


Abstract
A 1468 bp cDNA coding for the chicken homolog of the human MBD4 G/T mismatch DNA glycosylase was isolated and sequenced. The derived amino acid sequence (416 amino acids) shows 46% identity with the human MBD4 and the conserved catalytic region at the C-terminal end (170 amino acids) has 90% identity. The non-conserved region of the avian protein has no consensus sequence for the methylated DNA binding domain. The recombinant proteins from human and chicken have G/T mismatch as well as 5-methylcytosine (5-MeC) DNA glycosylase activities. When tested by gel shift assays, human recombinant protein with or without the methylated DNA binding domain binds equally well to symmetrically, hemimethylated DNA and non-methylated DNA. However, the enzyme has only 5-MeC DNA glycosylase activity with the hemimethylated DNA. Footprinting of human MBD4 and of an N-terminal deletion mutant with partially depurinated and depyrimidinated substrate reveal a selective binding of the proteins to the modified substrate around the CpG. As for 5-MeC DNA glycosylase purified from chicken embryos, MBD4 does not use oligonucleotides containing mCpA, mCpT or mCpC as substrates. An mCpG within an A+T-rich oligonucleotide is a much better substrate than an A+T-poor sequence. The Km of human MBD4 for hemimethylated DNA is ∼10–7 M with a Vmax of ∼10–11 mol/h/µg protein. Deletion mutations show that G/T mismatch and 5-MeC DNA glycosylase are located in the C-terminal conserved region. In sharp contrast to the 5-MeC DNA glycosylase isolated from the chicken embryo DNA demethylation complex, the two enzymatic activities of MBD4 are strongly inhibited by RNA. In situ hybridization with antisense RNA indicate that MBD4 is only located in dividing cells of differentiating embryonic tissues.
INTRODUCTION
Throughout embryonic development of vertebrates there are changes in the DNA methylation pattern of specific genes. These changes can possibly take place by a complex interplay between DNA methyltransferases, DNA demethylation activities and specific regulatory sequences and factors (1–3 and references therein). Recently we have identified in chicken embryos several components of a DNA demethylation complex consisting of RNA (4,5), RNA helicase (6) and a G/T mismatch DNA glycosylase combined with 5-methylcytosine (5-MeC) DNA glycosylase activity (7). One of the roles of the CpG-rich RNA present in the DNA demethylation complex is to target the DNA demethylation (8). The RNA helicase in the presence of the CpG-rich RNA and ATP potentiates the activity of 5-MeC DNA glycosylase (7). In contrast the same recombinant RNA inhibits the G/T mismatch DNA glycosylase activity. Recently, a family of methylated DNA binding proteins was isolated and sequenced (9). One of them, MBD4 (also known as MED-1) was shown to have G/T mismatch DNA glycosylase and endonuclease activities (10,11). Here we show that a human MBD4 and a related chicken protein have both the G/T mismatch and 5-MeC DNA glycosylase activities. Unlike the 5-MeC DNA glycosylase isolated from the DNA demethylation complex, both glycosylase activities of MBD4 are strongly inhibited by RNA.
MATERIALS AND METHODS
Construction and screening of a chicken cDNA library
The construction of the chicken cDNA library in phage λ was carried out as previously described (6). The screening was carried out by conventional procedures, using synthetic labeled oligonucleotides as probes.
The following antisense DNA sequences were used. For the N-terminal region (human sequence) we used: 5′-TCATCTTCTCCCACTCTTTCCAATTCCATAGCAACATCTTCTTTGCGGAG-3′; 5′-TGTGAAGATAATTAGCAAGTGAACTTTTGGATCTGAACTTCAGTCCTTG-3′. For the C-terminal region we used: 5′-AATTTGTGGTCTTCAGGGTGCACCTGCTTCCACTCATTGACACAAAAAATTT-3′. Positive clones were characterized by restriction mapping and DNA sequencing. DNA sequencing was carried out with an ABI Prism 377 DNA sequencer from PE Biosystems.
Cloning of the human and chicken MBD4
For the human MBD4, a 1778 bp DNA fragment corresponding to the whole human MBD4 coding region was amplified by RT–PCR using total RNA prepared from human embryonic kidney cells, HEK 293, as template. The following oligonucleotides were used as primers: human MBD4, N-terminal: 5′-TTCAGGATCCGCAGCCGGACCCTGCTCG-3′; and C-terminal: 5′-TATACCCGGGTGAAAGCTGCAGAGTTTAAG-3′.
Following digestion with BamHI and SmaI, the cDNA fragment was inserted in-frame into the BamHI/SmaI sites of the expression vector pQE30 (Qiagen). The plasmid carrying the human MBD4 cDNA was named pRMBD4.
Upon screening a chicken cDNA library with the above-labeled probes, positive clones were tested for their hybridization with either the C- or N-terminal probes. All clones hybridized with the conserved C-terminal sequence and none with the variable N-terminal region. The longest cDNA was sequenced and inserted in-frame into the pQE30 plasmid vector. Following digestion of the vector with BamHI and SalI the cDNA in λ phage was digested with FokI and XhoI and inserted in-frame into the BamHI/FokI (blunted ends) and SalI/XhoI (compatible cohesive ends) of pQE30. The plasmid carrying the chicken MBD4 was named pcMBD4.
