

Abstract
Background and Objectives
Despite significant progress in biomedical research, the rate of success in oncology drug development remains inferior to that of other therapeutic fields. Mechanistic models provide comprehensive understanding of the therapeutic effects of drugs, which is crucial for designing effective clinical trials. This study was performed to acquire a better understanding of PI3K–AKT–TOR pathway modulation and preclinical to clinical translational bridging for a specific compound, apitolisib (PI3K/mTOR inhibitor), by developing integrated mechanistic models.
Methods
Integrated pharmacokinetic (PK)–pharmacodynamic (PD)–efficacy models were developed for xenografts bearing human renal cell adenocarcinoma and for patients with solid tumors (phase 1 studies) to characterize relationships between exposure of apitolisib, modulation of the phosphorylated Akt (pAkt) biomarker triggered by inhibition of the PI3K–AKT–mTOR pathway, and tumor response.
Results
Both clinical and preclinical integrated models show a steep sigmoid curve linking pAkt inhibition to tumor growth inhibition and quantified that a minimum of 35–45% pAkt modulation is required for tumor shrinkage in patients, based on platelet-rich plasma surrogate matrix and in xenografts based on tumor tissue matrix. Based on this relationship between targeted pAkt modulation and tumor shrinkage rate, it appeared that a constant pAkt inhibition of 61% and 65%, respectively, would be necessary to achieve tumor stasis in xenografts and patients.
Conclusions
These results help when it comes to evaluating the translatability of the preclinical analysis to the clinical target, and provide information that will enhance the value of future preclinical translational dose-finding and dose-optimization studies to accelerate clinical drug development.
Trial Registry
ClinicalTrials.gov NCT00854152 and NCT00854126.
Supplementary Information
The online version contains supplementary material available at 10.1007/s40268-024-00459-5.
Key Points
| Apitolisib (PI3K/mTOR inhibitor) was used to develop integrated mechanistic models to characterize relationships between drug exposure, pAkt biomarker modulation, and tumor response in both preclinical xenografts and clinical phase 1 studies. |
| Both clinical and preclinical integrated models show a steep sigmoid curve linking pAkt inhibition to tumor growth inhibition and quantify that a minimum 35–45% of pAkt modulation is needed for tumor shrinkage in patients and in xenografts. |
| Translational PK–PD modeling is a powerful tool for understanding how anticancer drugs interact with cancer signaling pathways. Current translational analyses enhance the value of preclinical studies in accelerating clinical drug development and improving future clinical trial designs and interpretations. |
Introduction
Despite significant progress in biomedical research, which led to a greater understanding of the molecular mechanisms of carcinogenesis and cancer growth, the rate of success in oncology drug development remains inferior to that of other therapeutic fields [1]. The PI3K–AKT–mTOR signaling pathway plays a critical role in cell growth, survival, and metabolism and is abnormally expressed in many types of cancers [2]. Despite significant resources invested in developing PI3K-targeted drugs, the number of approved inhibitors remains limited. Many trials evaluating these drugs have failed at various stages of clinical development [3] or even post-approval [4], making it necessary to improve the dose-finding and dose-optimization approach to maximize both safety and efficacy when targeting this pathway.
Dose-finding trials for cytotoxic chemotherapy drugs used to treat cancer have historically been designed to determine the maximum tolerated dose (MTD). In contrast, most modern oncology drugs are targeted therapies, such as targeted kinase inhibitors, which are designed to modulate specific targets with the resulting effect of suppressing molecular pathway(s) driving an oncologic disease [5]. Hence, these targeted therapies are considered “safer” compared with commonly used cytotoxic drugs and can often achieve maximal therapeutic effects at doses well below the MTD [5]. As a result, the Food and Drug Administration (FDA) created the Project Optimizing Patient Treatment through Integrative Multiomics Use (OPTIMUS), which is a research initiative aimed at developing new methods to use multiomics data (such as genomics, proteomics, and metabolomics) to improve the selection and dosing of cancer therapies [6]. The goal of the FDA project is to incorporate new biomarkers that can be used to predict how a patient will respond to a particular treatment and to move forward with a dose-finding and dose-optimization paradigm across oncology that emphasizes selection of a dose or doses that maximize not only the efficacy of a drug but its safety and tolerability, as well [6]. Quantitative integrated analyses that bridge preclinical research and clinical trials play a crucial role in regulatory decision-making, offering supportive evidence of efficacy when optimizing therapeutic outcomes.
Integrated pharmacokinetic (PK)–pharmacodynamic (PD)–efficacy models are mathematical models that integrate information from pharmacokinetics, the pharmacodynamic biomarker, and efficacy to predict the relationship between drug concentration, target modulation, and the therapeutic response [7, 8]. The PK portion of these models describes how the body absorbs, metabolizes, distributes, and excretes a drug to provide information on how the drug concentration changes over time in the body. The PD portion of these models provides information on how the drug concentration alters biological target modulation, for example, the inhibition of a target protein. The efficacy portion of these models describes the relationship between the biological target modulation and the therapeutic outcome, for example, how the drug’s effects on the target protein translate into treatment outcomes, such as tumor shrinkage. Integrated PK–PD–efficacy models are important tools in translational drug development to bridge preclinical and clinical studies, as they allow researchers to predict the efficacious dose and to optimize dosing regimens. Despite the increased use of translational PK–PD modeling, there are few to no reported cases of a systematic examination of performance of translational PK–PD modeling using preclinical data on predicting patient response. In this study, we retrospectively employ an integrated PK–PD–efficacy-model approach to explore the exposure–response relationship of apitolisib in both preclinical and initial clinical phases to investigate relationships between drug exposure, biomarker modulation, and efficacy for apitolisib in xenograft mice and patients with cancer in phase 1 trials.
