the output signal. Flavin-based media, for example, can lead to highly fluorescent backgrounds, which can be reduced by eliminating yeast and beef extract (93). Also, several clostridial species, such as C. difficile and C. acetabutylicum, have a natural green auto-fluorescence that can increase the background signal (85).
SNAP-tag technology has also been adapted for C. difficile. The 20-kDa peptide tags are fused to target genes, and fusions are visualized using a fluorescent microscope by adding the cell-permeable fluorescent substrate TMR-star. This technology has been used to elucidate the role of the sigma factor cascade in the sporulation process in C. difficile and, furthermore, a split SNAP-tag has been employed to study protein interactions between the forespore protein SpoIIQ and the mother cell protein SpoIIIAH (77, 94). SNAP-tags have been proven to be versatile and have worked effectively on fixed cells, demonstrating the temporally regulated gene expression of the sigma factors involved in C. difficile spore formation and establishing the interactions of C. difficile SpoIIQ and SpoIIIA proteins and their control over forespore engulfment and integrity (77, 95).
Enzymatic reporter systems
The widely used β-GusA reporter assay has been used to study the regulation of the large toxins in C. difficile. To elucidate the function of the sigma factor TcdR, Mani et al. created gus-fusions in C. perfringens and then, in a follow-up publication, gus-fusions in C. difficile, the study of which showed that TcdR positively regulates tcdA and tcdB expression (30, 96).
An alternative reporter system used in C. difficile involves a luciferase gene fusion to the signal sequence of the secreted protein PPEP-1. PPEP-1 is a zinc metalloprotease that has been shown to be one of the most abundant secreted proteins of C. difficile (97). Two reporters have been constructed, AmyEopt and sLucopt (98). AmyEopt can be used to measure promoter activity in liquid or plate-based assays due to the production of α-amylase, a codon optimized enzyme from B. subtilis, which results in starch degradation. sLucopt is a codon-optimized luciferase based on NanoLuc (99) and has been shown to be detectable in culture supernatants when fused to an inducible promoter (98). Both reporters are assayed under aerobic conditions once the samples have been taken from the anaerobically grown culture (98).
Finally, the E. faecalis alkaline phosphatase gene, phoZ, has been successfully used in C. difficile for the qualitative and quantitative measurement of gene expression using an alkaline phosphatase assay. PhoZ was shown to be active anaerobically and is therefore presumed to fold correctly under these conditions, which makes it a useful reporter gene for anaerobic bacteria (100).
A promoter probe plasmid based on pJIR418 has also been constructed for use in C. perfringens. This plasmid, pPSV, was constructed by deletion of the catP gene from pJIR418 followed by the addition of a promoterless catP gene downstream of the multiple cloning site (101). The catP gene has also been used as a reporter system in the promoter probe shuttle vector pTCATT, which contains a promoterless catP gene flanked by transcriptional terminators. However, PCR-mediated regeneration of the 5′ terminus of the catP gene is required during the construction of promoter fusions in pTCATT, limiting its usefulness (102). Other studies have attempted to develop reporter systems for use in C. perfringens. One system involved the construction of a plasmid that contains the luxAB genes from Vibrio fischeri under the control of the plc promoter. Luciferase activity and bioluminescence were obtained from this plasmid in C. perfringens (103). In another study, reporter plasmids using the E. coli gusA gene cloned into pJIR750 were constructed, and β-GusA production was used successfully to monitor the sporulation-specific regulation of the cpe gene in C. perfringens (104). More recently, high-level expression of the C. perfringens NanI sialidase was achieved by using a ferredoxin promoter-based plasmid, pFF, in C. perfringens (105). This system was particularly useful for purification of NanI since the C. perfringens expression strain secreted the enzyme, as does the wild type, and seemed to efficiently express the protein, resulting in a 60-fold increased yield in comparison to protein isolation from an E. coli host. These results suggest that in C. perfringens the pFF plasmid may be very useful for the expression of AT-rich genes from other clostridia and other bacteria (105).
Allelic Exchange and Homologous Recombination
Homologous recombination has been used extensively as a mutagenesis tool for many bacterial species and may involve either single- or double crossover events. Since two crossovers are required for allelic exchange, it is a less efficient process than obtaining mutants by a single crossover event using insertional mutagenesis. However, the disadvantage of single crossover events is that wild-type genes can be regenerated from the mutants by further homologous recombination events. This issue is not a problem with allelic exchange or double crossovers, which result in inherently stable mutants. It is worth noting that the efficiency with which mutants are obtained by allelic exchange depends on the size of the homologous regions that flank the gene of interest, and it is recommended that there be at least 2 kb of flanking DNA present on either side of the gene to be replaced (106). Additionally, a large number of mutants can be generated using homologous recombination, in contrast to a limited number that can be generated with insertional mutagenesis, because of the small number of antibiotic cassettes available, which are required for insertional mutagenesis.
Homologous recombination and allelic exchange methods for the clostridia have also been developed and improved significantly during the past 2 decades. In early studies, allelic exchange was used successfully for genetic analysis in C. perfringens (22), and the first defined genetic mutants constructed in the pathogenic clostridia were the plc and pfoA toxin gene double-crossover mutants that were constructed in C. perfringens (22, 24). Since that time many C. perfringens mutants have been constructed by allelic exchange, too many to list here, and homologous recombination has become a standard genetic tool for the construction of mutants in this bacterium.
Early homologous recombination studies of C. difficile involved the construction of mutants by single crossover events using suicide plasmids that were introduced by conjugation from E. coli (31). Subsequently, two improved systems (107, 108) were developed for C. difficile, both of which are based on the use of negative selection markers. The first system uses cycloserine deaminase (107) as a counter selection marker and relies on the presence of the codA gene on a pseudo-suicide vector (107). Single crossovers are selected on the basis that cells that have the plasmid, which contains a heterologous codA gene from E. coli, integrated into their chromosome are able to grow much faster than cells carrying the resistance gene on a plasmid. This is because the plasmid used in this approach is inherently unstable in C. difficile and is lost easily from cells. The faster-growing colonies are selected and tested (via PCR) to determine if a single crossover event has occurred. They are then plated on C. difficile minimal medium containing 5-fluorocytosine. Only clones in which a second crossover event has occurred, resulting in the loss of the codA gene, are able to grow, because the cells still carrying the codA gene are killed when 5-fluorocytosine is converted to toxic 5-fluorouracil (97) by CodA. Double crossover events result either in a reversion back to wild type or in the construction of the desired mutant and loss of the plasmid. This system can be used for the construction of DNA deletions or point mutations. Genes can also be complemented by going through another cycle of single and double crossover events, using a different plasmid which contains the intact gene of interest. In this case, genes are reinserted into their original position on the chromosome. It is hence advisable to introduce a watermark or sequence signature into the gene to differentiate it from the wild type (107). This allelic exchange system has successfully been used in a number of studies, including work characterizing two sortase substrates of C. difficile and their mechanism, involving cyclic di-GMP, of anchoring proteins to the peptidoglycan layer (109). Another study investigated a dipicolinic acid release mechanism during C. difficile spore germination (110).
The second system for allelic exchange in C. difficile is based on creating a pyrE mutant, carrying a 3′ deletion of the gene, using allele-coupled exchange (111) in the strain of choice and then using a heterologous pyrE gene as a negative/counter selection marker. The pyrE gene encodes orotate phosphoribosyltransferase, which is involved in pyrimidine biosynthesis. This enzyme is essential for bacterial growth in the absence of exogenous pyrimidines and also acts on 5-fluoro-orotate to generate a toxic derivative of this compound. The pyrE mutants are auxotrophs for uracil. A functional pyrE gene from Clostridium sporogenes is supplied on an allelic exchange plasmid as the counterselection (108). pyrE-mutants were generated in C. difficile 630Δerm and R20291 (108) and were then used to generate deletion mutants using a process similar to that described for the codA methodology. Single crossover clones are selected on the basis of faster-growing colonies once the vector has integrated into the chromosome, since the vector is unstable due to a frameshift mutation in the oriR. Double crossover mutants in which the plasmid has been excised from the chromosome are then selected on C. difficile minimal medium supplemented with 5-fluoro-orotate and uracil. Once 5-fluoro-orotate-resistant uracil auxotrophs have been isolated and shown to have lost the plasmid, they can easily be restored to prototrophy by conjugative transfer of a plasmid that contains part of the pyrE gene that can be utilized to repair the pyrE gene in the chromosome using allelic exchange. The resultant transconjugants are streaked on C. difficile minimal medium without the addition of uracil, so only bacteria with a repaired pyrE gene are able to grow. It is also possible to complement the mutation at the same time as repairing the pyrE gene by cloning a copy of the disrupted gene into the plasmid either under the control of its native promoter or through the use of a constitutive promoter for overexpression. Using allele-coupled exchange, the complementation gene is then integrated simultaneously during pyrE repair at the pyrE locus (108). This technique is very powerful and rapid, allowing for the generation of complementation strains within 5 to 6 days, opposed to obtaining an allelic exchange mutant in 2 to 3 weeks. This approach has been used in several studies; for example, it was used to generate a complete pathogenicity locus deletion in C. difficile R20291 to study the role of binary toxin in the absence of the overpowering potency of TcdA and TcdB (112). It was used in a recent study that suggested that the introduction of trehalose into our diets has favored the emergence of so-called hypervirulent C. difficile ribotypes, such as RT027 (113). Ribis et al. created the first published quadruple mutant in C. difficile, using the pyrE-system, in their work studying the roles of putative engulfment regulators, IID (CD0126), IIP (CD2469), and IIM (CD1221), and the known engulfment regulator and putative endopeptidase, IIQ (CD0125) (88).