Construction of deletion mutants of human MBD4 and production of recombinant proteins
N-terminal truncated proteins were obtained by PCR amplification using the full-length pRMBD4 DNA as template. The oligonucleotide hMBD4-C (see above) was used as antisense C-terminal primer. For the N-terminal primers the following sense oligonucleotides were used: (hMBD4-ΔN273), 5′-TTCAGGATCCGAAAGTGAACCTGTTGCACA-3′, (hMBD4-ΔN378), 5′-TTCAGGATCCGGCTCTGAAATGGACAACAA-3′, and (hMBD4-ΔN433), 5′-TTCAGGATCCGCCTTTAAGAAATGGACACC-3′.
The three amplified fragments were digested with BamHI and SmaI, then inserted in-frame at the BamHI/SmaI sites of pQE30. The recombinant deletion mutants were designated as pRMBD4-ΔN273, pRMBD4-ΔN378 and pBMBD4-ΔN433, respectively.
For the construction of C-terminal deletion mutant, a 1.8 kb EcoRI–SmaI fragment of pRMBD4 was digested with MunI, followed by blunt-end treatment with T4 DNA polymerase. The sample was then digested with BamHI. The 1.62 kb BamHI–MunI (blunted) fragment was inserted in-frame at the BamHI/SmaI sites of pQE30. The recombinant deletion mutant was designated as pRMBD4-ΔC48. All constructs were fused at the N-terminus with a short sequence of six histidines and were tested by DNA sequencing.
Wild-type and mutants were transformed into Escherichia coli strain XL1-blue. For each recombinant protein, 1 liter of liquid culture derived from a single colony was grown at 37°C to an OD600 of 0.6. The culture was then treated with 1 mM IPTG at 37°C for 3 h. Cells were collected and broken by decompression with a French press cell. Recombinant proteins were purified from the inclusion bodies using a Ni-NTA agarose column (Qiagen) under denaturing conditions and then refolded by dialysis.
Enzymatic tests
5-MeC DNA glycosylase and G/T mismatch DNA glycosylase were tested with labeled synthetic oligonucleotides as previously described (12). All substrates used in the present work are listed in Table 1. The quality (methylation) of the synthetic methylated oligonucleotides was tested by chemical sequencing according to Maxam and Gilbert (13). All oligonucleotides were gel purified. Oligonucleotides were labeled at their 5′-ends with polynucleotide kinase and [γ-32P]ATP to a specific activity of ∼2 × 107 c.p.m./µg duplex and the substrate concentration for the assay was >5 × 10–8 M. Aliquots of 50–200 ng of recombinant proteins were used per 100 µl assay. Upon incubation for 30 min at 37°C, samples were diluted to 150 µl with water and treated as previously described (12). Where indicated, the samples were treated for 10 min at 95°C in 0.1 M NaOH. The alkaline treatment cleaves the abasic sugar resulting from the removal of the methylated base or the mispaired base by the glycosylase.
Table 1. Sequence of the lower modified strand (G/T mismatch or 5-MeC) of substrates tested in the glycosylase assays.

The arrowheads point to the position of cleavage upon removal of the abasic sugar at the G/T mismatch or the 5-MeCpX (X is either G, A, T or C). M represents 5-MeC. The first column indicates the size of the labeled DNA fragment generated by the cleavage at the abasic sugar. The second column gives the A+T content of the oligonucleotides.
The synthetic oligoribonucleotides 5′-GCUCCGGUCAC-3′ [complementary to the non-methylated strand of the substrate, oligo B (Table 1)] and 5′-GUGACCGGAGC-3′ (complementary to the methylated strand) were protected against ribonucleases present in the preparation of recombinant proteins by the methylation of the ribose at the O2 position. Upon separation on a 20% polyacrylamide–urea sequencing gel, the quantitative determination of the reaction products of the glycosylases was carried out by cutting out the specific radioactive bands and counting them for radioactivity. A slice of equivalent size was also cut out from the blanks, counted and subtracted from the test values.
Gel mobility shift assay
End-labeled oligonucleotides (10–7 M) were incubated in the glycosylase assay buffer with 100–200 ng of recombinant proteins at 37°C for 30 min. The non-specific competing DNA was from E.coli (50–250 ng/assay). The reaction product was separated on a native 5% polyacrylamide gel (acrylamide:bisacrylamide 29:1) prepared in 0.25× TBE (10× TBE is 0.89 M Tris base, 0.89 M boric acid, 0.02 M EDTA pH 8.0). Separation was carried out in the cold room at constant current (15 mA) in 0.25× TBE. Upon autoradiography bands were cut out of the gel, the labeled DNA was extracted and then analyzed on a 20% polyacrylamide denaturing gel.
Missing contact probing of recombinant protein with the labeled DNA substrate was carried out as described by Brunelle and Schleif (14) and Liang et al. (15). The partial depurination was done with formic acid and the partial depyrimidination was carried out with hydrazine. All reaction products were analyzed on a 20% polyacrylamide denaturing gel. Upon gel shift assay of the recombinant protein with the partial depurinated or depyrimidinated oligonucleotides, specific bands corresponding to the free and bound oligos were cut out of the gel, extracted and subjected to β-elimination with piperidine. The reaction product was analyzed on a 20% polyacrylamide DNA sequencing gel. Approximately 30 000 c.p.m. was loaded in each lane.