Phosphorylated Akt (pAkt) is a key PD biomarker in the PI3K–AKT–mTOR pathway, which is frequently dysregulated and hyperactivated in cancer cells [9]. Akt phosphorylates and activates downstream targets involved in cell survival, proliferation, and protein synthesis. For instance, it can activate the mammalian target of rapamycin (mTOR), a critical regulator of protein synthesis and cell growth [2]. The detection and measurement of pAkt levels serve as a biomarker to assess the activation status of this pathway [10, 11]. Apitolisib is a potent, selective dual inhibitor of PI3K and mTOR. Apitolisib has broad activity in xenograft efficacy tumor models [12], which is also demonstrated by collected pAkt protein. Clinical studies, reported previously by Dolly et al., have further established the PD evidence of apitolisib target modulation. Significant reductions in PI3K inhibition of ≥ 90% was observed in platelet-rich plasma PD markers, including pAkt at doses ≥ 16 mg in patients [13].
Dose optimization of targeted drugs can be informed by translational pharmacodynamic biomarkers that aim to define the degree of target and pathway modulation required for efficacy [10]. The suitability of these PD biomarkers is typically assessed and characterized at preclinical stages of drug development, providing an understanding of the relationship between drug concentrations and biomarker response as well as the biological relationship between modulation of the biomarker and downstream therapeutic effect. Often, in oncology, preclinical information regarding PD biomarker modulation is obtained from tumor tissue (site of action). Tumor tissue biomarkers provide a direct look at the extent and duration of target modulation. In contrast, in the clinic, collection of plasma biomarkers is far more common and serves as a surrogate of target modulation in a tumor. In this study, we aim to (1) develop an integrated PK–PD–efficacy model in xenograft mice to characterize both apitolisib’s ability to modulate pAkt in tumor tissue and to inhibit tumor growth; (2) utilize the same modeling methodology to characterize apitolisib PK, surrogate PD (pAkt modulation/response) in plasma, and efficacy (tumor growth inhibition) in patients with cancer in phase 1 clinical trials; and (3) evaluate the translatability of the preclinical analysis in xenografts to observations in patients with cancer. These objectives provide information that will enhance the value of future clinical and preclinical translational dose-finding and dose-optimization studies.
Methods
Xenograft Dataset
Mean plasma concentration–time profiles of apitolisib in nude mice after oral administration of 1, 5, and 10 mg/kg doses, composed of plasma concentrations from samples collected pre-dose and at 0.083, 0.25, 0.5, 1, 3, 6, 9, and 24 h post-dose (n = 27 mice per dose group), were previously published [14] and were used to provide pharmacokinetics (PK) for xenograft mice in this analysis.
The efficacy evaluation of apitolisib was conducted in female beige nude XID (bg.nu.xid Harlan) mice bearing subcutaneous 786-O human renal cell adenocarcinoma (RCC) xenografts. Mice were housed and maintained according to the animal use guidelines of Genentech, Inc, conforming to California state legal and ethical practices. Mice were inoculated subcutaneously in the right lateral thorax with tumor fragments from donor mice. The right flank was used to gain access to the subcutaneous space and deposit the tumor fragment. As tumors reach a volume in the range of 100–300 mm3, mice with similarly sized tumors were separated into groups. The mean tumor volume (TV) across all groups was 181 mm3 at the initiation of dosing. Apitolisib was administered once a day (QD) orally (PO) in 200 µL MCT (0.5% methylcellulose and 0.2% Tween 80) for 17 days. The apitolisib doses administered were the vehicle plus 0.008, 0.026, 0.085, 0.256, 1, 2.5, 5, 7, 8.5, 10, and 11 mg/kg (n = 6 per group). Tumor volume measurements were collected for each mouse pre-dose and at 72, 96, 168, 240, 264, 336, and 408 h from each treatment groups. Mice were euthanized if they lost greater than 20% from their initial body weight or if the tumors exceeded 2000 mm3. Tumor volumes (TV) were determined using digital calipers (Fred V. Fowler Company, Inc.) using Eq. 1:
| 1 |
The apitolisib preclinical PK–PD study where the pAkt biomarker was collected was also conducted in female beige nude XID (bg.nu.xid Harlan) mice bearing subcutaneous 786-O human renal cell adenocarcinoma xenografts. To evaluate pAkt modulation in 786-O xenografts, tumor cells were implanted as described previously for the efficacy studies. Mice were divided into four dose groups and received a single oral dose of vehicle (MCT), 0.3, 3, or 10 mg/kg apitolisib in 200 µL of MCT. Tumors were collected at 0.5, 2, 6, 12, 18, and 24 h post-dose from treatment groups (n = 3 per time point), and at 2, 6, and 12 h from the vehicle group (n = 6 per time point). Meso Scale Discovery (MSD) assay was used to quantify pAkt (serine 473) levels in tumor tissues, and %pAkt, defined as %pAkt relative to control (i.e., baseline), was calculated using Eq. 2:
| 2 |
where pAkt is the level of pAkt measured at a specific time point and pAkt0 is the pAkt at baseline, where there is no drug present. By definition, the %pAkt at control baseline is 100% according to Eq. 2.
Clinical Trials Dataset
Two phase 1 dose-escalation studies using 3 + 3 design were conducted in patients with advanced solid tumors or non-Hodgkin’s lymphoma [13]. Apitolisib was administered orally on day 1, followed by 1 week of washout. It was then dosed daily (QD) for 28 days (1 cycle), QD on a 3-weeks-on/1-week-off schedule, or once weekly (QW) for 28 days. Apitolisib doses that were administered are as follows: 30, 40, and 50 mg QD; 2, 4, 8, 16, 32, 40, 50, and 70 mg QD on a 3-weeks-on/1-week-off schedule; or 6, 12, 25, 50, 100, 150, and 200 mg QW. Trials were approved by the institutional review board or independent ethics committee. All patients provided written informed consent.
