

Abstract
Interleukin 10 (IL-10)-producing regulatory B-cells (Bregs) suppress inflammatory responses that mediate autoimmune diseases. However, it is unknown whether Bregs derive from a pre-existing dedicated B-cell lineage or if any B-cell can differentiate into Bregs in response to BCR or TLR activation. GL7+ B-cells are antigen-experienced differentiated B-cells while GL7−/lo are at an early stage of B-cell differentiation. While both GL7−/lo and GL7+ B cells can produce IL-10, differentiation of GL7− B-cells into Bregs does not require CD19- or Bcl6-induced signals, suggesting that BCR-induced proliferation or Ig class-switching is not necessary for generation of Breg cells. Of particular importance, we show that GL7− Breg cells are dramatically expanded in lupus-like mice and GL7− Bregs suppressed inflammatory responses in lupus-like mice by inducing expansion of Foxp3+Treg cells. Taken together, these results suggest that pre-existing GL7−IL-10+ cells are expanded during inflammation, differentiate into GL7+ Bregs and contribute to immune-regulation in lupus-like mice.
Keywords: GL7, Bregs, Tregs, Lupus, CD19
1. Introduction
Interleukin 10 (IL-10)-producing regulatory B-cells (Breg) play key roles in controlling autoimmunity and loss or reduction in Breg level is implicated in the etiology of several autoimmune diseases while aberrant elevation of Breg level suppresses immune responses to pathogen (Mauri and Bosma, 2012a). In addition, tumor-induced Bregs are implicated in carcinogenesis and promotion of breast cancer metastasis by converting resting CD4+ T cells to regulatory T cells (Tregs) (Tadmor et al., 2011; Mauri and Ehrenstein, 2008; Schioppa et al., 2011). B cell subsets including splenic marginal zone (MZ) (Brummel and Lenert, 2005; Lenert et al., 2005; Gray et al., 2007), less-mature CD21hiCD23hiIgMhi transitional 2-MZ precursor (T2-MZP) (Evans et al., 2007; Mizoguchi et al., 2002), CD1dhiCD5+ B10 (Yanaba et al., 2008a), plasmablasts (CD138+CD44hi) (Matsumoto et al., 2014) or CD138+MHC-IIloB220+ plasma cells (Shen et al., 2014) suppress autoimmune disease in the mouse through production of the anti-inflammatory cytokine, IL-10 (Dillilo et al., 2010; Mauri and Ehrenstein, 2008; Mizoguchi and Bhan, 2006; Yanaba et al., 2008b; Wang et al., 2014). Defective Breg cell development is associated with chronic inflammation, suggesting that therapeutic targeting of these cells can be used to suppress immune-mediated inflammatory diseases. However, therapeutic use of Bregs is constrained by the dearth of knowledge of the developmental origin of Breg cells.
Breg development is generally thought to be driven by innate and adaptive immune signals emanating from BCR or TLR activation of B cells (Neves et al., 2010; Mauri and Bosma, 2012a). In fact, studies in experimental autoimmune encephalomyelitis (EAE) and collagen-induced arthritis suggest that major sources of Bregs are resident B10 cells and proliferating CD138+CD44hi plasmablasts in the LN (Matsumoto et al., 2014). On the other hand, lupus, EAE or inflammatory bowel is exacerbated in mice deficient in Irf4 or Prdm1 due to inability to produce Breg cells, suggesting that Bregs might also derive from antibody-producing plasma cells (Shen et al., 2014). Nonetheless, there is still considerable debate as to whether Bregs that suppress autoimmune diseases derive from pre-existing dedicated B cell lineage or if any activated B cell has the potential to differentiate into a Breg cell upon exposure to specific environmental or pathogenic stimuli.
The GL7hi B cell is more efficient in producing antibodies or presenting antigen than its GL7−/lo counterpart, (Cervenak et al., 2001) suggesting that GL7−/lo cells are at an early stage of B cell differentiation toward plasma cells (Laszlo et al., 1993; Cervenak et al., 2001). In this study, we have examined whether GL7− and GL7+ B cells, two different stages of B cells differentiated toward plasma cells, are to similar extent induced into Breg. We show here that both GL7− and GL7+ B cells can become Bregs. We further show that the differentiation of GL7− B cells into Breg cells does not require CD19 or Bcl6. Interestingly, GL7− Breg cells expand dramatically in lupus-like mice, suggesting that non-activated pre-existing GL7− cells may be an important source Breg cells in SLE and possibly other autoimmune diseases.
2. Methods and materials
2.1. Mice
Seven-to-nine-week-old C57BL/6, Balb/c (Huafukang Corp., Beijing, China), female MRL/MpJ/lpr/lpr (MRL/lpr) mice, age-matched MRL/MpJ/+/+ (MRL/++) mice, CD19-Cre mice (Nanjing Biomedical Research Institute of Nanjing University, Nanjing, China), and IL-10 reporter mouse model IL10-IRES-EGFP tiger mice (Institute of Immunology, Zhejiang University School of Medicine, Hangzhou 310013, China) were bred in our animal facilities under specific pathogen-free conditions. Care, use, and treatment of mice in this study were in strict agreement with international guidelines for the care and use of laboratory animals. This study was approved by the Animal Ethics Committee of the Beijing Institute of Basic Medical Sciences.
2.2. Cytometric analysis and intracellular cytokine staining
All cell experiments were strictly prepared on ice, unless otherwise stated in other specific procedures. Cells (1 × 106 cells/sample) were washed with fluorescence-activated cell sorting staining buffer (phosphate-buffered saline, 2% fetal bovine serum or 1% bovine serum albumin, 0.1% sodium azide). All samples were incubated with anti-Fc receptor Ab (clone 2.4G2, BD Biosciences, San Jose, CA), prior to incubation with other Abs diluted in fluorescence-activated cell sorting buffer supplemented with 2% anti-Fc receptor Ab. For intracellular cytokine staining, 50 ng/ml PMA and 1 μg/ml ionomycin (Sigma–Aldrich, St. Louis, MO) were added and then 10 μg/ml brefeldin A and 2 μM monensin were added 3 h later. After 3 h, cells were collected and fixed for 50 min with 1 ml fixation buffer (IC Fixation and Permeabilization kit, eBioscience, San Diego, CA). After washing, the fixed cells were stained. The samples were filtered immediately before analysis or cell sorting to remove any clumps. Dead cell exclusion/discrimination dyes were used to eliminate dead cells from analysis and sorted. For non-fixed cells, we used DAPI, SYTOX Blue, Green, or Red; for fixed cells, we use LIVE/DEAD Fixable Blue, Green, or Red. Choice of dead cell exclusion dye depended on the color combination of fluorochromes within the sample. The following antibodies were purchased from eBioscience: fluorescence-conjugated anti-mouse CD3 (clone no. 145-2C11), CD4 (clone no. GK1.5), B220 (clone no. RA3-6B2), CD19 (clone no. MB19-1), CD5 (clone no. 53-7.3), CD1d (clone no. 1B1), IL-10 (clone no.JES5-16E3), Foxp3 (clone no.NRRF-30), IFNγ (clone no.XMG1.2), IL-17 (clone no. 17F3), CD21 (clone no.4E3) and CD23 (clone no. B3B4), GL7 (clone no. GL-7), IgG (Cat no. 11-4011-85) and IgM (clone no. RMM-1) antibodies. Data collection and analyses were performed on a FACS Calibur flow cytometer using CellQuest software.
2.3. Cell sorting
Splenic B220+B and CD4+T cells were isolated using mouse B220 and CD4 microbeads (Miltenyi Biotec, Germany), respectively. The purity of sorted B220+B and CD4+T cells was shown to be > 95% by flow cytometric analysis (Supplemental Fig. 1). Multicolor flow cytometry was performed by gating on eGFP+ cells or GL7 on the surface of B cells. The lymphocytes from the spleen of IL-10 reporter mouse model IL10-IRES-EGFP tiger mice were used to directly sort eGFP+ and eGFP− cells or stimulated for 3 days with 1 μg/ml LPS (Sigma L2630 from Escherichia Coli 0111:B4) to sort GL7−IL-10-eGFP+B or GL7+IL-10-eGFP+B cells. Multicolor flow cytometry was also performed by gating on B220+ cells that were either CD21hiCD23+ (marginal zone precursor, MZP B cells) or CD21hiCD23− (marginal zone, MZ B cells). The purity of sorted cells was shown to be >99% by flow cytometric analysis (Supplemental Fig. 1). All flow cytometry data were acquired with FACSCanto, FACSCantoII, or FACSAria (BD Biosciences), gated on live lymphocyte-sized cells on the basis of forward and side scatter, and analyzed using FlowJo software (Tree Star, Ashland, OR).
