

Abstract
Human DNA polymerase epsilon (pol ɛ) normally contains a 261-kDa catalytic subunit (p261), but from some sources it is isolated as a 140-kDa catalytic core of p261. This shortened form possesses normal or somewhat enhanced polymerase activity and its significance is unknown. We report here that caspase-3 and calpain can form p140 from p261 in vitro and in vivo and that during early stages of apoptosis induced in Jurkat cells by staurosporine or anti-Fas-activating antibody, p261 is cleaved into p140 by caspase-3. At later stages, activated calpain might also contribute to this conversion. The sites of cleavage by caspase-3 have been identified, and mutations at these ‘DEAD boxes’ resulted in cleavage-resistant enzyme. Cleavage at these sites separates the ‘N-terminal catalytic core’ from the ‘C-terminal’ regions described for p261. Cleavage does not occur during necrosis or following exposure to H2O2 or methanesulfonic acid methyl ester. p140 is unlikely to be able to functionally replace p261 in vivo, since it does not bind to PCNA or the other pol ɛ subunits.
INTRODUCTION
Human DNA polymerase epsilon (pol ɛ) is composed of a catalytic subunit of molecular weight 261 000 (p261) (1), and three smaller subunits, p59 (2), p17 and p12 (3). Human p261 shares 39% overall identity with the Saccharomyces cerevisiae pol ɛ catalytic subunit, Pol2p, with three distinctive homology regions: the ‘N-terminal domain’, amino acid residues 1–267 with 26.8% identity; the ‘core catalytic domain’, residues 268–1166 (which include the conserved pol-α family motifs) with 63.0% identity; the ‘C-terminal domain’, residues 1167–2285 with 25.0% identity (1). The last is separated from the remainder of the molecule by a protease-sensitive site (4), and binds the three smaller subunits.
Yeast pol ɛ has been implicated in chromosome DNA replication (5–7), DNA repair (8,9), and cell-cycle checkpoint control in which its C-terminus is required for sensing DNA damage (10–12). Human pol ɛ has been crosslinked to newly-synthesized, photolabeled chromosomal DNA in SV40-infected mammalian cells along with pol α and pol δ (13), indicating an involvement in DNA replication. In addition, pol ɛ has been identified as a repair factor in human fibroblasts (14,15), as well as the DNA polymerase component of the recombination complex, RC-1, from calf thymus (16). Pol ɛ can also function as a repair factor in an in vitro reconstituted nucleotide excision repair complex (17).
Human pol ɛ from HeLa cells was initially observed to contain the p261 catalytic subunit (18); however, a smaller form, p140, is normally observed in enzyme from calf thymus, chicken thymus and Xenopus laevis embryos (19,20) and has also been observed in yeast (21). p140 is derived from p261 and contains catalytic activity in in vitro polymerase assays (4,20). Trypsinization of purified HeLa p261 results in two polypeptide fragments with molecular masses of 122 and 136 kDa. p122 possessed polymerase activity and was resistant to further proteolysis (4,20). Trypsin treatment of p140 purified from calf thymus also produces p122. Therefore, both p140 and p122 contain the catalytic core of p261 (4).
In this paper, we demonstrate that in Jurkat cells, p140 appears only during apoptosis. Both caspase-3 and calpain can mediate the cleavage of pol ɛ p261 to produce p140, but the involvement of calpain occurs much later than that of caspase-3. Both caspase-3 and calpain cleavage occur at the junction between the previously proposed p261 catalytic domain and the C-terminal domains, and therefore effectively separate the catalytic core from the binding sites of the pol ɛ accessory subunits and PCNA.
MATERIALS AND METHODS
Antibodies, inhibitors and cell culture
Purified mouse monoclonal IgGs against DNA pol ɛ p261 (3C5.1) and p59 (3A5.6) were used for immunoblotting as previously described (2). Purified recombinant human caspase-3, monoclonal antibody against poly(ADP-ribose)polymerase (PARP) and polyclonal antibody against caspase-3 were from PharMingen. Anti-Fas-activating mouse monoclonal antibody (CH-11) was from Upstate Biotechnology. Monoclonal antibody against the common 28-kDa calpain II subunit of human m-calpain and µ-calpain was from Chemicon. Staurosporine, oligomycin and calpain inhibitors I and II (CI-I and CI-II) were from Sigma. Calpain inhibitor ZLLY-CHN2 and general caspase inhibitor ZVAD-fmk were from Enzyme Systems Products.
Jurkat T cells and IMR-90 normal diploid lung fibroblasts were from ATCC. Jurkat T cells were grown in RPMI 1640 plus 10% heat-inactivated fetal bovine serum (FBS) and IMR-90 cells were grown in DMEM plus 10% FBS, both in 5% CO2 in a humidified environment at 37°C.
Yeast two-hybrid screening
The yeast two-hybrid screening system (Clontech Laboratories) was used according to the accompanying manual, except that the yeast strain was PJ69-4A. The DNA binding domain vectors were constructed by inserting the p261 cDNA sequences coding for amino acids 1108–2256 (between two BspEI sites), 1301–2285 (between two SmaI sites) or 1603–2285 (between EcoRI and SmaI sites) from pBluescript-pol ɛ (2) into the pGBT9 vector. A human keratinocyte cDNA library in pACT2 (Clontech Laboratories) was co-transformed into the yeast cells with the DNA binding domain construct containing p261 cDNA coding for amino acids 1108–2256. Yeast transformants (8.9 × 106) were first plated onto SC–Trp–Leu–Ade plates and then colonies surviving the –Ade selection were replica-plated onto SC–Trp–Leu–His plates containing 1 mM 3-aminotriazole. Colonies surviving both the –Ade and –His selections were further tested by the liquid β-galactosidase activity assay as described in the manual. Plasmid DNAs were rescued from yeast cells, amplified in Escherichia coli TOP10 cells (Invitrogen), and restriction enzyme digested to obtain the activation domain construct. The cDNA insert in the activation domain construct was subsequently sequenced using an automated ABI373 DNA Analysis System (PE Applied Biosystem, Inc.) at the UC Berkeley Sequencing Facility.
