


Abstract
Reverse transcription of the Human Immunodeficiency Virus type I (HIV-1) RNA genome is primed by a cellular tRNA–lys3 molecule that binds to the primer binding site (PBS). The PBS is predicted to be part of an extended RNA structure, consisting of a small U5-PBS hairpin and a large U5-leader stem. In this study we stabilized the U5-leader stem of HIV-1 to study its role in reverse transcription. We tested in vitro synthesized wild-type and mutant templates in primer annealing, initiation and elongation assays. Stabilization of the stem inhibits the initiation of reverse transcription, but not the annealing of the tRNA primer onto the PBS. These results suggest that stabilization of the stem results in occlusion of a sequence motif that is involved in an additional interaction with the tRNA–lys3 primer and that is needed to trigger the initiation of reverse transcription. The stable structure was also found to affect the elongation of reverse transcription, causing the RT enzyme to pause upon copying 7–8 bases into the extended base paired stem. The stabilizing mutations were also introduced into proviral constructs for replication studies, demonstrating that the mutant viruses have a reduced replication capacity. Analysis of a revertant virus demonstrated that opening of the stabilized U5-leader stem can restore both virus replication and reverse transcription.
INTRODUCTION
Reverse transcription of retroviral genomes is primed by a cellular tRNA molecule that anneals to an 18 nt primer binding site (PBS) that is located in the 5′ untranslated leader region of the viral RNA genome (Fig. 1A) (1,2). For several retroviruses, this part of the genomic RNA has been suggested to fold a complex secondary structure (3–11). In the genome of Human Immunodeficiency Virus type 1 and 2 (HIV-1 and -2), the PBS is predicted to be part of an extended RNA structure (10,12). This structure consists of a small U5-PBS hairpin that contains part of the PBS, and a large stem region formed by sequences of the upstream U5 region and the downstream leader region, the U5-leader stem (Fig. 1A and B). Several sequences in the U5-PBS-leader region have been proposed to interact with the tRNA–lys3 primer to stimulate reverse transcription (13–17). We previously demonstrated that the small U5-PBS hairpin of HIV-1 is involved in the correct placement of the tRNA primer onto the PBS (18,19), and the A-rich loop of this hairpin has been suggested to interact with the U-rich anticodon of the tRNA–lys3 molecule (15,20,21). In the avian Rous Sarcoma Virus (RSV), an extended stem region similar to the U5-leader stem of HIV-1 is involved in the initiation of reverse transcription (22,23). A sequence motif in the U5 region was identified that may interact with the TΨC arm of the tRNA–trp primer, but the structure of the U5-leader stem also seems important for efficient initiation of reverse transcription in RSV.
Figure 1.

Schematic of the HIV-1 5′ untranslated leader RNA. (A) Outline of the RNA secondary structure motifs in the HIV-1 leader RNA. The tRNA–lys3 primer binds to the PBS (marked in gray) that is part of an extended RNA structure. This structure consists of a small U5-PBS hairpin that occludes part of the PBS, and a large stem region formed by upstream U5 and downstream leader sequences. This U5-leader stem is highlighted (shaded) and is the topic of this study. The position of several primers used in the reverse transcription assays is shown. (B) Shown is the wild-type U5-leader stem and several mutants thereof that were designed to increase the thermodynamic stability. The U5-leader stem of mutant s1 was stabilized by deletion of the left arm and the introduction of two U residues (marked by a black box). In mutant s2, the right arm was deleted and replaced by two residues (UC) (marked by a black box). The double mutant s1/2 contains both mutation s1 and s2. The thermodynamic stability of the structures is indicated at the bottom (ΔG in kcal/mol) and was calculated using the Zuker algorithm (38). Mutations observed in a revertant virus of mutant s2 are shown on the right. A 9 nt segment was deleted (marked by an open box) and replaced by a 7 nt sequence (dark gray box) that most likely results from duplication of the upstream sequence (duplication marked by an arrow, position of one non-identical nucleotide indicated by an asterisk). This mutation results in refolding of the U5-leader region and the thermodynamic stability of this alternative structure is shown for reference (ΔG = –32.3 kcal/mol). The position and intensity of the RT pauses observed in reverse transcription assays is indicated by dots. Open dots represent RT pauses observed with PBS-bound primers (lys21 and tRNA–lys3 primer) (Fig. 3), closed dots represent RT pauses in assays initiated from the downstream AUG primer (Fig. 4).
In the present study we introduced specific mutations to stabilize the predicted U5-leader stem in HIV-1 to analyze the role of this structure in reverse transcription. For instance, the occlusion of important U5 sequence motifs in a stabilized U5-leader stem could interfere with tRNA–lys3 annealing and/or initiation of reverse transcription. Furthermore, stable RNA structure in the template has been reported to interfere with the elongation of reverse transcription (24–26). Therefore, we measured tRNA annealing and both initiation and elongation of reverse transcription on the stabilized U5-leader stem templates in in vitro reverse transcription assays. The same mutations were also introduced in proviral constructs to study the replication capacity of the mutant viruses. Stabilization of the U5-leader stem impaired virus replication and partially inhibited initiation of reverse transcription, which was not caused by reduced binding of the tRNA–lys3 primer. The stable RNA structure also interfered with the elongation of reverse transcription, causing RT to pause upon copying 7–8 bases into the extended base paired stem. Analysis of a revertant virus demonstrated that opening of the stabilized U5-leader stem can restore both virus replication and reverse transcription.