2.4. In vitro cell cultures
B220+ B-cells were cultured in RPMI 1640 medium containing 10% FBS, 2 mM glutamine, penicillin (100 IU/ml), streptomycin (100 μg/ml), and 50 mM 2-mercapthoethanol. Cells were stimulated for 1–3 days with or without 1 μg/ml LPS plus or not 1 μg/ml Bcl-6 inhibitor (79-6, Cat no. 197345-50MG, Calbiochem, EMD Milipore, USA). For co-culture of GL7−IL-10-eGFP+B or GL7+IL-10-eGFP+B and CD4+ T cells, GL7−IL-10-eGFP+B or GL7+IL-10-eGFP+B cells were sorted by FACS. CD4+T cells at a concentration of 1 × 106 cells/ml were stimulated for 3 days with 3 μg/ml anti-mouse CD28 antibody in 10 μg/ml anti-mouse CD3 antibody-precoated 96-well plates (final volume 200 μl) in the presence of 1 μ 106 cells/ml GL7−IL-10-eGFP+B or GL7+IL-10-eGFP+B.
2.5. 2,6,10,14-Tetramethylpentadecane were in vivo injected into mice
0.5 ml 2,6,10,14-Tetramethylpentadecane (Cat no. 138462500, Fisher Scientific Corp., USA) per mice were injected i.p. into 8-week-old female or male IL-10 reporter mouse model (IL10-IRES-EGFP tiger mice). On day 1, 7, 14 and 21 after injection, lymphocytes were separated from spleen, lymph node (LN) and peripheral blood mononuclear cells (PBMC) by Ficoll separation solution and analyzed by FACS.
2.6. Treatment of lupus with GL7+IL-10+ and GL7−IL-10+ regulatory B cells
Splenic B cells were isolated from 8-week-old female or male IL-10 reporter mouse model (IL10-IRES-EGFP tiger mice) using B220 microbeads and stimulated for 3 days by 1 μg/ml LPS. Cells were stained with anti-mouse GL7 antibody, GL7−IL-10(EGFP)+ and GL7+IL-10(EGFP)+ were sorted by FACS. 5 × 105 Cells per mouse were injected into 8-month-old female lupus-like MRL/lpr mice.
2.7. Hematoxylin and eosin (H&E) staining
Kidney specimens were fixed in formalin for 24 h, dehydrated by successive incubation in 70%, 80%, and 90% ethanol for 3 h, and then 100% ethanol I for 2 h, 100% ethanol II for 2 h, and vitrified with xylene I and xylene II for 20 min. After immersing in paraffin I and II for 40 min, the specimens were embedded and sliced. Staining was performed as follows: hematoxylin staining for 15 min, followed by hydrochloric acid alcohol solution for 35 s, eosin staining for 10 min followed by 90% ethanol for 40 s. Neutral balsam was then used for mounting and the section was observed and photographed by light microscope.
2.8. Immunohistochemistry staining
Whole kidneys were perfusion- and post-fixed in 4% buffered formalin for 24 h and embedded in paraffin. 1 μm tissue sections were deparaffinized and rehydrated. Antigen retrieval was performed by incubation with proteinase type XXIV (5 mg/ml, Sigma, Steinheim, Germany and St. Louis, MO, USA) for 15 min at 37 “C or by microwave antigen retrieval in citrate buffer, pH 6.1 for 25 min. After a 30 min block in 5% horse serum, the tissue was incubated with HRP-conjugated rabbit anti-mouse IgG antibody in several dilutions (1:100–1:5000 in 5% horse serum). Color development for light microscopy was used for nuclear staining. The section was observed and photographed by light microscope.
2.9. Statistics
Statistics were generated using t-test in GraphPad Prism (version 5.0, GraphPad Software Inc., USA) and values are represented as mean ± standard error of the mean (SEM). Results were considered statistically significant at p < 0.05.
3. Results
3.1. GL7−Breg cells are the predominant Bregs in the spleen of lupus-like mice
The GL7hi B cell is more efficient in producing antibodies and antigen-presentation compared to its GL7−/lo counterpart (Cervenak et al., 2001), suggesting that GL7−/lo and GL7hi B cell represent an early and late stages of B cell differentiation toward plasma cells, respectively (Laszlo et al., 1993; Cervenak et al., 2001). We first examined the levels of GL7− or GL7+ Breg cells at various time points during 3 days of in vitro stimulation of B220+ B cells by the T cell-independent TLR ligand, lipopolysaccharide (LPS) (Nutt et al., 2011). We show here that LPS induced the expansion of Bregs in a time-dependent manner (Fig. 1A). By day 1 (d1) 77% of the Bregs were GL7−IL-10+B and 55% by d2 (Fig. 1A; lower panel). Analysis of phenotypic markers (e.g. CD38, CD5, CD1d) of IL-10+B cells revealed that IL-10-expresssing GL7− and GL7+ Breg cells were CD5+CD1d−CD38+ (Fig. 1B and C) suggesting that these both subtypes are phenotypically similar. We also used the MRL/lpr mouse strain that is prone to late-onset SLE (Theofilopoulos and Dixon, 1985) to examine kinetics of Breg generation. Both GL7−IL-10+B cells and GL7+IL-10+B cells increased significantly in the spleen of MRL/lpr mice with lupus-like symptom. Consistent with our in vitro results (Fig. 1A), majority of the Bregs were GL7−IL-10+B cells (Fig. 1D–F). Taken together, these results suggest that majority of the Bregs in vitro or during an inflammatory disease are GL7−IL-10+ B cells. This result is at variance with a recent report that proliferating immature plasma cells are the predominant source of IL-10 during EAE (Matsumoto et al., 2014). This difference may derive in part from the fact that while we analyzed Bregs in the spleen of lupus-like mice, analysis of Bregs in the EAE mice was on lymph node cells.
Fig 1.
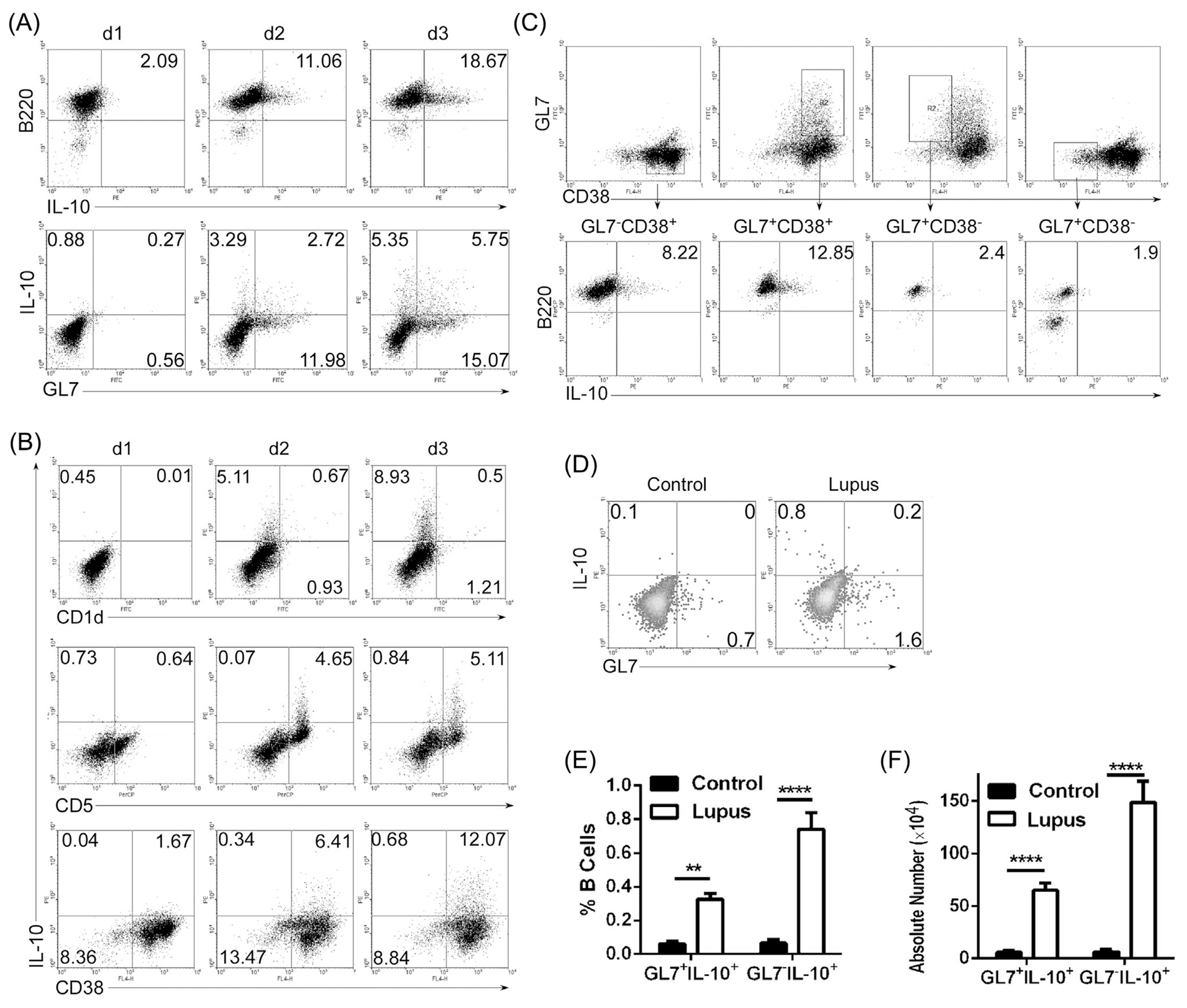
GL7−IL-10+ B cells are expanded in LPS-stimulated B cells and spleen of mice with lupus-like disease. (A–C) Splenic B cells were stimulated for 1–3 days with LPS (1 μg/ml) and analyzed by the intracellular cytokine-staining assay. Cells were gated for B220+ or GL7+ B cells (A, C) or CD1d+, CD5+, CD38+ B cells (B) and quadrants indicate percentage of IL-10-expressing cells. (D–F) Splenic lymphocytes from lupus-like MRL/lpr (Lupus) mice were analyzed by the intracellular cytokine-staining assay. Quadrants indicate percentages of IL-10-expressing GL7− or GL7+ B cells. Statistical analysis of the percentages (E) or absolute number (F) of IL-10-expressing GL7− or GL7+ B cells. Data represent at least four independent experiments. (E, F) Data were analyzed by Student’s t-test (two tailed). Error bars, s.e.m. **P < 0.01, ****P < 0.0001.