ClosTron and TargeTron
Two related technologies based on the exploitation of a mobile group II intron were developed for insertional mutagenesis in the clostridia. Mobile group II introns are found in bacterial genomes and are site-specific retroelements. They use a mobility mechanism termed retrohoming to create insertions in DNA, and they do this by inserting an excised intron lariat RNA directly into a DNA target site, which is then reverse transcribed by an intron-encoded enzyme protein (114). The DNA target site is primarily recognized by base pairing of intron RNA, and these bases can be modified to allow intron insertion into any specific DNA target (4). As a result, a mobile group II intron from Lactococcus lactis, L1.LtrB, was developed commercially (Sigma-Aldrich) and was used in the development of TargeTron and ClosTron technologies. The use of the TargeTron system in a clostridial species was first described in 2005, when it was used to inactivate the plc gene in C. perfringens (115). TargeTron was also used as a tool to introduce the simian immunodeficiency virus p27 gene into the pfoA gene on the C. perfringens chromosome, thereby simultaneously inactivating pfoA and introducing the simian immunodeficiency virus p27 gene, which was subsequently shown to be expressed within the mouse gastrointestinal tract (116).
ClosTron technology is an insertional mutagenesis tool similar to TargeTron, but it incorporates the use of a retrotransposition-activated marker (RAM), based on the ermB gene, for selective purposes, with successful insertion indicated by the acquisition of resistance to erythromycin (117). The presence of a RAM makes mutant selection much more efficient and less laborious. The retargeted region is designed using an online retargeting algorithm and is then incorporated into the plasmid of choice for subsequent introduction into the clostridial strain (118).
Although the acquisition of a resistance marker in the bacterial genome via ClosTron or similar systems makes mutant selection simpler, it can also be a drawback. In particular, this means that a second mutation using the same marker cannot be obtained. Furthermore, the use of a strong promoter, driving the transcription of the RAM, can lead to unwanted polar effects. While the ClosTron system is very efficient and easy to use, it can, on occasion, be nonspecific, requiring the empirical testing of several insertion sites. To be able to construct double mutants, it is necessary either to use another marker or to remove the initial marker, both of which have been attempted. No other RAM marker other than erythromycin resistance has been described for the clostridia, but markerless ClosTrons or ClosTrons with markers that are not retroposition activated have been used successfully (115, 119). The alternative and preferred option is marker removal. Flp recombinase has been used successfully in C. acetobutylicum to remove the retargeted ermB gene from a ClosTron mutant (120). The ability of the Tn4451/3 site-specific recombinase TnpX to recognize and excise specific DNA fragments has also been exploited for antibiotic resistance marker recycling. Specifically, TnpX was used to remove the ermB gene from a marked chromosomal C. perfringens mutant, enabling the construction of a double mutant by allowing the removal and subsequent reuse of ermB to construct the second mutation (81).
Transposon-Mediated Random Mutagenesis
Transposon mutagenesis has been established in C. perfringens for several decades, although the early systems were not optimized and lacked efficiency (22). The most successful early mutagenesis method utilized the conjugative enterococcal tetracycline-resistance transposon Tn916 (121). In these studies, C. perfringens cells were transformed with a suicide plasmid carrying Tn916, and transformants were selected on medium containing tetracycline. Hybridization analysis showed that Tn916 had inserted at different sites on the chromosome but that multiple insertion events were common. However, mutants that had a single Tn916 insertion and that abrogated the ability to produce perfringolysin O were detected. The insertion site was shown to be located within the virS sensor histidine kinase gene, leading to the identification of the VirSR two-component signal transduction system (121). Subsequent studies involved the isolation of Tn916 insertions in several toxin genes (122), and conjugative transfer or mobilization of Tn916 from E. faecalis and E. coli, respectively, has been used to isolate auxotrophic mutants of C. perfringens, but again, multiple insertion events were observed (123). Tn916 mutagenesis also has been used to isolate mutants that have an altered response to oxidative stress (124). In summary, the use of Tn916 for mutagenesis in C. perfringens has been useful, but it is hampered by the presence of multiple insertion and deletion events and subsequently has not been widely adopted.
Other transposon mutagenesis options include transposon mutagenesis of cloned genes in E. coli and subsequent transformation and screening in C. perfringens. This approach has been used to identify the replication protein of the C. perfringens plasmid pCW3 (125). Other workers (126) developed an EZ-Tn5-based random mutagenesis system that involved the electroporation of a transposome complex into C. perfringens and selection (erythromycin resistance) for the insertion of a modified Tn5 element. The erythromycin-resistant transformants appeared to contain random single insertions. These experiments led to the mutagenesis and identification of the agrB gene, which is involved in the regulation of perfringolysin O production by quorum-sensing (126). Similarly, the bacteriophage Mu-based transposon delivery system has been successfully adapted for C. perfringens (127). Electroporation was used to introduce a Mu transpososome complex, and a strain library that contained single transposon inserts was obtained. Finally, the Himar1 mariner transposon system has been adapted for C. perfringens and used to show that the sagA gene is required for gliding motility in C. perfringens (128). This method utilizes a replicating plasmid that carries an inducible mariner element, but it requires a strain with galK and galT mutations to ensure that the replicating delivery plasmid can subsequently be selected against in the presence of galactose.
Despite in-depth studies of transposons in C. difficile (129), there is only one system for random mutagenesis in this organism. Transposons such as Tn5397 and Tn916 (33, 130, 131) have been well characterized, but they either lead to multiple insertion events or show a strong bias to particular target sites. However, the mariner transposon system has been successfully developed for C. difficile (132, 133). The transposable element of the mariner system Himar1 inserts randomly into a TA target site, which lends itself for use in AT-rich organisms such as the clostridia. Furthermore, only the cognate Himar1 transposase is required for transposition to occur. As mentioned earlier, no transformation system has yet been developed successfully for C. difficile, which means that the transposon vectors cannot rely on a plasmid suicide mechanism for their introduction into cells. To overcome this problem, Cartman and Minton (132) developed a pseudo-suicide vector by creating a segregationally unstable plasmid, using the replicon pBP1. This method allowed them to obtain transposition events at a frequency of 4.5 × 10–4 per cell. Drawbacks of this method were the lack of effective control of the time of transposition and lack of plasmid loss, which meant that the size of the library that could be obtained was limited. Library size is important for experiments such as transposon-directed insertion site sequencing, which can be used to determine essential genes under specific conditions. Subsequently, the system was improved by integrating the previously described anhydrous tetracycline system (74) with the transposon vector, generating a plasmid that has tetracycline-dependent conditional replication. The new transposon vector not only has a conditional replication phenotype, but it also has tight control over transposition of the ermB transposon. This tetracycline-inducible system consists of two promoters (133); one promoter drives expression of tetR (74), and the other promoter drives expression of the codon-optimized Hinmar1 transposase. Upon induction, Dembek et al. showed near complete plasmid loss within 13 generations, whereas 40% of bacteria in the uninduced culture retained the plasmid. (133). The transposon system has been demonstrated in C. difficile 630Δerm and in the epidemic strain R20291. Transposon libraries have been created during in vitro growth and during sporulation, followed by transposon-directed insertion site sequencing, which resulted in the identification of 404 and 798 essential genes, respectively, for these conditions (133).
