

SUMMARY
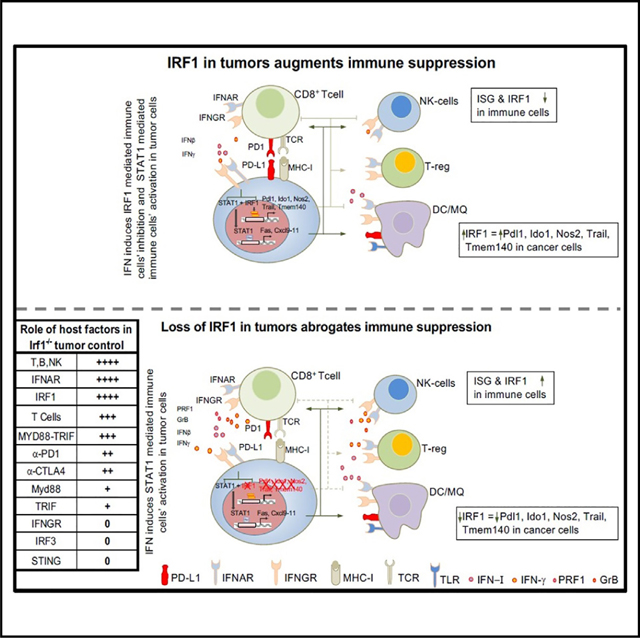
Type I interferon (IFN-I) and IFN-γ foster antitumor immunity by facilitating T cell responses. Paradoxically, IFNs may promote T cell exhaustion by activating immune checkpoints. The downstream regulators of these disparate responses are incompletely understood. Here, we describe how interferon regulatory factor 1 (IRF1) orchestrates these opposing effects of IFNs. IRF1 expression in tumor cells blocks Toll-like receptor- and IFN-I-dependent host antitumor immunity by preventing interferon-stimulated gene (ISG) and effector programs in immune cells. In contrast, expression of IRF1 in the host is required for antitumor immunity. Mechanistically, IRF1 binds distinctly or together with STAT1 at promoters of immunosuppressive but not immunostimulatory ISGs in tumor cells. Overexpression of programmed cell death ligand 1 (PD-L1) in Irf1−/− tumors only partially restores tumor growth, suggesting multifactorial effects of IRF1 on antitumor immunity. Thus, we identify that IRF1 expression in tumor cells opposes host IFN-I- and IRF1-dependent antitumor immunity to facilitate immune escape and tumor growth.
In brief
Proper lymphocyte activation is required for antitumor immunity. Purbey et al. show that, while IRF1 in immune cells is required for antitumor immunity, tumor cell IRF1 suppresses Toll-like receptor- and interferon-induced antitumor lymphocyte activation and regulates select immunosuppressive and MHC genes in interferon-stimulated tumor cells.
Graphical abstract
INTRODUCTION
The generation of T cell responses against tumor cells is critical for spontaneous and immune checkpoint blockade (ICB)-induced antitumor immunity.1–3 By directly activating cytotoxic T cells (CTLs),4 type I interferon (IFN-I) and type II IFN (IFN-γ) are critical for activation of antitumor responses.1,4–8 IFN-γ is mainly produced by T helper cells, cytotoxic T cells, and natural killer (NK) cells,4,7,9 while IFN-I is typically produced by dendritic cells (DCs) within the tumor microenvironment (TME).6,10 Activation of pattern recognition receptor (PRRs) pathways, including Toll-like receptors (TLRs)11,12 and cytosolic DNA13,14 or dsRNA15 sensors, can activate DCs and other TME cells to produce IFN-I spontaneously or upon ICB within growing tumors; this IFN-I is critical for enhancing tumor antigen presentation and cross-presentation by DCs to prime and activate the endogenous anti-tumor T cell-mediated immune pathway.16
While IFNs may directly promote antitumor immunity through immune cell activation, tumor-cell-intrinsic IFN signaling may promote immune dysfunction by enhancing immune checkpoints on tumor cells, resulting in T cell exhaustion.1,8,17–20 In particular, tumor cell loss of the receptor for IFN-γ (IFNγR) and/or IFN-I (IFNAR) allows improved clearance of tumors by ICB.21,22 Several factors activated by IFNs have been identified that can contribute to how IFN signaling in tumor cells can counteract protective immunity, including programmed cell death ligand 1 (PD-L1),21,22 indoleamine 2,3-dioxygenase 1 (IDO1),23 receptor interacting serine/threonine kinase 1 (RIPK1),24 and non-classical major histocompatibility complex (MHC) I (Qa-1b) through NKG2A/CD94,21,25 but whether an IFN-inducible factor can regulate several immunosuppressive genes/functions is unknown.
One candidate interferon-stimulated gene (ISG) that may regulate the effects of IFNs in both immune cells and tumor cells is the transcription factor interferon regulatory factor 1 (IRF1).26 IRF1 is expressed in multiple immune cell types, including antiviral T cells, NK cells, and the conventional DC1 (cDC1) subpopulation of DCs.27–30 However, IRF1 is also critical for the expression of PD-L1 on cancer cells upon IFN stimulation,31,32 which suppresses T cell responses.25,33 Thus, a better understanding of the positive and negative effects exerted by IRF1 during an anti-tumor immune response is warranted.
Herein, we dissect the roles of IRF1 defined by its loss in tumor cells or within host cells of the tumor microenvironment. We show that tumor cell loss of IRF1 expression unleashes antitumor immunity by activating ISG expression and effector programs in tumor-infiltrating immune cells. Eradication of IRF1-deficient tumors required adaptive immunity, TLR signaling, and IFN-I signaling in the host. Surprisingly, enhanced antitumor immunity to IRF1-deficient tumors did not require host IRF3- or IFN-γ signaling, but it did require host IRF1. Mechanistically, unlike the loss of IFN receptors in tumor cells, which eliminates the entire ISG program,21 the loss of IRF1 in tumor cells is more selective, as Irf1−/− cells showed reduced expression of ISGs with immune-suppressive but not -activating functions. Taken together, these studies demonstrate a novel pathway by which tumor-cell-intrinsic IRF1 is a potent suppressor of IFN-I-mediated and IRF1-dependent activation of T cell-mediated anti-tumor immunity in the TME.
RESULTS
Expression of IRF1 in tumor cells is required for tumor growth
To characterize the role of IRF1 in tumor cells, we generated Irf1−/− clones of highly immunogenic (MC38) and less immunogenic (B16F10 and YUMM2.1) tumor cells and implanted syngeneic C57BL/6 mice (Figures 1A and 1B). We also mutated Sting in MC38 cells, as Sting−/− tumor cell lines showed defective IFN-I response, enhanced growth, and diminished responsiveness to ICB.34–36 Several Irf1−/− and Sting−/− tumor cell lines were generated, and the mutations were verified by sequencing (Figures S1A–S1D). Immunoblot confirmed undetectable IRF1 protein in mutant clones (Figure 1B). The in vitro behaviors of the WT, Irf1−/−, and Sting−/− MC38 clones were similar with regard to proliferation and growth rate (Figure S1E) or sensitivity to radiation-induced cell death and clonogenic survival (Figures S1F and S1G).
Figure 1. Increased infiltration of adaptive immune cells is associated with growth impairment of immunogenic Irf1−/− tumor.

(A) Schematic representation of syngeneic tumor model.
(B) Immunoblot using anti-IRF1 antibody (top panel) anti-β-tubulin (bottom panel) loading control showing loss of IRF1 expression in various CRISPR-Cas937 mutant tumor clones.
(C) Upper panel shows growth kinetics of a representative experiment of WT (C1), Irf1−/− (I1), and Sting−/− (S3) MC38 clones in wild-type (C57BL/6J) hosts. Lower panel represents weight of tumors at termination of experiment in upper panel.
(D) Upper panel shows tumor growth kinetics (average tumor volume) of additional WT (C3), Sting−/− (S1), and Irf1−/− (I3 and I4) MC38 clones in wild-type (C57BL/6J) hosts. Lower panel shows weight of tumors from panel above on day 21.
(E) Upper panel shows growth kinetics of one each of WT and Irf1−/− B16F10 clones. Lower panel represents weight of tumors on day 18 from the experiment shown above.
(F) Upper panel shows growth kinetics of one WT and two Irf1−/− YUMM2.1 clones. Lower panel represents weight of tumors on day 32 from the experiment above. For (C)–(F), p values were determined. Error bars represent mean ± standard error. *p ≤ 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001 by Mann-Whitney U/Wilcoxon rank-sum test or Student’s paired t test as appropriate.
(G) Schematic representation of experimental approach for (H)–(J).
(H) Histology of tumor center and tumor boundary from WT (top left, bottom left) and Irf1−/− (top right, bottom right) from WT (bottom left) and Irf1−/− (bottom right) MC38 tumors grown in WT mice.
(I) Flow cytometry analysis of tumors for CD8+ and NK cells in the Irf1−/− MC38 tumors from WT (C57BL/6J) mice. Upper and lower panels represent immune cells/gram tumors and percent tumor CD45+ cells, respectively. p values were determined by a Mann-Whitney U/Wilcoxon rank-sum test or Student’s paired t test as appropriate. *p ≤ 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001.
(J) Representative immunofluorescence photomicrographs of tumor boundary (top panel) and tumor center (bottom panel) of WT (upper) and Irf1−/− (lower) MC38 tumors in WT mice.
We next tested whether WT, Irf1−/−, and Sting−/− MC38 clones displayed different in vivo growth properties in wild-type (WT) (C57BL/6J) mice following subcutaneous implantation. Sting−/− tumors grew to a similar extent as WT tumors (Figures 1C and 1D). However, tumor growth of Irf1−/− (I1 clone) MC38 clones was greatly diminished (Figures 1C and 1D), as previously reported.31 Injection sites of Irf1−/− MC38 injection developed palpable tumors initially (typically 6–9 days), which gradually diminished, and many mice displayed evidence of complete regression (Figures 1C and 1D).
We next evaluated growth of Irf1−/− B16F10 and YUMM2.1 tumors, which exhibit lower tumor mutational burden, basal MHC I expression, and poor immunogenicity (Figures S1H and S1I).21,38 Like Irf1−/− MC38, Irf1−/− B16F10 and YUMM2.1 displayed impaired tumor growth in WT mice (Figure 1E for B16F10; Figure 1F for YUMM2.1 clones). Thus, tumor-cell-intrinsic expression of IRF1 is required for optimal tumor growth across multiple tumor types regardless of tumor immunogenicity.
Tumor cell IRF1 expression impairs immune cell recruitment to tumors
We examined histology of tumors 12 days post implantation (Figure 1G). WT MC38 tumor cells displayed large tumor islands with rare scattered immune cells at the tumor boundary and throughout the tumor center (Figure 1H, left top and bottom panels). Irf1−/− tumors displayed small tumor islands with numerous apoptotic tumor cells among large areas of fibrotic stroma. Large, dense clusters of immune cells were seen at the tumor boundary and within the tumor parenchyma (Figure 1H).
Flow cytometric analysis of tumor-infiltrating immune cells from WT mice revealed increased numbers of total CD45+ immune cells in Irf1−/− MC38 tumors compared to WT MC38 tumors, confirming the histologic observations (Figure 1I, upper boxplots). Further phenotyping showed that CD3+ T cells accounted for most of the increased immune cells infiltrating in Irf1−/− MC38 tumors, with greater CD8+ cytotoxic T cells (CTLs) than CD4+ helper T cells (TH cells). Small increases in NK1.1+ NK or NK T cells were also noted, while myeloid cells showed little difference (Figure 1I).
Since CTL infiltration from the tumor boundary into the tumor parenchyma is critical for tumor regression in ICB therapy,39 we next identified the location of CD4+ and CD8+ T cells within the tumor. Multiplexed immunofluorescent (IF) histology confirmed that CD4+ and CD8+ T cells were present at the tumor boundary of WT MC38 tumors, with sparse lymphocytes present at the center of the tumor (Figure 1J, upper two panels). Irf1−/− MC38 cells displayed pronounced infiltration of CD4+ and CD8+ T lymphocytes at the tumor margin and within the center of the tumor (Figure 1J, bottom 2 panels).
We next addressed whether Irf1−/− YUMM2.1 tumors also display enhanced immune cell infiltration upon deletion of Irf1. WT and Irf1−/− YUMM2.1 tumors were isolated from WT mice 14 days after implantation (Figure S2A). Flow cytometry revealed decreased immune cell infiltrates in WT YUMM2.1 tumors when compared to WT MC38 tumors (Figure S2B, top left panel, vs. Figure 1I, top left panel). Like MC38 tumors, however, Irf1−/− YUMM2.1 tumors displayed enhanced infiltration of many immune cells, including total CD45+ immune cells, total T cells, TH cells, CTLs, NK, and NK T cells, when compared to WT YUMM2.1 tumors (Figure S2B), while myeloid cells (CD11b+ Ly6Chi) were unchanged. IF staining of YUMM2.1 tumors revealed lymphocytes were present in higher quantities at both the tumor boundary and tumor center in the Irf1−/− YUMM2.1 tumors, similar to MC38 Irf1−/− tumors (Figure S2C).
Tumor-cell-intrinsic IRF1 expression impairs antitumor immunity and ICB responses
Next, WT and Irf1−/− mutant tumors were grown in immunodeficient mice (Figures 2A and 2B, left panels). In contrast to WT mice, the most immunodeficient mouse strains, Rag2−/−γC−/− and NOD/Scid mice, displayed similar growth of WT and Irf1−/− MC38 tumors (Figure 2A, right panel). Growth of Irf1−/− MC38 tumors in Rag1−/− and Cd8−/− mice was intermediate to WT mice and Rag2−/−γC−/− or NOD/Scid mice, indicating that both CD8 T cells and NK cells contribute to the generation of enhanced antitumor immunity (Figure 2B).
Figure 2. Generation of adaptive immune memory and ICB control of Irf1−/− tumors.

(A) Schematic representation (top left panel) and of experimental design and table (bottom left panel) of immunological defects in indicated immunodeficient mice. Growth kinetics of WT and Irf1−/− MC38 tumors in Rag2−/− γC−/− (middle panel) and NOD/Scid (right panel) mice.
(B) Experimental schema (top left panel) and table of immune deficiencies (bottom left panel). Right panel shows weight of WT and Irf1−/− MC38 tumors in various mouse strains after 21 days.
(C) Experimental scheme for panels (D) and (E).
(D) Effect of anti-PD-1 treatment on WT and Irf1−/− MC38 tumors.
(E) Effect of anti-CTLA4 treatment on WT and Irf1−/− MC38 tumors. For (A)–(E), *p ≤ 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001 by Mann-Whitney U/Wilcoxon rank-sum test.
To determine whether Irf1−/− tumors generate adaptive immune memory, we tested whether mice that cleared Irf1−/− tumors could reject WT tumors in an antigen-specific manner. Indeed, we found that all mice that previously received and cleared Irf1−/− MC38 tumors rejected WT MC38 tumors (0 out of 10) implanted 2 months later, whereas all WT YUMM2.1 tumors grew normally in the mice that previously cleared Irf1−/− MC38 tumors (11 out of 11).
Mouse colorectal cancer cell lines (MC38) possess a high tumor mutation burden and express high levels of MHC I molecules, which make them amenable to ICB therapy (Figures S2D and S2E).21,40 ICB with anti-PD-1 antibody resulted in modestly decreased tumor size, while anti-CTLA-4 antibody led to complete clearance of about half of the WT MC38 tumors and reduced the size of the tumors that grew (Figures 2C–2E). To assess whether loss of IRF1 in tumors can result in enhanced ICB responses, we doubled the number of Irf−/− tumor cells injected to form mutant tumors, prior to treating mice with ICB antibodies. Either anti-PD-1 or anti-CTLA-4 therapy was able to completely eradicate Irf1−/− MC38 tumors despite the larger number of cells injected to initiate tumors (Figures 2D and 2E). These findings indicate that the presence of IRF1 in tumor cells suppresses adaptive immunity to tumors and limits the effectiveness of ICB responses.