MATERIALS AND METHODS
DNA constructs
A derivate of the full-length proviral HIV-1 clone pLAI was used to produce wild-type and U5-leader stem mutated viruses. This construct pLAI-R37 was described previously and contains a unique U5 region in the 5′ long terminal repeat (LTR) (9). The 3′LTR was truncated at the SacI site within the R region, and the chloramphenicol acetyltransferase (cat) gene and simian virus 40 (SV40) polyadenylation site were inserted at this position. Nucleotide numbers refer to positions on the genomic RNA transcript, with +1 being the capped G residue. For mutation of the U5-leader stem, the construct Blue-5′LTR was used (27), which contains a XbaI–ClaI fragment of HIV-1 encompassing the 5′LTR, PBS, leader and the 5′-end of the gag gene (positions –454 to +376) cloned into pBluescript (Stratagene). The U5-leader stem was mutated by oligonucleotide-directed in vitro mutagenesis with a Muta-Gene Phagemid In Vitro Mutagenesis Kit (Bio-Rad). Oligonucleotides used are: S1, 5′-+118TGTGTGACTCTGGT-TT-CCCTTTTAGTCAGTG+164-3′ and S2, 5′-+208GAAAGGGAAACCAGAG-TC-ACGCAGGACTCGGCT+249-3′ (deletion in-between dashes, inserted two nucleotides underlined). For sequence analysis, the 5′LTR-leader region was PCR amplified with the sense R region primer T7-1 (positions –54 to –34) with 5′-flanking T7 RNA polymerase promoter sequence and the antisense primer AUG (positions +348 to +368, with six additional nucleotides at its 5′-end). These PCR products were sequenced with the DYEnamicTM Direct cycle sequencing kit (Amersham) and an Applied Biosystems 373 DNA sequencer. Subsequently, the mutated XbaI–ClaI fragments were introduced into the proviral clone pLAI-R37, which again was verified by sequence analysis.
Cells, viruses and transfection
SupT1 T cells were grown in RPMI 1640 medium supplemented with 10% fetal calf serum at 37°C and 5% CO2. SupT1 cells (5 × 106 ) were transfected with 2 µg of the HIV-1 proviral constructs by electroporation (250 V, 960 µF). Fresh SupT1 cells (0.5 × 106 ) were added after transfection to support viral replication. Cells were split 1 to 10 twice a week. For the selection of revertant viruses, the transfected cells were passaged up to 9 weeks. At the peak of virus production (as witnessed by the appearance of large syncytia) a sample of the culture supernatant was used to infect fresh SupT1 cells. Initially 100 µl was used to infect the SupT1 cells, but we gradually used less culture supernatant (minimally 0.1 µl) per passage. At each passage, cells and supernatant samples were stored at –70°C. To study the increased replication capacity of the mutant s2 virus upon prolonged culturing, fresh SupT1 cells were infected with an equal amount of virus (10 ng CA-p24) sampled at weeks four and nine.
C33A cells were grown in Dulbecco’s modified Eagle’s medium containing 10% fetal calf serum at 37°C and 5% CO2. For the transient production of virions, C33A cells were transfected by the calcium phosphate method. Cells were grown in 20 ml of culture medium in a 75 cm2 flask to 60% confluency. Thirty micrograms of the proviral construct in 880 µl of water was mixed with 1 ml of 50 mM HEPES (pH 7.1), 250 mM NaCl, 1.5 mM Na2HPO4 and 120 µl of 2 M CaCl2, incubated at room temperature for 20 min, and added to the culture medium. The culture medium was changed after 16 h.
Analysis of the s2 revertant
Infected SupT1 were pelleted by centrifugation at 4000 r.p.m. for 4 min and washed with phosphate-buffered saline. The cells were resuspended in 10 mM Tris–HCl (pH 8.0), 1 mM EDTA, 0.5% Tween 20 and incubated with 200 µg of proteinase K per ml at 56°C for 1 h and at 95°C for 10 min to isolate total cellular DNA. The 5′LTR-leader region was PCR-amplified from cellular DNA with the sense R region primer T7-1 (positions –54 to –34) and the antisense primer AUG (positions +348 to +368). This PCR product was sequenced with the DYEnamicTM Direct cycle sequencing kit (Amersham) and an Applied Biosystems 373 DNA sequencer.
CA-p24 ELISA and RT enzyme assay
CA-p24 levels in the culture medium were determined by ELISA (28). RT assays were performed as described previously (29).