In situ hybridization (ISH) on tissue sections
The probe used for ISH was part of the cloned human MBD4 gene covering the coding sequence from nucleotides positions 1276 to 1890. The DNA fragment was amplified by PCR using primers (hMBD4-C and hMBD4-ΔN378 as mentioned above) containing a BamHI site at the 5′-end and a SmaI site at the 3′-end. The sequence was introduced into the vector pGEM-4Z (Promega) containing a T7 and SP6 promotor sequence flanking the polylinker. DIG-labeled RNA was obtained by using the SP6/T7 in vitro transcription kit (Roche Molecular Biochemicals). ISH was essentially performed as described by Schaeren-Wiemers and Gerfin-Moser (16). The concentration of the probe was 700 ng/ml hybridization mixture. The hybridization was done overnight at 58°C. The cover slides were removed from the sections in 5× SSC at 68°C and slides were washed for 1 h at 68°C in 0.2× SSC. Immunological detection of DIG-labeled hybrids was done as recommended by the manufacturer (Roche Molecular Biochemicals). Nuclear staining with Hoechst 33258 (DAPI) was performed on all slides.
Chemicals and enzymes
Polynucleotide kinase and DNA ligase were obtained from Roche Molecular Biochemicals. Restriction enzymes and ribonuclease-free BSA were purchased from Biofinex (Praroman, CH-1724, Switzerland). QIA express type IV kit, QIA quick gel extraction kit, Plasmid Maxi kit and Qiagen Lambda Maxi kit were obtained from Qiagen AG (Basel, Switzerland). Wizard Plus SV Minipreps DNA purification system was from Promega. [γ-32P]ATP triethylaminonium salt (3000 Ci/mmol) was from Amersham. Hydrazine was obtained from Kodak Eastman and piperidine from Merck.
RESULTS
Cloning of a chicken cDNA related to human MBD4
Since we have recently discovered that the chicken G/T mismatch DNA glycosylase also has 5-MeC DNA glycosylase activity (7), it was of interest to see whether the human MBD4 and its chicken homolog also both had enzymatic activities. Six positive clones were obtained from 200 000 phage plaques. They all hybridized very strongly with the probes covering the C-terminal part of MBD4 but no hybridization was obtained with the other probes. A 1468 bp long chicken cDNA was cloned and sequenced (EMBL accession no. AF257107). The amino acid alignment of the chicken sequence with human MBD4 is shown in Figure 1. At the N-terminal region of the chicken cDNA there is no sequence coding for the methylated DNA binding domain. The chicken cDNA has open reading frame coding for 416 amino acids whereas the human MBD4 codes for 580 amino acids. An in-frame stop codon was found upstream of the first ATG of the open reading frame, thus the chicken cDNA was believed to cover the whole coding region. The amino acid sequence upstream of the conserved catalytic region has no sequence identity with human MBD4 whereas the catalytic region at the C-terminus has 90% identity with the human sequence. Data from mass spectrometry analysis of peptides derived from the highly purified chicken embryo DNA demethylation complex showed the absence of MBD4-related proteins in this complex (D.Hess, unpublished results).
Figure 1.

Comparison of the deduced amino acid sequence of the chicken MBD4 (accession no. AF257107) with the human MBD4 (accession no. AF072250). The boxed sequence corresponds to the conserved region of mammalian mCpG binding proteins (methylated DNA binding domain).
The recombinant human MBD4 and its chicken homolog both have G/T mismatch DNA glycosylase and 5-MeC DNA glycosylase
The results shown in Figures 2 and 3 indicate that, in the standard assay for glycosylases, both the recombinant human (upper panels) and chicken MBD4 (lower panels) have the same substrate specificity as the purified chicken 5-MeC DNA glycosylase from chicken embryos (7,17). The activity of the G/T mismatch DNA glycosylase (Table 1, oligo A) is ∼30–40-times higher than the 5-MeC DNA glycosylase (Table 1, oligo B). Among the methylated DNA substrates, the hemimethylated DNA with a mCpG is the best substrate whereas symmetrically-methylated CpG is a poor substrate. As shown in Figure 2 (NM/M, lane 1) and Figure 3 (mCpG, lane 1) the recombinant 5-MCDG only partially cleaves the abasic sugar at the 3′ position. A complete cleavage of the abasic sugar at the 3′ and 5′ positions was only obtained upon alkaline hydrolysis (Figs 2 and 3, lane 2) yielding a fragment 25 bases long (Table 1, oligo A) for G/T mismatch DNA glycosylase and 28 bases long (Table 1, oligo B) for 5-MCDG. Upon denaturation and separation of the reaction product on DNA sequencing gels, the complete cleavage of the abasic sugar from the DNA resulted in an increase in intensity of the band and in a shift in the migration of the labeled oligonucleotide fragment [compare lanes 1 and 2 of NM/M (Fig. 2) and mCpG (Fig. 3)]. At the same time alkaline hydrolysis, depending on the sequences used in the assays, revealed other non-specific minor cuts (Figs 2–4, lanes 2). Lanes 3 of Figures 2–4 are the controls of oligonucleotides incubated only with BSA and denatured before loading onto the sequencing gel.
Figure 2.