Newer Technologies for Genetic Manipulation
New and improved technologies for genetic analysis and manipulation of the pathogenic clostridia are being reported all the time. Most C. difficile genomes contain prophages and lysogenic bacteriophages, which have been characterized in some detail (134–136), but to date, no lytic phages have been described. There has been one report of transduction, showing that a novel transposon, Tn6215, carrying an erythromycin resistance gene, could be transferred from a donor strain to a recipient strain and integrated into the recipient genome if the bacteriophage ΦC2 was integrated into the recipient genome. The study was unable to show transduction into another three recipient strains, which were all susceptible to the phage, suggesting that conditions for transduction need to be optimized further before it can be used as a reliable genetic tool (137).
Antisense RNA technology has successfully been used to repress gene expression in C. difficile and to show that the S-layer proteins and cell wall protein V are translocated across the cell membrane through an accessory Sec-system (SecA2). Most recently, CRISPR-gene editing technologies were successfully used in the clostridia. Initially developed in nonpathogenic clostridia, such as C. pasteurianum (138), C. beijerinckii (139), and C. cellulolyticum (140), this emerging technology was used in 2017 to generate deletion mutants in C. difficile (141). The authors developed a CRISPR-Cas9 system with a reported mutation frequency of 20 to 50%, exemplifying it by deleting selD, a selenophosphate synthetase that is essential for the specific incorporation of selenium into selenoproteins. They showed that deletion of selD leads to growth reduction and a lack of selenium incorporation, suggesting that the Stickland reaction could be a target for future antimicrobial therapy development. Since then, two more papers were published (142, 143), further developing and refining the technology for use in C. difficile. Wang et al. used a plasmid-based CRISPR-Cas9 system for mutagenesis, reporting 100% mutation efficiency and knocking out the sporulation master regulator Spo0A (142). The latest study by Hong et al. developed a CRISPR-Cpf1 system capable not only of targeted deletion from the C. difficile genome, including the largest deletion to date of 49.2 kb, but also of simultaneous deletion of at least two targets located on different parts of the genome. This new toolkit could greatly improve the genetic analysis of C. difficile and advance the development of new therapeutics and diagnostics by identifying new targets for these strategies (143).
CONCLUSIONS
The pathogenic clostridia are a diverse group of anaerobic, Gram-positive spore formers, all of which can cause human disease. Pathogenesis is toxin mediated, but many auxiliary virulence factors have been described and are still being discovered. Historically, it has been extremely difficult to study the clostridia at a molecular level owing to their anaerobic nature, but also because of a number of other difficulties such as the presence of multiple restriction modification systems, which preclude the uptake of foreign DNA into the bacterial cell. With the advent of next-generation sequencing and, consequently, the availability and analysis of whole-genome sequences, the development of clostridial molecular tools has taken an unprecedented leap. The technologies presented and reviewed in this article open up unique opportunities to study the pathogenic clostridia and their disease mechanisms in detail, with the objectives of improved diagnostics and the development of novel disease therapies.
There is, however, more work to be done. Recent years have seen a surge in interest in the nonpathogenic clostridia, particularly because of their utility in producing a range of biochemicals. Technologies have been developed that could be adapted for use in the pathogenic clostridia, such as a range of allelic exchange systems. Examples include the I-SceI system used in C. acetobutylicum and C. beijerinckii (144) and the MazF toxin-antitoxin system, which has been used in C. acetobutylicum and C. cellulolyticum (145, 146).
Additionally, there is an ever-growing interest in the healthy microbiota and in particular clostridial species within that microbiota (147, 148). To date, no mechanistic studies have been conducted, despite studies showing compelling data that suggest benefits to the host or microflora from certain of these species (147). The main reason for this information shortfall is the lack of available genetic tools. With the newest approaches, described in this article, we are likely to see this area of research develop rapidly in the very near future (38, 142).
In conclusion, the past 10 years have seen a rapid increase in mechanistic studies of the pathogenic clostridia owing to the development of novel, sophisticated tools, the outcomes of which are being translated into the clinic through improved diagnostics, prevention strategies, and novel treatments. The next decade will undoubtedly see even more significant advances in clostridial genetics and their subsequent application to provide a more detailed understanding of this important genus of bacteria.
ACKNOWLEDGMENTS
Research in S.A.K.’s laboratory is supported by grants from the British Society of Periodontology (RCFW20100) and DEBRA UK (Chapple1-20711). Research in J.I.R.’s laboratory is supported by grants from the National Health and Medical Research Council (GNT1082401, GNT1104502, and GNT1125349) and the Australian Research Council (DP160102680). Research in D.L.’s laboratory is supported by grants from the National Health and Medical Research Council (GNT1107812, GNT1107969, GNT1103024, and GNT1145760) and the Australian Research Council (DP150104670 and DP180102449).
We thank Andrew Dempster for help with Fig. 1.
REFERENCES
- 1.Brook I. 2012. Other Clostridium species, p 979–981. In Long SS (ed), Principles and Practice of Pediatric Infectious Diseases, 4th ed. Saunders, Philadelphia, PA. [PubMed] [Google Scholar]
- 2.Yutin N, Galperin MY. 2013. A genomic update on clostridial phylogeny: Gram-negative spore formers and other misplaced clostridia. Environ Microbiol 15:2631–2641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lawson PA, Citron DM, Tyrrell KL, Finegold SM. 2016. Reclassification of Clostridium difficile as Clostridioides difficile (Hall and O’Toole 1935) Prévot 1938. Anaerobe 40:95–99 10.1016/j.anaerobe.2016.06.008. [PubMed] [DOI] [PubMed] [Google Scholar]
- 4.Guo H, Karberg M, Long M, Jones JP III, Sullenger B, Lambowitz AM. 2000. Group II introns designed to insert into therapeutically relevant DNA target sites in human cells. Science 289:452–457 10.1126/science.289.5478.452. [PubMed] [DOI] [PubMed] [Google Scholar]
- 5.Sasi Jyothsna TS, Tushar L, Sasikala C, Ramana CV. 2016. Paraclostridium benzoelyticum gen. nov., sp. nov., isolated from marine sediment and reclassification of Clostridium bifermentans as Paraclostridium bifermentans comb. nov. Proposal of a new genus Paeniclostridium gen. nov. to accommodate Clostridium sordellii and Clostridium ghonii. Int J Syst Evol Microbiol 66:1268–1274 10.1099/ijsem.0.000874. [PubMed] [DOI] [PubMed] [Google Scholar]