Induction of p261 cleavage in vivo in Jurkat cells
Jurkat cells were fed fresh medium the day before experiments. Among controls, inhibitors of caspases (ZVAD-fmk) or calpain (ZLLY-CHN2, of CI-I and CI-II) were added to the culture 30 min before the induction of apoptosis or activation of calpain, respectively. Apoptosis was induced by the addition of staurosporine to 1.5 µM or anti-Fas-activating antibody to 0.5 µg/ml and incubation for 3.5 h. After washing twice with PBS, cells were harvested and one half used for DNA extraction and the other half for protein extraction as described below. For the induction of necrosis (22), cells were grown in RPMI 1640 medium with 2 mM pyruvate in place of glucose. Thirty minutes after inhibitor addition, oligomycin was added to cell cultures to a final concentration of 2.5 µM and then staurosporine was added after 45 min. For the activation of calpain, Jurkat cells were treated with 8 µg/ml calcium ionophore A23187 for 1 h before harvesting.
Immunoblotting
Cell pellets were resuspended in ice-cold 150 mM NaCl, 1% NP-40, 50 mM Tris–HCl, pH 8.0 in the presence of a mixture of protease inhibitors (19 µg/ml approtinin, 20 µM phenylmethylsulfonyl fluoride, 10 mM benzimide, 50 µM leupeptin, 5 µM pepstatin, 5 mM EGTA, 0.02 mM trans-epoxysuccinyl-l-leucylamido(4-guanidino)butane (E-64), 0.01 mM (2S,3S)-trans-epoxysuccinyl-l-leucylamido-3-methylbutane ethyl ester (E-64d). After 30 min on ice, cell lysates were passed through a 25 gauge needle several times and then incubated on ice for an additional 30 min. After centrifugation at 14 000 g for 15 min, supernatant aliquots containing 30 µg of protein were mixed with Laemmli gel loading buffer containing DTT (23), denatured at 65°C for 15 min and loaded onto denaturing 4–20% polyacrylamide gradient gels. SDS–PAGE and immunoblot analyses were done essentially as described (24) using the enhanced chemilumnescent kit from NEN Life Science Products. For multiple blottings, the nitrocellulose membrane was stripped in 63 mM Tris–HCl, pH 6.8, 2% SDS, 100 mM β-mercaptoethanol at 60°C for 30 min.
DNA fragmentation assay
Cells were lysed in 50 mM Tris–HCl, pH 7.4, 0.5% SDS, 100 mM NaCl, 2 mM EDTA, 0.1 mg/ml proteinase K, 0.1 mg/ml RNaseA. The lysates were incubated at 60°C overnight, then phenol–chloroform extracted, after which DNA was precipitated with ethanol and solvated in TE buffer. Five micrograms of DNA was loaded onto 0.5% agarose gels and resolved by electrophoresis. The gel was stained with 1 µg/ml ethidium bromide and the DNA migration pattern was photographed with the AlphaImager 2000 system (Alpha Innotech).
In vitro cleavage of p261 by bacterially-expressed caspases or purified m-calpain
Plasmid vectors pET15(b) and pET15(b) containing either the cDNA insert of the full-length human caspase-3 or of the p30 domain of murine caspase-1 were generous gifts from Dr J. Yuan (Harvard University, Boston, MA). Escherichia coli JM109 cell lysates containing recombinant human caspase-3 or recombinant murine caspase-1 were prepared according to published procedures (25). For caspase cleavage, 20 µl of partially purified HeLa DNA pol ɛ (5 µg total protein) was incubated with 20 µl of bacterial lysate in the presence of 25 mM DTT for 2 h at 37°C. For m-calpain cleavage, partially purified DNA pol ɛ (6 µg total protein) was incubated with 10 µg partially purified rabbit m-calpain (0.025 U, Sigma) in the presence of 1 mM CaCl2 in a total volume of 40 µl at 37°C for 1 h. For calpain inhibition, 14 µl partially purified human calpastatin (0.035 U, Sigma) was included. Reactions were stopped by the addition of EGTA to a final concentration of 6 mM. Cleavage reaction mixtures were resolved by 4–20% SDS–PAGE and immunoblotting was performed as described above.
Determination of caspase-3 cleavage sites in p261
The cDNA fragment corresponding to amino acids 830–1605 of p261 was cloned into the pET28(a) vector (Invitrogen) between two NcoI sites, to yield pET28(a)–pol ɛ. The cDNA sequence corresponding to amino acids 1–975 was cloned into the pBluescript SK vector (Stratagene) between two SacII sites to yield pBSSK–pol ɛ. Both corresponding peptides could be expressed under the control of a T7 promoter. The product from pET28 (a)–pol ɛ contains a unique DMED sequence at amino acids 1210–1213 (numbers refer to the full-length p261), whereas that from pBSSK–pol ɛ contains five DXXD sequences: DKMD, DQLD, DIED, DFFD and DPED at amino acids 42–45, 213–216, 316–319, 398–401 and 439–442, respectively. For the establishment of p261 sites which are substrate for caspase-3, D→A site-directed mutagenesis for the D1 of each of the six DXXD sequences was performed using the QuickChange Site-Directed Mutagenesis Kit (Stratagene). Mutations were confirmed by DNA sequencing. Both wild-type and mutant polypeptides labeled with [35S]methionine were synthesized in vitro with the TNT® T7 Quick Coupled Reticulocyte Lysate System (Promega) according to the manufacturer’s protocols in reaction volumes of 12.5 µl. Five microliters of the product was reacted with 1 µl of 0.1 µg/µl purified caspase-3 (PharMingen) in the presence of 50 mM DTT in 20 µl caspase-3 assay buffer (20 mM PIPES, 1 mM EDTA, 0.1% CHAPS, 10% sucrose, pH 7.2) at 37°C for 1 h. The reaction mixture was resolved by 4–20% SDS–PAGE and proteins were transferred onto nitrocellulose membranes and visualized by autoradiography.
Transfection, cell cycle analysis and DNA damage of IMR-90 cells
The cDNAs coding for amino acids 1–408 and 1–1408 of the N-terminal segment of p261 were cloned separately into the pMH vector (Roche Biochemicals). Normal human diploid fibroblast IMR-90 cells were transfected with the recombinant plasmid using the Fugene-6 transfection reagent (Roche Biochemicals) according to the manufacturer’s guidelines. Corresponding polypeptides were expressed as C-terminal HA-tagged fusions in transfected IMR-90 cells as verified by immunoblotting using an anti-HA monoclonal antibody (12CA5, Roche Biochemicals).