Tumor-cell-intrinsic IRF1 expression suppresses activation and effector T cell programs
Prior studies suggested that IRF1 loss in tumor cells does not affect numbers or subtypes of infiltrating T cell populations but does result in increased cytotoxicity by CTLs.31 However, our flow cytometry and IF analyses of MC38 and YUMM2.1 tumors revealed greater numbers of adaptive immune cells with different infiltration patterns in the Irf1−/− tumors compared to WT tumors. We performed single-cell (sc)RNA-seq analysis from tumors to gain greater insight into how loss of tumor cell IRF1 alters the antitumor immune response.41,42 Seurat-based integrated analysis and uniform manifold approximation and projection (UMAP) revealed 15 cell clusters (Figure S3A), which were further refined into 12 cell types (Figure S3B) based on marker gene expression.43,44 Quantitative analysis of distinct cell populations from WT and Irf1−/− tumors showed that WT tumors displayed an increased percentage of cancer cells and fibroblasts, while Irf1−/− tumors displayed the greatest increases in CD8T-NKT (2.14-fold) and NK (1.5-fold) populations (Figures 3A and 3B).
Figure 3. Enhanced effector T cell response in Irf1−/− tumors.

(A) UMAP showing 12 distinct cell populations in WT and Irf1−/− tumors.
(B) Percentage of indicated cell populations in WT and Irf1−/− tumors.
(C) Dot plot showing expression of select cell-type-specific and immune activation genes in cells from WT and Irf1−/− tumors.
(D) Differential expression (DE) of genes enriched in the CD8-NKT cell cluster.
(E) Gene Ontology analysis of cells in the CD8-NKT cluster from Irf1−/− tumors.
(F) Projection of T cells (black profile) from WT and Irf1−/− tumor scRNA-seq onto reference T cell atlas (colored distinct T cell states) from ProjectTILs.
(G) Proportion of various T cell states in WT and Irf1−/− tumors as defined by the reference T cell atlas. All data were generated by comparison of scRNA-seq data pooled from 4 WT and 6 Irf1−/− mouse tumors.
Broadly, Irf1−/− tumors showed enhanced expression of ISGs in almost all cells of the TME (Clcl5, Pdl1, Gbp5, Irf1, Irf8, Irf7, etc., Figure 3C). Additionally, within the CD8T-NKT cluster from Irf1−/− tumors, 866 of the 884 differentially expressed genes (DEGs) were overexpressed in the Irf1−/− tumors, corresponding to Gene Ontology (GO) pathways involved with T cell/lymphocyte activation and T cell differentiation.45 Several effector molecules, including Cd8, Prf1, Gzmb, Tnf, and Ccl5, were observed in a higher percentage of cells than in the same cluster from WT tumors (Figures 3C–3E).
The CD8-NKT cluster also showed increased expression of many of the genes representing T cell effector, proliferation, and memory functions (Gzmb, Gzmk, Prf1, Ccl5, Fasl, Satb1, Pdcd1, Irf7, etc.) in Irf1−/− tumors (Figures 3D and 3E). Cells within the CD8T-NKT cell cluster and the DC cluster expressed higher levels of several ISGs, while DCs also expressed higher levels of Ciita, Ccl5, and Il12b in Irf1−/− tumors (Figures 3C and 3E). These results suggest that tumor-cell-intrinsic IRF1 expression suppresses the overall activation state of multiple immune cell types, including innate and adaptive immune cells, in growing tumors.
To better understand the difference in T cell states between WT and Irf1−/− tumors, we projected our data to the T cell reference cell atlas, which was created using in silico purified T cell scRNA-seq data from many mouse tumors (Figure S3C, detailed in STAR Methods).46 Briefly, the reference T cell atlas defines nine T cell states (Figure S3C), including CD8_Tex (CD8+ terminally exhausted effector cells, characterized by expression of granzymes and inhibitory receptors, including Pdcd1, Ctla4, Lag3, Tigit, and Havcr2), CD8_Tpex (CD8+ precursor-exhausted cluster, with co-expression of Tcf7, Pdcd1, Ctla4, and Tox but low expression of Havcr2 or granzymes), CD8_NaiveLike (naive-like CD8+ T cells, which consist of naive as well as central memory cells), CD4_NaiveLike (naive-like CD4+ T cells co-expressing Tcf7 and Ccr7 but lacking cytotoxic/activation molecules including Pdcd1 and Tnfrsf9), CD8_EffectMemory (effector memory CD8+ T cells, co-expressing Tcf7 and granzymes, most prominently Gzmk, with low to intermediate expression of Pdcd1; an “early-activation” state of CD8+ T cells), Th1 (CD4+ Th1-like cells, expressing Ifngr1 and Fasl), Tfh (CD4+ follicular-helper population expressing Cxcr5, Tox, and Slamf6), and Treg (regulatory T cells expressing Foxp3). We projected WT and Irf1−/− tumor scRNA-seq data onto the T cell reference cell atlas, which revealed the presence of higher proportions of total T cells in Irf1−/− tumors (16.13%) compared to WT tumor (7.57%) (Figure S3D). Relative to WT tumors, Irf1−/− tumors showed a dramatic increase in CD8_EffectMemory (224%), CD8_naive like cells (200%), and Tfh (195%), as well as a moderate increase in Th1 (120%) cells (Figures 3F and 3G).46,47 In contrast, a substantial decreased percentage of Treg and CD8 early activated cells was observed in Irf1−/− tumors (Figures 3F and 3G).
Additionally, the aforementioned CD8_EffectMemory cluster showed increased expression of T cell activation genes (Figures S3E–S3G), including Pdcd1 (PD-1, programmed cell death protein 1), Lgals3, Isg20, Litaf, Klrk1 (NKG2D), Gzmb, and Fasl (Figure S3E) in Irf1−/− tumors.48–51 Similar proportions of CD8_Tex cells were found in WT and Irf1−/− tumors though these cells displayed increased expression of activation markers and ISGs in Irf1−/− tumors (Figure S3H).
In contrast to DEGs in immune cell clusters, DEGs in the tumor cell cluster suggested that loss of IRF1 in tumor cells resulted in decreased expression of IFN-I and IFN-γ inducible programs and genes involved in antigen presentation, while genes involved in ECM matrix remodeling were upregulated (Figures S3I and S2J). While slightly decreased, PD-L1 expression in Irf1−/− tumor cells was not identified as a DEG statistically (Figure 3C). These results demonstrate perturbation of a subset of ISGs due to loss of IRF1 in the cancer cells in vivo (Figures S3I and S3J).
Together, these results provide evidence that the Irf1−/− tumors impact IFN signatures in tumor-infiltrating cells. Thus, tumor cell expression of IRF1 impairs IFN-mediated activation of multiple populations of tumor-infiltrating immune cells including DCs, effector memory CD8+ T cells, and exhausted CD8+ T cells. Since both type I and II IFNs drive T cell-mediated antitumor immunity,4,5 these findings suggest that IRF1 expression in tumor cells impairs IFN-driven antitumor immune pathways.
Host IFN-I, but not IFN-γ, is critical for enhanced control of Irf1−/− tumors
We next wished to determine more directly whether IFN-I and/or IFN-γ were required for the enhanced regression of Irf1−/− tumors. Indeed, implanted Irf1−/− tumors in Ifnar−/− mice displayed largely restored growth when compared to those implanted in WT mice (Figures 4A and S4C). However, to our surprise, neither Ifngr−/− mice nor IFNγR blocking antibody restored Irf1−/− tumor growth (Figures 4A, S4C, and S4A). Treatment of Ifngr−/− mice with a blocking antibody against IFNAR restored Irf1−/− tumor growth (Figure S4A, right panel). Blockade of IFN-I in Ifngr−/− mice resulted in growth of Irf1−/− tumors that was similar to WT tumors implanted into WT mice (Figure S4A, right panel), suggesting a major non-redundant role of IFNAR signaling and a supporting role of IFN-γ in control of highly immunogenic Irf1−/− tumors.
Figure 4. Responsiveness to type I but not the type II interferon (IFN-γ) mediates Irf1−/− tumor control.

(A) Experimental design (left) for test the role of host IFN-I and IFN-II in WT and Irf1−/− MC38 tumors. Right dot plot represents weight of WT and Irf1−/− MC38 tumors from mice on day 20.
(B) Proportion of indicated cell populations among WT MC38 tumor grown in WT mice and Irf1−/− MC38 tumor grown in WT or Ifnar−/− mice.
(C) Dot plot showing expression of activation markers and ISGs in distinct cell types from Irf1−/− MC38 tumor grown in WT or Ifnar−/− mice.
(D) Projection of scRNA-seq data of T cells (black profile) from indicated tumors onto reference T cell atlas (colored distinct T cell states) from ProjectTILs.
(E) Percentage of T cells mapped to reference T cell states in the scRNA-seq data from indicated tumors.
(F) Proportion of various T cell states in the scRNA-seq data from the indicated tumors.
(G) Left side shows the experimental scheme of upcoming experimental testing. Middle boxplot shows weight of WT and Irf1−/− MC38 tumor from WT and Stinggt/gt mice, and right boxplot shows weight of WT and Irf1−/− tumors in indicated mice.
(H) Left side shows scheme of testing host TLR pathways in Irf1−/− tumors. First two dot plot panels show weight of WT (1st dot plot, day 23) and Irf1−/− (2nd dot plot, day 31) MC38 tumors in WT, Myd88−/−, and Trif−/− mice. Last boxplot shows weight of WT and Irf1−/− MC38 tumor in WT and Myd88−/−Trif−/− (double mutant) mice on day 17. For (A), (G), and (H) plots, *p ≤ 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001 by Mann-Whitney U/Wilcoxon rank-sum test.
We next performed scRNA-seq of Irf1−/− and WT MC38 tumors in Ifnar−/− mice and directly compared these data to scRNA-seq data from WT and Irf1−/− tumors in WT mice (described in Figure 3A). UMAP projection of Irf1−/− tumor cells in Ifnar−/− mice shows a similar pattern of cellularity as WT MC38 tumor cells grown in WT mice, demonstrating that the loss of IFNAR signaling impairs effector cell recruitment in Irf1−/− tumors (Figures S4B and 4B).
Quantitative analysis of Irf1−/− tumor cells in Ifnar−/− mice demonstrated a decrease in the percentage of CD8T-NKT cells and NK cells compared to Irf1−/− mutant tumors in WT mice (Figure 4B). In addition, the CD8T-NKT cluster possessed fewer cells expressing cytotoxicity effector genes, Nkg7, Ifng, Prf1, Gzmb, Ccl5, etc. (Figure 4C). Irf1−/− mutant tumors in Ifnar−/− mice displayed impaired ISG induction, including Irf1, Irf7, and Ccl5, in nearly all cell types when compared to Irf1−/− tumors in WT mice (Figure 4C). Further analysis showed a reduced percentage of T cells in Irf1−/− tumors from Ifnar−/− mice (9.7%) compared to Irf1−/− tumors from WT mice (16.1%) (Figures 4D and 4E). The CD8_EffectMemory cell cluster was most affected (7.5-fold decrease) in Irf1−/− tumors from Ifnar−/− mice (15.2% vs. 2%, Figure 4F). In contrast, an increase in regulatory T cells, CD8 naive-like cells, and Tfh and Th1 cells was observed in Irf1−/− tumors from Ifnar−/− mice compared to Irf1−/− tumors from WT mice (Figure 4F). This analysis supports a model whereby tumor cell IRF1 expression opposes IFN-I signaling that drives adaptive antitumor immunity.
TLR signaling is required for enhanced immunity to Irf1−/− tumor cells
Production of IFN-I by DCs is critical for antitumor immunity, and many PRR pathways may contribute to initiating this pathway, including cGAS-Sting (cytosolic DNA), RIG-I-MAVS (cytosolic dsRNA), or TLR-MyD88/TRIF (extracellular dsRNA, ssRNA, or DNA). STING, MAVS, and TRIF activation culminate in the activation of the transcription factor IRF3 to induce a robust IFN-I response. Unexpectedly, Irf1−/− tumors were strongly controlled in Stinggt/gt (Figure 4G, middle panel; Figure S4D, left panel) and Irf3−/− (Figure 4G, right panel; Figure S4D right panel) mice similarly to WT mice, suggesting that IRF3-dependent programs are dispensable for enhanced clearance of IRF1-deficient tumors (Figures 4G and S4D).
We next evaluated whether TLRs contribute to antitumor immune responses to Irf1−/− tumors. The TLR family uses two adaptors, MyD88 and TRIF, to signal activation. Thus, the role of multiple TLRs can be investigated by implanting Irf1−/− MC38 tumors in Myd88−/− or Trif−/− mice, respectively. Implanted Irf1−/− tumors in MyD88−/− or Trif−/− mice demonstrated a partial restoration of tumor growth, with Trif−/− mice showing a somewhat greater effect (Figure 4H, middle panel). Next, we tested whether both pathways worked together to control tumor growth using MyD88−/−/Trif−/− double knockout mice. Indeed, Irf1−/− tumors implanted in MyD88−/−/Trif−/− mice showed growth restoration to similar levels as WT mice, revealing that both adaptors work in concert to suppress tumor growth (Figure 4H, right panel). Even WT tumors grew larger in MyD88−/−/Trif−/− mice than in WT mice (Figure 4H, right panel), revealing that TLR signaling is critical for controlling WT tumor growth as well. Notably, we cannot explain why TRIF but not IRF3 (IRF3 is usually a key transcription factor target of TRIF signaling) contributed to the clearance of Irf1−/− tumors, but one possible explanation is redundancy between IRF3 and the closely related IRF7 family member in this setting.52–54
IRF1 in the host microenvironment is required for tumor control
Although IRF1 can be activated by TRIF signaling, its expression and activity are also induced by IFN-I and IFN-γ, and it is one of the key downstream regulators of antiviral immunity in CTLs.27,28 We therefore evaluated the immune response to WT and Irf1−/− tumors in Irf1−/− mice. Indeed, Irf1−/− mice displayed dramatically enhanced growth of both WT and Irf1−/− tumors, demonstrating a key role of IRF1 in promoting antitumor immunity in the TME. Notably, this role is in direct opposition to IRF1’s role in tumor cells in driving the suppression of IFN-I signaling in the TME and the antitumor immune responses that depend on IFN-I signaling in the TME. (Figure 4G, right panel).
The striking difference of tumor growth in Irf1−/− and WT mice suggests that IRF1 plays a larger role in antitumor immunity than its previously recognized role in regulating cDC1 activation in tumors.29 Indeed, IRF1 plays a critical role in cytotoxic T cell-mediated antiviral immunity and participates in IFN-I and IFN-γ signaling.27,28 Thus, we wished to evaluate immune cell recruitment in tumors implanted in Irf1−/− mice. Profiling of tumor-infiltrating lymphocytes (gating scheme shown in Figures S5A and S5B) revealed a 5-fold decrease in CD8+ T cells and a 10-fold decrease in NK cells without significant change in CD4+ T cells and NK T cells in tumors (Figures 5B–5E) and spleen (Figure S5C and S5D) isolated from Irf1−/− mice versus WT mice.
Figure 5. IRF1 expression in the host microenvironment is required for the clearance of Irf1−/− tumors.

(A) Experimental scheme for (B)–(E).
(B) Left panel shows total CD45+ cells/gram of tumors, and right panel displays CD45+CD3+, CD3+CD4+, and CD3+CD8+ T cells/gram of WT tumors in WT or Irf1−/− mice.
(C) Frequency of CD4+ and CD8+ T cells relative to CD3+ cells in WT tumors grown in WT (n = 3) and Irf1−/− mice (n = 4).
(D) Infiltration of “total NK1.1+,” “NK,” and “NKT” cells per gram of WT tumors from WT and Irf1−/− mice.
(E) Frequency of “total NK1.1”, “NK” and “NKT” cells relative to CD45+ cells infiltrated in WT tumors grown in WT (n = 3) and Irf1−/− (n = 4) mice.
(F) Left panel: schematic representation of experimental setup to monitor for (F)–(H). Right panel: weight of Irf1−/− MC38 (syngeneic; first 3 columns) and FSA (allogeneic; last 3 columns) subcutaneous tumors grown in WT (C57Bl6/J), NOD/Scid, or Irf1−/− mice for 22 days.
(G) Images of Irf1−/− MC38- or FSA-tumor-bearing mice (left panel) and harvested tumors (right panel) on day 16 in NOD/Scid mice.