- 6.Chumbler NM, Farrow MA, Lapierre LA, Franklin JL, Lacy DB. 2016. Clostridium difficile toxins TcdA and TcdB cause colonic tissue damage by distinct mechanisms. Infect Immun 84:2871–2877 10.1128/IAI.00583-16. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Navarro MA, McClane BA, Uzal FA. 2018. Mechanisms of action and cell death associated with Clostridium perfringens toxins. Toxins (Basel) 10:E212 10.3390/toxins10050212. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shimizu T, Ohtani K, Hirakawa H, Ohshima K, Yamashita A, Shiba T, Ogasawara N, Hattori M, Kuhara S, Hayashi H. 2002. Complete genome sequence of Clostridium perfringens, an anaerobic flesh-eater. Proc Natl Acad Sci U S A 99:996–1001 10.1073/pnas.022493799. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bruggemann H, Baumer S, Fricke WF, Wiezer A, Liesegang H, Decker I, Herzberg C, Martinez-Arias R, Merkl R, Henne A, Gottschalk G. 2003. The genome sequence of Clostridium tetani, the causative agent of tetanus disease. Proc Natl Acad Sci U S A 100:1316–1321 10.1073/pnas.0335853100. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sebaihia M, Wren BW, Mullany P, Fairweather NF, Minton N, Stabler R, Thomson NR, Roberts AP, Cerdeño-Tárraga AM, Wang H, Holden MT, Wright A, Churcher C, Quail MA, Baker S, Bason N, Brooks K, Chillingworth T, Cronin A, Davis P, Dowd L, Fraser A, Feltwell T, Hance Z, Holroyd S, Jagels K, Moule S, Mungall K, Price C, Rabbinowitsch E, Sharp S, Simmonds M, Stevens K, Unwin L, Whithead S, Dupuy B, Dougan G, Barrell B, Parkhill J. 2006. The multidrug-resistant human pathogen Clostridium difficile has a highly mobile, mosaic genome. Nat Genet 38:779–786 10.1038/ng1830. [PubMed] [DOI] [PubMed] [Google Scholar]
- 11.Sebaihia M, Peck MW, Minton NP, Thomson NR, Holden MTG, Mitchell WJ, Carter AT, Bentley SD, Mason DR, Crossman L, Paul CJ, Ivens A, Wells-Bennik MHJ, Davis IJ, Cerdeño-Tárraga AM, Churcher C, Quail MA, Chillingworth T, Feltwell T, Fraser A, Goodhead I, Hance Z, Jagels K, Larke N, Maddison M, Moule S, Mungall K, Norbertczak H, Rabbinowitsch E, Sanders M, Simmonds M, White B, Whithead S, Parkhill J. 2007. Genome sequence of a proteolytic (group I) Clostridium botulinum strain Hall A and comparative analysis of the clostridial genomes. Genome Res 17:1082–1092 10.1101/gr.6282807. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Myers GSA, Rasko DA, Cheung JK, Ravel J, Seshadri R, DeBoy RT, Ren Q, Varga J, Awad MM, Brinkac LM, Daugherty SC, Haft DH, Dodson RJ, Madupu R, Nelson WC, Rosovitz MJ, Sullivan SA, Khouri H, Dimitrov GI, Watkins KL, Mulligan S, Benton J, Radune D, Fisher DJ, Atkins HS, Hiscox T, Jost BH, Billington SJ, Songer JG, McClane BA, Titball RW, Rood JI, Melville SB, Paulsen IT. 2006. Skewed genomic variability in strains of the toxigenic bacterial pathogen, Clostridium perfringens. Genome Res 16:1031–1040 10.1101/gr.5238106. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hassan KA, Elbourne LD, Tetu SG, Melville SB, Rood JI, Paulsen IT. 2015. Genomic analyses of Clostridium perfringens isolates from five toxinotypes. Res Microbiol 166:255–263 10.1016/j.resmic.2014.10.003. [PubMed] [DOI] [PubMed] [Google Scholar]
- 14.Lacey JA, Allnutt TR, Vezina B, Van TTH, Stent T, Han X, Rood JI, Wade B, Keyburn AL, Seemann T, Chen H, Haring V, Johanesen PA, Lyras D, Moore RJ. 2018. Whole genome analysis reveals the diversity and evolutionary relationships between necrotic enteritis-causing strains of Clostridium perfringens. BMC Genomics 19:379 10.1186/s12864-018-4771-1. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cohen JE, Wang R, Shen RF, Wu WW, Keller JE. 2017. Comparative pathogenomics of Clostridium tetani. PLoS One 12:e0182909 10.1371/journal.pone.0182909. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brüggemann H, Brzuszkiewicz E, Chapeton-Montes D, Plourde L, Speck D, Popoff MR. 2015. Genomics of Clostridium tetani. Res Microbiol 166:326–331 10.1016/j.resmic.2015.01.002. [PubMed] [DOI] [PubMed] [Google Scholar]
- 17.Williamson CH, Sahl JW, Smith TJ, Xie G, Foley BT, Smith LA, Fernández RA, Lindström M, Korkeala H, Keim P, Foster J, Hill K. 2016. Comparative genomic analyses reveal broad diversity in botulinum-toxin-producing clostridia. BMC Genomics 17:180 10.1186/s12864-016-2502-z. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Eyre DW, Cule ML, Wilson DJ, Griffiths D, Vaughan A, O’Connor L, Ip CLC, Golubchik T, Batty EM, Finney JM, Wyllie DH, Didelot X, Piazza P, Bowden R, Dingle KE, Harding RM, Crook DW, Wilcox MH, Peto TEA, Walker AS. 2013. Diverse sources of C. difficile infection identified on whole-genome sequencing. N Engl J Med 369:1195–1205 10.1056/NEJMoa1216064. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.He M, Miyajima F, Roberts P, Ellison L, Pickard DJ, Martin MJ, Connor TR, Harris SR, Fairley D, Bamford KB, D’Arc S, Brazier J, Brown D, Coia JE, Douce G, Gerding D, Kim HJ, Koh TH, Kato H, Senoh M, Louie T, Michell S, Butt E, Peacock SJ, Brown NM, Riley T, Songer G, Wilcox M, Pirmohamed M, Kuijper E, Hawkey P, Wren BW, Dougan G, Parkhill J, Lawley TD. 2013. Emergence and global spread of epidemic healthcare-associated Clostridium difficile. Nat Genet 45:109–113 10.1038/ng.2478. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhou Y, Sugiyama H, Johnson EA. 1993. Transfer of neurotoxigenicity from Clostridium butyricum to a nontoxigenic Clostridium botulinum type E-like strain. Appl Environ Microbiol 59:3825–3831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Marvaud JC, Eisel U, Binz T, Niemann H, Popoff MR. 1998. TetR is a positive regulator of the tetanus toxin gene in Clostridium tetani and is homologous to BotR. Infect Immun 66:5698–5702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rood JI. 1997. Genetic analysis in Clostridium perfringens, p 65–71. In Rood JIMB, Songer JG, Titball RW (ed), The Clostridia: Molecular Biology and Pathogenesis. Academic Press Limited, London, United Kingdom. 10.1016/B978-012595020-6/50007-3. [DOI] [Google Scholar]
- 23.Scott PT, Rood JI. 1989. Electroporation-mediated transformation of lysostaphin-treated Clostridium perfringens. Gene 82:327–333 10.1016/0378-1119(89)90059-0. [DOI] [PubMed] [Google Scholar]
- 24.Awad MM, Bryant AE, Stevens DL, Rood JI. 1995. Virulence studies on chromosomal alpha-toxin and theta-toxin mutants constructed by allelic exchange provide genetic evidence for the essential role of alpha-toxin in Clostridium perfringens-mediated gas gangrene. Mol Microbiol 15:191–202 10.1111/j.1365-2958.1995.tb02234.x. [PubMed] [DOI] [PubMed] [Google Scholar]
- 25.Rood JI, Cole ST. 1991. Molecular genetics and pathogenesis of Clostridium perfringens. Microbiol Rev 55:621–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lyras D, Rood JI. 1998. Conjugative transfer of RP4-oriT shuttle vectors from Escherichia coli to Clostridium perfringens. Plasmid 39:160–164 10.1006/plas.1997.1325. [PubMed] [DOI] [PubMed] [Google Scholar]
- 27.Bannam TL, Rood JI. 1993. Clostridium perfringens-Escherichia coli shuttle vectors that carry single antibiotic resistance determinants. Plasmid 29:233–235 10.1006/plas.1993.1025. [PubMed] [DOI] [PubMed] [Google Scholar]
- 28.Sloan J, Warner TA, Scott PT, Bannam TL, Berryman DI, Rood JI. 1992. Construction of a sequenced Clostridium perfringens-Escherichia coli shuttle plasmid. Plasmid 27:207–219 10.1016/0147-619X(92)90023-4. [DOI] [PubMed] [Google Scholar]
- 29.Lyras D, Storie C, Huggins AS, Crellin PK, Bannam TL, Rood JI. 1998. Chloramphenicol resistance in Clostridium difficile is encoded on Tn4453 transposons that are closely related to Tn4451 from Clostridium perfringens. Antimicrob Agents Chemother 42:1563–1567 10.1128/AAC.42.7.1563. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mani N, Lyras D, Barroso L, Howarth P, Wilkins T, Rood JI, Sonenshein AL, Dupuy B. 2002. Environmental response and autoregulation of Clostridium difficile TxeR, a sigma factor for toxin gene expression. J Bacteriol 184:5971–5978 10.1128/JB.184.21.5971-5978.2002. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.O’Connor JR, Lyras D, Farrow KA, Adams V, Powell DR, Hinds J, Cheung JK, Rood JI. 2006. Construction and analysis of chromosomal Clostridium difficile mutants. Mol Microbiol 61:1335–1351 10.1111/j.1365-2958.2006.05315.x. [PubMed] [DOI] [PubMed] [Google Scholar]