Substrate specificity of 5-MeC DNA glycosylase. The upper panel is the reactions with human MBD4 and the lower panel, the reactions with the chicken homolog of human MBD4. Enzymatic reactions were carried out as described in Materials and Methods. The arrows point to the correct positions of the cleavage products which are 25 and 28 nt, respectively, for the G/T mismatch glycosylase and 5-MCDG (oligos A and B, respectively, Table 1). G/T, substrate with a G/T mismatch; NM, non-methylated DNA strand; M, methylated DNA strand. In lanes 1, the reaction product was denatured before loading onto the gel whereas in lanes 2, reaction products were treated with 0.1 M NaOH at 95°C for 10 min before loading onto the sequencing gel. In lanes 3, the substrate was incubated with BSA only and was denatured prior to loading onto the DNA sequencing gel (20% polyacrylamide–urea).
Figure 3.

Sequence specificity of 5-MeC DNA glycosylase. The upper panel represents the activity for human MBD4 and the lower panel the activity for the chicken homolog of MBD4. In lanes 1 the reaction product is denatured at 95°C before loading (this shows possible specific nicks at the 3′ position of the abasic sugar). In lanes 2 the reaction product is treated for 10 min at 95°C in 0.1 M NaOH before loading (this reaction removes the abasic sugar from the oligonucleotide, giving a shift in the migration of the band). In lanes 3 a blank is denatured at 95°C for 5 min before loading. The arrow points to the position of the reaction product which is 28 nt long for mCpG (see also Table 1). All substrates were hemimethylated and contained either one mCpG, mCpA, mCpT or mCpC (oligos B, C, D and E, respectively, Table 1).
Figure 4.
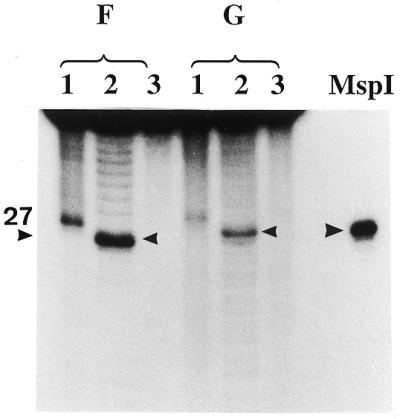
Influence of the A+T content of the oligonucleotide substrates on the activity of 5-MeC DNA glycosylase. Each assay had 50 ng of human recombinant MBD4 per 50 µl incubation mixture. Incubation was for 30 min at 37°C. Oligo F has 92% A+T whereas oligo G has 46% A+T. In lanes 1 the product of the reactions was denatured before loading onto the 20% acrylamide sequencing gel, whereas in lanes 2 the product of reaction was treated for 5 min at 95°C with 0.1 M NaOH before loading (cleavage and removal of the abasic sugar). Lanes 3 are the controls incubated without glycosylase. MspI is the 27 bp size marker resulting from cleavage of the substrate with MspI.
It was also of interest to further investigate whether the base composition surrounding the mCpG had any influence on the glycosylase activity. As can be seen in Figure 4, mCpG surrounded by an A+T-rich sequence (92%, oligo F, Table 1) is a better substrate than the one poorer in A+T (46%, oligo G, Table 1). Both substrates had the same specific radioactivity of 1.5 × 107 c.p.m./µg DNA duplex and the same concentration of substrate (10–7 M) was tested.
These results suggest that the activity level of 5-MeC DNA glycosylase depends on the base composition surrounding the methylated CpG.
The Km of the reaction for the hemimethylated DNA substrate (one hemimethylated CpG on a 50 bp long oligonucleotide, oligo B) with the human recombinant MBD4 is ∼10–7 M with a Vmax of ∼10–11 mol/h/µg protein (Fig. 5). This very low activity could possibly be explained by a big loss of catalytic activity occurring during the denaturation and renaturation of the enzyme isolated from inclusion bodies, or by the absence of cofactors.
Figure 5.

Determination of the Km and Vmax for hemimethylated DNA substrate incubated with recombinant human MBD4. Incubation conditions were as outlined in Materials and Methods. Reaction product was analyzed on a 20% urea–polyacrylamide DNA sequencing gel. Upon autoradiography, bands were cut out and counted for radioactivity. Controls were carried out with the DNA substrate only. An area corresponding to the reaction product was cut out, counted and subtracted from the test values. Results are expressed as a Lineweaver–Burk plot.
G/T mismatch and 5-MeC DNA glycosylase activities are localized in the same region, at the C-terminus of the recombinant enzyme
It has previously been shown (10) that G/T mismatch DNA glycosylase activity is situated at the C-terminal region of MBD4 where exonuclease activity is also located (11). The three N-terminal deletion mutants (Fig. 6A and B) were expressed in E.coli and tested for 5-MeC DNA glycosylase and G/T mismatch DNA glycosylase (Fig. 7). As shown in Figures 6 and 7, a deletion of up to 65% of the total length of MBD4 covering the non-conserved region gradually depressed the G/T mismatch DNA glycosylase activity without affecting the activity of 5-MeC DNA glycosylase. This resulted in a change in the ratio of G/T mismatch over 5-MeC DNA glycosylase from 37 to 8. Furthermore, a deletion of the last 48 amino acid residues at the C-terminus of MBD4 completely abolished the G/T mismatch and the 5-MeC DNA glycosylase activities. These results suggest that the two enzymatic actives are located in the same conserved region, but they are differently affected by the change of protein structures. In addition, there is no need of the methylated DNA binding domain for G/T mismatch and 5-MCDG activities.
Figure 6.