(H) Images of tumor-bearing mice (left panel) and harvested tumors (right panel) on day 22 in Irf1−/− mice. For (B)–(F) plots, *p ≤ 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001 by Mann-Whitney U/Wilcoxon rank-sum test.
Given the severity of the adaptive immune defect in clearance of syngeneic tumors, we assessed Irf1−/− mice for their ability to clear an allogeneic tumor. Both Irf1−/− and WT (C57BL/6J) mice rejected fibrosarcoma (FSA) tumors (C3H), while FSA tumors grew in NOD/Scid mice (Figures 5F–5H). These studies reveal that host IRF1 expression is critical for traditional antitumor immunity to tumor antigens, rather than causing a more severe, SCID-like defect.
IRF1 selectively regulates MHC class I and immunosuppressive ISGs in tumor cells
We next returned to the tumor cells and evaluated potential mechanisms by which tumor cell IRF1 regulates the suppression of antitumor immunity and IFN-I responses in the TME. Binding of IFNα/β to IFNAR or IFN-γ to IFNGR mainly results in activation of STAT and IRF transcription factors to induce the transcription of ISGs. Past studies of IFN-γ-treated cells have shown that IRF1 participates in the regulation of PD-L1, MHC molecules, and the antigen processing machinery.31,32,55–57 Consistent with previous reports, MC38 tumor cells express higher cell surface MHC I when compared to YUMM2.1 (Figure S2D). Basal PD-L1 expression was comparable in all cell lines tested regardless of IRF1 expression (Figure S6A). Upon IFN-γ stimulation, PD-L1 showed complete IRF1 dependence, whereas MHC I displayed only partial dependence (Figures S6A and S2E). We next performed RNA-seq to identify basal and IFN-γ-induced IRF1-regulated genes in tumor cells (Figures S6B, S2E, and 6A). We chose a 2-h time point for IFN-γ stimulation of cells based on the activation and protein induction profiles of STAT1 and IRF1, respectively, and to avoid genes that may be indirect targets of these factors.
Figure 6. IRF1 positively regulates a subset of IFN-γ-inducible genes that can promote the tumor progression.

(A) Scheme of in vitro model of WT and Irf1−/− tumor cells, IFN-γ stimulation, and RNA-seq data analysis.
(B) Volcano plot of expressed genes in MC38, B16F10, and YUMM2.1 cell lines induced with IFN-γ for 2 h (RPKM > 2, isoform removed, n = 11,502). Top-right quadrant shows 71 induced genes (red and gold dots are genes induced more than 3-fold; p < 0.05 from 5 control and 5 IFN-γ-treated datasets).
(C) Dot plot display of average percent expression of IFN-γ-induced genes in Irf1−/− mutant clones relative to WT clones: red dotted line shows <30% expression and purple dotted line shows >200% expression in IFN-γ-stimulated Irf1−/− vs. WT tumor cells considered IRF1-dependent genes. Important IRF1-regulated genes (both up and down) are labeled.
(D) Table detailing known function of important IFN-γ-induced genes and how they are regulated by IRF1.
(E) RPKM distribution of basal and IFN-γ-stimulated genes from 43 human melanoma cell lines (5 ng/mL IFN-γ for 6 h).
(F) Volcano plot of all the expressed genes in a representative human melanoma line (M238) induced with IFN-γ for 6 h (average RPKM > 2, isoform removed, n = 9,768). The top-right quadrant (red dots) shows genes induced >3-fold (n = 120) with p < 0.05 from two IFN-γ-treated datasets.
(G) A dot plot display of average percent expression of IFN-γ-induced genes in Irf1−/− M238 (human melanoma cell line) relative to WT M238 cells.
To identify basal IRF1-dependent genes, we compared RNA-seq data from 3 WT to 2 Irf1−/− clones of unstimulated MC38 cells. We found 39 upregulated genes and 45 downregulated genes (reads per kilobase per million mapped reads [RPKM] > 1, fold change [FC] > 3, p value < 0.05) in the Irf1−/− clones at basal level (Figure S6B). GO analysis of the downregulated genes in Irf1−/− cells revealed enrichment for MHC I antigen presentation (Figure S6B and S6C), including the known IFN-γ-induced Psmb8, Psmb9, and Tap2 genes,32 which could contribute to decreased basal MHC I protein expression in Irf1−/− cancer cells. The genes expressed more strongly in Irf1−/− clones at baseline displayed enrichment in epithelial to mesenchymal transition (Figure S6B and S6D).
Upon IFN-γ stimulation, we identified 71 IFN-γ-induced genes that include immune cell activators (Cxcl9 and Cxcl10) and suppressors (Pdl1) across all three WT cancer cell types (RPKM > 2, average FC > 3, p-adj < 0.05, Figure 6B), similar to previous studies.31,32 We classified twenty-eight (~40%) of the 71 IFN-γ-induced genes as IRF1 dependent (mean expression is <33% in Irf1−/− compared to that of the WT; Figure 6C). Six genes (blue) out of the remaining 43 induced genes showed >2-fold stronger induction in Irf1−/− over WT tumor cells. Indeed, several genes shown to negatively affect tumor growth were identified as positively regulated by IRF1, including Pdl1, Ido1, Nos2, Tnfsf10 (encodes TRAIL), and Tmem140 (Figures 6C, 6D, and S6E).58,59
In contrast, the expression of key IFN-inducible chemokines that signal through the CXCR3 receptor60 were either little influenced by IRF1 (Cxcl10 and Cxcl11) or were negatively regulated by IRF1 (Cxcl9) (Figures 6C and 6D). The expression of the death receptor, Fas, was negatively regulated by IRF1.61 Thus, Irf1−/− tumor cells exhibit selectively reduced expression of immunosuppressive ISGs with limited effects on key ISGs that can affect immune cell recruitment or effector pathways.
We next used a library of gene expression data of 43 human melanoma cell lines stimulated with IFN-γ to determine whether IRF1 affects gene expression similarly in human cancer. Many human orthologs of mouse ISGs, including IRF1, were expressed (n = 44, maxRPKM ~ 1) and induced (n = 42) by IFN-γ in most human melanoma cell lines (Figure 6E).62 We observed potent induction of 120 genes by IFN-γ in a representative human-melanoma-derived cell line, M238 (Figure 6F). Importantly, CRISPR deletion of Irf1−/− in the M238 line showed diminished expression of a similar small subset of ISGs, including Pdl1 (Figure 6G). These findings demonstrate that IRF1 regulates a subset of ISGs in mouse and human tumor cells upon stimulation with IFN.
Unique and co-operative binding of IRF-1 and STAT1 at promoters determines regulation of a distinct set of ISGs
Given the selective regulation of a subset of genes, we hypothesized that IRF1 may be uniquely required for the expression of these genes through selective engagement at their promoters and the activation of transcription. Since STAT1 and IRF1 can bind IFN-stimulated response elements (ISREs and GAS elements) and unique IRF1 binding motifs,63 we performed chromatin immunoprecipitation sequencing (ChIP-seq) for STAT1 and IRF1 transcription factors with untreated and IFN-γ-treated WT and Irf1−/− MC38 cells (Figures 7 and S7A). Genome-wide binding of both IRF1 and STAT1 increased strongly upon IFN-γ treatment of WT MC38 cells (Figure 7A). In Irf1−/− cells, most STAT1 binding sites were retained (Figure 7A, lower left panel). Although co-occupancy of genomic regions by IRF1 and STAT1 was frequently observed, most binding sites for each protein were unique (Figure 7B).
Figure 7. ChIP-seq highlights distinct and co-operative binding of IRF-1 and STAT-1 at promoters to regulate distinct set of ISGs.

(A) Bar graph represents number of genome-wide (left panels) and promoter (promoter = ±1 kb of transcription start site [TSS], right panels) ChIP-seq peaks for IRF1 (top graphs) and STAT1 (bottom graphs) in MC38 cells.
(B) Venn diagram showing overlap of IRF1 and STAT1 binding in IFN-γ-stimulated cells at genome-wide (left) and promoter regions (right).
(C) Scatterplot shows the distribution of IRF1 (Y axis ChIP-seq peak score) and STAT1 (X axis ChIP-seq peak score) promoter binding strength at IRF1-dependent (Pdl1, Trail, and Tmem140) and -independent (Cxcl9, Cxcl11, and Fas) genes.
(D) Scatterplot showing dependence of gene expression (Y axis) and STAT1 binding (X axis) on IRF1 for a subgroup of ISGs that showed promoter STAT1 binding in IFN-γ-stimulated WT MC38 cells highlighting the dependence of STAT1 binding on IRF1 in the expression of IRF1-dependent (Pdl1, Tmem140, etc.) and -independent (Cxcl9, Cxcl11, Fas, etc.) genes.
(E) Cartoon depicting context of PD-L1 in T cell function.
(F) Dot plot showing surface expression of PD-L1 on cancer cells from WT (n = 8) and Irf1−/− (n = 8) tumors on day 12 (red horizontal bar = mean).
(G) Dot plot showing weight of WT and Irf1−/− MC38 tumors in NOD/Scid, WT, or Pd1−/− mice.
(H) Top panel shows the expression of PD-L1 at baseline (control) and post-IFN-γ stimulation. Middle and bottom panels show stable overexpression of PD-L1 in the WT and Irf1−/− MC38 cells.
(I) Boxplot showing weight of tumors on day 23 from WT hosts injected with GFP or PD-L1 overexpressing WT and Irf1−/− MC38 cells. For (F), (G), and (I) plots, *p ≤ 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001 by Mann-Whitney U/Wilcoxon rank-sum test.
Pdl1, Tmem140, Cxcl11, and Fas were among the small number of genes whose promoters were bound by both IRF1 and STAT1 in WT cells, with Tnfsf10 serving as an example of an IRF1-dependent gene whose promoter was bound only by IRF1 (Figures 7C, S7C, and S7D). Importantly, IRF1 deficiency led to the loss of STAT1 binding at the promoters of the IRF1-dependent Pdl1 and Tmem140 genes (Figure 7D), suggesting that IRF1 and STAT1 collaborate to activate these genes in WT cells. In contrast, IRF1 deficiency led to moderately increased STAT1 binding at the promoters of the Fas, Cxcl11, and Cxcl9 genes, which are expressed at moderately increased levels in the absence of IRF1. These data suggest that IRF1 is a direct activator of several key IRF1-dependent genes, including Pdl1, Tmem140, and Tnfsf10, working in collaboration with STAT1 in some but not all instances. However, IRF1 may serve to limit STAT1-mediated activation of other genes, such as Fas, Cxcl11, and Cxcl9. These findings may help explain IRF1’s biological role in positively or negatively regulating key genes that suppress (Pdl1, Trail, Ido1, and Nos2) or promote (Cxcl9, Cxcl10, Cxcl11, and Fas) antitumor immunity, respectively.
Immune checkpoint PD-1/PD-L1 interaction is only partially required for the tumor-cell-intrinsic role of IRF1
Finally, as PD-L1 is a key immune checkpoint ligand that is selectively regulated by IRF1, we assessed whether IRF1 regulation of PD-1/PD-L1 signaling can explain the tumor-cell-intrinsic effects of IRF1 on tumor growth (Figure 7E). Unlike what was observed in tumor cells stimulated with IFN-γ in vitro, cell surface (Figure 7F) and mRNA (Figure 3C) expression of PD-L1 was only minimally impaired in Irf1−/− MC38 tumor cells compared to WT tumor cells by day 12 after mouse implantation. This finding suggests that compensatory pathways may lead to PD-L1 expression in growing tumors in vivo in the context of a robust host intratumoral immune response64 and that the critical role of IRF1 in regulating PD-L1 transcription observed early after IFN-γ stimulation in vitro may not entirely represent the mechanism by which tumor cell IRF1 expression regulates antitumor immunity in a complex tumor microenvironment in vivo.
To better understand the relevance of PD-L1 for our findings, we examined the impact of IRF1 deficiency in tumor cells implanted into Pdcd1−/− mice. WT MC38 tumors were significantly smaller in Pdcd1−/− mice (Figure 7G), consistent with an important role for the PD1-PDL1 interaction in limiting antitumor immunity. However, WT tumors grew larger in Pdcd1−/− mice than Irf1−/− tumors grown in WT mice. Moreover, Irf1−/− tumors were strongly controlled in Pdcd1−/− mice (Figure 7G). Given that Irf1−/− tumors retained enhanced immunogenicity when PD-L1-PD-1 signaling was eliminated, additional mechanisms other than IRF1’s regulation of PD-L1 expression may be involved in regulating antitumor immunity.
We next examined the extent to which stable overexpression of PD-L1 in Irf1−/− MC38 cells could allow enhanced tumor growth. For these experiments, a PD-L1 lentiviral expression plasmid was transduced into WT and Irf1−/− MC38 cells, resulting in PD-L1 expression levels comparable to those observed in IFN-γ-stimulated WT cells (Figure 7H). Consistent with prior experiments performed with IRF1-deficient B16F10 tumor cells,31 PD-L1 overexpression enhanced tumor growth (Figures 7I and S7E). However, these tumors were much smaller than those observed in WT MC38 cells with or without PD-L1 overexpression (Figure 7I), adding further support to a model in which both PD-L1-dependent and PD-L1-independent pathways are responsible for IRF1’s critical role in suppressing antitumor immunity.
DISCUSSION
While IRF1 promotes cytotoxic T cell and NK cell function, tumor-cell-intrinsic expression of IRF1 can contribute to T cell exhaustion by regulating the key checkpoint gene, PD-L1.31 Our study provides additional insights into these opposing roles of IRF1 in the tumor cells and TME. The presence of IRF1 in tumor cells blocks a powerful IFN-I and antitumor immune response capable of triggering effector lymphocyte-mediated tumor regression. Host TLR and IFN-I signaling, but not IFN-γ or IRF3, are required for optimal activation of antitumor immunity to WT and Irf1−/− tumors. In contrast, IRF1 expression in host cells is a critical driver of antitumor immunity, regardless of expression status of tumor cell IRF1, largely by regulating tumor-infiltrating lymphocytes.
The expression of IRF1 in tumor cells impairs IFN-dependent and effector programs in multiple immune cell types of the TME including DCs, effector memory T cells, and exhausted T cells. Mechanistically, loss of IFN-I signaling prevents adequate effector lymphocyte activation within the TME, including CD8+ T and NK cell subsets. While TLR signaling and IFN-I signaling participate in immunity to WT tumors, these signals are critical drivers of enhanced tumor regression observed in Irf1−/− tumors, promoting an outsized impact of these pathways in Irf1−/− versus WT tumors. Surprisingly, enhanced Irf1−/− tumor regression does not require host STING or other IRF3-activating pathways, pathways required for ICB responses in tumors that ordinarily express IRF1.13,14,65,66 Indeed, signaling through the NF-κB-IRF1 pathway in CD8α+/cDC1 was critical for activation of T cell-dependent antitumor immunity.6,29 Our results suggest that in Irf1−/− tumors, this NF-κB-IRF1 pathway in DCs activated directly by or in conjunction with TLR signaling may induce IFN-I that promotes lymphocyte-mediated tumor regression.
Unlike IFN-I, host IFN-γ signaling was not required for eradication of Irf1−/− tumors. This suggests the activation of TLR-mediated NF-κB and IFN-I was sufficient to generate, recruit, and activate T cells and NK cells with sufficient capacity to kill tumor cells. These findings differ from the findings of others demonstrating an important role of IFN-γ, such as in carcinogen-induced tumors and in the setting of ICB in advanced tumors.4,21,22,67,68 The increased tumor burden in those models may partially explain the differences observed on the requirement of IFN-γ. In our Irf1−/− tumor model, tumor cells lack Irf1 at the time of implantation, and tumor cells do not have time to develop mechanisms to evade immunity. However, in advanced tumors with large tumor burden, multiple immunosuppressive compensatory mechanisms exist, thus making host IFN-γ production necessary to overcome these immune evasion mechanisms.