- 32.Lyras D, O’Connor JR, Howarth PM, Sambol SP, Carter GP, Phumoonna T, Poon R, Adams V, Vedantam G, Johnson S, Gerding DN, Rood JI. 2009. Toxin B is essential for virulence of Clostridium difficile. Nature 458:1176–1179 10.1038/nature07822. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hussain HA, Roberts AP, Mullany P. 2005. Generation of an erythromycin-sensitive derivative of Clostridium difficile strain 630 (630Deltaerm) and demonstration that the conjugative transposon Tn916DeltaE enters the genome of this strain at multiple sites. J Med Microbiol 54:137–141 10.1099/jmm.0.45790-0. [PubMed] [DOI] [PubMed] [Google Scholar]
- 34.Collery MM, Kuehne SA, McBride SM, Kelly ML, Monot M, Cockayne A, Dupuy B, Minton NP. 2017. What’s a SNP between friends: the influence of single nucleotide polymorphisms on virulence and phenotypes of Clostridium difficile strain 630 and derivatives. Virulence 8:767–781 10.1080/21505594.2016.1237333. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Riedel T, Bunk B, Thürmer A, Spröer C, Brzuszkiewicz E, Abt B, Gronow S, Liesegang H, Daniel R, Overmann J. 2015. Genome resequencing of the virulent and multidrug-resistant reference strain Clostridium difficile 630. Genome Announc 3:e00276-15 10.1128/genomeA.00276-15. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.van Eijk E, Anvar SY, Browne HP, Leung WY, Frank J, Schmitz AM, Roberts AP, Smits WK. 2015. Complete genome sequence of the Clostridium difficile laboratory strain 630Δerm reveals differences from strain 630, including translocation of the mobile element CTn5. BMC Genomics 16:31 10.1186/s12864-015-1252-7. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Minton NP, Ehsaan M, Humphreys CM, Little GT, Baker J, Henstra AM, Liew F, Kelly ML, Sheng L, Schwarz K, Zhang Y. 2016. A roadmap for gene system development in Clostridium. Anaerobe 41:104–112 10.1016/j.anaerobe.2016.05.011. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Johnston CD, Skeete CA, Fomenkov A, Roberts RJ, Rittling SR. 2017. Restriction-modification mediated barriers to exogenous DNA uptake and incorporation employed by Prevotella intermedia. PLoS One 12:e0185234 10.1371/journal.pone.0185234. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mermelstein LD, Papoutsakis ET. 1993. In vivo methylation in Escherichia coli by the Bacillus subtilis phage phi 3T I methyltransferase to protect plasmids from restriction upon transformation of Clostridium acetobutylicum ATCC 824. Appl Environ Microbiol 59:1077–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pyne ME, Moo-Young M, Chung DA, Chou CP. 2013. Development of an electrotransformation protocol for genetic manipulation of Clostridium pasteurianum. Biotechnol Biofuels 6:50 10.1186/1754-6834-6-50. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lesiak JM, Liebl W, Ehrenreich A. 2014. Development of an in vivo methylation system for the solventogen Clostridium saccharobutylicum NCP 262 and analysis of two endonuclease mutants. J Biotechnol 188:97–99 10.1016/j.jbiotec.2014.07.005. [PubMed] [DOI] [PubMed] [Google Scholar]
- 42.Yang X, Xu M, Yang ST. 2015. Metabolic and process engineering of Clostridium cellulovorans for biofuel production from cellulose. Metab Eng 32:39–48 10.1016/j.ymben.2015.09.001. [PubMed] [DOI] [PubMed] [Google Scholar]
- 43.Yang X, Xu M, Yang ST. 2016. Restriction modification system analysis and development of in vivo methylation for the transformation of Clostridium cellulovorans. Appl Microbiol Biotechnol 100:2289–2299 10.1007/s00253-015-7141-9. [PubMed] [DOI] [PubMed] [Google Scholar]
- 44.Uzal FA, Freedman JC, Shrestha A, Theoret JR, Garcia J, Awad MM, Adams V, Moore RJ, Rood JI, McClane BA. 2014. Towards an understanding of the role of Clostridium perfringens toxins in human and animal disease. Future Microbiol 9:361–377 10.2217/fmb.13.168. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lewis BB, Carter RA, Ling L, Leiner I, Taur Y, Kamboj M, Dubberke ER, Xavier J, Pamer EG. 2017. Pathogenicity locus, core genome, and accessory gene contributions to Clostridium difficile virulence. MBio 8:e00885-17 10.1128/mBio.00885-17. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Joseph RC, Kim NM, Sandoval NR. 2018. Recent developments of the synthetic biology toolkit for Clostridium. Front Microbiol 9:154 10.3389/fmicb.2018.00154. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bradshaw M, Goodnough MC, Johnson EA. 1998. Conjugative transfer of the Escherichia coli-Clostridium perfringens shuttle vector pJIR1457 to Clostridium botulinum type A strains. Plasmid 40:233–237 10.1006/plas.1998.1366. [PubMed] [DOI] [PubMed] [Google Scholar]
- 48.Carter GP, Awad MM, Hao Y, Thelen T, Bergin IL, Howarth PM, Seemann T, Rood JI, Aronoff DM, Lyras D. 2011. TcsL is an essential virulence factor in Clostridium sordellii ATCC 9714. Infect Immun 79:1025–1032 10.1128/IAI.00968-10. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Carter GP, Larcombe S, Li L, Jayawardena D, Awad MM, Songer JG, Lyras D. 2014. Expression of the large clostridial toxins is controlled by conserved regulatory mechanisms. Int J Med Microbiol 304:1147–1159 10.1016/j.ijmm.2014.08.008. [PubMed] [DOI] [PubMed] [Google Scholar]
- 50.Awad MM, Cheung JK, Tan JE, McEwan AG, Lyras D, Rood JI. 2016. Functional analysis of an feoB mutant in Clostridium perfringens strain 13. Anaerobe 41:10–17 10.1016/j.anaerobe.2016.05.005. [PubMed] [DOI] [PubMed] [Google Scholar]
- 51.Vidor CJ, Watts TD, Adams V, Bulach D, Couchman E, Rood JI, Fairweather NF, Awad M, Lyras D. 2018. Clostridium sordellii pathogenicity locus plasmid pCS1-1 encodes a novel clostridial conjugation locus. MBio 9:e01761-17 10.1128/mBio.01761-17. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rabi R, Larcombe S, Mathias R, McGowan S, Awad M, Lyras D. 2018. Clostridium sordellii outer spore proteins maintain spore structural integrity and promote bacterial clearance from the gastrointestinal tract. PLoS Pathog 14:e1007004 10.1371/journal.ppat.1007004. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Carter GP, Douce GR, Govind R, Howarth PM, Mackin KE, Spencer J, Buckley AM, Antunes A, Kotsanas D, Jenkin GA, Dupuy B, Rood JI, Lyras D. 2011. The anti-sigma factor TcdC modulates hypervirulence in an epidemic BI/NAP1/027 clinical isolate of Clostridium difficile. PLoS Pathog 7:e1002317 10.1371/journal.ppat.1002317. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mackin KE, Carter GP, Howarth P, Rood JI, Lyras D. 2013. Spo0A differentially regulates toxin production in evolutionarily diverse strains of Clostridium difficile. PLoS One 8:e79666 10.1371/journal.pone.0079666. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kirk JA, Fagan RP. 2016. Heat shock increases conjugation efficiency in Clostridium difficile. Anaerobe 42:1–5 10.1016/j.anaerobe.2016.06.009. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Purdy D, O’Keeffe TA, Elmore M, Herbert M, McLeod A, Bokori-Brown M, Ostrowski A, Minton NP. 2002. Conjugative transfer of clostridial shuttle vectors from Escherichia coli to Clostridium difficile through circumvention of the restriction barrier. Mol Microbiol 46:439–452 10.1046/j.1365-2958.2002.03134.x. [PubMed] [DOI] [PubMed] [Google Scholar]
- 57.Fox ME, Lemmon MJ, Mauchline ML, Davis TO, Giaccia AJ, Minton NP, Brown JM. 1996. Anaerobic bacteria as a delivery system for cancer gene therapy: in vitro activation of 5-fluorocytosine by genetically engineered clostridia. Gene Ther 3:173–178. [PubMed] [Google Scholar]
- 58.Reynolds CB, Emerson JE, de la Riva L, Fagan RP, Fairweather NF. 2011. The Clostridium difficile cell wall protein CwpV is antigenically variable between strains, but exhibits conserved aggregation-promoting function. PLoS Pathog 7:e1002024 10.1371/journal.ppat.1002024. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kovacs-Simon A, Leuzzi R, Kasendra M, Minton N, Titball RW, Michell SL. 2014. Lipoprotein CD0873 is a novel adhesin of Clostridium difficile. J Infect Dis 210:274–284 10.1093/infdis/jiu070. [PubMed] [DOI] [PubMed] [Google Scholar]