PK sampling was done in the first cycle on day 1 (predose, 0.5, 1, 2, 3, 4, and 8 h post-dose), on days 2 (24 h post-dose) and 3 (48 h post-dose), on day 8 (pre-dose and 2 h post-dose), on day 15 (pre-dose and 0.5, 1, 2, 3, 4, and 8 h post-dose), and on days 22 and 29 (predose and 2 h post-dose); and on the first day (pre-dose) of each cycle thereafter. Blood samples for the surrogate PD marker (pAkt) were collected pre-dose and 1, 3, 8, and 24 h post-dose on day 1 to evaluate platelet-rich plasma (PRP). Apitolisib plasma concentrations were quantified using a liquid chromatography (LC)–mass spectrometry (MS)/MS assay with a lower limit of quantification (LLOQ) of 0.5 ng/mL (Tandem Labs, Inc., Salt Lake City, UT). Changes in pAkt in PRP in subset of patients (n = 37) were measured using the Meso Scale Discovery (MSD) assay, and the percent change in pAkt relative to baseline was calculated (Eq. 2).
Tumor lesions were measured using computed tomography or magnetic resonance imaging at baseline and at regular intervals afterward (after the first and second cycle and every two cycles subsequently). Longitudinal tumor size data, defined as the sum of the longest diameters of target lesions at each visit according to RECIST 1.0 [15] or, in the case of patients with malignant pleural mesothelioma, modified RECIST [16], were used for the estimation of tumor growth inhibition. Patients with at least baseline and one post-baseline tumor size measurements were defined as evaluable (n = 117), and data from patients who only had baseline tumor assessments were excluded from the analysis.
Pharmacokinetic Models
Mouse Pharmacokinetic Model
The pharmacokinetics of apitolisib appeared to be linear over the range of doses tested. A one-compartment model with first-order oral absorption was simultaneously fit to the mean plasma concentration–time data from mice following single PO administrations of 1, 5, and 10 mg/kg. The parameter estimates of the absorption rate constant (ka), the apparent oral clearance (CL/F), and the apparent volume of distribution (V/F) were used to simulate the apitolisib plasma concentrations for xenograft biomarker and tumor growth inhibition analysis.
Patient Population Pharmacokinetic Model
A nonlinear mixed-effects models (two-compartment model with oral absorption) was used to fit the concentration–time data of apitolisib from patients given oral doses of apitolisib. Apparent clearance (CL/F) after oral dosing, apparent volume of distribution parameters for the central (V1/F) and peripheral (V2/F) compartments, apparent and intercompartmental clearance (CLd/F), and absorption rate (ka) were estimated. Interindividual variability (IIV) in PK parameters were modeled as exponential random-effect models to constrain the individual parameter values positively, which were thus assumed to follow a log-normal distribution. The residual error model was a proportional error model.
Pharmacokinetic–Pharmacodynamic (PK–PD) Models Describing pAkt Modulation
In xenograft analysis, the relationship between pAkt in tumor tissue and apitolisib plasma concentrations was evaluated using an indirect response model that describes the inhibition of pAkt formation rate by apitolisib (Eq. 3):
| 3 |
where γ1 is the sigmoidicity factor, Imax is the maximum inhibition of %pAkt, kin is the rate of biomarker production, kout is the first-order rate constant describing biomarker loss, half maximal inhibitory concentration (IC50) is the apitolisib concentration producing 50% of Imax, t is time, and Cp is the apitolisib plasma concentration. At baseline, %pAkt is equal to 100% in the absence of apitolisib. Plasma concentrations of apitolisib were simulated using the mouse PK model described above.
Similar methodology was applied to clinical data. Patients’ individual PK parameters estimated by a population PK model were used to predict apitolisib plasma concentrations, and changes in pAkt in platelet-rich plasma (PRP) expressed as a percent of control (i.e., baseline) were simultaneously assessed for different dose levels using Eq. 3.