Deletion mutations of human MBD4. (A) The N- and C-terminal deletion mutants. FL represents the full-length protein. The relative activity of G/T mismatch and 5-MeC DNA glycosylase are indicated by +++, ++, +, ± and –. The radioactive bands corresponding to the specific reaction products were cut out from the gel and counted for radioactivity. The ratio of the two enzyme activities is given. (B) Silver stained gel of the mutant enzymes isolated on a Ni-NTA–agarose column.
Figure 7.
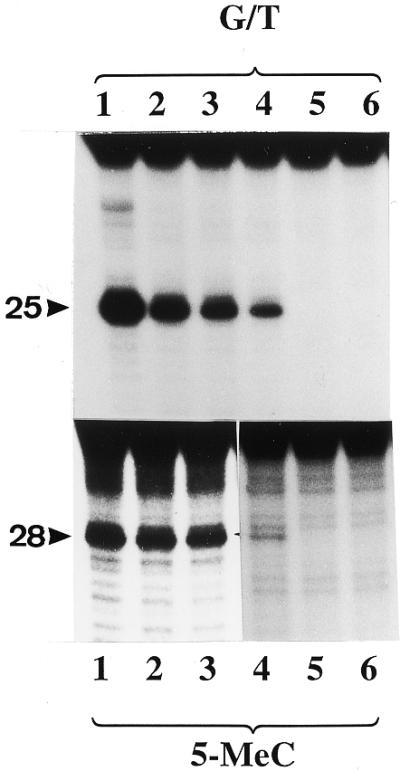
Analysis of N- and C-terminal mutants for G/T mismatch and 5-MeC DNA glycosylase activities. The enzyme preparations shown in Figure 6B were assayed for G/T mismatch and 5-MeC DNA glycosylase activities. The reaction product was treated with NaOH and separated on a 20% urea–polyacrylamide DNA sequencing gel. Lanes 1–5 correspond to full-length protein, ΔN273, ΔN378, ΔN433 and ΔC48, respectively, and lane 6 is a blank. The arrowheads point to the positions of the reaction products which are 25 and 28 nt long for the G/T mismatch glycosylase and 5-MCDG, respectively.
MBD4 and its deletion mutants bind equally well to methylated and non-methylated DNA
The difference in glycosylase activity observed in Figure 2 could possibly be the consequence of a differential binding of MBD4 to the different oligonucleotides substrates. Figure 8 shows that, at an oligonucleotide concentration of 10–7 M (Km for hemimethylated oligo B), regardless of whether the substrate is methylated or not, there is a binding of human MBD4. Chicken MBD4, which does not have the methylated DNA binding domain, also gives a specific band shift with hemimethylated DNA (Fig. 8, compare lanes 1 and 10). Similarly, the deletion mutants of human MBD4 lacking the methylated DNA binding domain also give a discrete binding with the methylated and non-methylated probes (Fig. 8, lanes 2, 3, 5, 6, 8 and 9). Under our incubation conditions, no selective binding of MBD4 to methylated oligonucleotides was observed. Since the above results could possibly represent a non-specific reaction of the DNA with other proteins present in the purified recombinant proteins (Fig. 8, lanes 1, 4, 7 and 10), it was of importance to determine whether the gel shift band assigned to the MBD4–DNA complex (indicated by the arrowheads in lanes 1, 4, 7 and 10) did indeed display 5-MeC DNA glycosylase activity. The two different bands shown in Figure 8 (lane 1) were cut out of the gel, the DNA extracted and analyzed on DNA sequencing gel. The resultant gel (Fig. 9) clearly shows that the upper band a of lane 1 has all the glycosylase activity whereas band b and the free DNA (band c) had no activity. Similar results were obtained for the deletion mutants (results not shown). These results clearly indicate that once the glycosylase or its mutants have reacted with the hemimethylated DNA, they form a stable complex that does not easily dissociate. For example, at concentrations of up to 250 ng of E.coli DNA/20 µl reaction mixture there is no displacement of the complex between the mutant recombinants and the DNA substrate (Fig. 7, lanes 2, 3, 5, 6, 8 and 9). However, one exception was observed for the wild-type protein making a complex with the non-methylated oligonucleotide. In this case no glycosylase activity could be detected (data not shown). These results corroborate those presented in Figure 2 (NM/NM, lanes 1, 2 and 3).
Figure 8.

Gel shift assay of recombinant MBD4 and its mutants with hemimethylated substrate (NM/M), symmetrically-methylated substrate (M/M) and non-methylated substrate NM/NM. Lanes a, human MBD4 (MW 60 000 Da); lanes b, mutant ΔN273; lanes c, mutant ΔN378. Lane ac is the chicken wild-type MBD4 (MW 40 000 Da). Gel shifts were carried out as outlined in Materials and Methods and the reaction product was analyzed in a 5% native polyacrylamide gel. The arrowheads point to the specific complex of the glycosylase and its mutants with the DNA substrate (see also Fig. 9).
Figure 9.