3.2. Differentiation of splenic B cells into GL7− Bregs does not require Bcl-6 or CD19 signaling
We next examined whether proliferation is essential for differentiation of B cells into Breg cells. The Bcl-6 transcription factor is highly expressed in MZ and germinal center (GC) B cells and induces the proliferation of GC B cells while antagonizing their differentiation into plasma cells (Fairfax et al., 2007; Klein and Dalla-Favera, 2008). We show here that Bcl-6 inhibitor (Fig. 2A) in cultures of LPS-stimulated B cells effectively suppressed the generation of GL7+ Breg cells with marginal effects on GL7−IL-10+B cells (Fig. 2B). These data suggest that while Bcl6 signaling is required for efficient generation of GL7+IL-10+ B cells, it is not required for the differentiation of GL7− Breg cells.
Fig. 2.
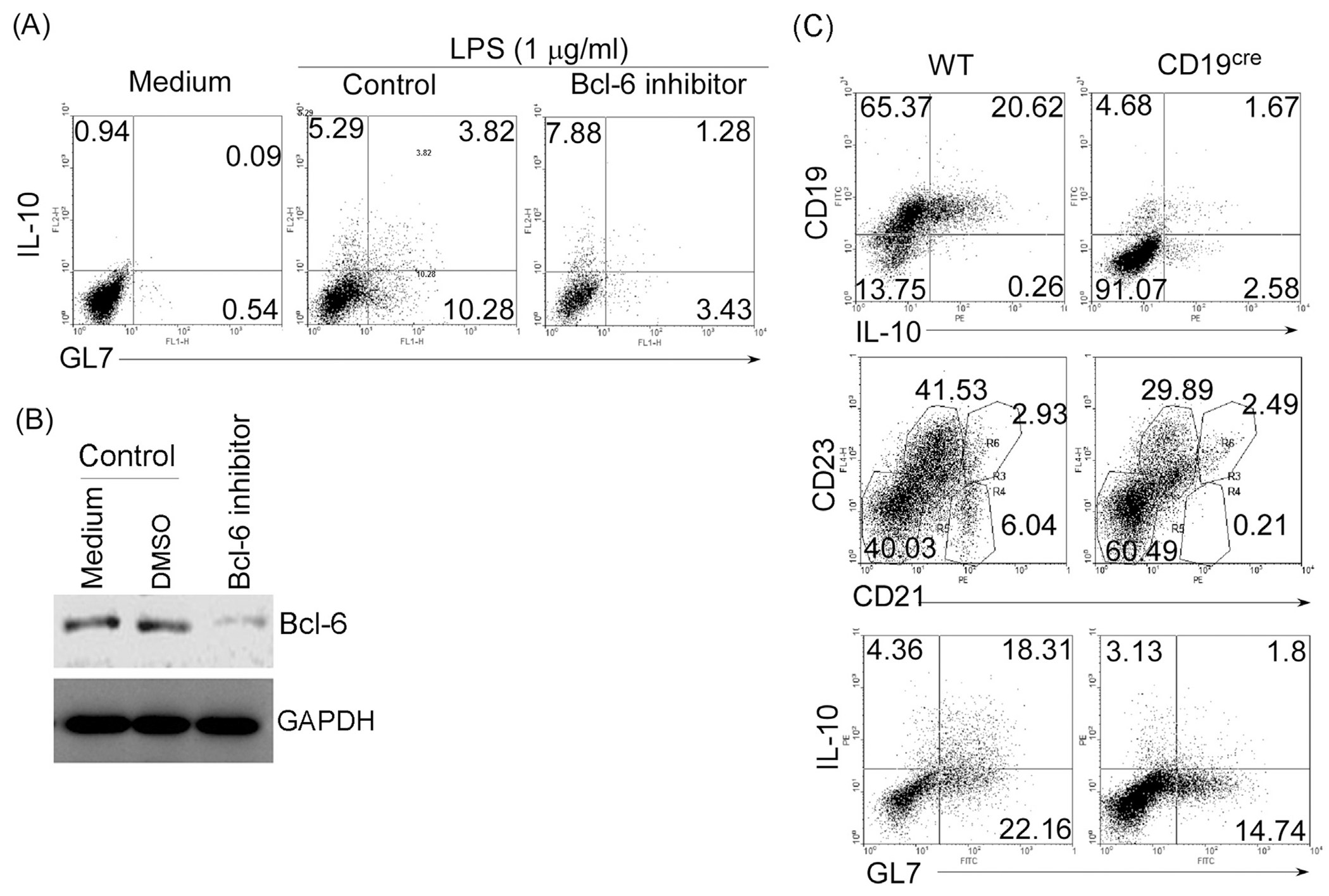
Expansion of GL7−IL-10+ Bregs does not require Bcl-6 or CD19 signal. (A, B) Splenic B cells were stimulated for 3 days with LPS (1 μg/ml) in medium containing PBS, control DMSO or Bcl-6 inhibitor (1 μg/ml) and analyzed by intracellular cytokine-staining assay (A) or Western blot (B). Quadrants indicate percentage ofIL-10-expressing GL7− or GL7+ B cells. (C) Splenic B cells from wild-type and CD19-deficient (CD19cre) mice were stimulated for 3 days with LPS (1 μg/ml) and analyzed by the intracellular cytokine-staining assay. Quadrants indicate percentages of IL-10-expressing GL7− and GL7+ B cells. Data represent at least three independent experiments.
CD19 deficiency is implicated in exacerbation of autoimmunity due in part to reduced capacity to generate Breg cells (Watanabe et al., 2010) and marginal zone B cells (You et al., 2009). We therefore used B cells with targeted deletion of CD19 (CD19cre) to directly examine the influence of this BCR co-receptor that regulate B cell proliferation on the generation of GL7+ and GL7− Breg cells. We found that while CD19 deficiency in conventional or MZ (CD21hiCD23−) B cells reduced percentage of GL7+Bregs in response to LPS-stimulation, generation of GL7−Breg cells was not affected (Fig. 2C), suggesting that LPS-induced generation of GL7+IL-10+ B cells is dependent on CD19 but not GL7−IL-10+ B cells.
3.3. GL7− Breg cells derive from CD21hiB MZP B cells
We next isolated splenic B220+ B cells, simulated the cells with LPS, and examined whether GL7−IL-10+B and GL7+IL-10+B cells derive mainly from mature follicular (CD21lo), MZ (CD21hiCD23−) or MZP (CD21hiCD23+) cells. We show here that the percentage of GL7−IL-10+B and GL7+IL-10+B cells was significantly increased in both the MZP and MZ but not in the FB B cells compartment (Fig. 3A and B). We further show that while CD19 deficiency inhibited LPS-induced development of MZP cells into MZ or GL7+IL-10+B cells, it had less effect on expansion of GL7−IL-10+B (Fig. 3C). These data thus suggest that LPS-induced the expansion of GL7− Bregs mainly among cells in the MZP B compartment, while GL7+ Bregs derived mainly from MZ B cells.
Fig. 3.