Tumor-intrinsic expression of IRF1 was previously suggested to suppress CD8+ T cell-mediated antitumor immunity in WT hosts by regulating PD-L1, a key immunosuppressive ISG.31 In our studies, the effects of tumor-intrinsic IRF1 expression on suppressing host immunity only partially depended upon PD-L1, suggesting that other IRF1-dependent factors contribute to its ability to suppress antitumor immunity. Indeed, using a tumor cell culture model stimulated with IFN-γ, we find that due to differential requirements for IRF1 and STAT transcription factors at promoters, IRF1 controls several genes that may directly impact how tumor cells interact with immune cells. These genes include MHC I, genes associated with MHC I antigen presentation, and a subset of IFN-inducible genes shown to contribute to immune suppression, including Pdl1, Nos2, Trail, and Ido1. Expressions of key pro-immunogenic chemokines (Cxcl9, Cxcl10, and Cxcl11) and death receptors (Fas) were maintained upon IFN stimulation of Irf1−/− cancer cells. Reduced expression of IDO-1 in Irf1−/− cancer cells may impair IDO-1-mediated depletion of tryptophan in tumor cells that is shown to directly impair antitumor immunity.69,70 Similarly, reduced expression of NOS2 in Irf1−/− cancer cells can allow CD8+ T cell penetration into tumor epithelium.71 Maintained and elevated expression of Cxcl9–11 and Fas, respectively, in Irf1−/− cancer cells can allow effective recruitment and antitumor potency of antitumor T cells.60,61 Future studies are needed to determine whether these potential mechanisms identified using Irf1−/− tumor cells cultured in vitro with IFN-γ, including transcriptional regulation of Pdl1, contribute to the mechanisms by which tumor-cell-intrinsic IRF1 expression regulates antitumor immunity in vivo in growing tumors.
Finally, our studies support a key role of IRF1 in immune cells of the TME in cancer. The expression of IRF1 was identified in gene signatures of patients’ tumors with responses to immunotherapy and is correlated positively with improved survival in TCGA datasets.29,32,72 Importantly, a CD8 T cell-specific deletion of IRF2, a negative regulator of IRF1, was shown to limit T cell exhaustion and improve ICB responses in mice.30 We find that IRF1 in the host is required to control WT or Irf1−/− tumors, likely because of the function of IRF1 in regulating the development and function of immune effector cells including CD8+ T, NK, and cDC1 cells.73,74 Thus, our data suggest that decreasing IRF1 expression in tumor cells while maintaining IRF1 expression in the TME may represent a viable strategy to enhance antitumor immunity and improve ICB.
Limitations of the study
In this study, we highlight how IRF1 expression in tumor cells directly suppresses a key immune response pathway whereby TLR- and IFN-I-dependent activation of cytotoxic T cells and NK cells induces tumor cell regression, while IRF1 in the TME is required for optimal recruitment of effector lymphocytes to tumor. While we demonstrate that PD-L1 is partially responsible for the immunosuppressive role of tumor-intrinsic IRF1, further work is needed to identify mechanisms by which tumor cell IRF1 impairs antitumor immunity. Our in vitro model identified several possible targets downstream of IRF1 that could be targeted to improve immunotherapy. Finally, models of inducible deletion of IRF1 in tumor cells and cell-specific deletion of IRF1 in host immune cells could further refine the role of IRF1 in tumor-host interactions and feasibility of therapeutic targeting of IRF1 in immunotherapy settings.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for all reagents and resources should be directed to and will be fulfilled by the lead contact, Philip O. Scumpia (pscumpia@mednet.ucla.edu).
Materials availability
All unique reagents (e.g., recombinant DNA and knockout cell lines) generated in this study are available by contacting the lead contact.
Data and code availability
All RNA-seq, ChIP-seq and single-cell RNA-seq data have been deposited at GEO and are publicly available on 1st May 2024 or on the date of publication. Accession numbers are listed in the key resources table. Microscopy data reported in this paper will be shared by the lead contact upon request.
This paper does not report new or original codes. Code that has been used for analysis is publicly available, referred to and listed in the references section.
Any additional information needed to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| Anti-IRF1 | Cell Signaling | Cat# 8478S; RRID:AB_10949108 |
| Anti-STAT1 (for mouse and human) | Cell Signaling | Cat# 9172; RRID:AB_2198300 |
| p-STAT1 (Tyr701) (for human) | Cell Signaling | Cat# 9167; RRID:AB_561284 |
| Rabbit a-CD4 (For immunofluorescence) | Abcam | cat# AB183685; RRID:AB_2686917 |
| Rat a-CD8 (For Immunofluorescence) | eBioscience | Cat# 14–0808; RRID:AB_2572860 |
| CD45_PerCPcy5.5 | Biolegend | Cat# 103236; RRID:AB_893354 |
| CD3_APC | eBioScience | Cat# 17–0031-82; RRID:AB_469315 |
| CD4_APC-PE780 | eBioScience | Cat# 47004280; RRID:AB_1272219 |
| CD8_FITC | eBioScience | Cat# 11–0081-81; RRID:AB_464914 |
| NK1.1_PE | eBioScience | Ca# 12–5941-82; RRID:AB_466050 |
| PD1_BV421 | Biolegend | Ca# 135217; RRID:AB_2562568 |
| Cd45_APC | Biolegend | Ca# 109813; RRID:AB_389210 |
| Cd11b_APC-Alexafluor 750 (APC-Cy7) | eBioscience | Cat# 27–0112-81; RRID:AB_469343 |
| Cd11c_PE | Biolegend | Cat# 117308; RRID:AB_313776 |
| Ccr2_BV785 | Biolegend | Cat# 150621; RRID:AB_2721565 |
| Ly6C_PE/Dazzle594 | Biolegend | Cat# 128043; RRID:AB_2566576 |
| Ly6G_FITC | Biolegend | Cat# 127606; RRID:AB_1236488 |
| Live/Dead Fixable Viable Stain 510 | BD Horizon | Cat# BDB564406; RRID:AB_2869572 |
| anti-PD-L1-PE | Biolegend | Cat# 124308; RRID:AB_2073557 |
| anti-MHC-I_AlexaFluor-647 | Biolegend | Cat# 114612; RRID:AB_492931 |
| InVivoMAb anti-mouse PD-1 (CD279) | BioXcell | Cat# BE0146; RRID:AB_10949053 |
| InVivoMAb anti-mouse CTLA-4 (CD152), clone 9H10 | BioXcell | Cat# BE0131; RRID:AB_10950184 |
| InVivoMAb anti-mouse IFNAR-1, clone MAR1–5A3 | BioXcell | Cat# BE0241; RRID:AB_2687723 |
| InVivoMAb rat IgG2a isotype control | BioXcell | Cat# BE0089; RRID: AB 1107769 |
| inVivoMAb anti-mouse IFNγR (CD119), clone GR-20 | BioXcell | Cat# BE0029; RRID:AB 1107576 |
|
| ||
| Chemicals, Peptides, and Recombinant Proteins | ||
|
| ||
| murine IFN-y | Peprotech | Cat# 315–05 |
| Human IFN-y | Invivogen | Cat# rcyec-hifng |
| Collagenase-D | Roche | Cat# 11088866001 |
| DNase I | Roche | Cat# 10104159001 |
| SpCas9 2NLS Nuclease | Synthego | Cat# SpCas9 2NLS Nuclease |
| DSG Crosslinker | Covachem | Cat# 13301–1 |
| Thermo Scientific™ Pierce™ Formaldehyde Ampules, Methanol-free | Thermo Fisher | Cat# 52622 |
| Pierce Protease Inhibitor Mini Tablets, EDTA-Free | Thermo Fisher | Cat# A32955 |
| Linear Acrylamide (5 mg/mL) | Thermo Fisher | Cat# AM9520 |
| Proteinase K, recombinant, PCR grade | Thermo Fisher | Cat# EO0492 |
| Linear Acrylamide | Invitrogen | Cat# AM9520 |
| RNaseA | Thermo Fisher | Cat# EN0531 |
|
| ||
| Critical Commercial Assays | ||
|
| ||
| TruSeq Stranded mRNA Sample Prep Kit B | Illumina | Cat# RS-122–2102 |
| Kapa Hyper Prep Kit Ilumina Platform | Kapa Biosystems | Cat# KK8502 |
| NEXTflex™ ChIP-Seq Barcodes - 12 | BIOO Scientific | Cat# 514121 |
| AMPure XP purification beads for DNA purification | Beckman Coulter | Cat# A63881 |
| RNeasy Mini Kit (250 rxn) | Qiagen | Cat# 74106 |
| DNeasy Mini Kit | Qiagen | Cat# 69506 |
| SuperScript™ III Reverse Transcriptase | Invitrogen | Cat# 18080044 |
| Dynabeads Protein G | Invitrogen | Cat# 1004D |
| Tumor Dissociation Kit | Miltenyi Biotec | Cat# 130–096730 |
| Dead Cell Removal Kit | Miltenyi Biotec | Cat# 130–090-101 |
| gentleMACS™ C Tubes | Miltenyi Biotec | Cat# 130–093-237 |
| MS Columns | Miltenyi Biotec | Cat# 130–042-201 |
| CellTiter 96® AQueous One Solution Cell Proliferation Assay (MTS) |
Promega | Cat# G3580 |
| BCA assay | Thermo Fisher Scientific | Cat# 23227 |
| Supersignal West Pico chemiluminescence kit | Thermo Fisher Scientific | Cat# 34080 |
| Neon™ Transfection System 10 μL Kit | Thermo Fisher | Cat# MPK1096 |
|
| ||
| Deposited Data | ||
|
| ||
| All raw and analyzed bulk mRNA-seq, sc-RNA-seq and ChIP-seq datasets | This Paper | GEO Accession GSE246923 |
|
| ||
| Experimental Models: Cell Lines | ||
|
| ||
| MC38, mouse colon adenocarcinoma | C57BL/6 | Kerafast #ENH204-FP, RRID:CVCL_B288 |
| YUMM2.1 | C57BL/6 | Moreno et al.75 |
| B16-F10 mouse melanoma | C57BL/6 | ATCC #CRL-6475, RRID:CVCL_0159 |
| FSA (Fibrosarcoma) | C3H strain derived McBride, UCLA | Suit et al.76 |
| M238 (human Melanoma cell line) | Human melanoma derived, A. Ribas lab | Grasso et al.62 |
| HEK293T Cells | ATCC | CRL-11268, RRID:CVCL_1926 |
|
| ||
| Experimental Models: Organisms/Strains | ||
|
| ||
| C57BL/6 | Jackson Laboratory | Cat# 000664, RRID:IMSR_JAX:000664 |
| StingGt/Gt | Jackson Laboratory | Cat# 017537, RRID:IMSR_JAX:017537 |
| Ifnar −/− | G. Cheng, UCLA | Müller et al.77 |
| NOD Scid | Jackson Laboratory | Cat# 001303, RRID:IMSR_JAX:001303 |
| Rag2−/− γC−/− | Jackson Laboratory | Cat# 014593, RRID:IMSR_JAX:014593 |
| Rag1 −/− | Jackson Laboratory | Cat# 002216, RRID:IMSR_JAX:002216 |
| Cd8 −/− | Jackson Laboratory | Cat# 002665, RRID:IMSR_JAX:002665 |
| Irf1 −/− | Jackson Laboratory | Cat# 002762, RRID:IMSR_JAX:002762 |
| Ifngr −/− | Jackson Laboratory | Cat# 003288, RRID:IMSR_JAX:003288 |
| Irf3 −/− | G. Cheng, UCLA | Sato et al.78 |
| Myd88 −/− | G. Cheng, UCLA | Cat# 009088, RRID:IMSR_JAX:009088 |
| Trif −/− | Hoebe et al., 2003 | JAX stock #005037, RRID:IMSR_JAX:005037 |
| Myd88 Trif DKO | Geg Barton, UC Berkley | Yamamoto et al.79 |
|
| ||
| Oligonucleotides | ||
|
| ||
| mPD-L1_gateway cloning_forward (5′−3′) GGGGACAAGTTTGTACAAAAAAGCAGGCT tcgccaccATGAGGATATTTGCTGGCATTAT |
This paper | NA |
| mPD-L1_gateway cloning_reverse (5′−3′) GGGGACCACTTTGTACAAGAAAGCTGGGT cTCACGTCTCCTCGAATTGTG |
This paper | NA |
| mIRF1 guide sequence g1_Forward for pX459 CACCGATGCCTGTCTGTTCCGGAGC |
This paper | NA |
| mIRF1 guide sequence g1_Reverse for pX459 AAACGCTCCGGAACAGACAGGCATC |
This paper | NA |
| mIRF1 guide sequence g2_Forward for pX459 CACCGGCTCTGCTGTGCGGGTGTAC |
This paper | NA |
| mIRF1 guide sequence g2_Reverse for pX459 AAACGTACACCCGCACAGCAGAGCC |
This paper | NA |
| mSTING guide sequence g1_Forward for pX459 CACCGCCCCACAGTCCAATGGAAAG |
This paper | NA |
| mSTING guide sequence g1_Reverse for pX459 AAACCTTTCCATTGGACTGTGGGGC |
This paper | NA |
| mSTING guide sequence g2_Forward for pX459 CACCGTCAGGCACCCCACAGTCCAA |
This paper | NA |
| mSTING guide sequence g2_Reverse for pX459 AAACTTGGACTGTGGGGTGCCTGAC |
This paper | NA |
| hIRF1 guide sequence g1 for RNP (Synthego) CUCUAGGCCGAUACAAAGCA |
This paper | NA |
| hIRF1 guide sequence g2 for RNP (Synthego) GCCAGAUCCCAAGACGUGGA |
This paper | NA |
|
| ||
| Recombinant DNA | ||
|
| ||
| pSpCas9(BB)-2A-puro(pX459)-V2.0 plasmid | Addgene #62988 | Cong and Zhang80 |
| pX459V2_Irf1-gd1 | This paper | N/A |
| pX459V2_Irf1-gd2 | This paper | N/A |
| pX459V2_Sting-gd1 | This paper | N/A |
| pX459V2_Sting-gd2 | This paper | N/A |
| pLenti_puro_Pdl1 | This paper | N/A |
|
| ||
| Software and Algorithms | ||
|
| ||
| Seurat 4.1 | https://github.com/satijalab/seurat | Butler et al.81 |
| FlowJo | FlowJo | Version 10.8.1 |
| Cell Ranger v6.0.2 | 10X Genomics | Version 6.0.2 https://www.10xgenomics.com/support/software/cell-ranger |
| Seurat 4.1 | https://github.com/satijalab/seurat | Butler et al.81 |
| R/Rstudio | CRAN | Version 4.3.1 |
| GGplot2 | CRAN | Version 3.4.3 https://ggplot2.tidyverse.org |
| DoubletFinder | https://github.com/chris-mcginnis-ucsf/DoubletFinder | McGinnis et al.82 |
| cowplot | https://wilkelab.org/cowplot/ | Version 1.1.1 |
| ProjecTILs | https://github.com/carmonalab/ProjecTILs | Andreatta et al.46 |
| clusterProfiler | https://bioconductor.org/packages/release/bioc/html/clusterProfiler.html | Wu et al.83 PMID: 34557778 |
| Interferome database | http://www.interferome.org | Rusinova et al.84 |
| EnhancedVolcano v1.20.0 package in Bioconductor | https://bioconductor.org/packages/EnhancedVolcano | Blighe et al.85 |
| Hisat2 | http://daehwankimlab.github.io/hisat2/ | Kim et al.86 |
| DEseq2 | https://bioconductor.org/packages/release/bioc/html/DESeq2.html | Anders and Huber87 |
| BEDTools | https://github.com/arq5x/bedtools2 | Quinlan et al.88 |
| Jasper TF motif database | https://jaspar.genereg.net/ | Castro-Mondragon et al.89 |
| Homer | http://homer.ucsd.edu/homer/ | Heinz et al.90 |
| Pscan | http://159.149.160.88/pscan/ | Zambelli et al.91 |
| Jvenn | http://jvenn.toulouse.inra.fr/app/index.html | Bardou et al.92 |
| R-Studio, | https://www.rstudio.com | RStudio Team (2015). RStudio: Integrated Development for R. RStudio, Inc., Boston, MA |
| Bioconductor DE-seq | http://bioconductor.org/packages/release/bioc/html/DESeq.html | Anders and Huber87 |
| MS Office 2010 and 365 | https://www.microsoft.com/ | Microsoft Office |
| Samtools | https://github.com/samtools/samtools | Li et al.93 |
| SeqMonk | https://www.bioinformatics.babraham.ac.uk/projects/seqmonk/ | Simon Andrews (Head of Bioinformatics) Email: simon.andrews@babraham.ac.uk |
EXPERIMENTAL MODEL AND STUDY PARTICIPANT DETAILS
Mice
All animal experiments were performed according to the protocols approved by UCLA Animal Research Committee (ARC) and Institutional Animal Care and Use Committee (IACUS). WT C57BL/6, StingGt/Gt, NOD Scid, Irf1−/−, Rag2−/− gC−/−, Rag1−/−, Cd8−/−, Ifngr−/−, Pdcd1−/−, Irf3−/− mice were obtained from Jackson Laboratory. Irf3−/− and Ifnar−/− mice were obtained from G. Cheng (UCLA), and Myd88−/− Trif−/− mice from G. Barton (UC Berkley). All the mutant strains used in this study were backcrossed except Rag2−/− γC−/−, and NOD Scid to a C57BL/6 background as stated by Jackson inventory. Both male and female mice, age between 8 and 16 weeks were used in the experiments. Gender was matched and age difference among mice were not more than 1 week when compared multiple mutants. Catalog numbers and specific source of the mice are provided in the key resources table. Mice were bred and kept under defined-flora pathogen-free conditions at the AALAC-approved animal facility of the Division of Experimental Radiation Oncology, UCLA. These rooms were supplied with autoclaved cages and irradiated feed. The mice were continuously monitored by the UCLA Division of Laboratory Animal Medicine.