Synthesis of RNA templates
The wild-type and mutant pBlue-5′LTR plasmids were used as template for PCR amplification and subsequent in vitro transcription. The 5′LTR region was PCR amplified with the sense primer T7-2 (positions +1 to +20) with 5′-flanking T7 RNA polymerase promoter sequence, and the antisense primer AUG (positions +348 to +368). The PCR fragments were phenol extracted, precipitated and dissolved in water. The in vitro transcription reaction was performed in 10 µl transcription buffer (40 mM Tris–HCl pH 7.5, 2 mM spermidine, 10 mM DTT and 12 mM MgCl2) containing 0.5 µg DNA template, 0.06 µmol ATP, GTP, CTP and UTP, 10 U T7 RNA polymerase (Boehringer) and 20 U RNase inhibitor (Boehringer), and incubated for 4 h at 37°C. Upon DNase treatment and phenol extraction, the unincorporated free nucleotides were removed by passage through a Sephadex G-50 column. Subsequently, the RNA was ethanol precipitated and dissolved in renaturation buffer (10 mM Tris–HCl pH 7.5, 100 mM NaCl). The RNA was renatured by incubation at 85°C for 2 min, followed by slow cooling to room temperature, and stored at –20°C. The RNA concentration was subsequently measured by UV spectroscopy.
Reverse transcription assays
The in vitro synthesized RNA template (10 ng) was incubated either with 1.5 µg calf liver tRNA (6 pmol total tRNA, of which ∼1.2 pmol tRNA–lys3; Boehringer) or with 20 ng DNA primer in 12 µl annealing buffer (83 mM Tris–HCl pH 7.5, 125 mM KCl) at 85°C for 2 min, 65°C for 10 min, followed by cooling to room temperature over a 1 h period. The primer was extended by 1 nt by addition of 6 µl RT(–) buffer (9 mM MgCl2, 30 mM DTT, 150 µg/ml actinomycine D), 1 µl [α-32P]dCTP and 0.5 U HIV-1 RT (MRC) and reverse transcription was performed for 30 min at 37°C. cDNA synthesis was accomplished in the similar RT(+) buffer, containing all dNTPs (30 µM dATP, dGTP and dTTP and 1.5 µM dCTP), 0.3 µl [α-32P]dCTP and 0.5 U HIV-1 RT (MRC). In the PBS occupancy assay, the RNA template was incubated simultaneously with 1.5 µg calf liver tRNA and 20 ng of AUG primer and reverse transcription was performed in RT(+) buffer. The cDNA products were precipitated in 0.3 M sodium acetate pH 5.2 and 70% ethanol at –20°C, dissolved in formamide loading buffer and analyzed on a denaturing 6% polyacrylamide–urea sequencing gel. The antisense primers used are: polyA (positions +77 to +104), lys21 (positions +182 to +202) and AUG (positions +348 to +368, with six additional nucleotides at its 5′-end). Sequence reactions initiated from the BB-3 (positions +215 to +245) and AUG primer on the pLAI DNA template were performed using the Sequenase kit 2.0 (Amersham). These sequence reactions were included on the sequencing gels to determine the exact position of RT pause sites.
RESULTS
Design of the stabilized U5-leader stem mutants
To study the role of the U5-leader stem in reverse transcription, we introduced mutations that stabilize this base paired stem region. The U5 region is encoded by the LTR that is present at both the 5′- and 3′-end of the HIV-1 proviral genome. Mutations introduced into the U5 region of the 5′LTR will be inherited in both LTRs of the progeny. However, the presence of a wild-type 3′LTR may result in reappearance of the wild-type sequence by a recombination event. We therefore introduced the mutations in a derivative of the proviral clone pLAI in which part of the 3′LTR, including the polyadenylation signal and the complete U5 region, is deleted. An SV40 polyadenylation site was placed downstream of the HIV-1 sequences to allow efficient polyadenylation of the viral transcript. Transfection of cells with this vector results in the production of viruses with a mutant U5 region in the 5′LTR. Subsequent infection of the T cells and reverse transcription of the viral RNA genome will produce proviral genomes with a full-length 5′LTR and 3′LTR with the mutant U5 sequence.
The upper part of the U5-leader stem was stabilized in mutant s1 by deletion of the left arm (Fig. 1B, positions 132–149), which was replaced by two U residues to complement the two bulged A nucleotides on the opposite side of the stem at positions 215–216 (all nucleotide numbers relate to the wild-type HIV-1 RNA). This results in an extended stem of 16 consecutive base pairs and increases the thermodynamic stability as calculated for the complete PBS domain shown in Figure 1B, from –36.6 kcal/mol for wild-type to –40.5 kcal/mol for mutant s1. In mutant s2, the lower part of the U5-leader stem was stabilized by deletion of the right arm (positions 224–234), which was replaced by two residues (UC) that can base pair with the GA bulge on the left side at positions 123–124. An extended stem of 14 uninterrupted base pairs can form, which increases the thermodynamic stability of the PBS domain to –42.3 kcal/mol. The combination of both mutations in the double mutant s1/2 produces a stem of 23 consecutive base pairs and increases the thermodynamic stability to –49.6 kcal/mol (Fig. 1B).
Replication capacity of the mutant viruses
To study the replication potential of viruses with U5-leader stem mutations, we transfected wild-type and mutant proviral genomes into the SupT1 T cell line. These cells express the CD4-CXCR4 receptor and are fully susceptible for replication of the HIV-1 LAI strain. Virus replication was followed by measuring the CA-p24 level in the culture medium at several days post-transfection. Transfection with 2 µg of the proviral constructs demonstrated delayed replication of mutant s1 compared with the wild-type virus, whereas mutation s2 abolished virus replication (Fig. 2A). The double mutant s1/2 is also replication-impaired. Three independent transfections were performed, with similar results. Thus, stabilization of the U5-leader stem by mutation s2 affects virus replication more severely than stabilization by mutation s1.