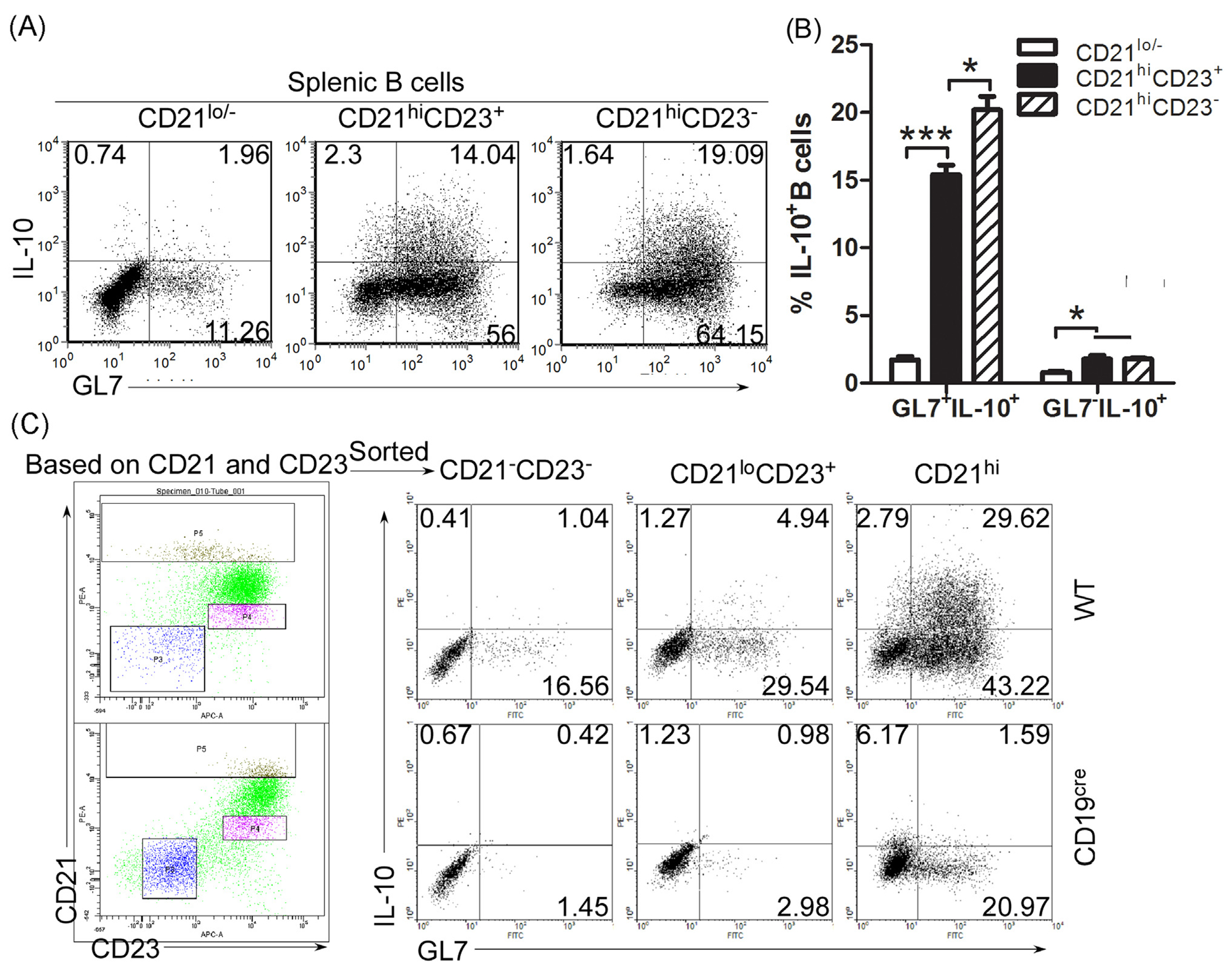
GL7− Breg cells derive from CD21hi MZP B cells. (A, B) Splenic CD21−/lo, CD21hiCD23+ (marginal zone precursor, MZP B cells) and CD21hiCD23− (marginal zone, MZ B cells) B cells from .from wild-type (A–C) or CD19-deficient mice (C) were stimulated for 3 days with LPS (1 μg/ml) and analyzed by the intracellular cytokine-staining assay. Quadrants indicate percentage of IL-10-expressing GL7− and GL7+ B cells (A, C). Statistical analysis of the percentages of GL7−IL-10+B and GL7+IL-10+B cells in CD21−/lo, CD21hiCD23+ and CD21hiCD23− B cells (B). Data represent 3 independent experiments. (B) Data were analyzed by Student’s t-test (two tailed). Error bars, s.e.m. *P < 0.05, ****P < 0.0001.
3.4. Pre-existing GL7−IL-10+Breg cells produce GL7+IL-10+Breg cells
Next, we examined whether GL7−IL-10+B and GL7+IL-10+B cells derived from pre-existing Breg cells or differentiated from IL-10− B cells. We isolated B220+ B cells from the spleen of IL-10 reporter mice (IL-10-IRES-EGFP tiger mice) and established the presence of GL7− B cells within both IL-10+ (IL-10-EGFP+) or IL-10− (IL-10-EGFP−) B compartments (Fig. 4A). Sorted IL-10-EGFP+ and IL-10-EGFP− B cells were stimulated with LPS and we found that EGFP+ or EGFP− B cells contained GL7− B cells and majority of the EGFP+ or EGFP− B cells after 2 days stimulation with LPS were GL7− (Fig. 4B). Interestingly, during the course of LPS stimulation, the GL7−IL-10+B and GL7+IL-10+B cells appeared to convert into GL7+IL-10+B cells and GL7+IL-10−B cells. Taken together, these results suggest that GL7+IL-10+B cells may in part derive from pre-existing GL7−Breg cells.
Fig. 4.
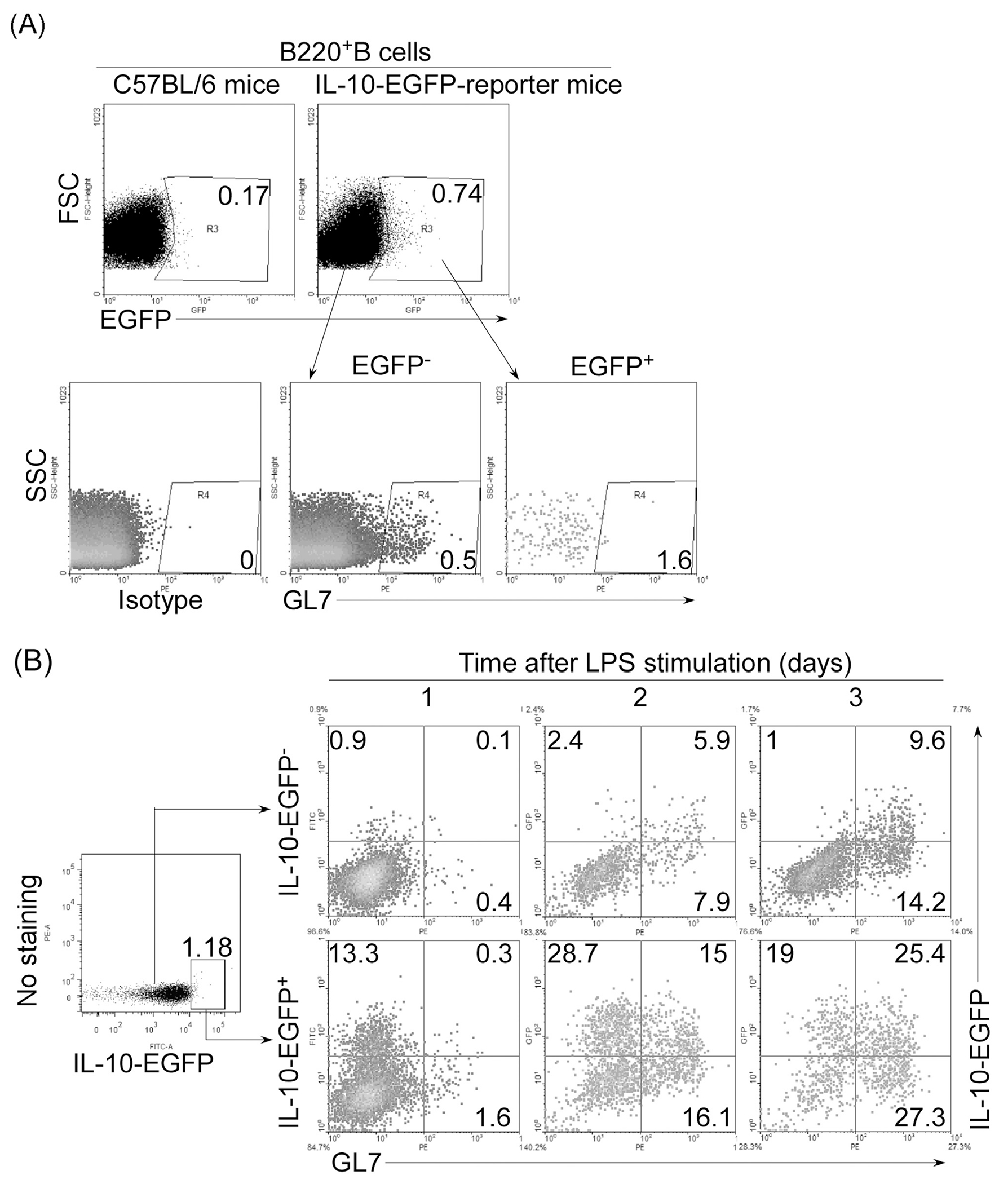
Breg cells derive from pre-existing GL7−Breg cells. (A) IL-10+ (EGFP+) or IL-10− (EGFP−) B220+ B cells sorted from the spleen of IL-10 reporter mouse model (IL10-IRES-EGFP tiger mice) and control C57BL/6 mice were analyzed by FACS. Quadrants indicate percentage of GL7− and GL7+ B cells. (B) The EGFP+ and EGFP− B cells were stimulated for 1–3 days by LPS (1 μg/ml) and analyzed by FACS. Quadrants indicate percentage of IL-10-expressing GL7−and GL7+ B cells.