Cell lines
Mouse cancer (MC38, YUMM2.1, B16-F10) and human melanoma (M238) cell lines were obtained from A. Ribas lab, UCLA. YUMM2.1, B16-F10 and MC38 cell lines were cultured in Dulbecco’s modified MEM (DMEM) media (Corning, NY) supplemented with 10% fetal bovine serum (Omega, CA) and 1X Penicillin/streptomycin (Gibco, #15140–122). MC38 cell line was maintained in DMEM with 10% fetal bovine serum, 2mM L-glutamine (Gibco, #25030–081), 0.1 mM non-essential amino acids (Gibco, #11140–050), 1 mM sodium pyruvate (Gibco, #11360–070), 10 mM HEPES (Gibco, #15630–080), and 1X pen/strep (Gibco, #15140–122).
METHOD DETAILS
Generation of Knock-out Tumor Cell lines using CRISPR/Cas9
Gene targeting of IRF1 and STING were carried out using CRISPR/Cas9 method as described by Zhang lab.1 Briefly, appropriate single guide RNAs (sgRNA) sequences (listed in the key resources table) for murine Irf1 and Tmem173 (Sting) were designed using Benchling tools (https://benchling.com). Briefly, the designed single guide RNAs (sgRNA) were cloned into pX459-V2.0 plasmid (Addgene Plasmid #62988). The confirmed guide RNA containing plasmids were used to transfect MC38, YUMM2.1, and B16-F10 cell lines using lipofectamine 3000 (Invitrogen). The transfected cells were transiently selected (48h) with 4ug/ml puromycin in the cell culture media. After brief selection, single cells clones were expanded in 96 well plates. The positive clones were confirmed by sanger sequencing followed by sequence analysis using manual curation, Synthego, and CRISPOR online tools. Expression of proteins was confirmed by immunoblot using specific antibodies against IRF1 (Cell Signaling #8478S) and STING (Cell Signaling #13647). Human IRF1 was mutated in M238 melanoma cell line by its electroporation (Neon, Invitrogen) with Cas9/Irf1 targeting guide RNA complex (Synthego).
In vitro proliferation and radiosensitivity
Three thousand cells of indicated Wt and mutant clones were seeded in 4 replicates in 4 independent micro plates on day 1 and MTS assay was performed on day2 (24h), day3 (48h), and day4 (72h) as per manufacturer instruction (Promega CellTiter 96 Aqueous, #G3582). To monitor radiosensitivity of clones, three thousand cells of indicated clones were seeded in 4 replicates in 4 independent micro plates on day 1. Three Plates were irradiated with 10GY of X-ray the next day and MTS was performed every 24h.
Clonogenic assay
Cells were irradiated at a dose rate of 1.705 Gy/min using an x-irradiator (Gulmay, 300 kV, HVL = 3mm Cu, 10 mA). Following irradiation (2 Gy, 4 Gy and 6Gy), a known numbers of cells were plated in triplicate in 100 mm tissue culture plates and incubated for 11 days in a humidified 5% CO2 atmosphere at 37C. The cells were fixed and stained using 0.5% crystal violet in a 70% ethanol solution. Colonies were enumerated; only colonies with at least 64 cells were counted. The experiments were repeated at least ten times.
In vivo mouse model
Tumor cells (0.5 × 106 or 1 × 106) were injected s.c. into the flanks of mice as indicated. Tumors were allowed to grow for about 2–3 weeks or longer depending on the experimental setup. Tumor volumes were measured along two axes (Length and width) and calculated as tumor volume = (width2 x length) x0.52, where width represents the shortest dimension and length represents the longest dimension.2 At the termination of the experiments, mice were humanely sacrificed, and tumors were excised, weight were also reported in most of the experiments. All WT and mutant mice were housed and bred in the same facility to maintain the microflora of the animals. Age and gender matched WT mice were used whenever mutant mice were tested for tumor growth.
Histologic evaluation of tumors
Tumors were fixed in 10% neutral buffered formalin (Fisher Scientific) for 24 h at room temperature followed by dehydration and embedding in paraffin. Tissue sections of 5uM were cut and mounted on slides for staining procedures. Hematoxylin and eosin (H&E) staining was performed according to standard procedures by the Translational Pathology Core Laboratory (TPCL) at UCLA and histologic evaluation was performed by a dermatopathologist (P.O. Scumpia) blinded to the identity of the samples.
Multiplex immunofluorescence
Formalin fixed tumors were sectioned and deparaffinized at 60°C followed by 3 times xylene washes (10 min each), then rehydrated in a gradient ethanol series for multiplex immunofluorescence (mIF). Antigen retrieval was performed with AR6 buffer (Akoya Biosciences) 97°C for 20 min. Endogenous peroxidases were blocked with 2.5% hydrogen peroxide in methanol for 30 min. The staining was performed with the OPAL-7 color manual kit following the manufacturer’s instructions (Akoya Biosciences) by trained technician at UCLA’s Translational Pathology Core Laboratory (TPCL). Briefly, A hydrophobic barrier pen was used to completely surround the tissue section on the slide and non-specific antigens were blocked by covering the tissue section with blocking buffer (PerkinElmer antibody diluent buffer) to cover the tissue section and incubation in a humidified chamber at room temperature for 10 min. Tissue sections were covered with primary antibodies at 1:200 dilution against CD8 (eBioscience, Rabbit, Cat #14–0808); and CD4 (Abcam, Rat, Cat # AB183685); and then were counterstained with the nuclear stain 4′,6-diamidino-2-phenylindole (DAPI) at room temperature. Sections were incubated sequentially with secondary antibodies at room temperature for 15 min, followed by tyramide-based labeling for 10 min, and the process repeated for each antibody. The slides were mounted with ProLong Gold Antifade (Invitrogen). The mIF images were acquired using a Vectra Polaris microscope (Akoya Bioscience) at 20× magnification.
In vitro stimulation of cells
WT and IRF1 mutant cells were treated with 100 units/ml of IFN-γ (mouse IFN-γ cat # 315–05 from Peprotec, human IFN-γ cat # rcyec-hifng, from Invivogen) for various time points before harvesting for RNA or protein isolation.
RNA-seq
Total RNA from cultured tumor cell lines was prepared using RNAeasy kit (QIAGEN). Strand-specific libraries were generated using 400 ng of total RNA using the Illumina TruSeq stranded RNA Sample Preparation Kit as described previously.3 cDNA libraries were single-end sequenced (50bp) on Illumina HiSeq 3000 machine.
ChIP-seq
ChIP-seq was performed as previously described3 with anti-IRF1 antibody (Cell Signaling, 8478S), anti-STAT1 antibody (Cell Signaling, 9172). Briefly, about 10 million cells from indicated conditions were used for cross-linking with 1mM DSG for 45 min and 1% formaldehyde for 10 min. Chromatin shearing was performed in a Covaris M220 Focused/Ultrasonicator with 15% duty factor, 300 cycles for 20 min to obtain chromatin fragment size range between 200 and 600 bp. Chromatin lysate were pre-cleared by incubating for 1h with magnetic protein G beads (Dynabeads protein G, Thermo Fisher #1004D) on a nutator followed by overnight incubation at 4°C with either anti-IRF1 or anti-STAT1 antibodies. Antibody bound chromatin were pooled using magnetic protein G beads by incubating for 4h in a nutator at 4°C. After multiple wash, antibodies/bead bound chromatin was de-crosslinked and digested with Proteinase K and RNase A for overnight at 60°C in a thermomixer. The digested DNA was purified using phenol chloroform and precipitated with sodium acetate, linear acrylamide (Invitrogen #AM9520) and absolute alcohol. 1ng of purified DNA was used for ChIP-seq libraries preparation using KAPA HyperPrep Kits (Roche) and barcode indices from NextFlex (PerkinElmer) as per manufacturer instructions. ChIP-seq libraries were sequenced in Illumina Hiseq 3000 machine at UCLA’s TCGB core.
PD-L1 over-expression in IRF1 mutant MC38 Cell
The coding region of the murine PD-L1 (PD-L1) gene was PCR amplified from cDNA of bone marrow derived macrophages’ RNA (C57BL/6J) using Pfu-phusion (NEB #E0553L), and mPD-L1 forward and reverse gateway cloning primers (listed in the key resources table). The procedure to clone using Gateway Technology (Invitrogen/Life Technology) is detailed in the manufacturer’s manual (Document Part Number 250522, Life technology). Briefly, the PCR amplified mPD-L1 was first clones into pDONR 221 plasmid using BP clonase enzyme mix (Invitrogen # 11789020). Sanger Sequencing confirmed clone was used for transfer the gene into pLenti-DEST plasmid (A lenti-viral plasmid, Addgene Plasmid #17452) using LR clonase enzyme mix (Life technology, # 11791020). Virus were produced in 293T max cell line by transfecting the PD-L1 construct and helper plasmids (VSVG, pMDL, pRSV-REV). Wild-type and IRF1 mutant MC38 cells were transduced with lentivirus, followed by selection with puromycin (5ug/ml) for 5 days. Cells were maintained in puromycin free media for about 4 days and the surface PD-L1 over expression was confirmed to be high and comparable in wild-type and Irf1−/− cells by flow cytometry using PE conjugated anti-mPD-L1 antibody (Biolegend, #124307-BL).
Tumor digestion for single Cell suspensions
MC38 and YUMM2.1 tumors were harvested from mice at defined time points. Tumors weight were noted followed by chopping into small pieces (2–4 mm size) and digestion with Miltenyi’s gentle MACS enzyme mix (mouse tumor dissociation kit, # 130–096-730) in a Miltenyi GentleMACS Octo Dissociator (Miltenyi Biotec Inc., San Diego, CA) as per manufacture’s recommended procedure (37C_mTDK_1). Cells were subsequently passed through a smart strainer (70uM) and washed with PBS and counted.
Flow cytometry analysis
Approximately 1 million cells were aliquoted into 2 tubes to stain with 2 panels of antibodies cocktails for 30 min on ice followed by 2 times wash and resuspension in 300 μl FACS buffer (1% FBS in PBS). Panel1 for T and NK cell include CD45_PerCPcy5.5, CD3_APC, CD4_APC-PE780, CD8_FITC, NK1.1_PE, PD1_421. 2nd panel for myeloid cells include Cd45_APC, Cd11b_APC-Alexafluor 750 (APC-Cy7), Cd11c_PE, Ccr2_BV785, Ly6C_PE/Dazzle594, Ly6G_FITC. Details of the antibodies are also provided in Key Resource Table. Live/dead cell discrimination was performed using Live/Dead Fixable Viable Stain 510 (BD Horizon). All cell events were collected on a flow cytometer (BD LSR II) and the total number of events of a given immune cell type were divided by the weight of the tumor.
For analysis of cell lines grown in vitro, sub-confluent cells either untreated or treated for 18 h with 100 ng/mL of IFNG (PeproTech) were harvested and single-cell suspensions were prepared before staining with antibodies anti-PD-L1-PE and anti-MHC-I_Alexa-Fluor-647.
Single Cell RNA-seq
Wt and Irf1−/− MC38 tumors (pooled 4–6) grown in WT mice or WT and Ifnar−/− mice from two independent clones of each type of tumor were harvested on day 12 were chopped into small pieces (2–4 mm size) with a scalpel before dissociation with Miltenyi’s gentle MACS enzyme mix (mouse tumor dissociation kit, # 130–096-730) in a Miltenyi GentleMACS Octo Dissociator (Miltenyi Biotec Inc., San Diego, CA) with pre-set 37C_mTDK_1 protocol. After dissociation, the cell suspension was filtered through 70uM MACS smart strainer and dead cells were removed using dead cell removal kit (Miltenyi Biotec Inc.). Cells were counted using Countess II Automated Cell Counter (Thermo Fisher Scientific) and hemocytometer for cell concentration and viability using Trypan Blue stain 0.4% (Invitrogen). Single cell gene expression libraries were created using Chromium Next GEM Single Cell 3’ (v3.1 Chemistry) (10x Genomics), Chromium Next GEM Chip G Single Cell Kit (10x Genomics), and Single Index Kit T Set A (10x Genomics) according to the manufacturer’s instructions. Briefly, cells were loaded to target 10,000 cells to form GEMs and barcode individual cells. GEMs were then cleaned cDNA and libraries were also created according to manufacturer’s instructions. Library quality was assessed using 4200 TapeStation System and D1000 ScreenTape (both from Agilent) and Qubit 2.0 (Invitrogen) for concentration and size distribution. Samples were sequenced using Novaseq S1 (Illumina) using 100 cycles (28 + 8+91). 200 M reads for each sample with about 50,000 reads per cell were targeted.
QUANTIFICATION AND STATISTICAL ANALYSIS
Tumor volume and weight Group difference analysis
Tumor volumes were determined by caliper measurements. Tumor volumes were measured along two axes (Length and width) and calculated as tumor volume = (width2 x length) x0.52, where width represents the shortest dimension and length represents the longest dimension.2 To quantify the precision of tumor volume means, we calculated the standard error of the mean (SEM) using Microsoft Excel. This involved deriving the sample standard deviation with the STDEV.S function, determining the sample size with the COUNT function, and dividing the standard deviation by the square root of the sample size, using the formula = STDEV.S(range)/SQRT(COUNT(range)). The SEM was presented alongside mean values as error bars. The significance of all two-way comparisons for volumetric and weight difference was determined by a two-sample two-tailed t test, or by a one-tailed t test when appropriate. For non-parametric data, a Mann-Whitney U (also known as Wilcoxon rank-sum) test was used in R package. If data had tied values, approximate instead of exact Wilcoxon test was performed by providing “exact = FALSE”.
RNA-seq analysis
Reads were aligned to the mouse genome (NCBI37/mm9 build) or human genome (GRCh37/hg19) with Hisat2 by allowing reads to be aligned once with up to two mismatches per read. SeqMonk (Babraham Bioinformatics) was used to quantify against the exons of Refseq genes and RPKM values for genes were calculated as described.3 Briefly, RPKM equals the number of reads mapped to exons of a gene divided by the exon length in kb times the total number of mapped reads in the dataset (in millions). All RPKMs used for the analysis represent averages of two to three independent biological replicates. Genes were called expressed if the RPKM reached 2 at least in one sample. Initially, we focused on the potently induced genes indicated by fold induction level 3-fold, and the induced expression level was consistently different from the basal level (p < 0.01), as determined by the DESeq2 package in R Bioconductor.4 DE genes were displayed using EnhancedVolcano plot package in R5. To determine the impact of Irf1−/− loss on the expression of a IFN-y induced gene, the maximum RPKM in the Irf1−/− samples was converted to a percentage after considering the WT maximum RPKM to be 100%.