Figure 2.

(A) Replication of wild-type (wt) and U5-leader stem mutant s1, s2 and s1/2. SupT1 cells were transfected with 2 µg of the proviral constructs. CA-p24 production was measured in the culture medium at several days post-transfection. (B) Improved replication kinetics of the mutant s2 virus upon prolonged culturing. SupT1 cells were infected with an equal amount of virus (10 ng) sampled at week four and week nine of the evolution experiment. CA-p24 production was measured in the culture medium at several days post-infection.
Evolution of the replication-impaired s2 mutant
The SupT1 cells transfected with the U5-leader stem mutants were cultured for a prolonged time to select for revertant viruses with increased replication capacity. Mutant s1 replicated too efficiently to allow for the selection of revertants within a reasonable time span. Neither did we obtain a revertant virus for the double mutant s1/2, probably because its replication defect is too severe. However, the s2 virus started to replicate slowly after 4 weeks. We passaged this virus for an additional 5 weeks and observed a further increase in replication kinetics. This is demonstrated upon infection of fresh SupT1 cells with an equal amount of virus isolated at week four and week nine (Fig. 2B).
The genome of the s2 revertant virus should be altered to restore efficient replication. We therefore isolated total DNA from infected cells at week nine, PCR-amplified the 5′LTR-leader region and performed population-based sequencing of the DNA fragment. A 9 nt segment including the s2 mutation was deleted and replaced by a 7 nt sequence that most likely results from duplication of the upstream sequence (positions +208 to +214) with 1 nt substitution, as is illustrated in Figure 1B. This mutation affects base pairing in this part of the stem region and reduces the stability of the U5-leader stem from ΔG = –42.3 kcal/mol to ΔG= –32.3 kcal/mol (Fig. 1B). These combined results indicate that virus replication is severely inhibited by a too stable U5-leader stem.
The U5-leader stem is involved in the initiation of reverse transcription
The U5-leader stem may influence the process of reverse transcription because important sequence motifs including the PBS are part of this extended RNA domain. Furthermore, a similar U5-leader stem in avian retroviruses is involved in reverse transcription. To test this, we performed in vitro reverse transcription reactions with the wild-type and mutant HIV-1 RNA templates. The position of the primers used in the different reverse transcription assays is shown in Figure 1A. We used in vitro transcribed RNA templates encompassing the complete untranslated leader region (positions +1 to +368) and the natural tRNA–lys3 primer to initiate reverse transcription. The tRNA primer was heat-annealed at 85°C and reverse transcription was initiated by the addition of [α-32P]CTP and HIV-1 RT enzyme. This results in the extension of the 76 nt tRNA primer by 1 nt (Fig. 3, lanes 5–8). The initiation products were quantified and corrected for the amount of input viral RNA template as determined by a regular reverse transcription reaction with the upstream DNA primer polyA (Fig. 3, lanes 1–4) The position of this primer is indicated in Figure 1A. The results (summarized in Table 1) indicate that the efficiency of reverse transcription is reduced for all mutant templates.
Figure 3.

Reverse transcription assays on the wild-type and U5-leader stem mutant templates. The amount of input viral RNA was quantified by DNA-primer extension with the polyA primer that produces a 104 nt product (lanes 1–4, relative position shown in Fig. 1A). The natural tRNA–lys3 primer was annealed onto the RNA templates and 1 nt was incorporated by addition of HIV-1 RT enzyme and dCTP (lanes 5–8). The 76 nt tRNA is extended by dCTP to produce a 77 nt radiolabeled product. Two PBS-primers, lys21 (lanes 9–12) and the natural tRNA–lys3 primer (lanes 13–16), were extended HIV-1 RT enzyme in the presence of all dNTPs. Extension of lys21 results in a 202 nt product and the tRNA primer produces a 257 nt tRNA–cDNA product on the wild-type template. These products are shorter for the s1 and s1/2 templates due to the U5-deletion. Several shorter cDNA products are visible that result from RT pausing. Stabilization of the stem in mutant s1, s2 and in particular s1/2 resulted in specific major pause sites (marked by arrows). To accurately determine the position of RT pausing, a sequence reaction was analyzed in parallel (lanes 17–20). The position of the pauses on the mutant templates is illustrated in Figure 1B.
Table 1. tRNA-primed reverse transcription on wild-type and mutant HIV-1 templates.
| tRNA binding (%) | 1 nt incorporation (%) | cDNA synthesis (%) | |
|---|---|---|---|
| wt | 100a | 100a | 100 a |
| s1 | 100 | 20 | 5 |
| s2 | 100 | 37 | 25 |
| s1/2 | 100 | 31 | 5 |
| s2 rev | ND | 250 | 250 |
aSet at 100%.
ND, not determined.