Cell cycle analysis was performed using a Beckman-Coulter XL flow cytometer. Synchronization of IMR-90 cells was performed essentially according to the method of Tobey et al. (26). For arresting cells in G0/G1 phase, cells were allowed to grow to confluence in 100 mm culture dishes, the medium was replaced with fresh medium and the culture was incubated for 4 days. The cells, which were primarily in G0/G1 as determined by flow cytometry analysis, were then allowed to grow into S phase by splitting 1:3 into new dishes and replacing the medium with DMEM + 20% FBS. The maximum fraction of cells in S phase was determined to be at 24 h post splitting as determined by flow cytometry analysis. For synchronization at early S phase, 40% confluent IMR-90 cells were first synchronized at G0/G1 by growing in DMEM + 0.25% FBS for 3 days, and then in fresh DMEM + 10% FBS for 7 h. Hydroxyurea was then added to a final concentration of 2 mM and the cells were allowed to grow for another 19 h. At this point, up to 90% of the cells were arrested in early S phase as determined by flow cytometry analysis. Cells were then released from arrest and allowed to grow in DMEM + 20% FBS.
DNA damage of log-phase IMR-90 cells was induced by the addition of H2O2 to the culture and incubation for 1 h or methanesulfonic acid methyl ester (MMS) and incubation for 4 h.
RESULTS
p261 is a substrate of calpain in vitro and in vivo
During a yeast two-hybrid screening using amino acids 1108–2256 HeLa pol ɛ p261 (the C-terminal domain plus 59 additional amino acids) as the ‘bait,’ the small subunit of calpain was identified in a positive clone. Calpains are calcium-activated neutral proteases that are expressed in a variety of mammalian tissues (27–30). Each calpain is a heterodimer of an 80-kDa large catalytic subunit (calpain I) and a common 28-kDa small subunit (calpain II). Calpain is activated by autoproteolytic cleavage of both subunits of the inactive proenzyme, with cleavage of the small subunit into a 18-kDa C-terminal fragment that remains associated with the large subunit (28). The peptide fragment identified in the screen contained the C-terminal region (residues 153–268) (Fig. 1A). This segment is the region for calcium binding and interaction with the 80-kDa catalytic subunit (31). Since the identified segment is present in activated calpain II, this result suggested that the C-terminal domain of pol ɛ can interact with activated calpain, and such interaction might reflect substrate recognition before or during a cleavage reaction by the calpain (32).
Figure 1.

Pol ɛ p261 interacts with the small subunit of calpain in a yeast two-hybrid screen. (A) Regions of the small subunit of calpain (residues 153–268) that were found to interact with p261 residues 1108–2256 during the initial yeast two-hybrid screen. (B) Residues of p261 that contribute to the interaction with calpain II as determined by further yeast two-hybrid assays using deletion constructs.
Both m-calpain and µ-calpain have been implicated in the process of nuclear protein turnover by their cleavage activities (33). Interestingly, they both can cleave the catalytic subunit of purified human DNA pol ɛ in in vitro cleavage experiments. Figure 2 demonstrates the result of p261 cleavage by partially purified rabbit m-calpain. (A similar result for µ-calpain cleavage is not shown.) One cleavage product had a molecular mass of 140 kDa, and was recognized by the monoclonal antibody, 3C5.1, that recognizes the N-terminal portion of p261. The p261 cleavage was inhibited by the calpain-specific inhibitor, calpastatin.
Figure 2.
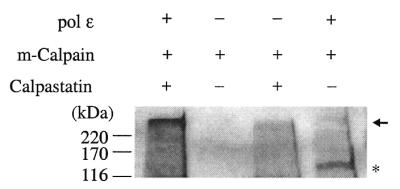
In vitro cleavage of p261 with purified rabbit m-calpain demonstrates that pol ɛ p261 is a substrate of calpain. Positions of full-length p261 and its cleavage product, p140, are denoted by an arrow and an asterisk, respectively. Positions of protein molecular mass markers are indicated to the left in kDa.
The size of the p261 cleavage product is indistinguishable by gel electrophoresis from the truncated, active form of DNA pol ɛ catalytic subunit (‘pol ɛ*’) (4,21) that has been isolated from a variety of sources. In contrast to p261, the p59 subunit of pol ɛ was not detectably cleaved by calpain (data not shown).
The residues in p261 that interact with calpain II were determined to reside between amino acids 1303 and 1603 by using p261 deletion constructs in further yeast two-hybrid assays (Fig. 1B). PEST sequences are presumably the recognition and cleavage sites for calpain (34–36) and two PEST sequences are present between residues 1108 and 1603. Cleavage by calpain after the first of these PEST sequences [1192-RQVTMAEASEDSPR-1205, PEST find score +0.85 (scores greater than –2.0 are significant)] would generate a predicted N-terminal product of 139 kDa, whereas cleavage after the second of these sequences (1232-RVLWESQEESQDLTPTVPWQEILGQPPALGTSQEEWLVWLR-1272, PEST score –0.94) would result in a 146-kDa N-terminal fragment. Both products would be recognized by the 3C5.1 monoclonal antibody. Cleavage at either or both of these sites would separate the core catalytic domain of p261 (residues 268–1166) from the C-terminal domain (residues 1167–2285). Since the accessory subunits (p59, p17 and p12) as well as PCNA interact with the C-terminal domain of p261 (3,6,7), calpain cleavage would also separate the catalytic region of pol ɛ from the binding sites for its accessory subunits.
p261 can be cleaved by calpain in vivo
When Jurkat cells were treated with the calcium ionophore A23187 to activate calpain in vivo, pol ɛ p261 was cleaved into a 140-kDa fragment that was recognized by antibody 3C5.1 (Fig. 3A). The treatment of Jurkat cells by A23187 did not induce apoptosis, as shown by the non-cleavage of 115-kDa PARP (Fig. 3B), a hallmark protein that is degraded by caspase-3 to produce a 86-kDa fragment during apoptosis (see below). The appearance of p140 correlated with the substantial activation of calpain, which was manifested by the autocleavage of calpain II from its 28-kDa proform to produce the 18-kDa activated form (Fig. 3C).
Figure 3.