ChIP-seq analysis
Reads were aligned to the mouse genome (NCBI37/mm9 build) with Hisat2. Uniquely mapped reads were used for peak calling over input as ref. 6. Peaks were called if the normalized signal was enriched more than 4-fold over input with a poison p value lower than 1e-4 and false discovery rate of 0.01. All peaks were annotated to Refseq genes based on the closest TSS using HOMER. Homer (makeUCSCfile command line) was used to generate bedgraph files of uniquely mapped reads for UCSC genome browser visualization. Pie chart (in Microsoft Excel) was used to represent genomic distribution of peaks and Venn diagram was used to represent overlap between IRF1 and STAT1 bound peaks. For comparative analysis of peaks, a master probe was generated with BEDTools7 to quantify reads in the peak regions in SeqMonk (The Babraham institute). Then RPKMs were generated using raw reads from SeqMonk normalized to the size of the peak (in kbps) and the depth of sample sequencing (in million reads). Position weight matrix motif for IRF1 and STAT1 were used from Jasper TF motif database.
Single Cell RNA-sequencing data analysis
Sequencing data were demultiplexed in Basespace and mapped, barcode collapsed, and filtered in the Cell Ranger software (10x Genomics). Reads were mapped to the mm10 Refseq mouse reference transcriptome. The output from Cell Ranger, a raw sparse matrix with digital expression of cell barcodes by genes, was used for downstream analysis. Using Seurat 4.1 in R (https://github.com/satijalab/seurat),8 quality control measures were imposed, where cells with fewer than 200 detected genes or more than 20% mitochondrial reads were excluded. To identify and remove doublets, the cell matrix was processed by DoubletFinder (https://github.com/chris-mcginnis-ucsf/DoubletFinder).9 The filtered dataset underwent normalization and scaling using the SCTransform method within the Seurat package (version 4.1), which stabilizes variance across features. Integration of multiple scRNA-seq datasets was carried out by employing the Harmony algorithm. The RunHarmony function was invoked with dims = 1:50 to perform the integration on the scaled and normalized expression data of the selected 3,000 features. Post-integration, we conducted principal component analysis (PCA) on the harmonized data, followed by the construction of a shared nearest neighbor (SNN) graph with the top 20 principal components. Clusters were identified using the Louvain algorithm with a resolution of 0.5 (optimized after trying several resolution). For visualization, we employed the Harmony embedding technique, which facilitated the creation of a Uniform Manifold Approximation and Projection (UMAP) plot to display the data in a two-dimensional space. Clusters were annotated based on expression of known cell type-specific markers using literature and various databases (PanglaoDB,10 Enrichr,11 human protein atlas12) and differentially expressed genes in distinct clusters. Genes were considered differentially expressed based on Wilcoxon rank-sum test with an adjusted p-value <0.05. Final analysis and plots were mostly created using R, Bioconductor packages and the libraries in ggplot2. Cluster profiler4.0 was used to show gene set enrichment for selected cell subsets. Interferome database13 (http://www.interferome.org) was used to categorize type-I and IFN-y induced gene program in CD8_Effectormemory cluster (Figure S3G). Data shown in all figures related to scRNA-seq experiments were data from one representative WT and one representative Irf1−/− clone as both pairs of clones sequenced generated nearly identical results.
ProjecTILs T cell subtype Classification and Annotation
To understand the difference in T cells’ states between Wt and Irf1−/− tumors, we projected our tumors’ single cell RNA-seq data to T cell reference atlas generated from mouse tumors (mouse TILs atlas) in ProjecTILs R package.14 The TILs atlas was created using mouse tumors’ single cell RNA-seq data. The reference T cell atlas defines 9 T cells states (Figure S3C), including CD8_Tex (a CD8+ terminally-exhausted effector cluster, characterized by high expression of granzymes, multiple inhibitory receptors, such as Pdcd1, Ctla4, Lag3, Tigit, Havcr2/TIM-3, etc. and Tox), CD8_Tpex (CD8+ precursor-exhausted cluster, with co-expression of Tcf7, Pdcd1, Ctla4, Tox but low expression of Havcr2 or granzymes), CD8_NaiveLike (a naive-like CD8+ T cells, which may consist of naive as well as central memory cells),CD4_NaiveLike (a naive-like CD4+ T cells co-expressing Tcf7 and Ccr7 while lacking cytotoxic molecules and activation features such as Pdcd1 and Tnfrsf9/4–1BB), CD8_EffectMemory (effector-memory CD8+ T cells, co-expressing Tcf7 and granzymes “most prominently Gzmk”, with low to intermediate expression of Pdcd1; an “early-activation” state of CD8+ T cells), Th1 (a cluster of CD4+ Th1-like cells, expressing IFN-gamma receptor 1 “Ifngr1” and Fasl), Tfh (a CD4+ follicular-helper population with a pronounced expression level of Cxcr5, Tox, and Slamf6), and Treg (a cluster of regulatory T cells, identified by Foxp3 expression). Briefly, a normalized expression matrix post doublet removal (Seurat logNorm data) was provided as input and non-T cells were filtered using TILPRED-1.0 before alignment and batch-effect correction. Additional parameters for “make.projection” were kept as the default. Cell states were predicted for each cell using a nearest-neighbor algorithm (ProjecTILs “cellstate.predict”) and overlaid onto the original mouse UMAP with TILPRED filtered cells. Projection of our data over the reference in UMAP space was plotted using “plot.projection” function with default setting in ProjectTILs. The contour lines display the density of projected query cells onto the reference map (Figures 3F and 4D). Relative percent distribution of various T cell subtypes/states in terms of reference T cell subtypes was plotted using “plot.statepred.composition” function in with parameter metric = “Percent” in ProjectTILs package in R.
Supplementary Material
Highlights.
IRF1 expression in tumor cells or the TME governs tumor progression or regression
Tumor cell IRF1 blocks host IFNAR and TLR activation of antitumor adaptive immunity
Tumor cell IRF1 controls only an immunosuppressive gene program induced by IFN-γ
IRF1 controls distinct IFN-γ-induced genes by binding promoters alone or with STAT1
ACKNOWLEDGMENTS
We thank R. Lo for critical review of the manuscript, Technology Center for Genomics and Bioinformatics (TCGB) core for the high-throughput sequencing, and Y. Li at UCLA Translational Pathology Core Laboratory for Opal immunofluorescence staining. We thank C. Garcia, A. Marin, T. Yik, J. Ting, and J. Olay for maintaining mouse colonies and genotyping, E. Ganapathy for flow cytometry setup, and T. Marcus for help with lentiviral overexpression system. This work was supported by UCLA’s institutional and Jonsson Comprehensive Cancer Center Seed grants (S.T.S. and P.O.S.), NIH grants K08 AR066545 (P.O.S.), R01 AR079470 (P.O.S.), VA Merit Award I01CX002194 (P.O.S.), the Howard Hughes Medical Institute (S.T.S.), and NIH U19AI067769 (W.H.M).
DECLARATION OF INTERESTS
K.M.C. reports being a shareholder in Geneoscopy and has received consulting fees from Geneoscopy, PACT Pharma, Tango Therapeutics, Flagship Labs 81, and the Rare Cancer Research Foundation. S.M.D. reports service on scientific advisory boards for LungLife AI and Early Diagnostics. A.R. has received honoraria from consulting with Amgen, Bristol Myers Squibb, and Merck; is or has been a member of the scientific advisory board and holds stock in Advaxis, Appia, Apricity, Arcus, Compugen, CytomX, Highlight, ImaginAb, ImmPact, ImmuneSensor, Inspirna, Isoplexis, Kite-Gilead, Lutris, MapKure, Merus, PACT, Pluto, RAPT Therapeutics, Synthekine, and Tango; and has received research funding from Agilent and from Bristol Myers Squibb through SU2C and patent royalties from Arsenal Bio. P.O.S. reports research support from Castle Biosciences, advisory board service for Castle Biosciences, and being a co-founder and shareholder of Tempo Therapeutics.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2024.114289.
REFERENCES
- 1.Ribas A, and Wolchok JD (2018). Cancer immunotherapy using checkpoint blockade. Science 359, 1350–1355. 10.1126/science.aar4060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mittal D, Gubin MM, Schreiber RD, and Smyth MJ (2014). New insights into cancer immunoediting and its three component phases–elimination, equilibrium and escape. Curr. Opin. Immunol 27, 16–25. 10.1016/j.coi.2014.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Koebel CM, Vermi W, Swann JB, Zerafa N, Rodig SJ, Old LJ, Smyth MJ, and Schreiber RD (2007). Adaptive immunity maintains occult cancer in an equilibrium state. Nature 450, 903–907. 10.1038/nature06309. [DOI] [PubMed] [Google Scholar]
- 4.Shankaran V, Ikeda H, Bruce AT, White JM, Swanson PE, Old LJ, and Schreiber RD (2001). IFNgamma and lymphocytes prevent primary tumour development and shape tumour immunogenicity. Nature 410, 1107–1111. 10.1038/35074122. [DOI] [PubMed] [Google Scholar]
- 5.Dunn GP, Bruce AT, Sheehan KCF, Shankaran V, Uppaluri R, Bui JD, Diamond MS, Koebel CM, Arthur C, White JM, and Schreiber RD (2005). A critical function for type I interferons in cancer immunoediting. Nat. Immunol 6, 722–729. 10.1038/ni1213. [DOI] [PubMed] [Google Scholar]
- 6.Diamond MS, Kinder M, Matsushita H, Mashayekhi M, Dunn GP, Archambault JM, Lee H, Arthur CD, White JM, Kalinke U, et al. (2011). Type I interferon is selectively required by dendritic cells for immune rejection of tumors. J. Exp. Med 208, 1989–2003. 10.1084/jem.20101158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ferris ST, Durai V, Wu R, Theisen DJ, Ward JP, Bern MD, Davidson JT 4th, Bagadia P, Liu T, Briseño CG, et al. (2020). cDC1 prime and are licensed by CD4(+) T cells to induce anti-tumour immunity. Nature 584, 624–629. 10.1038/s41586-020-2611-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Minn AJ, and Wherry EJ (2016). Combination Cancer Therapies with Immune Checkpoint Blockade: Convergence on Interferon Signaling. Cell 165, 272–275. 10.1016/j.cell.2016.03.031. [DOI] [PubMed] [Google Scholar]
- 9.O’Sullivan T, Saddawi-Konefka R, Vermi W, Koebel CM, Arthur C, White JM, Uppaluri R, Andrews DM, Ngiow SF, Teng MWL, et al. (2012). Cancer immunoediting by the innate immune system in the absence of adaptive immunity. J. Exp. Med 209, 1869–1882. 10.1084/jem.20112738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fuertes MB, Kacha AK, Kline J, Woo SR, Kranz DM, Murphy KM, and Gajewski TF (2011). Host type I IFN signals are required for antitumor CD8+ T cell responses through CD8alpha+ dendritic cells. J. Exp. Med 208, 2005–2016. 10.1084/jem.20101159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sistigu A, Yamazaki T, Vacchelli E, Chaba K, Enot DP, Adam J, Vitale I, Goubar A, Baracco EE, Remédios C, et al. (2014). Cancer cell-autonomous contribution of type I interferon signaling to the efficacy of chemotherapy. Nat. Med 20, 1301–1309. 10.1038/nm.3708. [DOI] [PubMed] [Google Scholar]
- 12.Javaid N, and Choi S. (2020). Toll-like Receptors from the Perspective of Cancer Treatment. Cancers 12, 297. 10.3390/cancers12020297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Woo SR, Fuertes MB, Corrales L, Spranger S, Furdyna MJ, Leung MYK, Duggan R, Wang Y, Barber GN, Fitzgerald KA, et al. (2014). STING-dependent cytosolic DNA sensing mediates innate immune recognition of immunogenic tumors. Immunity 41, 830–842. 10.1016/j.immuni.2014.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang H, Hu S, Chen X, Shi H, Chen C, Sun L, and Chen ZJ (2017). cGAS is essential for the antitumor effect of immune checkpoint blockade. Proc. Natl. Acad. Sci. USA 114, 1637–1642. 10.1073/pnas.1621363114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Canadas I, Thummalapalli R, Kim JW, Kitajima S, Jenkins RW, Christensen CL, Campisi M, Kuang Y, Zhang Y, Gjini E, et al. (2018). Tumor innate immunity primed by specific interferon-stimulated endogenous retroviruses. Nat Med 24, 1143–1150. 10.1038/s41591-018-0116-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Duong E, Fessenden TB, Lutz E, Dinter T, Yim L, Blatt S, Bhutkar A, Wittrup KD, and Spranger S. (2022). Type I interferon activates MHC class I-dressed CD11b(+) conventional dendritic cells to promote protective anti-tumor CD8(+) T cell immunity. Immunity 55, 308–323.e9. 10.1016/j.immuni.2021.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Baumeister SH, Freeman GJ, Dranoff G, and Sharpe AH (2016). Co-inhibitory Pathways in Immunotherapy for Cancer. Annu. Rev. Immunol 34, 539–573. 10.1146/annurev-immunol-032414-112049. [DOI] [PubMed] [Google Scholar]
- 18.Blank CU, Haining WN, Held W, Hogan PG, Kallies A, Lugli E, Lynn RC, Philip M, Rao A, Restifo NP, et al. (2019). Defining ‘T cell exhaustion. Nat. Rev. Immunol 19, 665–674. 10.1038/s41577-019-0221-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McLane LM, Abdel-Hakeem MS, and Wherry EJ (2019). CD8 T Cell Exhaustion During Chronic Viral Infection and Cancer. Annu. Rev. Immunol 37, 457–495. 10.1146/annurev-immunol-041015-055318. [DOI] [PubMed] [Google Scholar]
- 20.Trinchieri G. (2010). Type I interferon: friend or foe? J. Exp. Med 207, 2053–2063. 10.1084/jem.20101664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Benci JL, Johnson LR, Choa R, Xu Y, Qiu J, Zhou Z, Xu B, Ye D, Nathanson KL, June CH, et al. (2019). Opposing Functions of Interferon Coordinate Adaptive and Innate Immune Responses to Cancer Immune Checkpoint Blockade. Cell 178, 933–948.e14. 10.1016/j.cell.2019.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Benci JL, Xu B, Qiu Y, Wu TJ, Dada H, Twyman-Saint Victor C, Cucolo L, Lee DSM, Pauken KE, Huang AC, et al. (2016). Tumor Interferon Signaling Regulates a Multigenic Resistance Program to Immune Checkpoint Blockade. Cell 167, 1540–1554.e12. 10.1016/j.cell.2016.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Meireson A, Devos M, and Brochez L. (2020). IDO Expression in Cancer: Different Compartment, Different Functionality? Front. Immunol 11, 531491. 10.3389/fimmu.2020.531491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cucolo L, Chen Q, Qiu J, Yu Y, Klapholz M, Budinich KA, Zhang Z, Shao Y, Brodsky IE, Jordan MS, et al. (2022). The interferon-stimulated gene RIPK1 regulates cancer cell intrinsic and extrinsic resistance to immune checkpoint blockade. Immunity 55, 671–685.e10. 10.1016/j.immuni.2022.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dubrot J, Du PP, Lane-Reticker SK, Kessler EA, Muscato AJ, Mehta A, Freeman SS, Allen PM, Olander KE, Ockerman KM, et al. (2022). In vivo CRISPR screens reveal the landscape of immune evasion pathways across cancer. Nat. Immunol 23, 1495–1506. 10.1038/s41590-022-01315-x. [DOI] [PubMed] [Google Scholar]
- 26.Purbey PK, Scumpia PO, Kim PJ, Tong AJ, Iwamoto KS, McBride WH, and Smale ST (2017). Defined Sensing Mechanisms and Signaling Pathways Contribute to the Global Inflammatory Gene Expression Output Elicited by Ionizing Radiation. Immunity 47, 421–434.e3. 10.1016/j.immuni.2017.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kimura T, Nakayama K, Penninger J, Kitagawa M, Harada H, Matsuyama T, Tanaka N, Kamijo R, Vilcek J, Mak TW, et al. (1994). Involvement of the IRF-1 transcription factor in antiviral responses to interferons. Science 264, 1921–1924. 10.1126/science.8009222. [DOI] [PubMed] [Google Scholar]
- 28.Panda D, Gjinaj E, Bachu M, Squire E, Novatt H, Ozato K, and Rabin RL (2019). IRF1 Maintains Optimal Constitutive Expression of Antiviral Genes and Regulates the Early Antiviral Response. Front. Immunol 10, 1019. 10.3389/fimmu.2019.01019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ghislat G, Cheema AS, Baudoin E, Verthuy C, Ballester PJ, Crozat K, Attaf N, Dong C, Milpied P, Malissen B, et al. (2021). NF-kappaB-dependent IRF1 activation programs cDC1 dendritic cells to drive anti-tumor immunity. Sci. Immunol 6, eabg3570. 10.1126/sciimmunol.abg3570. [DOI] [PubMed] [Google Scholar]