3.5. GL7− Breg cells are expanded in mice with lupus-like disease
To test in vivo the time-dependent generation of GL7−IL-10+ and GL7+IL-10+Breg cells we injected the lupus-inducing compound, pristine, into IL-10 reporter mice and quantified the frequency of IL-10-expressing cells on day 1, 7, 14 and 21 after injection of the drug. Consistent with data presented in Fig. 1, GL7−IL-10+ B cells were preferentially expanded by pristane compared to GL7+IL-10+ B cells (Fig. 5A and B). Analysis of IL-10+B cells in the spleen, lymph node (LN) and peripheral blood mononuclear cells (PBMC) revealed that GL7−IL-10+B cells were induced mainly in the spleen, (Fig. 5C and D). These results suggest that GL7− Breg cells may be expanded in spleen before lupus onset.
Fig. 5.
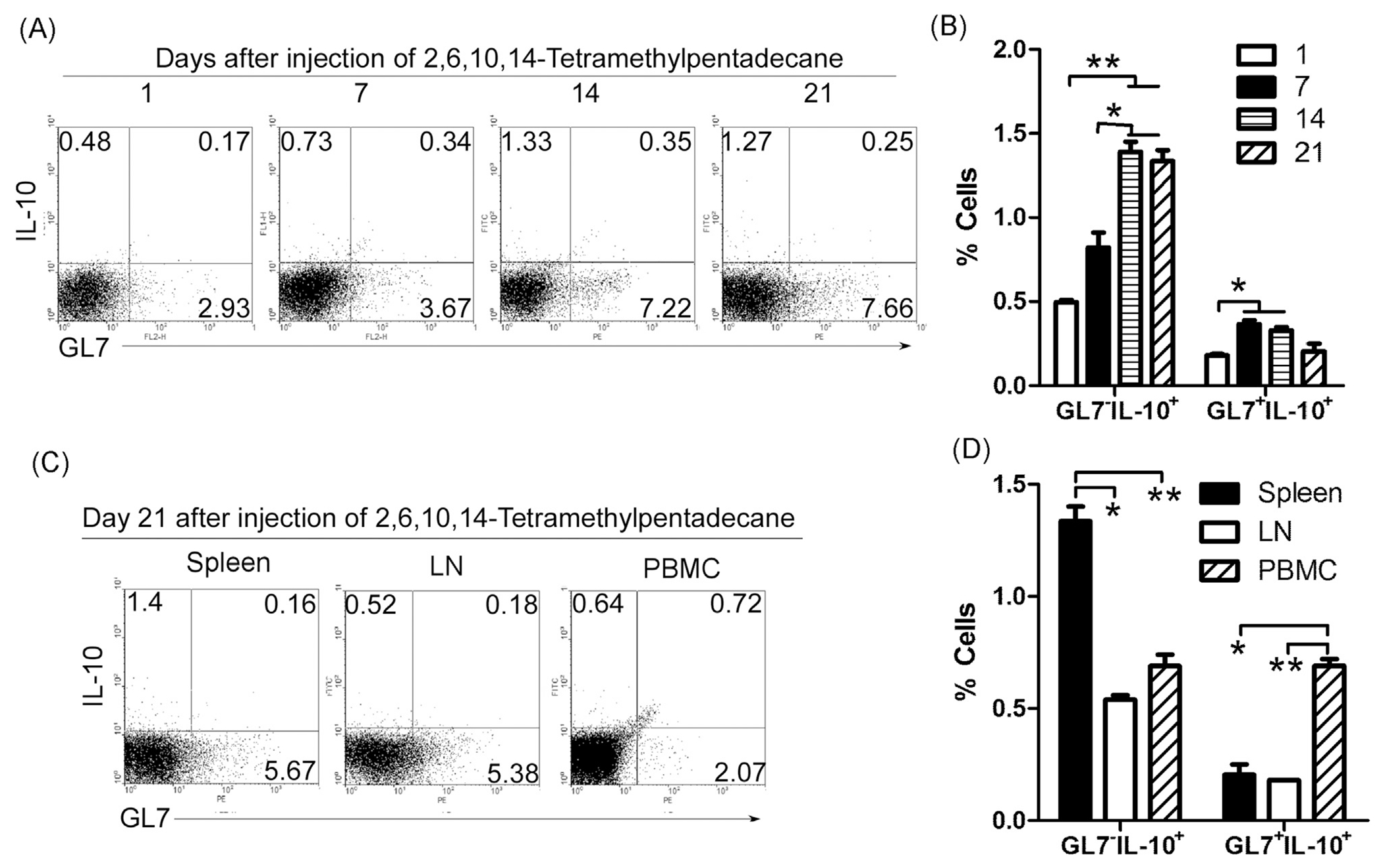
GL7− Breg cells are expanded in mice with lupus-like disease. Experimental lupus induced by the hydrocarbon oil 2,6,10,14-tetramethylpentadecane (TMPD; also known as pristane) displays many key immunological and clinical features of human SLE. 0.5 ml 2,6,10,14-tetramethylpentadecane per mice were injected i.p. into IL-10 reporter mice. On day 1, 7, 14 and 21 after injection, lymphocytes were isolated from spleen, lymph node (LN) and peripheral blood mononuclear cells (PBMC) and analyzed by FACS. Quadrants (A, C) indicate percentage of IL-10-expressing (EGFP+) GL7− and GL7+ B cells. (B, D) Statistical analysis of the percentages of GL7−IL-10+B and GL7+IL-10+B cells. Data represent at least three independent experiments and were analyzed by Student’s t-test (two tailed). Error bars, s.e.m. *P < 0.05, **P < 0.01.
3.6. GL7− Bregs suppress inflammatory responses by expanding Treg cells
To test the suppressive function of GL7+IL-10+ and GL7−IL-10+ B cells, we isolated splenic B cells from IL-10 reporter mice, stimulated the cells with LPS and then sorted for GL7−IL-10(EGFP)+ or GL7+IL-10(EGFP)+ B cells. We co-cultured the sorted B cells with syngeneic CD4+ T cells for 3 days in anti-CD3 coated plates containing anti-CD28 antibody. Both GL7+IL-10+ and GL7−IL-10+Breg cells suppressed CD4+ T cell proliferation (Fig. 6A) in part by up-regulating CD4+Foxp3+ Treg cells (Fig. 6B and C). We then examined whether GL7−IL-10+B cells possess immune suppressive functions in vivo. GL7−IL-10 (EGFP)+ and GL7+IL-10 (EGFP)+ cells were sorted as described and injected (i.v) into lupus-like MRL/lpr mice and we analyzed splenic lymphocytes by FACS 14 days after disease induction. In accordance with in vitro results, GL7+IL-10+ or GL7−IL-10+ Bregs cells up-regulated CD4+Foxp3+ Tregs (Fig. 7A), with concomitant reduction in inflammatory T and B cells such as IL-17-expressing Th17 cells, GL7+B cells, IgG+B cells (Fig. 7B). Interestingly, the suppressive effects of GL7− and GL7+ Bregs were associated with dramatic reduction in the size of the spleen of lupus-like mice that received the Breg cells (Fig. 7C). These results suggest that similar to GL7+ Breg, GL7− Bregs suppressed inflammatory responses by expanding Treg cells.
Fig. 6.