In vivo cleavage of pol ɛ p261 by activated calpain in Jurkat cells. Jurkat cells were treated with different doses of the calcium ionophore A23187 for 1 h and cell lysates were prepared as described in Materials and Methods. After SDS–PAGE and transfer, the nitrocellulose membrane was probed with monoclonal antibodies against pol ɛ p261 (A), PARP (B) or calpain II (C). Full-length proteins are indicated by an arrow and cleavage products by an asterisk [p140 in (A)] or an arrowhead [activated calpain II in (C)].
p261 is degraded into p140 during apoptosis
A p140 fragment of pol ɛ was also observed by immunoblotting during staurosporine-induced apoptosis (37) of Jurkat cells (Fig. 4A). This appearance started at about the same time as nucleosomal DNA ladder formation, i.e. ∼2 h after the addition of staurosporine (Fig. 4F). This time-course is similar to that reported for the proteolysis of the p140 subunit of the DNA replication factor C (DSEB/RF-C140) during apoptosis of Jurkat cells, which started at the same time as DNA ladder formation, but ∼1 h later than the onset of PARP cleavage (38). In our experiments p140 cleavage was detected about half an hour later than the detection of PARP cleavage (Fig. 4B). (This slight difference could be real or it could be apparent, due to differences in the relative abundances of the two antigens and/or their antibody affinities.)
Figure 4.

Time courses of protein cleavages in apoptotic Jurkat cells. After staurosporine treatment, cell lysates and DNA were prepared as described in Materials and Methods. Protein samples were separated by SDS–PAGE and transferred onto a nitrocellulose membrane which was then probed with antibodies against pol ɛ p261 (A), PARP (B), pol ɛ p59 (C), caspase-3 (D) or calpain II (E). The membrane was stripped between each probing. Positions of molecular weight markers are indicated to the left of each panel in kDa. Full-length proteins are denoted by an arrow and cleavage products by an asterisk [p140 in (A)], or an arrowhead [cleaved PARP, (B); activated caspase-3, (D); activated calpain II, (E)]. (F) DNA fragmentation during apoptosis. Chromosomal DNA was prepared from apoptotic Jurkat cells, separated by 2% agarose gel electrophoresis and stained with ethidium bromide. DNA markers are to the left in base pairs (bp).
The p59 subunit of pol ɛ was not observed to be degraded in the first 3 h after apoptosis induction (Fig. 4C). The p59 pol ɛ subunit was observed, however, to be degraded during the later stages of apoptosis (Fig. 4C). It has been predicted that degradation of p59 might occur due to its possession of an L2G box, which has been implicated in the E3-ubiquitin ligase/E6-AP mediated ubiquitination/degradation pathway (39).
After stripping and reprobing the same nitrocellulose membrane with a polyclonal anti-caspase-3 antibody (Fig. 4D), we also observed activation of caspase-3 that started 1.5 h after apoptosis induction, as shown by the appearance of the 17-kDa activated form of caspase-3. The fragmentation of both PARP (40,41) and DSEB/RF-C140 (38) in apoptotic Jurkat cells has been attributed to the activation of caspase-3.
Activation of calpain II, on the other hand, occurred at a much later stage of apoptosis, between 3 and 9 h after induction of apoptosis (Fig. 4E). Caspase-mediated calpain activation has also been observed only in later stages of drug 9-AC-induced apoptosis in HL-60 cells (42). In addition, the calpain-specific inhibitor, calpastatin, has been shown to be degraded by caspases in staurosporine or anti-Fas-reactive antibody activated apoptosis of Jurkat cells (43). Degradation of calpastatin, which produced a <2-fold increase of calpain activity in the study, could provide another mechanism for activating calpain during apoptosis. Since substantial disappearance of p261 corresponded to the time of activation of calpain II, complete degradation of p261 might require the participation of calpain at later stages of apoptosis.
The p140 fragment could also be detected during apoptosis induced in Jurkat cells by treatment with 0.5 µg/ml anti-Fas-activating antibody (44) (Fig. 5, top panel). Calpain inhibitors ZLLY-CHN2, CI-I and CI-II did not prevent apoptosis, as indicated by the degradation of PARP (Fig. 5, bottom panel) and the appearance of p140, confirming that calpain is not the cleaving enzyme during early stages of apoptosis. On the other hand, the general caspase inhibitor ZVAD-fmk prevented the occurrence of apoptosis, as indicated by intact PARP, and it likewise prevented the degradation of pol ɛ p261.
Figure 5.
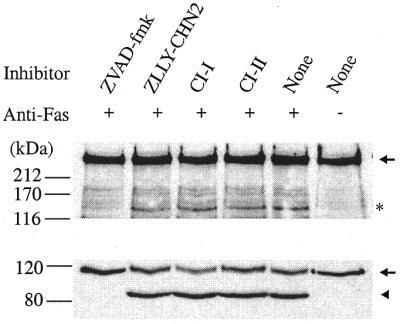
Cleavage of p261 during anti-Fas-activating antibody-induced apoptosis. Jurkat cells were pretreated with inhibitors for 30 min prior to 0.5 µg/ml anti-Fas-activating antibody treatment for 3.5 h. After SDS–PAGE and transfer, the membrane was probed with antibodies against pol ɛ (top panel) and PARP (bottom panel). Full-length proteins are denoted by an arrow and cleavage products by an asterisk (p140, top panel), or an arrowhead (cleaved PARP, bottom panel).
In control experiments (data not shown) the same levels of inhibitors as were used in Figure 5 were added to Jurkat cells and then after 30 min the cells were incubated with 4 µg/ml of A23187 for 2 h in order to induce calpain activation in the absence of apoptosis as in the experiment of Figure 3. The cells were then harvested and analyzed by immunoblotting. These results verified that in this case p140 was observed only in the absence of calpain inhibitors. The general caspase inhibitor, ZVAD-fmk, did not inhibit calpain II activation (autocleavage to generate the 18 kDa active form) and cleavage of p261 was also observed in its presence. On the other hand, no p140 was detected in the presence of any of the three calpain inhibitors, nor was there activation of calpain. These controls verify that at the levels used, the calpain inhibitors do inhibit the calpain-dependent cleavage of pol ɛ and that the level of ZVAD-fmk used does not inhibit the calpains. Hence, the data in Figure 5, along with these controls, suggest strongly that caspase(s) is(are) the p261-cleaving enzyme(s) during the early stages of apoptosis.