- 30.Lukhele S, Rabbo DA, Guo M, Shen J, Elsaesser HJ, Quevedo R, Carew M, Gadalla R, Snell LM, Mahesh L, et al. (2022). The transcription factor IRF2 drives interferon-mediated CD8(+) T cell exhaustion to restrict anti-tumor immunity. Immunity 55, 2369–2385.e10. 10.1016/j.immuni.2022.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shao L, Hou W, Scharping NE, Vendetti FP, Srivastava R, Roy CN, Menk AV, Wang Y, Chauvin JM, Karukonda P, et al. (2019). IRF1 Inhibits Antitumor Immunity through the Upregulation of PD-L1 in the Tumor Cell. Cancer Immunol. Res 7, 1258–1266. 10.1158/2326-6066.CIR-18-0711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Garcia-Diaz A, Shin DS, Moreno BH, Saco J, Escuin-Ordinas H, Rodriguez GA, Zaretsky JM, Sun L, Hugo W, Wang X, et al. (2017). Interferon Receptor Signaling Pathways Regulating PD-L1 and PD-L2 Expression. Cell Rep. 19, 1189–1201. 10.1016/j.celrep.2017.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lawson KA, Sousa CM, Zhang X, Kim E, Akthar R, Caumanns JJ, Yao Y, Mikolajewicz N, Ross C, Brown KR, et al. (2020). Functional genomic landscape of cancer-intrinsic evasion of killing by T cells. Nature 586, 120–126. 10.1038/s41586-020-2746-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zheng H, Wu L, Xiao Q, Meng X, Hafiz A, Yan Q, Lu R, and Cao J. (2023). Epigenetically suppressed tumor cell intrinsic STING promotes tumor immune escape. Biomed. Pharmacother 157, 114033. 10.1016/j.biopha.2022.114033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Harding SM, Benci JL, Irianto J, Discher DE, Minn AJ, and Greenberg RA (2017). Mitotic progression following DNA damage enables pattern recognition within micronuclei. Nature 548, 466–470. 10.1038/nature23470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kwon J, and Bakhoum SF (2020). The Cytosolic DNA-Sensing cGAS-STING Pathway in Cancer. Cancer Discov. 10, 26–39. 10.1158/2159-8290.CD-19-0761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, and Zhang F. (2013). Genome engineering using the CRISPR-Cas9 system. Nat. Protoc 8, 2281–2308. 10.1038/nprot.2013.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Meeth K, Wang JX, Micevic G, Damsky W, and Bosenberg MW (2016). The YUMM lines: a series of congenic mouse melanoma cell lines with defined genetic alterations. Pigment Cell Melanoma Res. 29, 590–597. 10.1111/pcmr.12498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJM, Robert L, Chmielowski B, Spasic M, Henry G, Ciobanu V, et al. (2014). PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 515, 568–571. 10.1038/nature13954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hos BJ, Camps MGM, van den Bulk J, Tondini E, van den Ende TC, Ruano D, Franken K, Janssen GMC, Ru A, Filippov DV, et al. (2019). Identification of a neo-epitope dominating endogenous CD8 T cell responses to MC-38 colorectal cancer. OncoImmunology 9, 1673125. 10.1080/2162402X.2019.1673125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Huang AC, Postow MA, Orlowski RJ, Mick R, Bengsch B, Manne S, Xu W, Harmon S, Giles JR, Wenz B, et al. (2017). T-cell invigoration to tumour burden ratio associated with anti-PD-1 response. Nature 545, 60–65. 10.1038/nature22079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Miller BC, Sen DR, Al Abosy R, Bi K, Virkud YV, LaFleur MW, Yates KB, Lako A, Felt K, Naik GS, et al. (2019). Subsets of ehausted CD8(+) T cells differentially mediate tumor control and respond to checkpoint blockade. Nat. Immunol 20, 326–336. 10.1038/s41590-019-0312-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Franzen O, Gan LM, and Bjorkegren JLM (2019). PanglaoDB: a web server for exploration of mouse and human single-cell RNA sequencing data. Database 2019. 10.1093/database/baz046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Uhlen M, Fagerberg L, Hallstrom BM, Lindskog C, Oksvold P, Mardinoglu A, Sivertsson A, Kampf C, Sjostedt E, Asplund A, et al. (2015). Proteomics. Tissue-based map of the human proteome. Science 347, 1260419. 10.1126/science.1260419. [DOI] [PubMed] [Google Scholar]
- 45.Kuleshov MV, Jones MR, Rouillard AD, Fernandez NF, Duan Q, Wang Z, Koplev S, Jenkins SL, Jagodnik KM, Lachmann A, et al. (2016). Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 44, W90–W97. 10.1093/nar/gkw377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Andreatta M, Corria-Osorio J, Müller S, Cubas R, Coukos G, and Carmona SJ (2021). Interpretation of T cell states from single-cell transcriptomics data using reference atlases. Nat. Commun 12, 2965. 10.1038/s41467-021-23324-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hoch T, Schulz D, Eling N, Gómez JM, Levesque MP, and Bodenmiller B. (2022). Multiplexed imaging mass cytometry of the chemokine milieus in melanoma characterizes features of the response to immunotherapy. Sci. Immunol 7, eabk1692. 10.1126/sciimmunol.abk1692. [DOI] [PubMed] [Google Scholar]
- 48.Mackay LK, Rahimpour A, Ma JZ, Collins N, Stock AT, Hafon M-L, Vega-Ramos J, Lauzurica P, Mueller SN, Stefanovic T, et al. (2013). The developmental pathway for CD103+CD8+ tissue-resident memory T cells of skin. Nat. Immunol 14, 1294–1301. 10.1038/ni.2744. [DOI] [PubMed] [Google Scholar]
- 49.Kaur M, Kumar D, Butty V, Singh S, Esteban A, Fink GR, Ploegh HL, and Sehrawat S. (2018). Galectin-3 Regulates gamma-Herpesvirus Specific CD8 T Cell Immunity. iScience 9, 101–119. 10.1016/j.isci.2018.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hudson WH, Gensheimer J, Hashimoto M, Wieland A, Valanparambil RM, Li P, Lin JX, Konieczny BT, Im SJ, Freeman GJ, et al. (2019). Proliferating Transitory T Cells with an Effector-like Transcriptional Signature Emerge from PD-1(+) Stem-like CD8(+) T Cells during Chronic Infection. Immunity 51, 1043–1058.e4. 10.1016/j.immuni.2019.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wensveen FM, Jelenčić V, and Polić B. (2018). NKG2D: A Master Regulator of Immune Cell Responsiveness. Front. Immunol 9, 441. 10.3389/fimmu.2018.00441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sharma S, tenOever BR, Grandvaux N, Zhou GP, Lin R, and Hiscott J. (2003). Triggering the interferon antiviral response through an IKK-related pathway. Science 300, 1148–1151. 10.1126/science.1081315. [DOI] [PubMed] [Google Scholar]
- 53.Fitzgerald KA, Rowe DC, Barnes BJ, Caffrey DR, Visintin A, Latz E, Monks B, Pitha PM, and Golenbock DT (2003). LPS-TLR4 signaling to IRF-3/7 and NF-kappaB involves the toll adapters TRAM and TRIF. J. Exp. Med 198, 1043–1055. 10.1084/jem.20031023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Han KJ, Su X, Xu LG, Bin LH, Zhang J, and Shu HB (2004). Mechanisms of the TRIF-induced interferon-stimulated response element and NF-kappaB activation and apoptosis pathways. J. Biol. Chem 279, 15652–15661. 10.1074/jbc.M311629200. [DOI] [PubMed] [Google Scholar]
- 55.Abou El Hassan M, Huang K, Eswara MBK, Xu Z, Yu T, Aubry A, Ni Z, Livne-Bar I, Sangwan M, Ahmad M, and Bremner R. (2017). Properties of STAT1 and IRF1 enhancers and the influence of SNPs. BMC Mol. Biol 18, 6. 10.1186/s12867-017-0084-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kriegsman BA, Vangala P, Chen BJ, Meraner P, Brass AL, Garber M, and Rock KL (2019). Frequent Loss of IRF2 in Cancers Leads to Immune Evasion through Decreased MHC Class I Antigen Presentation and Increased PD-L1 Expression. J. Immunol 203, 1999–2010. 10.4049/jimmunol.1900475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Castro F, Cardoso AP, Gonç alves RM., Serre K, and Oliveira MJ (2018). Interferon-Gamma at the Crossroads of Tumor Immune Surveillance or Evasion. Front. Immunol 9, 847. 10.3389/fimmu.2018.00847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Boukhaled GM, Harding S, and Brooks DG (2021). Opposing Roles of Type I Interferons in Cancer Immunity. Annu. Rev. Pathol 16, 167–198. 10.1146/annurev-pathol-031920-093932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jayaraman P, Parikh F, Lopez-Rivera E, Hailemichael Y, Clark A, Ma G, Cannan D, Ramacher M, Kato M, Overwijk WW, et al. (2012). Tumor-expressed inducible nitric oxide synthase controls induction of functional myeloid-derived suppressor cells through modulation of vascular endothelial growth factor release. J. Immunol 188, 5365–5376. 10.4049/jimmunol.1103553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tokunaga R, Zhang W, Naseem M, Puccini A, Berger MD, Soni S, McSkane M, Baba H, and Lenz HJ (2018). CXCL9, CXCL10, CXCL11/CXCR3 axis for immune activation - A target for novel cancer therapy. Cancer Treat Rev. 63, 40–47. 10.1016/j.ctrv.2017.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Nagata S, and Golstein P. (1995). The Fas death factor. Science 267, 1449–1456. 10.1126/science.7533326. [DOI] [PubMed] [Google Scholar]
- 62.Grasso CS, Tsoi J, Onyshchenko M, Abril-Rodriguez G, Ross-Macdonald P, Wind-Rotolo M, Champhekar A, Medina E, Torrejon DY, Shin DS, et al. (2021). Conserved Interferon-gamma Signaling Drives Clinical Response to Immune Checkpoint Blockade Therapy in Melanoma. Cancer Cell 39, 122. 10.1016/j.ccell.2020.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sekrecka A, Kluzek K, Sekrecki M, Boroujeni ME, Hassani S, Yamauchi S, Sada K, Wesoly J, and Bluyssen HAR (2023). Time-dependent recruitment of GAF, ISGF3 and IRF1 complexes shapes IFNalpha and IFNgamma-activated transcriptional responses and explains mechanistic and functional overlap. Cell. Mol. Life Sci. 80, 187. 10.1007/s00018-023-04830-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yamaguchi H, Hsu JM, Yang WH, and Hung MC (2022). Mechanisms regulating PD-L1 expression in cancers and associated opportunities for novel small-molecule therapeutics. Nat. Rev. Clin. Oncol 19, 287–305. 10.1038/s41571-022-00601-9. [DOI] [PubMed] [Google Scholar]
- 65.Thomas G, Micci L, Yang W, Katakowski J, Oderup C, Sundar P, Wang X, Geles KG, Potluri S, and Salek-Ardakani S. (2021). Intra-Tumoral Activation of Endosomal TLR Pathways Reveals a Distinct Role for TLR3 Agonist Dependent Type-1 Interferons in Shaping the Tumor Immune Microenvironment. Front. Oncol 11, 711673. 10.3389/fonc.2021.711673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Jiang Y, Zhang H, Wang J, Chen J, Guo Z, Liu Y, and Hua H. (2023). Exploiting RIG-I-like receptor pathway for cancer immunotherapy. J. Hematol. Oncol 16, 8. 10.1186/s13045-023-01405-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sharma P, Hu-Lieskovan S, Wargo JA, and Ribas A. (2017). Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell 168, 707–723. 10.1016/j.cell.2017.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gao J, Shi LZ, Zhao H, Chen J, Xiong L, He Q, Chen T, Roszik J, Bernatchez C, Woodman SE, et al. (2016). Loss of IFN-gamma Pathway Genes in Tumor Cells as a Mechanism of Resistance to Anti-CTLA-4 Therapy. Cell 167, 397–404.e9. 10.1016/j.cell.2016.08.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sittig SP, van Beek JJP, Flórez-Grau G, Weiden J, Buschow SI, van der Net MC, van Slooten R, Verbeek MM, Geurtz PBH, Textor J, et al. (2021). Human type 1 and type 2 conventional dendritic cells express indoleamine 2,3-dioxygenase 1 with functional effects on T cell priming. Eur. J. Immunol 51, 1494–1504. 10.1002/eji.202048580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Liu X, Shin N, Koblish HK, Yang G, Wang Q, Wang K, Leffet L, Hansbury MJ, Thomas B, Rupar M, et al. (2010). Selective inhibition of IDO1 effectively regulates mediators of antitumor immunity. Blood 115, 3520–3530. 10.1182/blood-2009-09-246124. [DOI] [PubMed] [Google Scholar]
- 71.Somasundaram V, Ridnour LA, Cheng RY, Walke AJ, Kedei N, Bhattacharyya DD, Wink AL, Edmondson EF, Butcher D, Warner AC, et al. (2022). Systemic Nos2 Depletion and Cox inhibition limits TNBC disease progression and alters lymphoid cell spatial orientation and density. Redox Biol. 58, 102529. 10.1016/j.redox.2022.102529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Shao YJ, Ni JJ, Wei SY, Weng XP, Shen MD, Jia YX, and Meng LN (2020). IRF1-mediated immune cell infiltration is associated with metastasis in colon adenocarcinoma. Medicine (Baltim.) 99, e22170. 10.1097/MD.0000000000022170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Matsuyama T, Kimura T, Kitagawa M, Pfeffer K, Kawakami T, Watanabe N, Kündig TM, Amakawa R, Kishihara K, Wakeham A, et al. (1993). Targeted disruption of IRF-1 or IRF-2 results in abnormal type I IFN gene induction and aberrant lymphocyte development. Cell 75, 83–97. [PubMed] [Google Scholar]
- 74.Ogasawara K, Hida S, Azimi N, Tagaya Y, Sato T, Yokochi-Fukuda T, Waldmann TA, Taniguchi T, and Taki S. (1998). Requirement for IRF-1 in the microenvironment supporting development of natural killer cells. Nature 391, 700–703. 10.1038/35636. [DOI] [PubMed] [Google Scholar]
- 75.Homet Moreno B, Zaretsky JM, Garcia-Diaz A, Tsoi J, Parisi G, Robert L, Meeth K, Ndoye A, Bosenberg M, Weeraratna AT, and Graeber TG (2016). Response to programmed cell death-1 blockade in a murine melanoma syngeneic model requires costimulation, CD4, and CD8 T cells. Cancer immunology research 4, 845–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Suit HD, and Suchato C. (1967). Hyperbaric oxygen and radiotherapy of a fibrosarcoma and of a squamous-cell carcinoma of C3H mice. Radiology 89, 713–719. [DOI] [PubMed] [Google Scholar]
- 77.Müller U, Steinhoff U, Reis LF, Hemmi S, Pavlovic J, Zinkernagel RM, and Aguet M. (1994). Functional role of type I and type II interferons in antiviral defense. Science 264, 1918–1921. [DOI] [PubMed] [Google Scholar]
- 78.Sato M, Suemori H, Hata N, Asagiri M, Ogasawara K, Nakao K, Nakaya T, Katsuki M, Noguchi S, Tanaka N, and Taniguchi T. (2000). Distinct and essential roles of transcription factors IRF-3 and IRF-7 in response to viruses for IFN-α/β gene induction. Immunity 13, 539–548. [DOI] [PubMed] [Google Scholar]
- 79.Yamamoto M, Sato S, Hemmi H, Hoshino K, Kaisho T, Sanjo H, Takeuchi O, Sugiyama M, Okabe M, Takeda K, and Akira S. (2003). Role of adaptor TRIF in the MyD88-independent toll-like receptor signaling pathway. Science 301, 640–643. [DOI] [PubMed] [Google Scholar]
- 80.Cong L, and Zhang F. (2015). Genome engineering using CRISPR-Cas9 system. Chromosomal mutagenesis, 197–217. [DOI] [PubMed] [Google Scholar]
- 81.Butler A, Hoffman P, Smibert P, Papalexi E, and Satija R. (2018). Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol 36, 411–420. 10.1038/nbt.4096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.McGinnis CS, Murrow LM, and Gartner ZJ (2019). DoubletFinder: Doublet Detection in Single-Cell RNA Sequencing Data Using Artificial Nearest Neighbors. Cell Syst. 8, 329–337.e4. 10.1016/j.cels.2019.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Wu T, Hu E, Xu S, Chen M, Guo P, Dai Z, Feng T, Zhou L, Tang W, Zhan L, et al. (2021). clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation (Camb) 2, 100141. 10.1016/j.xinn.2021.100141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Rusinova I, Forster S, Yu S, Kannan A, Masse M, Cumming H, Chapman R, and Hertzog PJ (2013). Interferome v2.0: an updated database of annotated interferon-regulated genes. Nucleic Acids Res. 41, D1040–D1046. 10.1093/nar/gks1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Blighe K, Rana S, and Lewis M. (2024). EnhancedVolcano: Publication-ready volcano plots with enhanced colouring and labeling. https://github.com/kevinblighe/EnhancedVolcano. [Google Scholar]
- 86.Kim D, Paggi JM, Park C, Bennett C, and Salzberg SL (2019). Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat Biotechnol 37, 907–915. 10.1038/s41587-019-0201-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Anders S, and Huber W. (2010). Differential expression analysis for sequence count data. Genome Biol. 11, R106. 10.1186/gb-2010-11-10-r106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Quinlan AR, and Hall IM (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Castro-Mondragon JA, Riudavets-Puig R, Rauluseviciute I, Lemma RB, Turchi L, Blanc-Mathieu R, Lucas J, Boddie P, Khan A, Manosalva Perez N, et al. (2022). JASPAR 2022: the 9th release of the open-access database of transcription factor binding profiles. Nucleic Acids Res 50, D165–D173. 10.1093/nar/gkab1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, and Glass CK (2010). Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 38, 576–589. 10.1016/j.molcel.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Zambelli F, Pesole G, and Pavesi G. (2009). Pscan: finding over-represented transcription factor binding site motifs in sequences from co-regulated or co-expressed genes. Nucleic Acids Res 37, W247–252. 10.1093/nar/gkp464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Bardou P, Mariette J, Escudie F, Djemiel C, and Klopp C. (2014). jvenn: an interactive Venn diagram viewer. BMC Bioinformatics 15, 293. 10.1186/1471-2105-15-293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, and Genome Project Data Processing, S. (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All RNA-seq, ChIP-seq and single-cell RNA-seq data have been deposited at GEO and are publicly available on 1st May 2024 or on the date of publication. Accession numbers are listed in the key resources table. Microscopy data reported in this paper will be shared by the lead contact upon request.