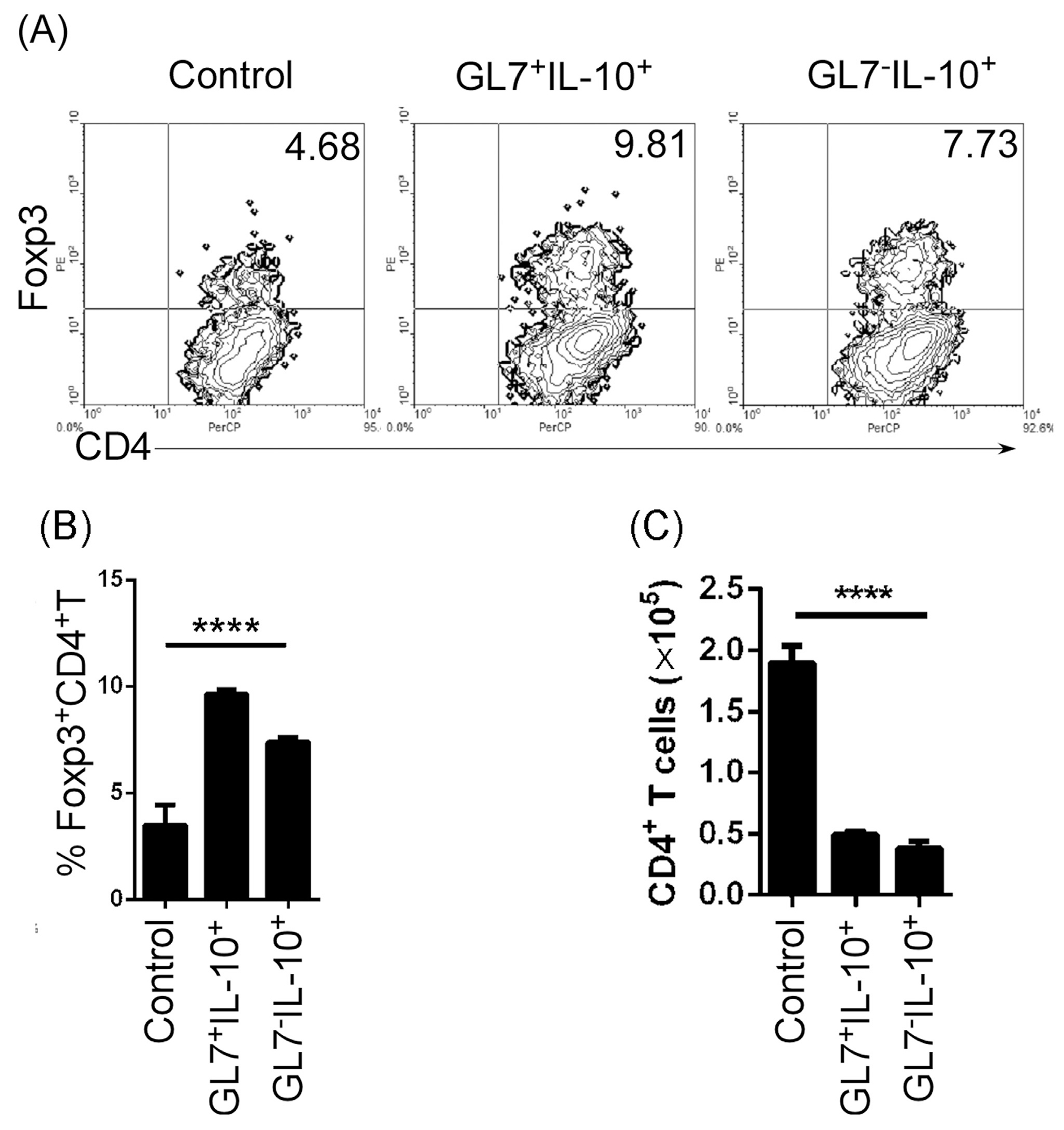
GL7− Breg cells suppressed CD4+T cell proliferation and up-regulated CD4+Foxp3+Treg cells. Splenic B cells from IL-10 reporter mice were stimulated for 3 days by LPS (1 μg/ml). GL7+IL-10− (control), GL7−IL-10 (EGFP)+ or GL7+IL-10(EGFP)+ B cells were sorted by FACS and co-cultured with purified CD4+T cells for 3 days in anti-CD3 pre-coated plates containing anti-mouse CD28 antibody. (A) Quadrants indicate percentage of Foxp3-expressing CD4+ Treg cells. (B) Percentages of CD4+Foxp3+Treg cells. (C) Absolute numbers of CD4+T cells in the cultures. Data were analyzed by Student’s t-test (two tailed). Error bars, s.e.m. ****P < 0.0001.
Fig. 7.
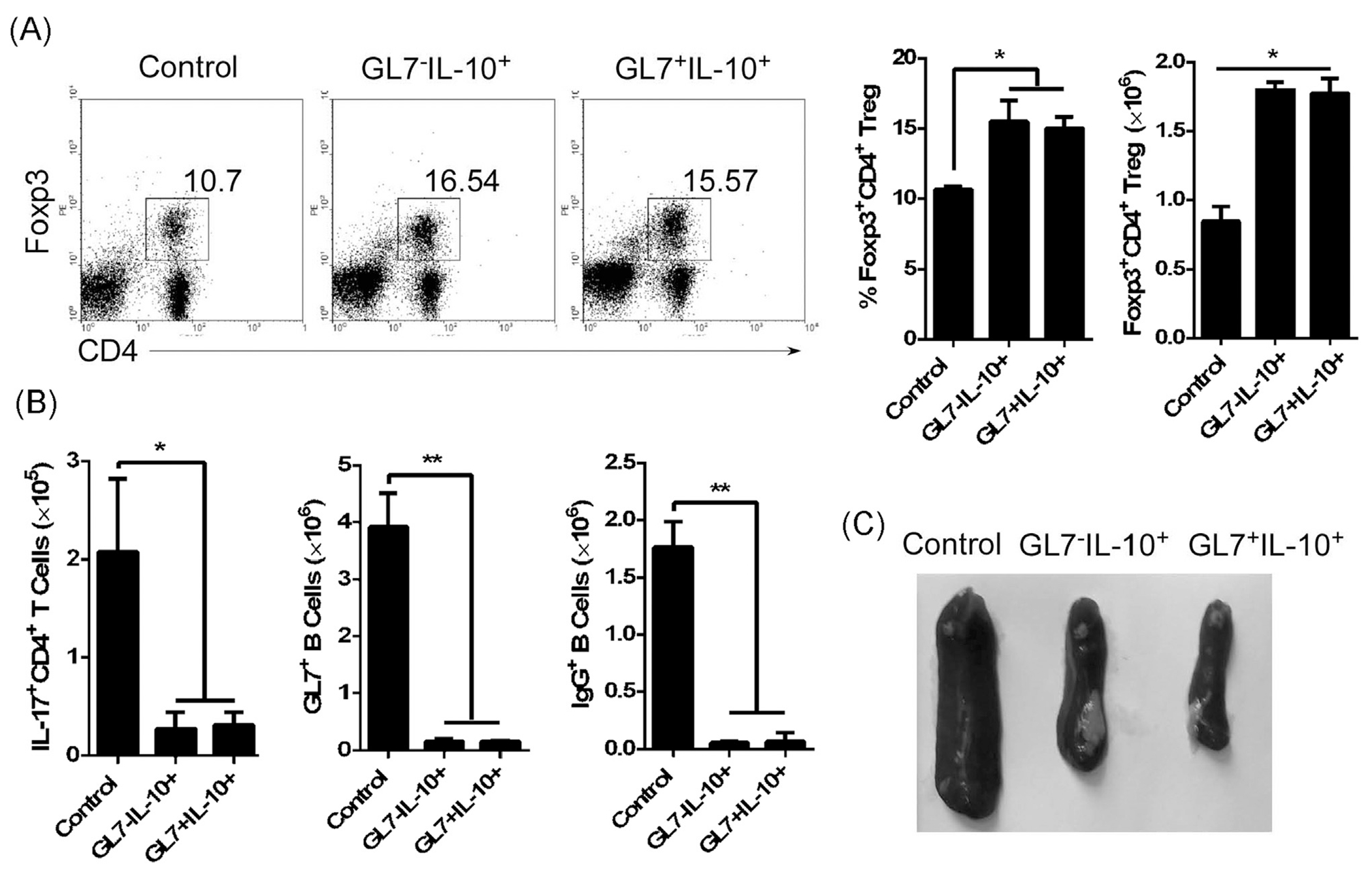
GL7− Breg cells suppressed inflammatory responses in lupus-like mice by upregulating CD4+Foxp3+Treg cells. We isolated B220+ B cells from IL-10 reporter mice, stimulated the cells with LPS (1 μg/ml) for 3 days, and purified IL-10-expressing cells by cell sorting. The GL7−IL-10(EGFP)+ and GL7+IL-10(EGFP)+ B cells (5 × 105/mouse) were injected into 8-month-old female lupus-like MRL/lpr mice with untreated 8-month-old female lupus-like MRL/lpr mice serving as control. (A) On day 14 after adoptive transfer, cells isolated from the spleen were subjected to intracellular cytokine staining assay. Quadrants indicate percentage of CD4+Foxp3+Treg cells (left panel) and statistical analysis of the absolute numbers of CD4+Foxp3+Treg cells per spleen is shown to the right. (B) Relative abundance of IL-17-expressing Th17, GL7+ B, IgG+ B cells in the spleen on day 14 after adoptive transfer. (C) Megalosplenia is reflected by the morphology of the spleen from mice that received GL7−IL-10+ (GL7−EGFP+)or GL7+IL-10+ (GL7+EGFP+) B cells. Data represent at least three independent experiments and were analyzed by Student’s t-test (two tailed). Error bars, s.e.m. *P < 0.05, **P < 0.01.
3.7. GL7− Bregs ameliorate autoimmune symptoms in lupus-like mice
A histological hallmark of SLE is membranous glomerulonephritis. We show here that untreated lupus-like MRL/lpr mice exhibited shrinkage of the balloon cavity and appearance of excessive inflammatory cells in the surrounding interstitial vasculature (Fig. 8, upper panel) while adoptive transfer of GL7+ and GL7− Breg cells reduced the level of infiltrating inflammatory cells and restored the structure of the glomerular region in MRL/lpr mice (Fig. 8, upper panel). We also noted large amounts of IgG deposits in kidney lesions of lupus-like MRL/lpr mice and these were markedly reduced in the glomerular region of MRL/lpr mice treated with GL7+ and GL7− Breg cells (Fig. 8, lower panel). These results suggest that like GL7+Breg, GL7− Bregs ameliorated autoimmune symptoms in lupus-like mice.