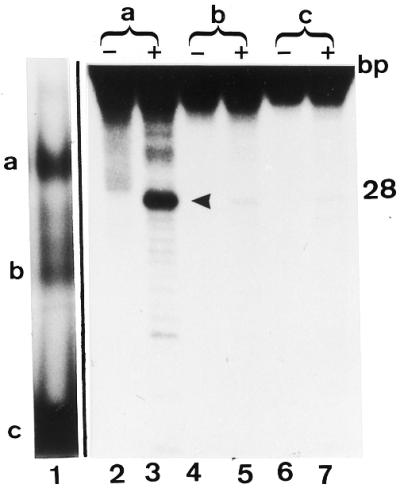
Which of the two gel shift bands a and b seen with the wild-type MBD4 contains 5-MCDG activity? Lane 1 is a gel shift assay of the human MBD4 with the hemimethylated substrate. Bands a and b are the protein DNA complex and c is the free DNA. Gel shift assays were carried out as outlined in Materials and Methods. Lanes 2–7 are the products of the reactions from bands a, b and c analyzed on a 20% acrylamide DNA sequencing gel. In the lanes marked –, the oligonucleotides extracted from bands a, b and c were denatured and analyzed on the sequencing gel whereas for the lanes marked with a +, the product of the reaction was subjected to alkaline hydrolysis before analyzing on the DNA sequencing gel. The size of the correct reaction product of 5-MCDG (arrowhead) is 28 nt.
Footprinting of MBD4 with partially depurinated and depyrimidinated oligonucleotides (missing contact probing of protein–DNA interactions)
Since the wild-type MBD4 and its mutants lacking the methylated DNA binding domain gave specific band shifts with the hemimethylated substrate (Fig. 8) it was of interest to see whether they bind to the same area on the DNA substrate. The basic principle of the assay is based on the partial depurination and depyrimidation of the DNA substrate. From the mixture of modified bases in the oligonucleotide, the protein only binds to those where the bases required for the specific amino acid–base contact are intact. This is just the reverse for DNA repair enzymes such as MBD4 which preferentially recruits those sites which are damaged. In this case, one would logically expect to see a random distribution of MBD4 on the modified DNA. This is clearly not the case. Figures 10 and 11 show that MBD4 preferentially recruits oligonucleotides with abasic sites around the target 5-mCpG. In addition, suppression of the methylated DNA binding domain (see mutant ΔN273) gave the same pattern of binding as the wild-type protein (Fig. 11). The strong signals observed for C in the upper non-methylated strand in the lanes B of A+G and C+T (Fig. 10, lanes 8, 11, 20 and 23) are most probably not only due to the chemical modifications of the probe; they may represent specific nicks caused by the single strand nuclease activity still present in the highly purified recombinant protein.
Figure 10.

Missing contact probing of MBD4 and its mutant Δ273 with partially depurinated and depyrimidinated hemimethylated substrate. Experiment was carried out as outlined in Materials and Methods. C is the control DNA substrate partially depurinated (A+G) or depyrimidinated (C+T), B is the modified substrate bound to the enzyme in the gel shift assay and F is the unbound free modified substrate migrating at the bottom of the gel (Fig. 9). The substrate was hemimethylated. The mCpG is located on the lower strand and the upper strand is non-methylated. Numbers in the lanes refer to the nucleotide positions shown in the DNA sequence of Figure 11 (oligo B).
Figure 11.

Trend analysis of the results shown in Figure 10. From the pool of randomly modified oligos (partial depurination and depyrimidination) MBD4 and its mutant have a selective affinity for some abasic sites. The stronger the affinity to such sites, the stronger are the signals in the B lanes when compared with the C and F lanes. The strongest signals are marked with a closed circle and the weaker one with an open circle, M represents 5-MeC. The numbers indicate the nucleotide position in the DNA substrate (oligo B) which is 50 bp long.
Effect of oligoribonucleotides on the activities of 5-MeC DNA glycosylase and G/T mismatch DNA glycosylase
As we have previously shown, DNA demethylation by the purified 5-MeC DNA glycosylase needs protein and RNA (4,5). It was also shown that oligoribonucleotides complementary to the methylated strand could stimulate the recombinant 5-MeC DNA glycosylase while inhibiting the G/T mismatch DNA glycosylase activity (7). In sharp contrast, the results shown in Figure 12 (upper panel, human MBD4; lower panel, chicken MBD4) clearly indicate that the same oligonucleotide inhibited very strongly both the G/T mismatch DNA glycosylase and the 5-MeC DNA glycosylase activities. The oligoribonucleotide complementary to the opposite strand also inhibited the G/T mismatch glycosylase activity and, to a lesser extent, 5-MeC DNA glycosylase.
Figure 12.
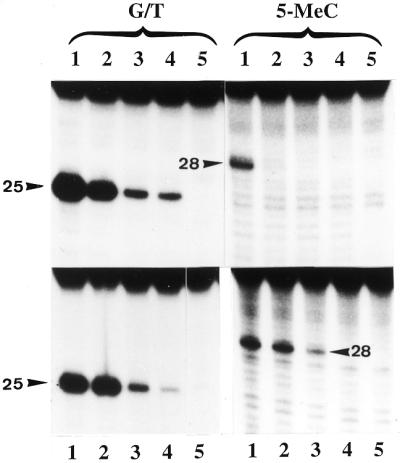
Effect of increasing concentrations of synthetic oligoribonucleotides on the activity of G/T mismatch and 5-MeC DNA glycosylases. In the upper panels the enzyme (human MBD4) was incubated with 0, 0.25, 0.5 and 1 µg of the oligo 5′-GUGACCGGAGC-3′ complementary to the target sequence whereas the lower panels are the enzyme (chicken MBD4) incubated with 0, 0.25, 0.5 and 1 µg of 5′-GCUCCGGUCAC-3′ complementary to the opposite strand. Lane 5 is the blank incubated without protein. The reaction product was subjected to alkaline hydrolysis and analyzed on a 20% acrylamide sequencing gel. The arrowheads point to the correct position of the cleavage products.