The reverse transcription defect may occur at the level of initiation, but the amount of tRNA primer that is annealed onto the PBS may also be reduced for the mutant templates. To discriminate between these two possibilities, the tRNA occupancy of the PBS was determined. The tRNA primer was annealed onto the RNA template and this complex was subsequently used for extension of the DNA primer AUG that is positioned downstream of the PBS (Fig. 1A). We used the AMV–RT enzyme to selectively extend the DNA primer because this enzyme is unable to extend the tRNA primer (30 and unpublished results). When the PBS is occupied by the tRNA primer, extension of the AUG primer will stop prematurely to produce a cDNA product of ∼175 nt, whereas free RNA templates will produce a full-length cDNA product of 374 nt on the wild-type template. Control reactions were performed with the upstream polyA primer and the AUG primer in the absence of tRNA (Fig. 4, lanes 1–4 and 5–8, respectively). Extension of the downstream AUG primer in the presence of the annealed tRNA exclusively yielded the 175 nt stop product with the wild-type and mutant templates (Fig. 4, lanes 9–12). The results are summarized in Table 1, and indicate that all templates are fully occupied by the tRNA–lys3 primer. These combined results indicate that the reverse transcription defect on the mutant templates results from reduced initiation.
Figure 4.

PBS occupancy of the wild-type and mutant templates. The tRNA–lys3 primer and downstream DNA primer AUG were simultaneously heat-annealed onto the wild-type and mutant templates. See Figure 1A for the position of the primers. Subsequently, the DNA primer was selectively extended by the addition of AMV–RT enzyme and dNTPs (lanes 9–12). When the PBS is occupied by tRNA, the cDNA initiated at the AUG primer stop prematurely to generate a 175 nt product. Free RNA templates will produce a full-length cDNA product of 374 nt (lanes 5–8). Control reactions were performed with the polyA primer (lanes 1–4). Besides the full-length cDNA product of 374 nt, extension of the AUG primer produces several shorter cDNAs that result from RT pausing. Stabilization of the U5-leader stem in mutant s1, s2 and in particular s1/2, resulted in new major pause sites (marked by arrows). To accurately determine the position of RT pausing, an AUG-primed sequence reaction was analyzed in parallel (lanes 13–16). The position of the pauses on the mutant templates is illustrated in Figure 1B.
Stabilization of the U5-leader stem causes the RT enzyme to pause
Stable stem–loop structures have been reported to interfere with elongation of reverse transcription. The mutations introduced in the U5-leader stem may also result in RT pauses. We therefore studied the elongation of reverse transcription. Reactions on the wild-type and mutant templates were initiated by the tRNA–lys3 primer or the DNA primer lys21, which is also complementary to the PBS. The tRNA and DNA primers were heat-annealed at 85°C onto the in vitro transcribed templates, and extended by the addition of all dNTPs and HIV-1 RT enzyme. Extension of the lys21 primer on the wild-type template produces a full-length cDNA product of 202 nt. Besides this product, several shorter cDNAs were detected that result from RT pausing (Fig. 3, lane 12). The same pause sites were visible for the mutant templates (lanes 9–11), with a predicted change in cDNA length for the s1 and s1/2 templates due to the deletion in the U5 region. Most importantly, new major stop products were observed for mutant s1 and s2, and in particular for mutant s1/2 (Fig. 3, indicated by arrows). To accurately determine the position of RT pausing, a sequence reaction was analyzed in parallel (lanes 17–20). The position of the RT pauses induced by the mutations in the U5-leader stem is summarized in Figure 1B. On the s1 template, the RT enzyme pauses at a position that overlaps the s1 mutation. However, the RT enzyme also paused on the left side of the stem in mutant s2, which is opposite the s2 mutation. This result strongly suggests that the pauses are induced by secondary structure in the RNA template, and not by the mutant template sequence. The intensity of the s1 pause products increased on the template of the s1/2 double mutant. This finding confirms the correlation between stable template RNA structure and RT pausing. We observed similar RT pauses on the mutant templates in tRNA-primed reverse transcription (Fig. 3, lanes 13–16). Due to the difference in length of the tRNA (76 nt) versus lys21 (21 nt) primer, these products migrate more slowly in the gel. Because of the initiation and elongation defects observed for the mutant templates, a strongly reduced level of full-length tRNA–cDNA product is obtained. Mutant s1 and s1/2 produce only 5% of the wild-type cDNA level and mutant s2 yields 25% cDNA synthesis (Table 1).
To probe the effect of the stabilized U5-leader stem on reverse transcription in the region downstream of the PBS, we performed assays with the downstream AUG primer (Fig. 4, lanes 5–8). Extension of this primer also yielded new RT pauses that are specific for the mutant templates (stops indicated by arrows in Fig. 4). A sequence reaction primed by AUG was analyzed in parallel to accurately determine the position of RT pausing (Fig. 4, lanes 13–16). The position of the RT pauses induced by the mutations in the U5-leader stem is summarized in Figure 1B. On the mutant s2 template, the RT enzyme paused 2–4 nt after copying the s2 mutation. The RT enzyme also paused on the right side of the stem in mutant s1, opposite the position of the s1 mutation. The intensity of the s2 pause products increased on the s1/2 template. These results demonstrate that the pauses are induced by secondary structure in the mutant RNA templates. The combined results with the mutant templates and the primers lys21, tRNA and AUG indicate that structure-induced RT pauses occur upon penetrating the stable stem region for 7–8 bp (Fig. 1B). Extension of the lys21 primer on the s1/2 template results in a pausing pattern similar to that on the s1 template, whereas extension of the AUG primer on the s1/2 template results in a pausing pattern similar to that on the s2 template. Apparently, the RT enzyme pauses upon copying the first 7–8 bp of the stabilized stem, and the subsequent pause sites are not observed. This observation strengthens the finding that the RT pauses are induced by the secondary structure, and not specific sequences, in the mutant RNA templates.