This paper does not report new or original codes. Code that has been used for analysis is publicly available, referred to and listed in the references section.
Any additional information needed to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| Anti-IRF1 | Cell Signaling | Cat# 8478S; RRID:AB_10949108 |
| Anti-STAT1 (for mouse and human) | Cell Signaling | Cat# 9172; RRID:AB_2198300 |
| p-STAT1 (Tyr701) (for human) | Cell Signaling | Cat# 9167; RRID:AB_561284 |
| Rabbit a-CD4 (For immunofluorescence) | Abcam | cat# AB183685; RRID:AB_2686917 |
| Rat a-CD8 (For Immunofluorescence) | eBioscience | Cat# 14–0808; RRID:AB_2572860 |
| CD45_PerCPcy5.5 | Biolegend | Cat# 103236; RRID:AB_893354 |
| CD3_APC | eBioScience | Cat# 17–0031-82; RRID:AB_469315 |
| CD4_APC-PE780 | eBioScience | Cat# 47004280; RRID:AB_1272219 |
| CD8_FITC | eBioScience | Cat# 11–0081-81; RRID:AB_464914 |
| NK1.1_PE | eBioScience | Ca# 12–5941-82; RRID:AB_466050 |
| PD1_BV421 | Biolegend | Ca# 135217; RRID:AB_2562568 |
| Cd45_APC | Biolegend | Ca# 109813; RRID:AB_389210 |
| Cd11b_APC-Alexafluor 750 (APC-Cy7) | eBioscience | Cat# 27–0112-81; RRID:AB_469343 |
| Cd11c_PE | Biolegend | Cat# 117308; RRID:AB_313776 |
| Ccr2_BV785 | Biolegend | Cat# 150621; RRID:AB_2721565 |
| Ly6C_PE/Dazzle594 | Biolegend | Cat# 128043; RRID:AB_2566576 |
| Ly6G_FITC | Biolegend | Cat# 127606; RRID:AB_1236488 |
| Live/Dead Fixable Viable Stain 510 | BD Horizon | Cat# BDB564406; RRID:AB_2869572 |
| anti-PD-L1-PE | Biolegend | Cat# 124308; RRID:AB_2073557 |
| anti-MHC-I_AlexaFluor-647 | Biolegend | Cat# 114612; RRID:AB_492931 |
| InVivoMAb anti-mouse PD-1 (CD279) | BioXcell | Cat# BE0146; RRID:AB_10949053 |
| InVivoMAb anti-mouse CTLA-4 (CD152), clone 9H10 | BioXcell | Cat# BE0131; RRID:AB_10950184 |
| InVivoMAb anti-mouse IFNAR-1, clone MAR1–5A3 | BioXcell | Cat# BE0241; RRID:AB_2687723 |
| InVivoMAb rat IgG2a isotype control | BioXcell | Cat# BE0089; RRID: AB 1107769 |
| inVivoMAb anti-mouse IFNγR (CD119), clone GR-20 | BioXcell | Cat# BE0029; RRID:AB 1107576 |
|
| ||
| Chemicals, Peptides, and Recombinant Proteins | ||
|
| ||
| murine IFN-y | Peprotech | Cat# 315–05 |
| Human IFN-y | Invivogen | Cat# rcyec-hifng |
| Collagenase-D | Roche | Cat# 11088866001 |
| DNase I | Roche | Cat# 10104159001 |
| SpCas9 2NLS Nuclease | Synthego | Cat# SpCas9 2NLS Nuclease |
| DSG Crosslinker | Covachem | Cat# 13301–1 |
| Thermo Scientific™ Pierce™ Formaldehyde Ampules, Methanol-free | Thermo Fisher | Cat# 52622 |
| Pierce Protease Inhibitor Mini Tablets, EDTA-Free | Thermo Fisher | Cat# A32955 |
| Linear Acrylamide (5 mg/mL) | Thermo Fisher | Cat# AM9520 |
| Proteinase K, recombinant, PCR grade | Thermo Fisher | Cat# EO0492 |
| Linear Acrylamide | Invitrogen | Cat# AM9520 |
| RNaseA | Thermo Fisher | Cat# EN0531 |
|
| ||
| Critical Commercial Assays | ||
|
| ||
| TruSeq Stranded mRNA Sample Prep Kit B | Illumina | Cat# RS-122–2102 |
| Kapa Hyper Prep Kit Ilumina Platform | Kapa Biosystems | Cat# KK8502 |
| NEXTflex™ ChIP-Seq Barcodes - 12 | BIOO Scientific | Cat# 514121 |
| AMPure XP purification beads for DNA purification | Beckman Coulter | Cat# A63881 |
| RNeasy Mini Kit (250 rxn) | Qiagen | Cat# 74106 |
| DNeasy Mini Kit | Qiagen | Cat# 69506 |
| SuperScript™ III Reverse Transcriptase | Invitrogen | Cat# 18080044 |
| Dynabeads Protein G | Invitrogen | Cat# 1004D |
| Tumor Dissociation Kit | Miltenyi Biotec | Cat# 130–096730 |
| Dead Cell Removal Kit | Miltenyi Biotec | Cat# 130–090-101 |
| gentleMACS™ C Tubes | Miltenyi Biotec | Cat# 130–093-237 |
| MS Columns | Miltenyi Biotec | Cat# 130–042-201 |
| CellTiter 96® AQueous One Solution Cell Proliferation Assay (MTS) |
Promega | Cat# G3580 |
| BCA assay | Thermo Fisher Scientific | Cat# 23227 |
| Supersignal West Pico chemiluminescence kit | Thermo Fisher Scientific | Cat# 34080 |
| Neon™ Transfection System 10 μL Kit | Thermo Fisher | Cat# MPK1096 |
|
| ||
| Deposited Data | ||
|
| ||
| All raw and analyzed bulk mRNA-seq, sc-RNA-seq and ChIP-seq datasets | This Paper | GEO Accession GSE246923 |
|
| ||
| Experimental Models: Cell Lines | ||
|
| ||
| MC38, mouse colon adenocarcinoma | C57BL/6 | Kerafast #ENH204-FP, RRID:CVCL_B288 |
| YUMM2.1 | C57BL/6 | Moreno et al.75 |
| B16-F10 mouse melanoma | C57BL/6 | ATCC #CRL-6475, RRID:CVCL_0159 |
| FSA (Fibrosarcoma) | C3H strain derived McBride, UCLA | Suit et al.76 |
| M238 (human Melanoma cell line) | Human melanoma derived, A. Ribas lab | Grasso et al.62 |
| HEK293T Cells | ATCC | CRL-11268, RRID:CVCL_1926 |
|
| ||
| Experimental Models: Organisms/Strains | ||
|
| ||
| C57BL/6 | Jackson Laboratory | Cat# 000664, RRID:IMSR_JAX:000664 |
| StingGt/Gt | Jackson Laboratory | Cat# 017537, RRID:IMSR_JAX:017537 |
| Ifnar −/− | G. Cheng, UCLA | Müller et al.77 |
| NOD Scid | Jackson Laboratory | Cat# 001303, RRID:IMSR_JAX:001303 |
| Rag2−/− γC−/− | Jackson Laboratory | Cat# 014593, RRID:IMSR_JAX:014593 |
| Rag1 −/− | Jackson Laboratory | Cat# 002216, RRID:IMSR_JAX:002216 |
| Cd8 −/− | Jackson Laboratory | Cat# 002665, RRID:IMSR_JAX:002665 |
| Irf1 −/− | Jackson Laboratory | Cat# 002762, RRID:IMSR_JAX:002762 |
| Ifngr −/− | Jackson Laboratory | Cat# 003288, RRID:IMSR_JAX:003288 |
| Irf3 −/− | G. Cheng, UCLA | Sato et al.78 |
| Myd88 −/− | G. Cheng, UCLA | Cat# 009088, RRID:IMSR_JAX:009088 |
| Trif −/− | Hoebe et al., 2003 | JAX stock #005037, RRID:IMSR_JAX:005037 |
| Myd88 Trif DKO | Geg Barton, UC Berkley | Yamamoto et al.79 |
|
| ||
| Oligonucleotides | ||
|
| ||
| mPD-L1_gateway cloning_forward (5′−3′) GGGGACAAGTTTGTACAAAAAAGCAGGCT tcgccaccATGAGGATATTTGCTGGCATTAT |
This paper | NA |
| mPD-L1_gateway cloning_reverse (5′−3′) GGGGACCACTTTGTACAAGAAAGCTGGGT cTCACGTCTCCTCGAATTGTG |
This paper | NA |
| mIRF1 guide sequence g1_Forward for pX459 CACCGATGCCTGTCTGTTCCGGAGC |
This paper | NA |
| mIRF1 guide sequence g1_Reverse for pX459 AAACGCTCCGGAACAGACAGGCATC |
This paper | NA |
| mIRF1 guide sequence g2_Forward for pX459 CACCGGCTCTGCTGTGCGGGTGTAC |
This paper | NA |
| mIRF1 guide sequence g2_Reverse for pX459 AAACGTACACCCGCACAGCAGAGCC |
This paper | NA |
| mSTING guide sequence g1_Forward for pX459 CACCGCCCCACAGTCCAATGGAAAG |
This paper | NA |
| mSTING guide sequence g1_Reverse for pX459 AAACCTTTCCATTGGACTGTGGGGC |
This paper | NA |
| mSTING guide sequence g2_Forward for pX459 CACCGTCAGGCACCCCACAGTCCAA |
This paper | NA |
| mSTING guide sequence g2_Reverse for pX459 AAACTTGGACTGTGGGGTGCCTGAC |
This paper | NA |
| hIRF1 guide sequence g1 for RNP (Synthego) CUCUAGGCCGAUACAAAGCA |
This paper | NA |
| hIRF1 guide sequence g2 for RNP (Synthego) GCCAGAUCCCAAGACGUGGA |
This paper | NA |
|
| ||
| Recombinant DNA | ||
|
| ||
| pSpCas9(BB)-2A-puro(pX459)-V2.0 plasmid | Addgene #62988 | Cong and Zhang80 |
| pX459V2_Irf1-gd1 | This paper | N/A |
| pX459V2_Irf1-gd2 | This paper | N/A |
| pX459V2_Sting-gd1 | This paper | N/A |
| pX459V2_Sting-gd2 | This paper | N/A |
| pLenti_puro_Pdl1 | This paper | N/A |
|
| ||
| Software and Algorithms | ||
|
| ||
| Seurat 4.1 | https://github.com/satijalab/seurat | Butler et al.81 |
| FlowJo | FlowJo | Version 10.8.1 |
| Cell Ranger v6.0.2 | 10X Genomics | Version 6.0.2 https://www.10xgenomics.com/support/software/cell-ranger |
| Seurat 4.1 | https://github.com/satijalab/seurat | Butler et al.81 |
| R/Rstudio | CRAN | Version 4.3.1 |
| GGplot2 | CRAN | Version 3.4.3 https://ggplot2.tidyverse.org |
| DoubletFinder | https://github.com/chris-mcginnis-ucsf/DoubletFinder | McGinnis et al.82 |
| cowplot | https://wilkelab.org/cowplot/ | Version 1.1.1 |
| ProjecTILs | https://github.com/carmonalab/ProjecTILs | Andreatta et al.46 |
| clusterProfiler | https://bioconductor.org/packages/release/bioc/html/clusterProfiler.html | Wu et al.83 PMID: 34557778 |
| Interferome database | http://www.interferome.org | Rusinova et al.84 |
| EnhancedVolcano v1.20.0 package in Bioconductor | https://bioconductor.org/packages/EnhancedVolcano | Blighe et al.85 |
| Hisat2 | http://daehwankimlab.github.io/hisat2/ | Kim et al.86 |
| DEseq2 | https://bioconductor.org/packages/release/bioc/html/DESeq2.html | Anders and Huber87 |
| BEDTools | https://github.com/arq5x/bedtools2 | Quinlan et al.88 |
| Jasper TF motif database | https://jaspar.genereg.net/ | Castro-Mondragon et al.89 |
| Homer | http://homer.ucsd.edu/homer/ | Heinz et al.90 |
| Pscan | http://159.149.160.88/pscan/ | Zambelli et al.91 |
| Jvenn | http://jvenn.toulouse.inra.fr/app/index.html | Bardou et al.92 |
| R-Studio, | https://www.rstudio.com | RStudio Team (2015). RStudio: Integrated Development for R. RStudio, Inc., Boston, MA |
| Bioconductor DE-seq | http://bioconductor.org/packages/release/bioc/html/DESeq.html | Anders and Huber87 |
| MS Office 2010 and 365 | https://www.microsoft.com/ | Microsoft Office |
| Samtools | https://github.com/samtools/samtools | Li et al.93 |
| SeqMonk | https://www.bioinformatics.babraham.ac.uk/projects/seqmonk/ | Simon Andrews (Head of Bioinformatics) Email: simon.andrews@babraham.ac.uk |