Integrated PK–PD–Efficacy Models
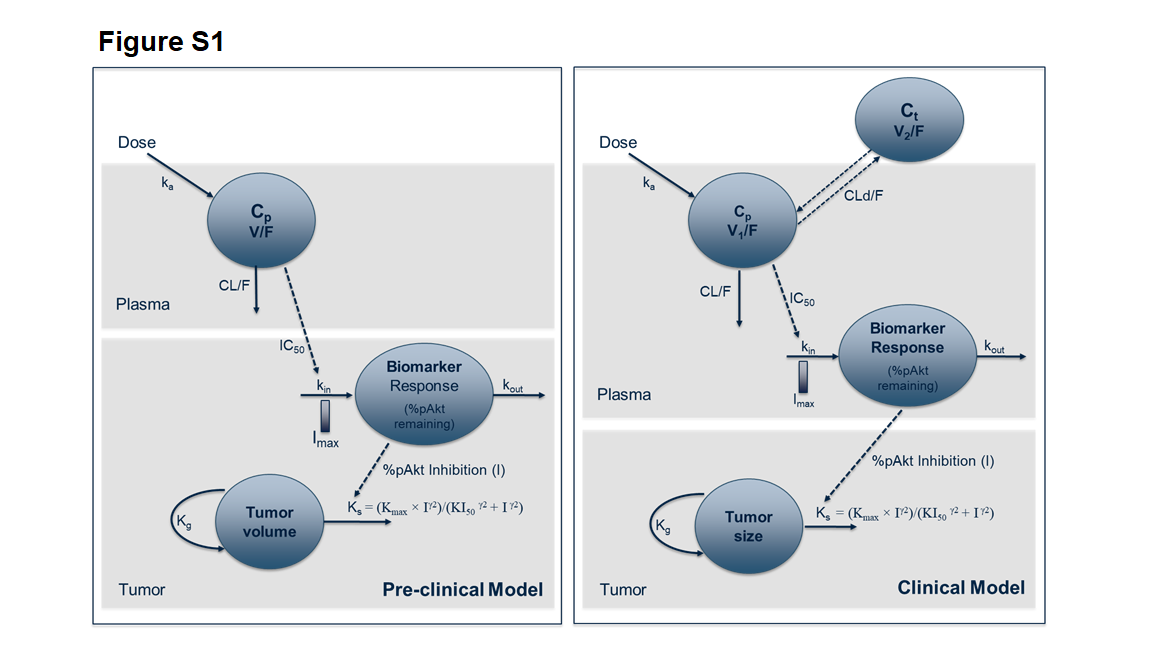
Mechanistic models were developed to integrate and characterize the relationship between plasma concentrations of apitolisib, modulation of pAkt biomarkers triggered by inhibition of the PI3K–AKT–mTOR pathway, and tumor response in xenografts and in patients. These integrated models (Supplementary Fig. S1) assume that the decrease in pAkt (i.e., inhibition of the phosphorylation of Akt) is representative of the inhibition of the PI3K–AKT–mTOR pathway, which is assumed to be the primary driver of tumor growth. The changes in %pAkt were predicted for each dose using the PK–PD model (Eq. 3) parameter estimates. Next, the %pAkt were related to the tumor metrics using Eq. 4:
| 4 |
where “Tumor” is the tumor volume for xenografts or tumor size for patients, Kg is the net tumor growth rate constant, and Ks is the tumor shrinkage rate constant, and “Tumor” at time 0 was set as the baseline tumor observations prior to the application of apitolisib, as well as Eq.
| 5 |
where Ks is the tumor shrinkage rate constant, I is the percent inhibition of Akt phosphorylation, Kmax is the maximum value of Ks, KI50 is I where Ks is 50% of Kmax, and γ2 is the sigmoidicity factor of the sigmoid Kmax equation.
In the xenograft-mice-integrated PK–PD–efficacy model, apitolisib plasma concentrations, %pAkt, and the corresponding I in tumor tissue were predicted based on the parameters determined by the mouse PK and mouse PK–PD models (Eq. 3). Tumor volumes from all dose groups (ranging from 0.008 to 11 mg/kg) were fitted simultaneously to obtain parameter estimates describing the PD–efficacy relationship from the integrated PK–PD–efficacy model.
In the integrated PK–PD–efficacy model used to describe clinical data of patients with cancer, individual patients’ PK parameters and individual patients’ dosing records were used to predict the apitolisib plasma concentrations; therefore, any dosing reduction or dosing interruption for specific patients was captured. For pAkt, only a subset of patients had pAkt data collected. In those patients with pAkt data, individual PK parameters and individual IC50 estimates along with the PK–PD model described by Eq. 3 were used to predict %pAkt and the corresponding I in the integrated model. For patients for whom pAkt data were not collected, individual PK parameters and a typical estimated IC50 was used for this purpose. Tumor size data from all patient dose groups (ranging from 2 to 200 mg) were fitted simultaneously to estimate parameters describing the PD–efficacy relationship from the integrated PK–PD–efficacy model developed using patient data.
Model Evaluation
Model evaluation was based on the inspection of graphical diagnostics, such as goodness of fit plots, changes in the objective function value (OFV) provided by NONMEM, relative standard errors (RSE), and plausibility of the parameters estimate. If more than one interindividual variability (IIV) term was estimated, correlations between IIV terms were evaluated. A comprehensive description of models’ development, assumptions, and limitations is provided in Supplementary Information (Online Resource 1).
Software
Models were developed using NONMEM version 7.4 (ICON Development Solutions, Ellicott City, MD), with the first-order conditional estimation (FOCE) and the INTERACTION option [17]. Data management and further processing of NONMEM output were performed using R version 4.2.3 [18].
Results
Pharmacokinetic Models
Mouse Pharmacokinetic Model
The final PK model that characterized the mouse concentration–time data well was a linear one-compartment model with first-order absorption and oral clearance (data not shown). The estimated parameters (RSE%) were 0.387 (9.5%) L/h/kg for apparent oral clearance (CL/F), 1.13 (17.3%) L/kg for apparent volume of distribution after oral dosing (V/F), and 1.1 (36.4%) h−1 for the absorption rate constant (ka).