Cleavage of p261 is not found during necrosis or following treatment with the DNA damaging agents, H2O2 or MMS
To investigate whether pol ɛ p261 degradation occurred during necrosis of Jurkat cells, necrosis was induced by depleting cellular ATP with oligomycin followed by incubation with staurosporine for 3 h (22). Apoptosis was induced in the same experiment by treating with only staurosporine. p140 was not observed during necrosis, but was observed during the apoptosis induction (Fig. 6A, top panel). This is similar to the observation that nuclear replication factor MCM3 is cleaved during apoptosis but not necrosis (45). As observed for apoptosis induced by the anti-Fas-activating antibody (Fig. 5), the general caspase inhibitor ZVAD-fmk inhibited apoptosis, as shown by the intact PARP (Fig. 6A, bottom panel), and also prevented p261 cleavage. On the other hand, calpain inhibitor ZLLY-CHN2 failed to prevent the onset of apoptosis, as indicated by PARP cleavage and p261 cleavage, confirming again that caspase(s) is(are) the responsible protease(s).
Figure 6.

No p140 was detected during necrosis or following cellular DNA damage. (A) pol ɛ p261 cleavage is not detected during necrosis. Jurkat cells were induced to undergo apoptosis or necrosis as described in Materials and Methods. Immunoblots show that the cleavage of p261 [(top panel, (A)] occurs only in apoptotic cells as manifested by the PARP cleavage [(bottom panel, (A)]. For cellular DNA damage, IMR-90 cells were exposed to H2O2 for 1 h (B) or MMS for 4 h (C). Immunoblots were probed for p140 generation [(top panels of (B) and (C)] or PARP degradation (bottom panels). Full-length proteins are indicated by an arrow and clevage products in (A) are shown by an asterisk (p140) or an arrowhead (PARP).
We also did not observe p140 following substantial cellular DNA damage in normal diploid human fibroblast IMR-90 cells which was induced by exposure to H2O2 (Fig. 6B, top panel) or MMS (Fig. 6C, top panel). Under both conditions no PARP cleavage was detected even at the highest doses of H2O2 or MMS (bottom panels, Fig. 6B and C), indicating that apoptosis was also not induced. Similar results were also obtained when Jurkat cells were subjected to DNA damage with H2O2 and UV (data not shown).
p140 is not detected throughout the cell cycle initiated from G0 or from early S phase
It has been speculated that p140 could play a role in DNA replication, as it has been isolated as the only form of pol ɛ from chicken or calf thymus and X.laevis embryos (20), and it has 2–3 times more activity than the full-length p261 in in vitro assays (4). However, we did not observe p140 during the S phase of the cell cycle in which DNA synthesis is abundant (Fig. 7A). Normal human fibroblast IMR-90 cells were utilized in this experiment, which were released into the cell cycle from synchronized G0/G1 phase. We did observe, however, that the amount of the p261 changed with the cell cycle: a minimum or almost undetectable amount was present during the G0/G1 phase and maximum amount during the S phase (Fig. 7A), in agreement with previous findings that DNA pol ɛ is expressed in a pattern that corresponds with cell proliferation (46). In addition, no p140 was detected when IMR-90 cells were released from early S phase of the cell cycle (Fig. 7B). Therefore, combined with the observation that no p140 was detected following cellular DNA damage, the possibility that p140 is involved in either DNA replication or DNA damage repair in vivo appears remote.
Figure 7.

No p140 was detected throughout the cell cycle initiating from G0 or early S phase. IMR-90 cells were synchronized and released from G0 (A) or early S phase (B), extracts were prepared and immunoblots using monoclonal antibody 3C5.1 were performed as described in Materials and Methods. Full-length p261 is indicated by an arrow.
Cleavage of p261 into p140 by caspase-3 in vitro and localization of the two sites of cleavage
When purified human DNA pol ɛ was incubated with recombinant human caspase-3, cleavage occurred that produced a 140-kDa protein recognized by antibody 3C5.1 in an immunoblot. However, cleavage was not catalyzed by recombinant murine caspase-1. This is not surprising since caspase-3 has been regarded as the major ‘executor’ enzyme for the cleavage of cellular proteins during apoptosis (47–49).
Three in vitro cleavage products formed from p261 by caspase-3 were detected with the 3C5.1 monoclonal antibody recognizing the N-terminal portion of pol ɛ p261 of apparent molecular weights of 240, 140 and 90 kDa, with p140 being the major product (Fig. 8). Given the substrate specificity of caspase-3, i.e. the DXXD sequence, we located six DXXD sequences in full-length p261: DKMD (residues 42–45), DQLD (residues 213–216), DIED (residues 316–319), DFFD (residues 398–401), DPED (residues 439–442) and DMED (residues 1210–1213).
Figure 8.
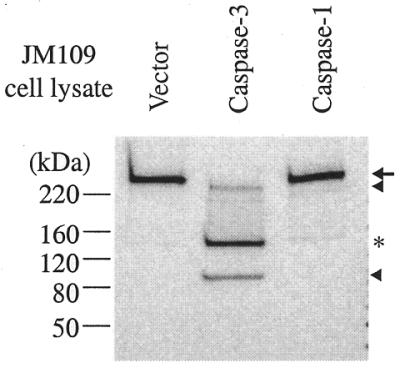
Caspase-3 cleaves pol ɛ p261 in vitro. Bacterial extracts (20 µl) containing recombinant caspase-3, caspase-1 or extracts from cells harboring the parental vector were incubated with 20 µl of partially purified HeLa pol ɛ for 2 h. Western blot analysis indicates three cleavage products of 240 and 90 kDa (noted by arrowheads) and a major product of 140 kDa (asterisk). Full-length p261 is marked with an arrow.
Cleavage at the DMED site (residues 1210–1213) after Asp-1213 would generate an N-terminal fragment of p261 of 139.4 kDa, corresponding to the estimated size of the p140 degradation product of p261 observed in apoptotic Jurkat cells. Therefore we cloned a cDNA fragment corresponding to amino acids 830–1605 of the full-length p261 into the pET28(a) vector under the control of the T7 promoter. We also made an Asp→Ala mutation on the second Asp residue (corresponding to Asp-1213 of the full-length p261) in the only DMED sequence of this cloned fragment. Both wild-type and mutant polypeptides were finally synthesized in vitro using a coupled cell-free transcription and translation system and incubated with purified recombinant caspase-3 (Fig. 9A). Caspase-3 cleaved the wild-type polypeptide into fragments of 60, 50 and 37 kDa. However, no cleavage products corresponding to the 50- and 37-kDa sizes were present for the mutant polypeptide fragment, suggesting that a caspase-3 cleavage site was at residue Asp-1213 of the full-length p261. The appearance of the 60-kDa band is probably due to the non-specific cleavage at a non-‘dead box’ sequence, since the same extent of cleavage also occurred in the mutant substrate (Fig. 9A). Therefore, one caspase-3 cleavage site is at the Asp1213 of the full-length p261.