The s2 revertant template (Fig. 1B) was also studied in reverse transcription assays (Fig. 5). The tRNA-extension defect of the s2 mutant (Fig. 5, lane 7) was much improved in the s2 revertant (lane 8). The RT pauses observed on the s2 template with the lys21 primer (indicated on the left) are resolved on the s2 revertant template. Interestingly, tRNA-primed cDNA synthesis on the s2 revertant template was 2.5-fold more efficient than cDNA synthesis on the wild-type template. We therefore also studied initiation of reverse transcription in the 1 nt incorporation assay (lanes 10–12). This demonstrates that the initiation defect of mutant s2 is restored by the reversion-based mutation. In fact, the initiation efficiency on the s2 revertant template is 2.5-fold more efficient compared with the wild-type template. These results are summarized in Table 1. Thus, destabilization of the stem from ΔG = –36.6 for the wild-type template to ΔG = –32.3 for the s2 revertant (Fig. 1B) enhances the initiation of reverse transcription. These results suggest that the wild-type RNA structure may have a negative effect on the process of reverse transcription.
Figure 5.
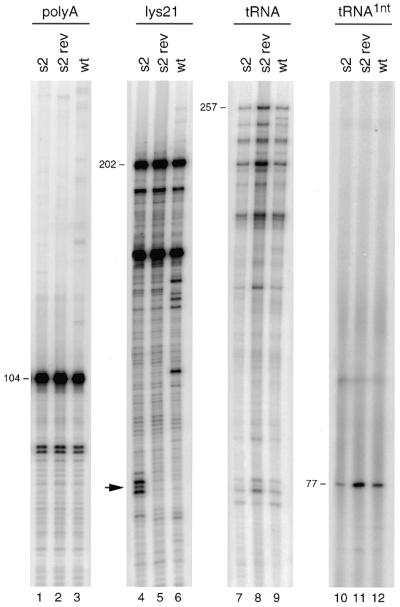
Reverse transcription on the s2 revertant template. The amount of input viral RNA was quantified by reverse transcription with the polyA primer (lanes 1–3). The DNA primer lys21 (lanes 4–6) and tRNA–lys3 primer (lanes 7–9) were annealed onto the RNA templates and reverse transcription was initiated by the addition of HIV-1 RT enzyme and dNTPs. In addition, initiation of reverse transcription was measured in the 1 nt incorporation assay (lanes 10–12).
Mutation of the U5-leader region does not affect viral gene expression
The replication defect of mutant s2 is more severe than that of mutant s1 (Fig. 2A) whereas the reverse transcription defect of mutant s2 is less pronounced than that of mutant s1 (Table 1). This suggests that mutation s2 affects additional steps in the viral replication cycle, either as part of the viral RNA or proviral DNA genome. For instance, several studies reported the presence of transcriptional enhancers in this leader region of the HIV-1 genome (31–34). We therefore tested the effect of the mutations on viral gene expression. C33A cells (human cervix carcinoma cells that are not susceptible for HIV-1 infection) were transfected with the wild-type and mutant proviral vectors and the level of viral gene expression was measured. Virus production was monitored by measuring the amount of CA-p24 and virion-associated RT activity in the culture medium. No significant differences in virus production were observed between the wild-type and mutant constructs (Fig. 6). In addition, a normal amount of viral RNA and viral proteins was measured in transfected cells by northern blot and western blot analysis, respectively (results not shown). These combined results demonstrate that stabilization of the U5-leader stem does not affect viral gene expression (e.g. transcription and translation) and virion assembly. The additional viral function(s) affected by mutation s2 currently remain unknown.
Figure 6.

Virus production in cells transiently transfected with the wild-type and mutant proviruses. C33A cells were transfected with the wild-type and mutant proviral constructs and virus production was measured after three days by CA-p24 ELISA. In addition we measured the virion-associated RT activity in the culture medium. Both parameters of virus production were set at 1 for the wild-type construct.
DISCUSSION
The untranslated leader region of the HIV-1 RNA genome encodes the extended U5-leader stem structure that encompasses sequences involved in reverse transcription. Mutations were introduced in this structure to study its role in viral replication and reverse transcription. Stabilization of this structure by mutation s1 on the left side of the stem resulted in delayed viral replication, whereas replication was completely impaired by mutation s2 on the right side of the stem, and the double mutant s1/2. Through prolonged culturing of mutant s2 we obtained a revertant virus with improved replication capacity. Analysis of this revertant showed a 9 nt deletion and a 7 nt insertion near the position of the s2 mutation. These changes open the stabilized stem structure, but do not restore the wild-type sequence or the wild-type structure in this part of the stem. This suggests that the stability of the U5-leader stem is important for viral replication.