Patient Population Pharmacokinetic Model
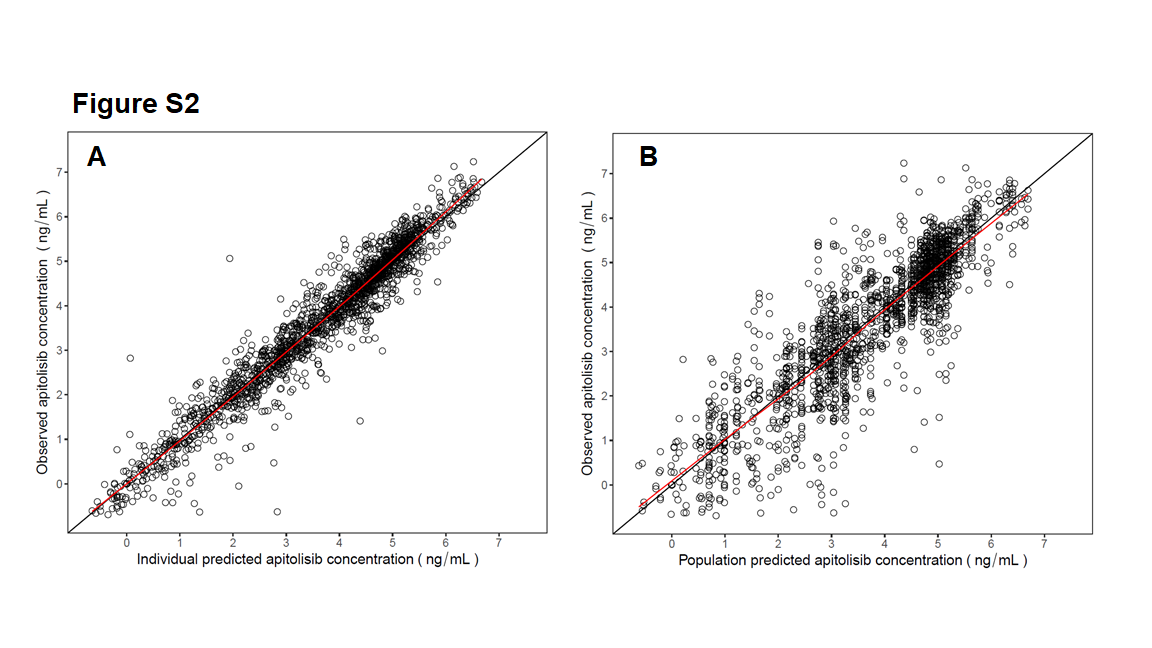
A linear two-compartment PK model with first-order absorption and elimination best described the apitolisib plasma concentration–time data in patients (n = 146; 1835 PK observations). Population typical values (RSE%) were 21.3 (4.28%) L/h for apparent oral clearance (CL/F), 210.6 (3.51%) L for apparent volume of the central compartment (V1/F), 639.1 (15.1%) L for apparent volume of distribution of the peripheral compartment (V2/F), 5.93 (8.53%) L/h for apparent intercompartmental clearance (CLd/F), and 3.35 (16.8%) h−1 for absorption rate constant (ka). Also, based on observed data, a delay was detected in apitolisib absorption, which was captured in the PK model by a lagged time estimate of 0.465 (0.43%) h. Estimates of IIV (RSE%) [shrinkage%] were 0.17 (17.6%) [13.7%] for CL/F, 0.0639 (23.2%) [23.2%] for V1/F, 0.208 (33.5%) [44.2%] for CLd/F, 0.881 (44.8%) [40.5%] for V2/F, and 2.09 (16.4%) [13.2%] for ka. The goodness-of-fit plots from model fitting showed that the predictions from the final model were generally consistent with the observed data (Supplementary Figs. S2 and S3).
Assuming a reference body weight of 70 kg, body-weight-normalized PK parameters were 0.304 L/h/kg for apparent oral clearance (CL/F), 3.01 L/kg for apparent volume of the central compartment (V1/F), 9.13 L/kg for apparent volume of distribution of the peripheral compartment (V2/F), and 0.085 L/h/kg for apparent intercompartmental clearance (CLd/F). As expected, a faster elimination rate constant (CL/V) was observed in xenografts (0.3 h−1) compared with patients (0.1 h−1).
PK–PD Models Describing pAkt Modulation
Observed mean %pAkt versus time data for xenograft mice and for patients and PK–PD models fitted lines (average individual predictions for patients) are presented in Fig. 1A and B. The levels of pAkt were measured in tumors after a single oral administration of apitolisib at 0.3, 3, and 10 mg/kg in 786-O tumor-bearing mice (Fig. 1A). A dose-dependent decrease in pAkt was observed at the three doses. Suppression of the pAkt was pronounced at 30 min post-dose and sustained for 6–12 h at the 3 and 10 mg/kg doses, respectively. In contrast, the pAkt reduction was very weak and recovered 1–2 h post-dose at the 0.3 mg/kg dose level. In general, higher apitolisib doses resulted in greater and more sustained suppression of the pAkt in tumor tissue of xenografts. In patients, pAkt levels were measured in platelet-rich plasma over a large range of doses (2–70 mg; Fig. 1B). A dose-dependent decrease in pAkt in plasma was observed with increasing doses of apitolisib. Peak suppression of the platelet pAkt was 90% observed at doses ≥ 12 mg.
Fig. 1.

Mean observed and predicted %pAkt relative to baseline versus time. A Tumor tissue mean %pAkt relative to baseline from 786-O xenografts following oral administration of 0.3, 3, and 10 mg/kg apitolisib. B Surrogate plasma mean %pAkt relative to baseline from patients with advanced solid tumors (phase 1 studies) following oral administration of 2–70 mg apitolisib, stratified by dose. Observed mean %pAkt relative to baseline data is presented as dots, and the model predicted is presented as a solid line
In general, the PK–PD models were able to capture the %pAkt inhibition (Fig. 1). Estimates of PD parameters from fitting the %pAkt data to the PK–PD models described by Eq. 3 are shown in Table 1 for xenografts and Table 2 for patients. Model estimates of kin and kout were comparable when comparing xenograft mice to patients, suggestive of there being similar biomarker production and elimination rates between humans and xenograft mice with human-origin tumors. In contrast, model estimated IC50 differed, being 43-fold higher when the estimate was obtained from %pAkt measurements from xenograft tumor tissue (403 µg/L) compared with %pAkt measurement from surrogate plasma for a typical patient (9.32 µg/L). The unbound fraction of apitolisib was similar between mice (fraction unbound 29.4%) and humans (fraction unbound 38.8%), suggesting similar free concentrations of apitolisib in plasma and tumor upon equilibration, which does not appear to provide an explanation for the differences in IC50. Overall, the PK–PD models described by Eq. 3 could adequately characterize the relationship between apitolisib plasma concentrations and pAkt reductions in tumors from xenograft mice and platelet-rich plasma in patients.
Table 1.