Figure 9.

Determination of caspase-3 cleavage sites. Both wild-type and D→A mutant polypeptides corresponding to the full-length p261 residues 830–1605 (A) and 1–975 (B) were synthesized in vitro and then reacted with purified caspase-3. Reaction mixtures were separated by SDS–PAGE, transferred onto a nitrocellulose membrane and visualized by autoradiography. Full-length polypeptides are denoted by an arrow and cleavage products that are not present in the mutant are by arrowheads. (C) The two caspase-3 cleavage sites are located at the junctions between the three proposed domains (1).
Using the same strategy, we have also determined that another cleavage site was located immediately after the first DXXD sequence, DQLD (residues 213–216) (Fig. 9B). cDNA sequence corresponding to amino acids 1–975 of the full-length p261 was cloned into the pBluescript vector pBSSK and the polypeptide was synthesized in vitro using the coupled transcription and translation system. Caspase-3 cleavage resulted in two fragments of 28 and 104 kDa and a D216A mutation prevented such cleavage (Fig. 9B). Thus another cleavage site of caspase-3 is determined to be at Asp216 of p261. Cleavage at Asp-216 would generate two fragments of 25 and 237 kDa from the full-length p261.
For the remaining four DXXD sequences, D→A mutations were also made at the second Asp residues to generate D45A, D319A, D401A and D442A mutations. Mutant substrates were then subjected to caspase-3 cleavage. However, these mutations did not prevent caspase-3 cleavages (data not shown). Therefore, the two caspase-3 cleavage sites of native p261 were determined to be at Asp-216 and Asp-1213. However, it is not definitively established whether the p140 observed during Jurkat cell apoptosis resulted from a single caspase-3 cleavage at Asp-1213, or two simultaneous cleavages at Asp-216 and Asp-1213 of p261. However, the presence of a small amount of a product, p240, could indicate cleavage at Asp-216 prior to cleavage at Asp-1213 to form p140.
Interestingly, these two cleavage sites are located at the junctions between the three putative proposed domains (1) of p261 (Fig. 9). Cleavage at both sites would separate the N-terminal domain (residues 1–267) and the C-terminal domain (residues 1167–2285) from the core catalytic domain (residues 268–1166) of p261, while a single cleavage at Asp1213 would separate the core catalytic domain from the C-terminal domain as achieved with calpain cleavage. In either case, the cleavage dissociates the pol ɛ catalytic core domain from the accessory subunits, which interact with the C-terminal domain of p261 (3,6,7).
DISCUSSION
The cleavage pattern of p261 resembles very closely that of other nuclear protein cleavages by caspase-3, i.e. cleavage between an anchoring domain (DNA, RNA or subunit binding domains) and a functional or catalytic domain. Cleavage of PARP separates its DNA binding from its automodification and catalytic domain (40); cleavage of DSEB/RF-C140 separates its DNA and nucleotide binding domain from the PCNA and RF-C association domain (38); cleavage of the catalytic subunit of DNA-dependent protein kinase (DNA-PKc) separates its leucine zipper DNA binding domain from the catalytic domain (50). Such cleavage of these nuclear proteins that are involved in DNA replication and repair might reflect redundant steps to assure the completion of the apoptotic process. In addition, cleavage of these DNA replication and repair enzymes occurs in parallel with the nucleosomal DNA fragmentation, indicating that DNA degradation and nuclear protein cleavage occur independently of one another and are not apparently causally linked. However, the exact implication of p261 cleavage (or any of these individual cleavages) in the apoptosis process still remains to be determined.
Much of the information regarding the biological function of pol ɛ is from S.cerevisiae, in which disruption of the POL2 gene, which encodes the large subunit of pol ɛ (Pol2p), is lethal and a temperature-sensitive allele triggers arrest with a terminal dumbbell morphology, indicating a failure to complete DNA replication (5–7). The C-terminus of Pol2p, which has a conserved ‘zinc finger’ motif, is involved in the S phase cell cycle checkpoint control (10) by functioning as a sensor for DNA damage (11,12). While the catalytic domain of Pol2p is not required for viability, the C-terminal domain is essential for DNA replication, DNA repair and the viability of yeast cells (51). (However, cells lacking the N-terminus of Pol2p have serious defects in DNA replication; 52.) Since the C-terminal domain binds to PCNA and the other pol ɛ subunits, including those with histone-fold motifs (3), this domain might be required to maintain a replicative or repair complex.
The C-terminal domain of human pol ɛ p261 is very sensitive to proteolytic degradation: upon trypsin treatment, it is completely degraded, leaving behind the more protease resistant p122 form of pol ɛ, which retains the catalytic activity (4). (p122 can also be derived from the p140 form of pol ɛ from calf thymus by trypsin treatment, but p122 has not been purified from cells, nor did we observe p122 in this study.) Assuming that after caspase-3 and/or calpain cleavage, the C-terminus can be degraded rapidly by other proteases in the cell, its loss would further assure cell death, if it were essential for the viability of mammalian cells like its yeast counterpart. Since the catalytic domain of p261 is protease-resistant (4), it may resist cleavage during apoptosis and purification processes from various sources. Nonetheless, it is unlikely that the p140 fragment, though it possesses polymerase activity in in vitro polymerase assays, could functionally substitute for p261 in vivo in any cell processes since it cannot assemble into the pol ɛ holoenzyme. In this regard, we have highly over-expressed the N-terminal segments (1–408 or 1–1408) of p261 in transfected IMR-90 normal human fibroblasts and did not observe any changes in cell morphology, cell growth or passage through the cell cycle (data not shown).
The purification exclusively of p140 (i.e. no p261) from chicken or calf thymus is probably due to the fact that thymocytes are very susceptible to apoptosis. More immature T cells are produced than are needed, and cells that fail the selection process are continually being eliminated, presumably by the apoptotic process (53,54). Furthermore, calpain has been shown to be activated during early stages of thymocyte apoptosis independent of caspase activations (55,56). Early involvement of calpain in apoptosis would facilitate the complete cleavage of p261 in apoptotic thymocytes. For the same reason, since apoptosis is an integral mechanism of embryonic development and differentiation, activation of caspase-3 or even calpain presumably contributes to the cleavage of p261 in some embryonic cells. Hence, it is not surprising that p140 was purified as the only form of the catalytic subunit of pol ɛ from X.laevis embryos (20). Recently a cysteine protease, similar in sequence to calpain, has been identified by examining the yeast genome (57). This protease could account for the purification of p140 with p261 from yeast cells (20,21).