To study the putative role of the U5-leader stem in the process of reverse transcription, we performed in vitro reverse transcription assays. Normal amounts of tRNA–lys3 primer can be annealed onto the PBS of the mutant templates, but initiation of reverse transcription is reduced compared with the wild-type template. Apparently, the primer–template duplex formed on the stabilized U5-leader stem templates is either not recognized or extended by the HIV-1 RT enzyme. This result is consistent with the idea that additional interactions exist between tRNA–lys3 and the U5-leader region (13–17), and the introduced mutations in this region may interfere with these interactions. The U5-leader sequences involved may either be deleted in the mutant templates, or occluded within the stabilized U5-leader stem, thus making them less available for base pairing with tRNA–lys3. Alternatively, the structure of the wild-type U5-leader stem may play an active role in initiation of reverse transcription, as was demonstrated for RSV (22). Interestingly, we demonstrate that full reverse transcription activity can be restored in a revertant of mutant s2 by a deletion-insertion event that opens the stem, but does not restore the wild-type sequence and structure. This finding is consistent with the idea that s2-stabilization results in occlusion of a sequence that interacts with tRNA–lys3 during initiation of reverse transcription. In fact, the initiation efficiency of this s2 revertant significantly exceeds that of the wild-type template, indicating that the U5-leader stem of the wild-type template is partially inhibitory. These results suggest that the wild-type U5-leader stem may play a regulatory role in reverse transcription by controlling the timing and/or efficiency of the initiation step. This mechanism may preclude premature initiation of reverse transcription in the virus-producing cell, thereby restricting reverse transcription to viral RNA genomes that are packaged in virion particles. Because this in vitro reverse transcription assay seems rather sensitive to test the contribution of accessory U5-leader sequences, we are currently using this approach to map in more detail the HIV-1 RNA sequences that interact with the tRNA–lys3 primer.
Stabilization of the U5-leader stem also interferes with the elongation of reverse transcription. Somewhat to our surprise, the RT enzyme does not pause at the base of the stabilized stem region, but rather upon penetrating the structure for 7–8 bp. This consensus stop position was observed with three mutant templates and three different primers. Previous studies indicated that the position of structure-induced pause sites is rather diverse. RT has been reported to pause at the base of stem–loop structures (24,25,35), but RT pausing has also been reported to occur either 6–10 nt ahead (25,26) or 4–8 nt behind (26) a stable structure in the RNA template. In addition, the formation of an alternative structure in the RNA template or nascent cDNA was demonstrated to result in RT pausing (36). Pausing ahead of a stable stem region can occur at the base of the helix, which may reflect the inability of the RT enzyme to open the first base pair. However, pause sites 6–10 nt ahead of secondary structure have also been reported, which may reflect the collision of the elongating RT, which covers 7 nt upstream of the cDNA extension point, with the stem. RT pausing 4–8 nt behind a secondary structure in the RNA template was suggested to be caused by formation of stable secondary structure in the nascent cDNA. Because enzymatic probing experiments (37) indicate that up to 25 template nucleotides behind the polymerization site are protected by the RT enzyme, the formation of stable cDNA structure within the RT enzyme may trigger pausing of the polymerase. We currently have no mechanistic explanation for the RT stops observed in this study after copying exactly 7–8 bp into the extended base paired stem. These combined results demonstrate that the rules of RT pausing are not unambiguous, and may depend on the exact secondary and tertiary structure of the base paired region and/or its folding kinetics.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Dr D. Stammers for the gift of purified HIV-1 RT enzyme that was obtained through the MRC AIDS Reagent Project, Damian Purcell for providing the Mfold server, and Wim van Est for photography work. This work was supported by the Netherlands Foundation for Chemical Research with financial aid from the Netherlands Organization for Scientific Research.