Parameter estimates following fitting of the PD–PK model describing pAkt modulation for xenograft mice (786-O)
| Parameter | Unit | Estimate | RSE% |
|---|---|---|---|
| IC50 | μg/L | 403 | 14.7 |
| kin | %/h | 88,698 | 0.8 |
| kout* | 1/h | 886.98 | – |
| Imax | – | 1 (fix) | – |
| γ1 | – | 1 (fix) | – |
| Proportional error | – | 0.353 | 5.6 |
γ1, sigmoidicity factor; Imax, maximum inhibition of %pAkt; kin, rate of biomarker production; kout, first-order rate constant describing biomarker loss; IC50, half maximal inhibitory concentration; t, time; Cp, plasma concentration
*kout = kin/%pAkt(0), where %pAkt(0) is the baseline %pAkt at time zero = 100%
Table 2.
Parameter estimates following fitting of the PK–PD model describing pAkt modulation for patients with advanced solid tumors (phase 1 studies)
| Parameter | Unit | Estimate | RSE% | Shrinkage% |
|---|---|---|---|---|
| IC50 | μg/L | 9.32 | 12.7 | – |
| kin | %/h | 88,699 | 3.8 | – |
| kout* | 1/h | 886.99 | – | – |
| Imax | – | 1 (fix) | – | – |
| γ1** | – | 1 (fix) | – | – |
| Proportional error | – | 0.672 | 9.5 | – |
| IIV IC50 | Variance | 0.296 | 44.3 | 15.5 |
γ1, sigmoidicity factor; Imax, maximum inhibition of %pAkt; kin, rate of biomarker production; kout, first-order rate constant describing biomarker loss; IC50, half maximal inhibitory concentration; t, time; and Cp, plasma concentration
*kout = kin/%pAkt(0), where %pAkt(0) is the baseline %pAkt at time zero = 100%
**Sigmoidicity parameter estimate was 0.868–0.937; therefore it was fixed to 1 in the final model
Integrated PK–PD–Efficacy Models
The relationship between inhibition of the PI3K pathway and efficacy was investigated using a PK–PD–efficacy model that describes the relations between apitolisib plasma concentrations, pAkt modulation, and tumor growth inhibition. The model structures used to fit the preclinical and clinical data are presented in Supplementary Fig. S1. Model codes are provided in Supplementary Information (Online Resource 2).
Two integrated models were independently developed for xenografts (n = 64; 381 tumor volume observations) and patients (n = 117; 417 tumor size observations). Individual baseline tumor data were used for model development (Eq. 4). Median baseline tumor observations were 173.5 mm3 and 130.0 mm for xenograft mice and the phase 1 patient population, respectively. The described integrated PK–PD–efficacy models were used to fit tumor data collected from individual xenograft mice and patients. The goodness-of-fit (GOF) plots support the use of both integrated models to fit preclinical and clinical data. The models’ GOF plots indicated a good model fit, as shown in plots of individual observed versus predicted tumor data presented in Figs. S4 and S5. Additionally, GOF plots of conditional weighted residuals versus time- and population-predicted tumor data also indicated a good model fit, as shown in Figs. S6 and S7 for the preclinical and clinical models, respectably. A visual predictive check of the final models shows that the models captured the central tendency, extreme values, and variability in the observed preclinical and clinical tumor data relatively well (Figs. S8 and S9).
The estimated PD parameters are presented in Table 3 (xenografts) and Table 4 (patients). The net growth rate constant was 98.7-fold higher in xenograft mice’s subcutaneous tumors compared with tumors in patients, which is consistent with prior publication of growth rate detection comparing diameter and volume in measuring tumor size [19]. Specific to the relationship between target modulation and anti-tumor efficacy, Kmax was more than 200-fold lower in patients when compared with xenograft mice. In contrast, KI50 and sigmoidicity factors were similar in xenografts and patients, suggesting that the shape, but not magnitude, of the relationship between %pAkt and Ks was similar (Fig. 2A). When Ks is normalized as a percentage of Kmax, this similarity in the relationship between target modulation and antitumor effect can be more clearly seen (Fig. 2B). In both xenograft mice and humans, Kmax > Kg, suggesting that apitolisib given at high enough concentrations can cause tumor regressions. Based on the relationship between %pAkt inhibition and the tumor shrinkage rate constant (Ks; Fig. 2A), a similar constant pAkt inhibition of 61% and 65% would be necessary to achieve tumor stasis (where Ks = Kg) in 786-O xenografts and patients, respectively.
Table 3.
Integrated PK–PD–efficacy model parameter estimate for xenografts (786-O)
| Parameter | Unit | Estimate | RSE% | Variability% | Shrinkage% |
|---|---|---|---|---|---|
| Kg | 1/h | 0.0057 | 3.9 | – | – |
| Kmax | 1/h | 0.0194 | 43.2 | – | – |
| KI50 | %pAKT inhibition | 67.2 | 12.6 | – | – |
| γ2 | – | 9.4 | 208.5 | – | – |
| Proportional error | – | 0.248 | 5.1 | – | – |
| IIV Kg | Variance | 0.0276 | 29.6 | 16.6 | 22.6 |
| IIV Kmax | Variance | 0.0529 | 54.6 | 23.0 | 45.8 |
IIV, interindividual variability; Kg, net tumor growth rate constant; Kmax, maximum value of Ks; KI50, I where Ks is 50% of Kmax; RSE, relative standard error; γ2, sigmoidicity factor of the sigmoid Kmax equation
Table 4.