- 60.de la Riva L, Willing SE, Tate EW, Fairweather NF. 2011. Roles of cysteine proteases Cwp84 and Cwp13 in biogenesis of the cell wall of Clostridium difficile. J Bacteriol 193:3276–3285 10.1128/JB.00248-11. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Heap JT, Pennington OJ, Cartman ST, Minton NP. 2009. A modular system for Clostridium shuttle plasmids. J Microbiol Methods 78:79–85 10.1016/j.mimet.2009.05.004. [PubMed] [DOI] [PubMed] [Google Scholar]
- 62.Walter BM, Rupnik M, Hodnik V, Anderluh G, Dupuy B, Paulič N, Žgur-Bertok D, Butala M. 2014. The LexA regulated genes of the Clostridium difficile. BMC Microbiol 14:88 10.1186/1471-2180-14-88. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Martin MJ, Clare S, Goulding D, Faulds-Pain A, Barquist L, Browne HP, Pettit L, Dougan G, Lawley TD, Wren BW. 2013. The agr locus regulates virulence and colonization genes in Clostridium difficile 027. J Bacteriol 195:3672–3681 10.1128/JB.00473-13. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Dapa T, Unnikrishnan M. 2013. Biofilm formation by Clostridium difficile. Gut Microbes 4:397–402 10.4161/gmic.25862. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zhang Z, Korkeala H, Dahlsten E, Sahala E, Heap JT, Minton NP, Lindström M. 2013. Two-Component Signal Transduction System CBO0787/CBO0786 Represses Transcription from Botulinum Neurotoxin Promoters in Clostridium botulinum ATCC 3502. PLoS Pathog 9(3): e1003252. 10.1371/journal.ppat.1003252. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Little GT, Willson BJ, Heap JT, Winzer K, Minton NP. 2018. The butanol producing microbe Clostridium beijerinckii NCIMB 14988 manipulated using forward and reverse genetic tools. Biotechnol J 13:e1700711 10.1002/biot.201700711. [PubMed] [DOI] [PubMed] [Google Scholar]
- 67.Liew F, Henstra AM, Köpke M, Winzer K, Simpson SD, Minton NP. 2017. Metabolic engineering of Clostridium autoethanogenum for selective alcohol production. Metab Eng 40:104–114 10.1016/j.ymben.2017.01.007. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hartman AH, Liu H, Melville SB. 2011. Construction and characterization of a lactose-inducible promoter system for controlled gene expression in Clostridium perfringens. Appl Environ Microbiol 77:471–478 10.1128/AEM.01536-10. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hendrick WA, Orr MW, Murray SR, Lee VT, Melville SB. 2017. Cyclic Di-GMP binding by an assembly ATPase (PilB2) and control of type IV pilin polymerization in the Gram-positive pathogen Clostridium perfringens. J Bacteriol 199:e00034-17 10.1128/JB.00034-17. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Obana N, Nakamura K, Nomura N. 2016. Role of RNase Y in Clostridium perfringens mRNA decay and processing. J Bacteriol 199:e00703-16. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kumar RS, Hendrick W, Correll JB, Patterson AD, Melville SB, Ferry JG. 2013. Biochemistry and physiology of the β class carbonic anhydrase (Cpb) from Clostridium perfringens strain 13. J Bacteriol 195:2262–2269 10.1128/JB.02288-12. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Banerjee A, Leang C, Ueki T, Nevin KP, Lovley DR. 2014. Lactose-inducible system for metabolic engineering of Clostridium ljungdahlii. Appl Environ Microbiol 80:2410–2416 10.1128/AEM.03666-13. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Nariya H, Miyata S, Kuwahara T, Okabe A. 2011. Development and characterization of a xylose-inducible gene expression system for Clostridium perfringens. Appl Environ Microbiol 77:8439–8441 10.1128/AEM.05668-11. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Fagan RP, Fairweather NF. 2011. Clostridium difficile has two parallel and essential Sec secretion systems. J Biol Chem 286:27483–27493 10.1074/jbc.M111.263889. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Corrigan RM, Foster TJ. 2009. An improved tetracycline-inducible expression vector for Staphylococcus aureus. Plasmid 61:126–129 10.1016/j.plasmid.2008.10.001. [PubMed] [DOI] [PubMed] [Google Scholar]
- 76.Govind R, Dupuy B. 2012. Secretion of Clostridium difficile toxins A and B requires the holin-like protein TcdE. PLoS Pathog 8:e1002727 10.1371/journal.ppat.1002727. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Pereira FC, Saujet L, Tomé AR, Serrano M, Monot M, Couture-Tosi E, Martin-Verstraete I, Dupuy B, Henriques AO. 2013. The spore differentiation pathway in the enteric pathogen Clostridium difficile. PLoS Genet 9:e1003782 10.1371/journal.pgen.1003782. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Dembek M, Willing SE, Hong HA, Hosseini S, Salgado PS, Cutting SM. 2017. Inducible expression of spo0A as a universal tool for studying sporulation in Clostridium difficile. Front Microbiol 8:1793 10.3389/fmicb.2017.01793. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Permpoonpattana P, Phetcharaburanin J, Mikelsone A, Dembek M, Tan S, Brisson MC, La Ragione R, Brisson AR, Fairweather N, Hong HA, Cutting SM. 2013. Functional characterization of Clostridium difficile spore coat proteins. J Bacteriol 195:1492–1503 10.1128/JB.02104-12. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Purcell EB, McKee RW, McBride SM, Waters CM, Tamayo R. 2012. Cyclic diguanylate inversely regulates motility and aggregation in Clostridium difficile. J Bacteriol 194:3307–3316 10.1128/JB.00100-12. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Adams V, Bantwal R, Stevenson L, Cheung JK, Awad MM, Nicholson J, Carter GP, Mackin KE, Rood JI, Lyras D. 2014. Utility of the clostridial site-specific recombinase TnpX to clone toxic-product-encoding genes and selectively remove genomic DNA fragments. Appl Environ Microbiol 80:3597–3603 10.1128/AEM.04285-13. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Bantwal R, Bannam TL, Porter CJ, Quinsey NS, Lyras D, Adams V, Rood JI. 2012. The peptidoglycan hydrolase TcpG is required for efficient conjugative transfer of pCW3 in Clostridium perfringens. Plasmid 67:139–147 10.1016/j.plasmid.2011.12.016. [PubMed] [DOI] [PubMed] [Google Scholar]
- 83.Lyras D, Rood JI. 2000. Transposition of Tn4451 and Tn4453 involves a circular intermediate that forms a promoter for the large resolvase, TnpX. Mol Microbiol 38:588–601 10.1046/j.1365-2958.2000.02154.x. [DOI] [PubMed] [Google Scholar]
- 84.Ransom EM, Ellermeier CD, Weiss DS. 2015. Use of mCherry Red fluorescent protein for studies of protein localization and gene expression in Clostridium difficile. Appl Environ Microbiol 81:1652–1660 10.1128/AEM.03446-14. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Buckley AM, Jukes C, Candlish D, Irvine JJ, Spencer J, Fagan RP, Roe AJ, Christie JM, Fairweather NF, Douce GR. 2016. Lighting up Clostridium difficile: reporting gene expression using fluorescent LOV domains. Sci Rep 6:23463 10.1038/srep23463. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Ransom EM, Williams KB, Weiss DS, Ellermeier CD. 2014. Identification and characterization of a gene cluster required for proper rod shape, cell division, and pathogenesis in Clostridium difficile. J Bacteriol 196:2290–2300 10.1128/JB.00038-14. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ribis JW, Ravichandran P, Putnam EE, Pishdadian K, Shen A. 2017. The conserved spore coat protein SpoVM is largely dispensable in Clostridium difficile spore formation. MSphere 2:e00315-17 10.1128/mSphere.00315-17. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ribis JW, Fimlaid KA, Shen A. 2018. Differential requirements for conserved peptidoglycan remodeling enzymes during Clostridioides difficile spore formation. Mol Microbiol 110:370–389 10.1111/mmi.14090. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Anjuwon-Foster BR, Tamayo R. 2017. A genetic switch controls the production of flagella and toxins in Clostridium difficile. PLoS Genet 13:e1006701 10.1371/journal.pgen.1006701. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Ransom EM, Kaus GM, Tran PM, Ellermeier CD, Weiss DS. 2018. Multiple factors contribute to bimodal toxin gene expression in Clostridioides (Clostridium) difficile. Mol Microbiol 110:533–549 10.1111/mmi.14107. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Drepper T, Eggert T, Circolone F, Heck A, Krauss U, Guterl JK, Wendorff M, Losi A, Gärtner W, Jaeger KE. 2007. Reporter proteins for in vivo fluorescence without oxygen. Nat Biotechnol 25:443–445 10.1038/nbt1293. [PubMed] [DOI] [PubMed] [Google Scholar]