REFERENCES
- 1.Telesnitsky A. and Goff,S.P. (1997) In Coffin,J.M., Hughes,S.H. and Varmus,H.E., (eds), Reverse Transcriptase and the Generation of Retroviral DNA. Retroviruses, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [PubMed]
- 2.Marquet R., Isel,C., Ehresmann,C. and Ehresmann,B. (1995) Biochimie, 77, 113–124. [DOI] [PubMed] [Google Scholar]
- 3.Hoglund S., Ohagen,A., Goncalves,J., Panganiban,A.T. and Gabuzda,D. (1997) Virology, 233, 271–279. [DOI] [PubMed] [Google Scholar]
- 4.Baudin F., Marquet,R., Isel,C., Darlix,J.L., Ehresmann,B. and Ehresmann,C. (1993) J. Mol. Biol., 229, 382–397. [DOI] [PubMed] [Google Scholar]
- 5.Harrison G.P. and Lever,A.M.L. (1992) J. Virol., 66, 4144–4153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Damgaard C.K., Dyhr-Mikkelsen,H. and Kjems,J. (1998) Nucleic Acids Res., 26, 3667–3676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McBride M.S. and Panganiban,A.T. (1996) J. Virol., 70, 2963–2973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Clever J., Sassetti,C. and Parslow,T.G. (1995) J. Virol., 69, 2101–2109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Das A.T., Klaver,B., Klasens,B.I.F., van Wamel,J.L.B. and Berkhout,B. (1997) J. Virol., 71, 2346–2356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Berkhout B. (1996) Prog. Nucleic Acid Res. Mol. Biol., 54, 1–34. [DOI] [PubMed] [Google Scholar]
- 11.Sakuragi J.-I., Fukasawa,M., Shibata,R., Sakai,H., Kawamura,M., Akari,H., Kiyomasu,T., Ishimoto,A., Hayami,M. and Adachi,A. (1991) Virology, 185, 455–459. [DOI] [PubMed] [Google Scholar]
- 12.Berkhout B. and Schoneveld,I. (1993) Nucleic Acids Res., 21, 1171–1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Arts E.J., Ghosh,M., Jacques,P.S., Ehresmann,B. and Le Grice,S.F.J. (1996) J. Biol. Chem., 271, 9054–9061. [DOI] [PubMed] [Google Scholar]
- 14.Arts E.J., Stetor,S.R., Li,Y., Rausch,J.W., Howard,K.J., Ehresmann,B., North,T.W., Wohrl,B.M., Goody,R.S., Wainberg,M.A. and Le Grice,S.F.J. (1996) Proc. Natl Acad. Sci. USA, 93, 10063–10068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Isel C., Ehresmann,C., Keith,G., Ehresmann,B. and Marquet,R. (1995) J. Mol. Biol., 247, 236–250. [DOI] [PubMed] [Google Scholar]
- 16.Wakefield J.K., Kang,S.-M. and Morrow,C.D. (1996) J. Virol., 70, 966–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wakefield J.K. and Morrow,C.D. (1996) Virology, 220, 290–298. [DOI] [PubMed] [Google Scholar]
- 18.Beerens N., Klaver,B. and Berkhout,B. (2000) J. Virol., 74, 2227–2238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Beerens N. and Berkhout,B. (2000) J. Biol. Chem., 275, 15474–15481. [DOI] [PubMed] [Google Scholar]
- 20.Liang C., Li,X., Rong,L., Inouye,P., Quan,Y., Kleiman,L. and Wainberg,M.A. (1997) J. Virol., 71, 5750–5757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Isel C., Keith,G., Ehresmann,B., Ehresmann,C. and Marquet,R. (1998) Nucleic Acids Res., 26, 1198–1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cobrinik D., Soskey,L. and Leis,J. (1988) J. Virol., 62, 3622–3630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cobrinik D., Aiyar,A., Ge,Z., Katzman,M., Huang,H. and Leis,J. (1991) J. Virol., 65, 3864–3872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Suo Z. and Johnson,K.A. (1997) Biochemistry, 36, 12459–12467. [DOI] [PubMed] [Google Scholar]
- 25.Klasens B.I.F., Huthoff,H.T., Das,A.T., Jeeninga,R.E. and Berkhout,B. (1999) Biochim. Biophys. Acta, 1444, 355–370. [DOI] [PubMed] [Google Scholar]
- 26.Harrison G.P., Mayo,M.S., Hunter,E. and Lever,A.M.L. (1998) Nucleic Acids Res., 26, 3433–3442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Klaver B. and Berkhout,B. (1994) J. Virol., 68, 3830–3840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Back N.K.T., Nijhuis,M., Keulen,W., Boucher,C.A.B., Oude Essink,B.B., van Kuilenburg,A.B.P., Van Gennip,A.H. and Berkhout,B. (1996) EMBO J., 15, 4040–4049. [PMC free article] [PubMed] [Google Scholar]
- 29.Willey R.L., Smith,D.H., Lasky,L.A., Theodore,T.S., Earl,P.L., Moss,B., Capon,D.J. and Martin,M.A. (1988) J. Virol., 62, 139–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Oude Essink B.B., Das,A.T. and Berkhout,B. (1996) J. Mol. Biol., 264, 243–254. [DOI] [PubMed] [Google Scholar]
- 31.Lenz C., Scheid,A. and Schaal,H. (1997) J. Virol., 71, 2757–2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liang C., Li,X., Quan,Y., Laughrea,M., Kleiman,L., Hiscott,J. and Wainberg,M.A. (1997) J. Mol. Biol., 272, 167–177. [DOI] [PubMed] [Google Scholar]
- 33.Van Lint C., Amella,C.A., Emiliani,S., John,M., Jie,T. and Verdin,E. (1997) J. Virol., 71, 6113–6127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.El Kharroubi A., Piras,G., Zensen,R. and Martin,M.A. (1998) Mol. Cell. Biol., 18, 2535–2544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tzfati Y., Fulton,T.B., Roy,J. and Blackburn,E.H. (2000) Science, 288, 863–867. [DOI] [PubMed] [Google Scholar]
- 36.Suo Z. and Johnson,K.A. (1998) Biochemistry, 36, 14778–14785. [DOI] [PubMed] [Google Scholar]
- 37.Wohrl B.M., Tantillo,C., Arnold,E. and Le Grice,S.F. (1995) Biochemistry, 34, 5343–5356. [DOI] [PubMed] [Google Scholar]
- 38.Zuker M. (1989) Science, 244, 48–52. [DOI] [PubMed] [Google Scholar]