Integrated PK–PD–efficacy model parameter estimate for patients with advanced solid tumors (phase 1 studies)
| Parameter | Unit | Estimate | RSE% | Variability% | Shrinkage% |
|---|---|---|---|---|---|
| Kg | 1/week | 0.0097 | 20.7 | – | – |
| Kmax | 1/week | 0.0142 | 43.9 | – | – |
| KI50 | %pAKT inhibition | 58.0 | 34.3 | – | – |
| γ2 | – | 6.52 | 101.4 | – | – |
| Proportional error | – | 0.141 | 2.3 | – | – |
| IIV Kg | Variance | 0.668 | 34.9 | 81.7 | 37.9 |
| IIV Kmax | Variance | 0.377 | 44.6 | 61.4 | 55.8 |
| IIV KI50 | Variance | 0.0225 (fix) | – | – | – |
| IIV γ2 | Variance | 0.0225 (fix) | – | – | – |
IIV, interindividual variability; Kg, net tumor growth rate constant; Kmax, maximum value of Ks; KI50, I where Ks is 50% of Kmax; RSE, relative standard error; γ2, sigmoidicity factor
Fig. 2.

Preclinical and clinical population integrated PK–PD–efficacy model estimated relationship between tumor shrinkage rate constant (Ks) versus %pAkt inhibition in a xenograft (tumor tissue) and in a typical patient with advanced solid tumors (surrogate plasma). A The steep sigmoid curve describes the relationship between the tumor shrinkage rate constant and %pAkt inhibition in xenografts (tumor tissue site, red line) and typical patient (surrogate plasma site, blue line). B Tumor shrinkage rate constant normalized to Kmax versus %pAkt inhibition in xenografts (tumor tissue site, red line) and typical patients (surrogate plasma site, blue line)
Discussion
The PI3K–AKT–mTOR signaling pathway is essential for numerous fundamental cellular functions such as cell differentiation, growth, proliferation, mobility, and metabolism [2, 10]. Somatic alterations and genetic amplifications causing activation of this pathway are frequently found in cancer; as a result, there has been a significant effort to develop therapeutics that target critical members of the pathway [10]. Successful development of these drugs requires pharmacodynamic biomarkers that aim to define the degree and duration of target and pathway inhibition [10], such as pAkt (phosphorylated Akt), which is a protein biomarker that is often used as a marker of cellular signaling through the PI3K–AKT–mTOR pathway. Drugs that target this pathway, such as apitolisib, lead to the inhibition of pAkt phosphorylation and the induction of apoptosis in cancer cells [12]. In this study, preclinical and clinical apitolisib concentration–response relationships were explored to investigate the utility of the pAkt biomarker in assessing the extent of PI3K–AKT–mTOR pathway inhibition in terms of tumor growth inhibition. More importantly, we present a case study of a formal assessment on the real-world translatability of preclinical translational PK–PD analyses to clinical response in patients using common endpoints that are collected at the preclinical and early clinical stages of drug development (refer to Supplementary Information, Online Resource 1: Analysis objectives). Formal assessment on the translatability of preclinical PK–PD is lacking in literature.
Levels of the biomarker, pAkt, can be measured in both tumor tissue and plasma. However, measurement of pAkt in the tumor tissue is typically considered more relevant to therapy and serves to better inform cancer prognosis. Development of targeted therapeutics for cancer at the preclinical research stage is conducted on the xenograft models to determine drug efficacy and identify active doses based on assessment of the drug’s in vivo concentration and target engagement at the site of action. At this stage, the extent and duration of target modulation typically is quantified by measuring tumor tissue biomarkers. However, once the drug moves into the clinical stage, often target modulation and response is assessed by surrogate plasma biomarkers, which are less invasive.
PD biomarkers provide invaluable insights into the interaction of novel therapies with their intended targets; however, use of these biomarkers requires a deep understanding of their relationship to drug concentrations and clinical outcomes. Over the past few decades, there has been a significant rise in preclinical understanding of drug concentrations and biomarker response as evident in numerous publications [20–22]. This development is driven by advancements in technologies and methodologies for measuring drug levels and biomarkers in various biological matrices. One of the key factors driving the interest in preclinical understanding of drug concentrations and biomarker response is the need to improve the efficacy and safety of drugs in clinical development and design more effective dosing regimens that minimize the risk of adverse events. Based on our knowledge, this is the first report to quantitatively examine the relationship between target modulation and efficacy in humans for a PI3K compound using a translational biomarker (pAkt). Additionally, this is the first attempt to quantitatively assess similarities and differences between the biomarker response observed in the preclinical and clinical phases to assess the translatability of preclinical PD and efficacy data within the PI3K–AKT–mTOR pathway. In this study, we focused on a translational PD biomarker in the pathway, pAkt, and developed an integrated sequential PK–PD–efficacy model to facilitate the interpretation of efficacy studies conducted from the preclinical to early phases of clinical development for a specific PI3K/mTOR inhibitor compound (apitolisib). These integrated models can be used to predict the optimal dosing regimen for a drug, considering not only the PK properties of the drug but also the relationship between drug concentrations, pAkt levels, and tumor growth inhibition.
The model used to characterize the relationship between apitolisib and pAkt levels is an indirect response model. Indirect response models are used when the response measured is the product of an indirect mechanism, such as the inhibition or stimulation of the formation (kin) or loss (kout) of the mediator controlling the physiological effect [23, 24]. Hence, in the present study, the levels of pAkt in tumor tissue (preclinically) and plasma (clinically) that were monitored after upstream inhibition of PI3K by apitolisib were characterized using indirect response models to quantify the inhibition of the production of pAkt in tumor tissue or surrogate plasma. The quantified kin estimates were comparable between xenograft mice and humans, suggesting comparable rates of production and loss (kout is derived from kin) for pAkt (Tables 1 and 2) in surrogate plasma from patients versus tumors from xenograft mice. Notably, the estimated IC50 was ~40-fold lower in a typical patient, being 9.32 µg/L compared with 403 µg/L in xenografts when the PD biomarker was assessed in surrogate plasma rather than from tumor tissue. The higher pAkt IC50 estimates in xenograft study could be partially due to the RCC tumor type (786-O cell line) being less responsive to apitolisib treatment compared with other tumor types such as MCF-7 in in vitro studies conducted using cancer cell lines [12, 25]. In patients in whom the plasma pAkt biomarker was collected, individual IC50 ranged from 4.03 to 26.1 µg/L (Supplementary Table S1); notably, within this group, the individual IC50 estimate for a patient with metastatic renal cell carcinoma was 19.12 µg/L, which was on the higher end.