Developmental expression pattern of MBD4 RNA in the chicken embryo
Recently we described that transcription of one of the RNA clones (RNA clone 1) which is associated with the DNA demethylation complex is developmental-stage-specific but not tissue-specific (18). Here we investigated by ISH whether or not transcription of the G/T mismatch/5-MeC DNA glycosylase is also developmentally regulated. Figure 13 shows the results of ISH on sections through the eye of a 5-day-old chicken embryo. Figure 13A shows that the expression of the glycosylase is restricted to the same tissues where the RNA helicase and the CpG rich RNA were expressed (7,18). In the cells of the lens which produce the crystalline and which are mitotically inactive there is no trace of the glycosylase mRNA, whereas in the neuroepithelia of the optical cup specific transcripts were detected. As we have shown in the past this tissue consists of proliferating cells (18). Furthermore, in other differentiated tissues like the skeletal muscle in the somites there is no expression of the glycosylase mRNA (data not shown).
Figure 13.

The chicken MBD4 RNA is only transcribed in mitotically active tissues. (A) Section through the eye of a 5-day-old chicken embryo. Transcripts are present in the neuroepithelia of the optical cup (oc) but not in the cells of the lens (arrows) producing the crystalline. (C) Section adjacent to (A) probed with sense RNA (negative control). (B and D) DAPI staining of the corresponding sections.
DISCUSSION
MBD4 was first discovered as a member of the family of mammalian methyl-CpG binding proteins (9). This protein is expressed in many different somatic tissues as well as in ovary and testis (9). MBD4 is localized within the foci of heavily methylated satellite DNA and this localization is disrupted in cells that have a reduced level of CpG methylation (9). MBD4 (MED1) was independently discovered by Bellacosa et al. (11) using the yeast two hybrid system with the DNA mismatch repair protein MLH1 used as a bait. Later on Hendrich et al. (10) found that neither glycosylase nor endonuclease activities could be detected with substrates containing either symmetrically-methylated CpGs or hemimethylated CpG sites. One possible reason for the discrepancy between their results and ours could be the sensitivity of their assay. With the fluorometric assay they could not possibly detect the activity of 5-MeC DNA glycosylase which is about 30-times lower than the G/T mismatch glycosylase. The small but significant activity of 5-MeC DNA glycosylase detected in recombinant MBD4 is most probably not an experimental artefact since the deletion mutants of MBD4 affect the G/T mismatch and 5-MeC DNA glycosylase activities very differently. In the mutants with increasing size of deletions at the N-terminus there is a sharp decrease in the G/T mismatch DNA glycosylase activity. Up to the deletion ΔN378, however, there is basically no major change in the activity of 5-MeC DNA glycosylase (Fig. 8). The low activity of 5-MeC DNA glycosylase present in the recombinant MBD4 may have different reasons. For example, the denaturation and renaturation of the protein isolated from bacterial inclusion bodies may partially irreversibly inactivate 5-MeC DNA glycosylase activity. Alternatively, one cannot exclude specific covalent modifications and/or specific interactions of MBD4 with other proteins which could possibly control the activity of 5-MeC DNA glycosylase. As shown in Figure 4 the base composition surrounding the mCpG has an influence on the catalytic activity of 5-MeC DNA glycosylase. An A+T-rich sequence is a much better substrate than an A+T-poor one. In this case it is conceivable that for the glycosylase reaction it is necessary to have a partial melting of the DNA duplex. This would allow disruption of the hydrogen bonds between G and MeC for the glycosylase reaction to occur. The presence of the methylated DNA binding domain does not seem to be important for the activity of 5-MCDG present in MBD4. In addition, as seen in Figure 8 both the wild-type and the mutant protein lacking the methylated DNA binding domain bind to the DNA substrate. This, together with the results of the missing contact probing (Figs 10 and 11) of the proteins with the DNA showing similar footprints on the DNA substrate, strongly suggest that sequences different from the methylated DNA binding domain control the specificity of the binding of the protein to the CpGs. In addition, as also seen in Figure 8 there is the formation of a complex between recombinant MBD4 and the non-methylated oligonucleotide. However, the formation of the complex did not result in any trace of glycosylase activity (Fig. 2, NM/NM, lanes 1 and 2).
One of the proposed functions of MBD4 (MED1) is to counteract the mutability of 5-MeC (19,20). Recently it has been found that MBD4 is mutated in human carcinomas such as in the hereditary non-polyposis colorectal cancers which are characterized by microsatellite instability (19). Frameshift mutations affecting MBD4 were found in over 40% of the microsatellite unstable sporadic colon cancers (19,20). Based on these results it was proposed that MBD4 could have a tumor suppressor role similar to that for mismatch repair genes (including MSH2, MSH3, MSH6, MLH1, PMS1 and PMS2) that are mutated or down-regulated by aberrant DNA methylation (20 and references therein).