Fig. 8.
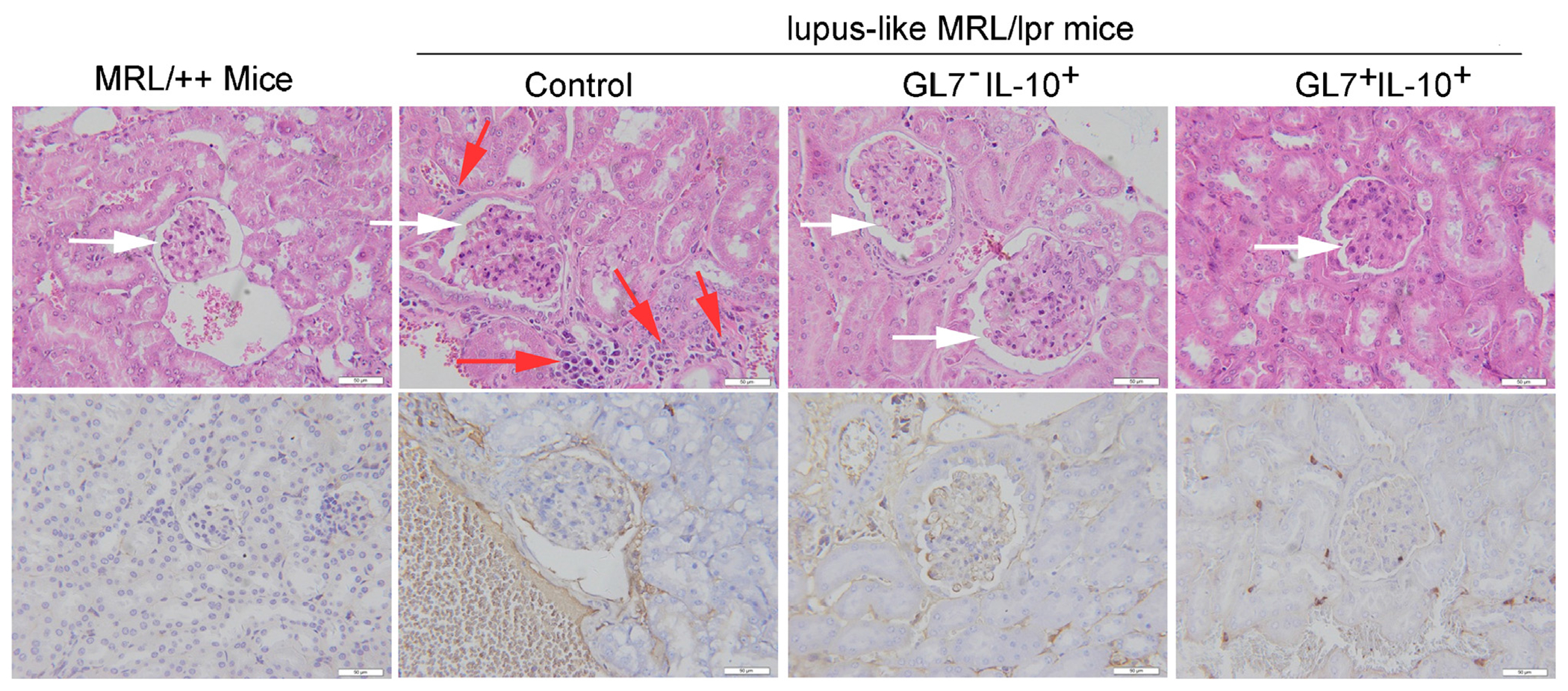
GL7− Breg cells ameliorate autoimmune symptoms in lupus-like mice. 8-month-old female lupus-like MRL/lpr mice were injected with control cells, GL7−IL-10+ (EGFP)+ or GL7+IL-10(EGFP)+ as described above and kidneys were harvested 30 days after adoptive transfer. H&E-staining (upper panel) and immunochemical staining (anti-IgG Ab) (lower panel) of paraffin embedded kidney sections. Original scale bars at 50 μM; white arrows show glomeruli; red arrows show infiltrated inflammatory cells that appeared in the surrounding interstitial vasculature. Data represent at least three independent experiments. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
4. Discussion
In this study, we have used a B cell subset at early stage of development (GL7− B cells) and an antigen-experienced B cell type at a late stage of development (GL7+ B cells) to study factors that mediate Breg generation and/or expansion. We also examined whether Bregs constitute a distinct B cell lineage, developmental stage or induced in response to BCR/TLR signaling or inflammation.
We show that the differentiation of GL7+ B cells into Breg cells requires BCR and Bcl-6 signaling, while differentiation of GL7− B cells into Breg cells does not (Fig. 2A), suggesting that BCR-induced proliferation or Ig class-switching is not necessary for generation of Breg cells. We further show that signals induced expansion of GL7− Breg cells mainly from MZP B cells while LPS-induced expansion of GL7+ Breg cells derived mainly from MZ B cells (Fig. 3). Our results are consistent with recent reports suggesting that B cells at different stages of development, from immature cells to terminally differentiated plasma cells, can acquire suppressive functions and that Bregs are not a terminally differentiated B cells (Fillatreau et al., 2002; Matsumoto et al., 2014). Thus, Bregs can exhibit suppressive functions at different stages of differentiation (Mauri and Menon, 2015).
Experimental evidence concerning the role of Breg cells in the suppression of inflammatory responses has been confounded by the description of multiple Breg cell subsets (Rosser and Mauri, 2015), making it necessary to understand stimuli that induce the differentiation of different regulatory B cells. Here, we have demonstrated that T-cells-independent antigens induced two different regulatory B-cell populations (GL7+1L-10+and GL7−1L-10+) from splenic marginal zone B-cell population. Thus, when autoantigens breach the mucosal barrier and enter the general circulation, MZ B cells might provide a second line of protection via IL-10 and bridege the temporal gap prior to induction of suppressive follicular (FO) B cells. Data presented here is in line with previous studies indicating that the number and suppressive ability of Breg cells increase in response to inflammation (Fillatreau et al., 2002; Rincon-Arevalo et al., 2015; Rosser and Mauri, 2015; Mauri and Ehrenstein, 2008; Mauri and Bosma, 2012a).
An important function of Breg is to suppress CD4+ T cell proliferation and induce differentiation of Tregs (Mauri and Bosma, 2012b). Consistent with these axioms we have shown here that splenic GL7− Breg cells in vitro and in vivo suppressed CD4+ T cell proliferation and increased the percentage of CD4+Foxp3+ Treg cells (Figs. 6 and 7). Both GL7+ and GL7− IL-10-producing B cells express CD5 (Fig. 1). Our previous study has shown that the interaction of CD5 on the surface of Bregs and CD72 on the surface of Tregs plays a critical role in maintaining regulatory T and B cell homeostasis (Xing et al., 2015; Zheng et al., 2014). Thus, both GL7+ and GL7− IL-10-producing B cells maybe increase CD4+Foxp3+ Treg by CD5 expression.
Of particular importance, we show that GL7− Breg cells are dramatically expanded in lupus-like mice and GL7− Bregs suppressed inflammatory responses in lupus-like mice by inducing expansion of Foxp3+ Treg cells. Adoptive transfer of GL7-Bregs also induced the reduction of reduced GL7+B, IgG+B and IL-17+CD4+Th17 cells in lupus-like MRL/lpr mice and suppressed megalosplenia (Fig. 7). In parallel with their suppressive functions, GL7− Breg cells reduced IgG deposition, infiltration of inflammatory cells into the kidneys and restored the structure of the glomerular region in MRL/lpr mice (Fig. 8). These results showing that GL7− IL-10-producing B cells possess potent suppressive functions in an autoimmune disease by inducing the expansion of GL7+ Bregs, thus suggest that Breg cells may constitute a bona fide distinct B cell lineage. Our data further suggest that while differentiation into the Breg phenotype does not require BCR signaling, their expansion is enhanced by reception of BCR signals and isotype switching. These results furthermore proves that IL-10 expression is independent of GL7 expression, apical in regulatory function and the most examined mechanism of Breg action (Asseman et al., 1999).
In conclusion, GL7− Breg cells are pre-existing Bregs, CD19-independent and expanded during inflammation and contributes to immune regulation in lupus-like mice.
Supplementary Material
Acknowledgements
This study was supported by National Basic Research Program 973 Grants (2013CB530506, 2015CB553704), National Nature and Science Funds (81471529, 81401332, 81272320, 81471540, 81472647 and 81202292), the Key Program of the Beijing Natural Science Foundation (7141007) and Service Industry Scientific Research of National Health and Family Planning Commission of China (2015SQ00192).