The cleavage of pol ɛ catalytic subunit was not complete in the Jurkat cells induced for apoptosis. This incomplete cleavage might be a peculiarity of the Jurkat cell model systems. Alternatively, pol ɛ has been implicated not only in DNA replication and repair, but recently also in chromosome maintenance involving interaction of maintenance factors with its two subunits (p17 and p12) with histone fold motifs (3), and cell cycle control at the S phase checkpoint through its interaction with MDM2 (58). Given this multiplicity of functions, we expect that pol ɛ might exist in several different complexed states. Perhaps not all types of pol ɛ complex are subject to the cleavage. On the other hand, for tissues from which only p140 was obtained, no protease inhibitors specific for caspase-3 and/or calpain were used to prevent p261 degradation during cell lysis or subsequent purification steps. Therefore, the presence of only p140 in the final purification products could be due partially to the degradation of p261 by caspase-3 and/or calpain in vivo in apoptotic cells, and partially to the degradation of the remaining p261 during the final purification process.
Acknowledgments
ACKNOWLEDGEMENTS
The authors would like to thank Dr H. Asahara at UC Berkeley for providing purified HeLa pol ɛ, Ms Ann Fischer for expert tissue culture help, and Mr Hector A. Nolla for help with flow cytometry analysis. We are also grateful to Dr J. Yuan at Harvard University for providing the plasmid constructs for expressing recombinant caspases. This work was supported by the NIH through training grant T32-ES07075 to W.L., center grant P30ES08196 and grant 1RO1GM30415.
REFERENCES
- 1.Kesti T., Frantti,H. and Syvaoja,J.E. (1993) J. Biol. Chem., 268, 10238–10245. [PubMed] [Google Scholar]
- 2.Li Y., Asahara,H., Patel,V.S., Zhou,S. and Linn,S. (1997) J. Biol. Chem., 272, 32337–32344. [DOI] [PubMed] [Google Scholar]
- 3.Li Y., Pursell,Z.F. and Linn,S. (2000) J. Biol. Chem., 275, 23247–23252. [DOI] [PubMed] [Google Scholar]
- 4.Kesti T. and Syvaoja,J.E. (1991) J. Biol. Chem., 266, 6336–6341. [PubMed] [Google Scholar]
- 5.Araki H., Hamatake,R.K., Johnston,L.H. and Sugino,A. (1991) Proc. Natl Acad. Sci. USA, 88, 4601–4605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Araki H., Ropp,P.A., Johnson,A.L., Johnston,L.H., Morrison,A. and Sugino,A. (1992) EMBO J., 11, 733–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Morrison A., Araki,H., Clark,A.B., Hamatake,R.K. and Sugino,A. (1990) Cell, 62, 1143–1156. [DOI] [PubMed] [Google Scholar]
- 8.Budd M.E. and Campbell,J.L. (1995) Mol. Cell. Biol., 15, 2173–2179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Holmes A.M. and Haber,J. (1999) Cell, 96, 415–424. [DOI] [PubMed] [Google Scholar]
- 10.Navas T.A., Zhou,Z. and Elledge,S.J. (1995) Cell, 80, 29–39. [DOI] [PubMed] [Google Scholar]
- 11.Navas T.A., Sanchez,Y., Desany,B., Jones,W., Alcasabas,A. and Elledge,S.J. (1995) Mol. Biol. Cell, 65, 5A. [Google Scholar]
- 12.Navas T.A., Sanchez,Y. and Elledge,S.J. (1996) Genes Dev., 10, 2632–2643. [DOI] [PubMed] [Google Scholar]
- 13.Zlotkin T., Kaufmann,G., Jiang,Y., Lee,M.Y., Uitto,L., Syvaoja,J., Dornreiter,I., Fanning,E. and Nethanel,T. (1996) EMBO J., 15, 2298–2305. [PMC free article] [PubMed] [Google Scholar]
- 14.Nishida C., Reinhard,P. and Linn,S. (1988) J. Biol. Chem., 263, 501–510. [PubMed] [Google Scholar]
- 15.Randahl H., Elliot,G.C. and Linn,S. (1988) J. Biol. Chem., 263, 12228–12234. [PubMed] [Google Scholar]
- 16.Jessberger R., Chui,G., Linn,S. and Kemper,B. (1996) Mutat. Res., 80, 859–868. [DOI] [PubMed] [Google Scholar]
- 17.Aboussekha A., Biggerstaff,M., Shivji,M.K.K., Vilpo,J.A., Moncollin,V., Podust,V.N., Protic,M., Hubscher,U., Egly,J.-M. and Wood,R.D. (1995) Cell, 80, 859–868. [DOI] [PubMed] [Google Scholar]
- 18.Chui G. and Linn,S. (1995) J. Biol. Chem., 270, 7799–7808. [DOI] [PubMed] [Google Scholar]
- 19.Crute J.J., Wahl,A.F. and Bambara,R.A. (1986) Biochemistry, 25, 26–36. [DOI] [PubMed] [Google Scholar]
- 20.Uitto L., Hallen,J., Hentunen,T., Hoyhtya,M. and Syvaoja,J.E. (1995) Nucleic Acids Res., 23, 244–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hamatake R.K., Hasegawa,H., Clark,A.B., Benek,K., Kunkel,T. and Sugino,A. (1990) J. Biol. Chem., 265, 4072–4083. [PubMed] [Google Scholar]
- 22.Leist M., Single,B., Castoldi,A.F., Kuhnle,S. and Nicotera,P. (1997) J. Exp. Med., 185, 1481–1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Laemmli E.K. (1970) Nature, 227, 680–685. [DOI] [PubMed] [Google Scholar]