- 92.Christie JM, Gawthorne J, Young G, Fraser NJ, Roe AJ. 2012. LOV to BLUF: flavoprotein contributions to the optogenetic toolkit. Mol Plant 5:533–544 10.1093/mp/sss020. [PubMed] [DOI] [PubMed] [Google Scholar]
- 93.Molitor B, Kirchner K, Henrich AW, Schmitz S, Rosenbaum MA. 2016. Expanding the molecular toolkit for the homoacetogen Clostridium ljungdahlii. Sci Rep 6:31518 10.1038/srep31518. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Serrano M, Crawshaw AD, Dembek M, Monteiro JM, Pereira FC, Pinho MG, Fairweather NF, Salgado PS, Henriques AO. 2016. The SpoIIQ-SpoIIIAH complex of Clostridium difficile controls forespore engulfment and late stages of gene expression and spore morphogenesis. Mol Microbiol 100:204–228 10.1111/mmi.13311. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Fimlaid KA, Jensen O, Donnelly ML, Siegrist MS, Shen A. 2015. Regulation of Clostridium difficile spore formation by the SpoIIQ and SpoIIIA proteins. PLoS Genet 11:e1005562 10.1371/journal.pgen.1005562. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Mani N, Dupuy B. 2001. Regulation of toxin synthesis in Clostridium difficile by an alternative RNA polymerase sigma factor. Proc Natl Acad Sci U S A 98:5844–5849 10.1073/pnas.101126598. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Hensbergen PJ, Klychnikov OI, Bakker D, van Winden VJ, Ras N, Kemp AC, Cordfunke RA, Dragan I, Deelder AM, Kuijper EJ, Corver J, Drijfhout JW, van Leeuwen HC. 2014. A novel secreted metalloprotease (CD2830) from Clostridium difficile cleaves specific proline sequences in LPXTG cell surface proteins. Mol Cell Proteomics 13:1231–1244 10.1074/mcp.M113.034728. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Oliveira Paiva AM, Friggen AH, Hossein-Javaheri S, Smits WK. 2016. The signal sequence of the abundant extracellular metalloprotease PPEP-1 can be used to secrete synthetic reporter proteins in Clostridium difficile. ACS Synth Biol 5:1376–1382 10.1021/acssynbio.6b00104. [PubMed] [DOI] [PubMed] [Google Scholar]
- 99.Hall MP, Unch J, Binkowski BF, Valley MP, Butler BL, Wood MG, Otto P, Zimmerman K, Vidugiris G, Machleidt T, Robers MB, Benink HA, Eggers CT, Slater MR, Meisenheimer PL, Klaubert DH, Fan F, Encell LP, Wood KV. 2012. Engineered luciferase reporter from a deep sea shrimp utilizing a novel imidazopyrazinone substrate. ACS Chem Biol 7:1848–1857 10.1021/cb3002478. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Edwards AN, Pascual RA, Childress KO, Nawrocki KL, Woods EC, McBride SM. 2015. An alkaline phosphatase reporter for use in Clostridium difficile. Anaerobe 32:98–104 10.1016/j.anaerobe.2015.01.002. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Matsushita C, Matsushita O, Koyama M, Okabe A. 1994. A Clostridium perfringens vector for the selection of promoters. Plasmid 31:317–319 10.1006/plas.1994.1035. [PubMed] [DOI] [PubMed] [Google Scholar]
- 102.Bullifent HL, Moir A, Titball RW. 1995. The construction of a reporter system and use for the investigation of Clostridium perfringens gene expression. FEMS Microbiol Lett 131:99–105 10.1111/j.1574-6968.1995.tb07761.x. [DOI] [PubMed] [Google Scholar]
- 103.Phillips-Jones MK. 1993. Bioluminescence (lux) expression in the anaerobe Clostridium perfringens. FEMS Microbiol Lett 106:265–270 10.1111/j.1574-6968.1993.tb05974.x. [DOI] [PubMed] [Google Scholar]
- 104.Zhao Y, Melville SB. 1998. Identification and characterization of sporulation-dependent promoters upstream of the enterotoxin gene (cpe) of Clostridium perfringens. J Bacteriol 180:136–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Takamizawa A, Miyata S, Matsushita O, Kaji M, Taniguchi Y, Tamai E, Shimamoto S, Okabe A. 2004. High-level expression of clostridial sialidase using a ferredoxin gene promoter-based plasmid. Protein Expr Purif 36:70–75 10.1016/j.pep.2004.03.004. [PubMed] [DOI] [PubMed] [Google Scholar]
- 106.Faulds-Pain A, Wren BW. 2013. Improved bacterial mutagenesis by high-frequency allele exchange, demonstrated in Clostridium difficile and Streptococcus suis. Appl Environ Microbiol 79:4768–4771 10.1128/AEM.01195-13. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Cartman ST, Kelly ML, Heeg D, Heap JT, Minton NP. 2012. Precise manipulation of the Clostridium difficile chromosome reveals a lack of association between the tcdC genotype and toxin production. Appl Environ Microbiol 78:4683–4690 10.1128/AEM.00249-12. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Ng YK, Ehsaan M, Philip S, Collery MM, Janoir C, Collignon A, Cartman ST, Minton NP. 2013. Expanding the repertoire of gene tools for precise manipulation of the Clostridium difficile genome: allelic exchange using pyrE alleles. PLoS One 8:e56051 10.1371/journal.pone.0056051. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Peltier J, Shaw HA, Couchman EC, Dawson LF, Yu L, Choudhary JS, Kaever V, Wren BW, Fairweather NF. 2015. Cyclic diGMP regulates production of sortase substrates of Clostridium difficile and their surface exposure through ZmpI protease-mediated cleavage. J Biol Chem 290:24453–24469 10.1074/jbc.M115.665091. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Francis MB, Sorg JA. 2016. Dipicolinic acid release by germinating Clostridium difficile spores occurs through a mechanosensing mechanism. MSphere 1:e00306-16 10.1128/mSphere.00306-16. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Heap JT, Ehsaan M, Cooksley CM, Ng YK, Cartman ST, Winzer K, Minton NP. 2012. Integration of DNA into bacterial chromosomes from plasmids without a counter-selection marker. Nucleic Acids Res 40:e59 10.1093/nar/gkr1321. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Bilverstone TW, Kinsmore NL, Minton NP, Kuehne SA. 2017. Development of Clostridium difficile R20291ΔPaLoc model strains and in vitro methodologies reveals CdtR is required for the production of CDT to cytotoxic levels. Anaerobe 44:51–54 10.1016/j.anaerobe.2017.01.009. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Collins J, Robinson C, Danhof H, Knetsch CW, van Leeuwen HC, Lawley TD, Auchtung JM, Britton RA. 2018. Dietary trehalose enhances virulence of epidemic Clostridium difficile. Nature 553:291–294 10.1038/nature25178. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Lambowitz AM, Zimmerly S. 2004. Mobile group II introns. Annu Rev Genet 38:1–35 10.1146/annurev.genet.38.072902.091600. [PubMed] [DOI] [PubMed] [Google Scholar]
- 115.Chen Y, McClane BA, Fisher DJ, Rood JI, Gupta P. 2005. Construction of an alpha toxin gene knockout mutant of Clostridium perfringens type A by use of a mobile group II intron. Appl Environ Microbiol 71:7542–7547 10.1128/AEM.71.11.7542-7547.2005. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Chen Y, Caruso L, McClane B, Fisher D, Gupta P. 2007. Disruption of a toxin gene by introduction of a foreign gene into the chromosome of Clostridium perfringens using targetron-induced mutagenesis. Plasmid 58:182–189 10.1016/j.plasmid.2007.04.002. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Heap JT, Pennington OJ, Cartman ST, Carter GP, Minton NP. 2007. The ClosTron: a universal gene knock-out system for the genus Clostridium. J Microbiol Methods 70:452–464 10.1016/j.mimet.2007.05.021. [PubMed] [DOI] [PubMed] [Google Scholar]
- 118.Kuehne SA, Minton NP. 2012. ClosTron-mediated engineering of Clostridium. Bioengineered 3:247–254 10.4161/bioe.21004. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Kuehne SA, Cartman ST, Heap JT, Kelly ML, Cockayne A, Minton NP. 2010. The role of toxin A and toxin B in Clostridium difficile infection. Nature 467:711–713 10.1038/nature09397. [PubMed] [DOI] [PubMed] [Google Scholar]