Footnotes
Conflict of interest
The authors declare no conflict of interest.
Appendix A. Supplementary data
Supplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.molimm.2016.01.011.
References
- Asseman C, Mauze S, Leach MW, Coffman RL, Powrie F, 1999. An essential role for interleukin 10 in the function of regulatory T cells that inhibit intestinal inflammation. J. Exp. Med 190, 995–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brummel R, Lenert P, 2005. Activation of marginal zone B cells from lupus mice with type A(D) CpG-oligodeoxynucleotiedes. J. Immunol 174, 2429–2434. [DOI] [PubMed] [Google Scholar]
- Cervenak L, Magyar A, Boja R, Laszlo G, 2001. Differential expression of GL7 activation antigen on bone marrow B cell subpopulations and peripheral B cells. Immunol. Lett 78, 89–96. [DOI] [PubMed] [Google Scholar]
- Dillilo DJ, Matsushita T, Tedder TF, 2010. B10 cells and regulatory B cells balance immune responses during inflammation autoimmunity, and cancer. Ann. N. Y. Acad. Sci 1183, 38–57. [DOI] [PubMed] [Google Scholar]
- Evans JG, Chavez-Rueda KA, Eddaoudi A, Meyer-Bahlburg A, Rawlings DJ, Ehrenstein MR, Mauri C, 2007. Novel suppressive function of transitional 2 B cells in experimental arthritis. J. Immunol 178, 7868–7878. [DOI] [PubMed] [Google Scholar]
- Fairfax KA, Corcoran LM, Pridans C, Huntington ND, Kallies A, Nutt SL, Tarlinton DM, 2007. Different kinetics of Blimp-1 induction in B cell subsets revealed by reporter gene. J. Immunol 178, 4104–4111. [DOI] [PubMed] [Google Scholar]
- Fillatreau S, Sweenie CH, McGeachy MJ, Gray D, Anderton SM, 2002. B cells regulate autoimmunity by provision of IL-10. Nat. Immunol 3, 944–950. [DOI] [PubMed] [Google Scholar]
- Gray M, Miles K, Salter D, Savill J, 2007. Apoptotic cells protect mice from autoimmune inflammation by the induction of regulatory B cells. Proc. Natl. Acad. Sci. U. S. A 104, 14080–14085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein U, Dalla-Favera R, 2008. Germinal centres: role in B-cell physiology and malignancy. Nat. Rev. Immunol 8, 22–33. [DOI] [PubMed] [Google Scholar]
- Laszlo G, Hathcock KS, Dickler HB, Hodes RJ, 1993. Characterization of a novel cell-surface molecule expressed on subpopulations of activated T and B cells.J. Immunol 150, 5252–5262. [PubMed] [Google Scholar]
- Lenert P, Brummel R, Field EH, Ashman RF, 2005. TLR-9 activation of marginal zone B cells in lupus mice regulates immunity through increased IL-10 production. J. Clin. Immunol 25, 29–40. [DOI] [PubMed] [Google Scholar]
- Matsumoto M, Baba A, Yokota T, Nishikawa H, Ohkawa Y, Kayama H, Kallies A, Nutt SL, Sakaguchi S, Takeda K, Kurosaki T, Baba Y, 2014. Interleukin-10-producing plasmablasts exert regulatory function in autoimmune inflammation. Immunity 41, 1040–1051. [DOI] [PubMed] [Google Scholar]
- Mauri C, Bosma A, 2012a. Immune regulatory function of B cells. Annu. Rev. Immunol 30, 221–241. [DOI] [PubMed] [Google Scholar]
- Mauri C, Bosma A, 2012b. Immune regulatory function of B cells. Annu. Rev. Immunol 30, 221–241. [DOI] [PubMed] [Google Scholar]
- Mauri C, Ehrenstein MR, 2008. The ‘short’ history of regulatory B cells. Trends Immunol. 29, 34–40. [DOI] [PubMed] [Google Scholar]
- Mauri C, Menon M, 2015. The expanding family of regulatory B cells. Int. Immunol 27, 479–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizoguchi A, Bhan AK, 2006. A case for regulatory B cells. J. Immunol 176, 705–710. [DOI] [PubMed] [Google Scholar]
- Mizoguchi A, Mizoguchi E, Takedatsu H, Blumberg RS, Bhan AK, 2002. Chronic intestinal inflammatory condition generates IL-10-producing regulatory B cell subset characterized by CD1d upregulation. Immunity 16, 219–230. [DOI] [PubMed] [Google Scholar]
- Neves P, Lampropoulou V, Calderon-Gomez E, Roch T, Stervbo U, Shen P, Kuhl AA, Loddenkemper C, Haury M, Nedospasov SA, Kaufmann SH, Steinhoff U, Calado DP, Fillatreau S, 2010. Signaling via the MyD88 adaptor protein in B cells suppresses protective immunity during Salmonella typhimurium infection. Immunity 33, 777–790. [DOI] [PubMed] [Google Scholar]
- Nutt SL, Taubenheim N, Hasbold J, Corcoran LM, Hodgkin PD, 2011. The genetic network controlling plasma cell differentiation. Semin. Immunol 23, 341–349. [DOI] [PubMed] [Google Scholar]
- Rincon-Arevalo H, Sanchez-Parra CC, Castano D, Yassin L, Vasquez G, 2015. Regulatory B cells and mechanisms. Int. Rev. Immunol, 10.3109/08830185.2015.1015719. [DOI] [PubMed] [Google Scholar]
- Rosser EC, Mauri C, 2015. Regulatory B cells: origin, phenotype, and function. Immunity 42, 607–612. [DOI] [PubMed] [Google Scholar]
- Schioppa T, Moore R, Thompson RG, Rosser EC, Kulbe H, Nedospasov S, Mauri C, Coussens LM, Balkwill FR, 2011. B regulatory cells and the tumor-promoting actions of TNF-a during squamous carcinogenesis. Proc. Natl. Acad. Sci. U. S. A 108, 10662–10667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen P, Roch T, Lampropoulou V, O’Connor RA, Stervbo U, Hilgenberg E, Ries S, Dang VD,Jaimes Y, Daridon C, Li R,Jouneau L, Boudinot P, Wilantri S, Sakwa I, Miyazaki Y, Leech MD, McPherson RC, Wirtz S, Neurath M, Hoehlig K, Meinl E, Grutzkau A, Grun JR, Horn K, Kuhl AA, Drner T, Baror A, Kaufmann SH, Anderton SM, Fillatreau S, 2014. IL-35-producing B cells are critical regulators of immunity during autoimmune and infectious diseases. Nature 507, 366–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tadmor T, Zhang Y, Cho HM, Podack ER, Rosenblatt JD, 2011.The absence of B lymphocytes reduces the number and function of T-regulatory cells and enhances the anti-tumor response in a murine tumor model. Cancer Immunol. Immunother 60, 609–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theofilopoulos AN, Dixon FJ, 1985. Murine models of systemic lupus erythematosus. Adv. Immunol 37, 269–390. [DOI] [PubMed] [Google Scholar]
- Wang RX, Yu CR, Dambuza IM, Mahdi RM, Dolinska MB, Sergeev YV, Wingfield PT, Kim SH, Egwuagu CE, 2014. Interleukin-35 induces regulatory B cells that suppress autoimmune disease. Nat. Med 20, 633–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe R, Ishiura N, Nakashima H, Kuwano Y, Okochi H, Tamaki K, Sato S, Tedder TF, Fujimoto M, 2010. Regulatory B cells (B10 cells) have a suppressive role in murine lupus: CD19 and B10 cell deficiency exacerbates systemic autoimmunity. J. Immunol 184, 4801–4809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing C, Ma N, Xiao H, Wang X, Zheng M, Han G, Chen G, Hou C, Shen B, Li Y, Wang R, 2015. Critical role for thymic CD19+CD5+CD1dhiIL-10+ regulatory B cells in immune homeostasis. J. Leukoc. Biol 97, 547–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanaba K, Bouaziz JD, Haas KH, Poe JC, Fujimoto M, Tedder TF, 2008a. A regulatory B cell subset with a unique CD1dhiCD5+ phenotype controls T cell-dependent inflammatory responses. Immunity 28, 639–650. [DOI] [PubMed] [Google Scholar]
- Yanaba K, Bouaziz ZD, Matsushita T, Magro CM, St Clair EW, Tedder TF, 2008b. B-lymphocyte contributions to human autoimmune disease. Immunol. Rev 223, 284–299. [DOI] [PubMed] [Google Scholar]
- You Y, Zhao H, Wang Y, Carter RH, 2009. Primary and secondary effects of CD19 deficiency on cells of the marginal zone.J. Immunol 182, 7343–7347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng M, Xing C, Xiao H, Ma N, Wang X, Han G, Chen G, Hou C, Shen B, Li Y,Wang R, 2014. Interaction of CD5 and CD72 is involved in regulatory T and B cell homeostasis. Immunol. Invest 43, 705–716. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.