- 24.Harlow E. and Lane,D. (1988) Antibodies: A Laboratory Manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 25.Li H., Bergeron,L., Cryns,V., Pasternack,M.S., Zhu,H., Shi,L., Greenberg,A. and Yuan,J. (1997) J. Biol. Chem., 272, 21010–21017. [DOI] [PubMed] [Google Scholar]
- 26.Tobey R.A., Valdez,J.G. and Crissman,H.A. (1988) Exp. Cell Res., 179, 400–416. [DOI] [PubMed] [Google Scholar]
- 27.Murachi T. (1989) Biochem. Int., 18, 263–294. [PubMed] [Google Scholar]
- 28.Croall D.E. and Demartino,G.N. (1991) Physiol. Rev., 71, 813–847. [DOI] [PubMed] [Google Scholar]
- 29.Sorimachi H., Saido,T.C. and Suzuki,K. (1994) FEBS Lett., 343, 1–5. [DOI] [PubMed] [Google Scholar]
- 30.Deshpande R.V., Goust,J.-M., Chakrabarti,A.K., Barbosa,E., Hogan,E.L. and Banik,N.L. (1995) J. Biol. Chem., 270, 2497–2505. [DOI] [PubMed] [Google Scholar]
- 31.Elce J.S., Davies,P.L., Hegadorn,C., Maurice,D.H. and Arthur,J.S.C. (1997) Biochem. J., 326, 31–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Noguchi M., Sarin,A., Aman,M.J., Nakajima,H., Shores,E.W., Henkart,P.A. and Leonard,W.J. (1997) Proc. Natl Acad. Sci. USA, 94, 11534–11539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mellgren R.L. (1991) J. Biol. Chem., 266, 13920–13924. [PubMed] [Google Scholar]
- 34.Rechsteiner M. and Rogers,S.W. (1996) Trends Biochem. Sci., 21, 267–271. [PubMed] [Google Scholar]
- 35.Molinari M., Vilei,E.M., Carafoli,E. and Angali,J. (1994) Experientia, 50, A53. [Google Scholar]
- 36.Molinari M., Anagli,J. and Carafoli,E. (1995) J. Biol. Chem., 270, 2032–2035. [DOI] [PubMed] [Google Scholar]
- 37.Weil M., Jacobson,M.D., Coles,H.S.R., Davies,T.J., Gardner,R.L., Raff,K.D. and Raff,M.C. (1996) J. Cell Biol., 133, 1053–1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ubeda M. and Harbener,J.F. (1997) J. Biol. Chem., 272, 19562–19568. [DOI] [PubMed] [Google Scholar]
- 39.Kuhne C. and Banks,L. (1998) J. Biol. Chem., 273, 34302–34309. [DOI] [PubMed] [Google Scholar]
- 40.Lazebnik Y.A., Earnshaw,W.C., Kaufman,S.H., Desnoyers,S. and Poirier,G.G. (1994) Nature, 371, 346–347. [DOI] [PubMed] [Google Scholar]
- 41.Nicholson D.W., Ali,A., Thornberry,N.A., Viallancourt,J.P., Ding,C.K., Gallant,M., Garean,Y., Griffin,P.R., Labelle,M., Lazebnik,Y.A., Munday,N.A., Raju,S.M., Smulson,M.E., Yamin,T.-T., Yu,V.L. and Miller,D.K. (1995) Nature, 376, 37–43. [DOI] [PubMed] [Google Scholar]
- 42.Wood D.E. and Newcomb,E.W. (1999) J. Biol. Chem., 274, 8309–8315. [DOI] [PubMed] [Google Scholar]
- 43.Wang K.K.W., Posmantur,R., Nadimpalli,R., Nath,R., Mohan,P., Nixon,A.R., Talanian,R.V., Keegan,M., Herzog,L. and Allen,H. (1998) Arch. Biochem. Biophys., 356, 187–196. [DOI] [PubMed] [Google Scholar]
- 44.Trauth B.C., Clas,C., Peters,A.M.J., Matzku,S., Moeller,P., Falk,W., Debatin,K.-M. and Krammer,P.H. (1989) Science, 245, 301–305. [DOI] [PubMed] [Google Scholar]
- 45.Schwab B.L., Leist,M., Knippers,R. and Nicotera,P. (1998) Exp. Cell. Res., 238, 415–421. [DOI] [PubMed] [Google Scholar]
- 46.Tuusa J., Uitto,L. and Syvaoja,J.E. (1995) Nucleic Acids Res., 23, 2178–2183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nicholson D.W. and Thornberry,N.A. (1997) Trends Biochem. Sci., 22, 299–306. [DOI] [PubMed] [Google Scholar]
- 48.Thornberry N.A. (1998) Chem. Biol., 5, 97–103. [DOI] [PubMed] [Google Scholar]
- 49.Thornberry N.A. (1999) FASEB J., 13, 1335.10428758 [Google Scholar]
- 50.Song Q., Less-Miller,S.P., Kumar,S., Zhang,N., Chan,D.W., Smith,G.C.M., Jackson,S.P., Alnemri,E.S., Litwack,G., Khanna,K.K. and Lavin,M.F. (1996) EMBO J., 15, 3238–3246. [PMC free article] [PubMed] [Google Scholar]
- 51.Kesti T., Flick,K., Keranen,S., Syvaoja,J.E. and Wittenberg,C. (1999) Mol. Cell, 3, 697–685. [DOI] [PubMed] [Google Scholar]
- 52.Dua R., Levy,D.L. and Campbell,J.L. (1998) J. Biol. Chem., 273, 30046–30055. [DOI] [PubMed] [Google Scholar]
- 53.Krammer P.H., Behrmann,I., Daniel,P., Dhein,J. and Debatin,K.-M. (1994) Curr. Opin. Immunol., 6, 279–289. [DOI] [PubMed] [Google Scholar]
- 54.Von Boehmer H. (1994) Cell, 76, 219–228. [DOI] [PubMed] [Google Scholar]
- 55.Squier M.K.T., Miller,A.C.K., Malkinson,A.M. and Cohen,J.J. (1994) J. Cell Physiol., 159, 229–237. [DOI] [PubMed] [Google Scholar]
- 56.Squier M.K.T. and Cohen,J.J. (1997) J. Immunol., 158, 2690–2697. [Google Scholar]
- 57.Bowmann S., Churcher,C., Badcock,K., Brown,D., Chillingworth,T., Conner,R., Dedman,K., Devin,K., Gentles,S., Hamlin,N., Hunt,S., Jagels,K., Lye,G., Moule,S., Odell,S., Pearson,D., Rajadream,M., Rice,P., Skelton,J., Walsh,S., Whitehead,S. and Barrel,B. (1997) Nature, 387, 90–93. [PubMed] [Google Scholar]
- 58.Vlatkovic N., Guerrera,S., Li,Y., Linn,S., Haines,D.S. and Boyd,M.T. (2000) Nucleic Acids Res., 28, 3581–3586. [DOI] [PMC free article] [PubMed] [Google Scholar]