- 120.Heap JT, Kuehne SA, Ehsaan M, Cartman ST, Cooksley CM, Scott JC, Minton NP. 2010. The ClosTron: mutagenesis in Clostridium refined and streamlined. J Microbiol Methods 80:49–55 10.1016/j.mimet.2009.10.018. [PubMed] [DOI] [PubMed] [Google Scholar]
- 121.Lyristis M, Bryant AE, Sloan J, Awad MM, Nisbet IT, Stevens DL, Rood JI. 1994. Identification and molecular analysis of a locus that regulates extracellular toxin production in Clostridium perfringens. Mol Microbiol 12:761–777 10.1111/j.1365-2958.1994.tb01063.x. [PubMed] [DOI] [PubMed] [Google Scholar]
- 122.Awad MM, Rood JI. 1997. Isolation of alpha-toxin, theta-toxin and kappa-toxin mutants of Clostridium perfringens by Tn916 mutagenesis. Microb Pathog 22:275–284 10.1006/mpat.1996.0115. [PubMed] [DOI] [PubMed] [Google Scholar]
- 123.Kaufmann PLY, Meile L. 1996. Conjugative transposition of Tn916 from Enterococcus faecalis and Escherichia coli into Clostridium perfringens. Syst Appl Microbiol 19:35–39 10.1016/S0723-2020(96)80006-3. [DOI] [Google Scholar]
- 124.Briolat V, Reysset G. 2002. Identification of the Clostridium perfringens genes involved in the adaptive response to oxidative stress. J Bacteriol 184:2333–2343 10.1128/JB.184.9.2333-2343.2002. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Bannam TL, Teng WL, Bulach D, Lyras D, Rood JI. 2006. Functional identification of conjugation and replication regions of the tetracycline resistance plasmid pCW3 from Clostridium perfringens. J Bacteriol 188:4942–4951 10.1128/JB.00298-06. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Vidal JE, Chen J, Li J, McClane BA. 2009. Use of an EZ-Tn5-based random mutagenesis system to identify a novel toxin regulatory locus in Clostridium perfringens strain 13. PLoS One 4:e6232 10.1371/journal.pone.0006232. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Lanckriet A, Timbermont L, Happonen LJ, Pajunen MI, Pasmans F, Haesebrouck F, Ducatelle R, Savilahti H, Van Immerseel F. 2009. Generation of single-copy transposon insertions in Clostridium perfringens by electroporation of phage mu DNA transposition complexes. Appl Environ Microbiol 75:2638–2642 10.1128/AEM.02214-08. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Liu H, Bouillaut L, Sonenshein AL, Melville SB. 2013. Use of a mariner-based transposon mutagenesis system to isolate Clostridium perfringens mutants deficient in gliding motility. J Bacteriol 195:629–636 10.1128/JB.01288-12. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Mullany P. 2014. Functional metagenomics for the investigation of antibiotic resistance. Virulence 5:443–447 10.4161/viru.28196. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Wang H, Smith MC, Mullany P. 2006. The conjugative transposon Tn5397 has a strong preference for integration into its Clostridium difficile target site. J Bacteriol 188:4871–4878 10.1128/JB.00210-06. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Hussain HA, Roberts AP, Whalan R, Mullany P. 2010. Transposon mutagenesis in Clostridium difficile. Methods Mol Biol 646:203–211 10.1007/978-1-60327-365-7_13. [PubMed] [DOI] [PubMed] [Google Scholar]
- 132.Cartman ST, Minton NP. 2010. A mariner-based transposon system for in vivo random mutagenesis of Clostridium difficile. Appl Environ Microbiol 76:1103–1109 10.1128/AEM.02525-09. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Dembek M, Barquist L, Boinett CJ, Cain AK, Mayho M, Lawley TD, Fairweather NF, Fagan RP. 2015. High-throughput analysis of gene essentiality and sporulation in Clostridium difficile. MBio 6:e02383 10.1128/mBio.02383-14. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Sangster W, Hegarty JP, Stewart DB Sr. 2015. Phage tail-like particles kill Clostridium difficile and represent an alternative to conventional antibiotics. Surgery 157:96–103 10.1016/j.surg.2014.06.015. [PubMed] [DOI] [PubMed] [Google Scholar]
- 135.Meader E, Mayer MJ, Steverding D, Carding SR, Narbad A. 2013. Evaluation of bacteriophage therapy to control Clostridium difficile and toxin production in an in vitro human colon model system. Anaerobe 22:25–30 10.1016/j.anaerobe.2013.05.001. [PubMed] [DOI] [PubMed] [Google Scholar]
- 136.Nale JY, Spencer J, Hargreaves KR, Buckley AM, Trzepińński P, Douce GR, Clokie MR. 2015. Bacteriophage combinations significantly reduce Clostridium difficile growth in vitro and proliferation in vivo. Antimicrob Agents Chemother 60:968–981 10.1128/AAC.01774-15. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Goh S, Hussain H, Chang BJ, Emmett W, Riley TV, Mullany P. 2013. Phage ɸC2 mediates transduction of Tn6215, encoding erythromycin resistance, between Clostridium difficile strains. MBio 4:e00840-13 10.1128/mBio.00840-13. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Pyne ME, Bruder MR, Moo-Young M, Chung DA, Chou CP. 2016. Harnessing heterologous and endogenous CRISPR-Cas machineries for efficient markerless genome editing in Clostridium. Sci Rep 6:25666 10.1038/srep25666. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Wang Y, Zhang ZT, Seo SO, Choi K, Lu T, Jin YS, Blaschek HP. 2015. Markerless chromosomal gene deletion in Clostridium beijerinckii using CRISPR/Cas9 system. J Biotechnol 200:1–5 10.1016/j.jbiotec.2015.02.005. [PubMed] [DOI] [PubMed] [Google Scholar]
- 140.Xu T, Li Y, Shi Z, Hemme CL, Li Y, Zhu Y, Van Nostrand JD, He Z, Zhou J. 2015. Efficient genome editing in Clostridium cellulolyticum via CRISPR-Cas9 nickase. Appl Environ Microbiol 81:4423–4431 10.1128/AEM.00873-15. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.McAllister KN, Bouillaut L, Kahn JN, Self WT, Sorg JA. 2017. Using CRISPR-Cas9-mediated genome editing to generate C. difficile mutants defective in selenoproteins synthesis. Sci Rep 7:14672 10.1038/s41598-017-15236-5. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Wang S, Hong W, Dong S, Zhang ZT, Zhang J, Wang L, Wang Y. 2018. Genome engineering of Clostridium difficile using the CRISPR-Cas9 system. Clin Microbiol Infect 24:1095–1099 10.1016/j.cmi.2018.03.026. [PubMed] [DOI] [PubMed] [Google Scholar]
- 143.Hong W, Zhang J, Cui G, Wang L, Wang Y. 2018. Multiplexed CRISPR-Cpf1-mediated genome editing in Clostridium difficile toward the understanding of pathogenesis of C. difficile infection. ACS Synth Biol 7:1588–1600 10.1021/acssynbio.8b00087. [PubMed] [DOI] [PubMed] [Google Scholar]
- 144.Zhang N, Shao L, Jiang Y, Gu Y, Li Q, Liu J, Jiang W, Yang S. 2015. I-SceI-mediated scarless gene modification via allelic exchange in Clostridium. J Microbiol Methods 108:49–60 10.1016/j.mimet.2014.11.004. [PubMed] [DOI] [PubMed] [Google Scholar]
- 145.Al-Hinai MA, Fast AG, Papoutsakis ET. 2012. Novel system for efficient isolation of Clostridium double-crossover allelic exchange mutants enabling markerless chromosomal gene deletions and DNA integration. Appl Environ Microbiol 78:8112–8121 10.1128/AEM.02214-12. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Zhang J, Ni C, Yang Z, Piontek A, Chen H, Wang S, Fan Y, Qin Z, Piontek J. 2015. Specific binding of Clostridium perfringens enterotoxin fragment to Claudin-b and modulation of zebrafish epidermal barrier. Exp Dermatol 24:605–610 10.1111/exd.12728. [PubMed] [DOI] [PubMed] [Google Scholar]
- 147.Buffie CG, Bucci V, Stein RR, McKenney PT, Ling L, Gobourne A, No D, Liu H, Kinnebrew M, Viale A, Littmann E, van den Brink MR, Jenq RR, Taur Y, Sander C, Cross JR, Toussaint NC, Xavier JB, Pamer EG. 2015. Precision microbiome reconstitution restores bile acid mediated resistance to Clostridium difficile. Nature 517:205–208 10.1038/nature13828. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Studer N, Desharnais L, Beutler M, Brugiroux S, Terrazos MA, Menin L, Schürch CM, McCoy KD, Kuehne SA, Minton NP, Stecher B, Bernier-Latmani R, Hapfelmeier S. 2016. Functional intestinal bile acid 7α-dehydroxylation by Clostridium scindens associated with protection from Clostridium difficile infection in a gnotobiotic mouse model. Front Cell Infect Microbiol 6:191 10.3389/fcimb.2016.00191. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]