What are the actual biological functions of 5-MeC DNA glycosylase activity associated with MBD4? Since the enzyme prefers hemimethylated DNA over fully methylated CpG pairs one can speculate that in addition to its role in DNA G/T mismatch repair it has either a surveillance function in the genome by removing all random asymmetrically methylated CpGs in the replicating DNA, or it could, in conjunction with other factors, remove specific MeC on specific replicating genes creating non-methylated sites. These two possibilities are not mutually exclusive and experiments to investigate them are underway. The concerted actions of MBD4 and 5-MCDG (7) could then have a dual role in embryos: the repair of DNA and the demethylation of specific genes in the differentiating tissues. However, it is unlikely that these two enzymes could be responsible for the genome wide demethylation in blastocytes (21), since the glycosylase would create a very large number of abasic sites which could, if not immediately repaired, destabilize the DNA structure and jeopardize its integrity.
Acknowledgments
ACKNOWLEDGEMENTS
We would like to thank Mrs Y. C. Jost for typing the manuscript. Many thanks also to Mr P. Müller for the synthesis of the oligonucleotides and to Mrs I. Obergfoell for the photographic work. This work was sponsored by Novartis AG, Basel, Switzerland.
DDBJ/EMBL/GenBank accession no. AF257107
REFERENCES
- 1.Russo V.E.A., Martienssen,R.S. and Riggs,A.D. (1986) Epigenetic Mechanisms of Gene Regulation. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 2.Laird P.W. (2000) In Jones,P.A. and Vogt,P.K. (eds), DNA Methylation and Cancer. Springer Verlag, pp. 119–134.
- 3.Jost J.P., Oakeley,E.J. and Schwarz,S. (1999) In Cheng,X. and Blumenthal,R.M. (eds), S-Adenosyl Methionine Dependent Methyltransferases: Structures and Functions. World Scientific.
- 4.Frémont M., Siegmann,M., Gaulis,S., Mathies,R., Hess,D. and Jost,J.P. (1997) Nucleic Acids Res., 25, 2375–2380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jost J.P., Siegmann,M., Thiry,S., Jost,Y.C., Benjamin,D. and Schwarz,S. (1999) FEBS Lett., 449, 251–254. [DOI] [PubMed] [Google Scholar]
- 6.Jost J.P., Schwarz,S., Hess,D., Angliker,H., Fuller-Pace,F.V., Stahl,H., Thiry,S. and Siegmann,M. (1999) Nucleic Acids Res., 27, 3245–3252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhu B., Zheng,Y., Hess,D., Angliker,H., Schwarz,S., Siegmann,M., Thiry,S. and Jost,J.P. (2000) Proc. Natl Acad. Sci. USA, 97, 5135–5139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jost J.P., Frémont,M., Siegmann,M. and Hofsteenge,J. (1997) Nucleic Acids Res., 25, 4545–4550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hendrich B. and Bird,A. (1998) Mol. Cell. Biol., 18, 6538–6547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hendrich B., Hardeland,U., Ng,H.H., Jiricny,J. and Bird,A. (1999) Nature, 401, 301–304. [DOI] [PubMed] [Google Scholar]
- 11.Bellacosa A., Cicchillitti,L., Schepis,F., Riccio,A., Yeung,A.T., Matsumoto,Y., Golemis,E.A., Genuardi,M. and Neri,G. (1999) Proc. Natl Acad. Sci. USA, 96, 3969–3974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jost J.P., Siegmann,M., Sun,L. and Leung,R. (1995) J. Biol. Chem., 270, 9734–9739. [DOI] [PubMed] [Google Scholar]
- 13.Maxam A.M. and Gilbert,W. (1980) Methods Enzymol., 65, 499–560. [DOI] [PubMed] [Google Scholar]
- 14.Brunelle A. and Schleif,R.F. (1987) Proc. Natl Acad. Sci. USA, 84, 6673–6676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liang H., Pawlak,A. and Jost,J.P. (1991) In Jost,J.P and Saluz,H.P. (eds), A Laboratory Guide to In Vitro Studies of Protein–DNA Interactions. Birkhäuser Verlag, pp. 93–109.
- 16.Schaeren-Wiemers N. and Gerfin-Moser,A. (1993) Histochemistry, 100, 431–440. [DOI] [PubMed] [Google Scholar]
- 17.Jost J.P. (1993) Proc. Natl Acad. Sci. USA, 90, 4684–4688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schwarz S., Bourgeois,C., Soussaline,F., Homsy,C., Podestà,A. and Jost,J.P. (2000) Eur. J. Cell Biol., 79, 488–494. [DOI] [PubMed] [Google Scholar]
- 19.Riccio A., Aaltonen,L.A., Godwin,A.K., Loukola,A., Percesepe,A., Salovaara,R., Masciullo,V., Gennordi,M., Paravatou-Petsotas,M., Bassi,D.E., Ruggeri,B.A., Klein-Szanto,A.J.P., Testa,J.R., Neri,G. and Bellacosa,A. (1999) Nature Genet., 23, 266–268. [DOI] [PubMed] [Google Scholar]
- 20.Bader S., Walker,M., Hendrich,B., Bird,A., Bird,C., Hooper,M. and Wyllie,A. (1999) Oncogene, 18, 8044–8047. [DOI] [PubMed] [Google Scholar]
- 21.Mayer W., Niveleau,A., Walter,J., Fundele,R. and Haaf,T. (2000) Nature, 403, 501–502. [DOI] [PubMed] [Google Scholar]