The higher potency observed in patient surrogate plasma may be attributed to differences in assay conditions that likely favor greater inhibition in the plasma or differences in drug concentrations between normal and tumor tissues due to variances in tissue architecture, and hemodynamics may also contribute to this phenomenon [26]. These differences underline the complexity of accurately defining the pharmacokinetic–pharmacodynamic relationship of molecularly targeted drugs based on observed data alone, as well as the important issues of extrapolation of preclinical models to predict effects in patients and differences in analytical sensitivity/methodology between platelet-rich plasma and tumor biopsies [26]. Model-informed drug development can identify and characterize these differences and their correlation to treatment effect; therefore, disease-mechanism-based biomarkers that are indicative of a relevant biological aspect of cancer can serve as additional surrogates of antitumor activity during the drug-development process and bridge preclinical studies efficacy and clinical response.
The relationship between pAkt modulation and its impact on tumor growth was investigated preclinically and clinically by developing integrated PK–PD–efficacy models that assume that tumor growth inhibition has dependence on the PI3K–AKT–mTOR signaling pathway (Supplementary Fig. S1). Apitolisib is a dual PI3K/mTOR inhibitor that targets two crucial points in the same pathway that could lead to higher efficacy compared with a PI3K inhibitor alone by possibly overcoming mTOR negative feedback due to inhibition of pathway [27]. In our current integrated models used for both the preclinical and clinical analyses, pAkt inhibition serves as surrogate for both PI3K and mTORC1 inhibition by apitolisib; hence, the models utilized have some limitations. Specifically, the role/contribution of mTORC2 inhibition to tumor growth inhibition is not accounted for separately and is “lumped” in with PI3Kinase inhibition by the models used. Further, preclinical study was conducted in xenografts with renal cancer carcinoma (786-O cell lines), and relationships between pAkt modulation and tumor inhibition may differ when compared with patients with different cancer types enrolled in the phase 1 studies utilized in this analysis.
Individual longitudinal tumor profiles were adequately described by both preclinical and clinical integrated PK–PD–efficacy models. Numerical and graphical model diagnostics were used to evaluate different components of the integrated PK–PD–efficacy models, which verified adequacy of the underlying structural models to capture observed preclinical and clinical data. A steep sigmoid curve describing the relationship between inhibition of Akt phosphorylation and tumor growth inhibition, represented by the rate constant Ks, was observed in xenograft mice and patients despite differences in species and matrices where pAkt inhibition was evaluated. In both species, a threshold of approximately 35–45% pAkt inhibition is required prior to rapid increases in tumor growth inhibition with increasing pAkt modulation (Fig. 2). The commonality of curve shape has implications in clinical trial design, as pharmacodynamic endpoints can be appropriately selected in clinical trials based on similarity in observed pharmacodynamic thresholds required prior to onset of an antitumor effect. A similar steep relationship (sigmoidicity factor ~9) was estimated for target modulation (Gli mRNA inhibition) and vismodegib efficacy (i.e., the rate constant describing the measure of antitumor effect) in the preclinical models of the hedgehog pathway, and approximately a minimum of 50% Gli mRNA inhibition was needed for tumor growth inhibition [28]. Finally, of note, the net growth rate constant was considerably lower in patients compared with xenograft mice. This is consistent with previous publications of the tumor growth rate constant in humans versus preclinical xenograft models [28, 29].
Previous study delves into the translational relevance of preclinical murine subcutaneous tumor models in predicting clinical response [30]. Through a retrospective pharmacokinetic–pharmacodynamic analysis of eight agents, including both molecularly targeted and cytotoxic ones, with known clinical response data, the research sheds light on the correlation between simulated tumor growth inhibition in xenografts/allografts using human exposures and actual clinical response. A significant correlation was found between tumor growth inhibition (TGI) driven by human pharmacokinetics and clinical response, contrasting with TGI observed at maximum tolerated doses in mice. Agents inducing over 60% TGI in preclinical models, at clinically relevant exposures, exhibited a higher likelihood of eliciting clinical response. These findings establish a framework for effectively utilizing murine subcutaneous tumor models for selecting promising agents for clinical advancement.
In summary, modeling can play a critical role in bridging the gap between xenograft and human studies and in facilitating the development of more effective cancer treatments. The complexity of accurately defining the treatment exposure–response relationship based on different matrices of origin (tumor tissue versus surrogate plasma) due to analytical sensitivity and methodology is challenging, specifically at lower drug exposures. The preclinically and clinically developed integrated PK–PD–efficacy models bring together the information from PK, PD, and efficacy studies to provide a more complete understanding of the relationship between drug dose, drug concentration, and therapeutic response. The primary objective of our analysis was to examine a real-world translational analysis and assess both insights gained and shortcomings of the analysis. The overall goal of this analysis was to better characterize preclinical to clinical translation, with the aim of improving predictivity and use of translational biomarkers such as pAkt, which reflect the biological response of a drug. These PD biomarkers can be used to monitor the treatment efficacy and facilitate informed decision-making on dose finding and dosing-regimen optimization, to better inform future clinical trial design and interpretation based on preclinical studies.
Supplementary Information
Below is the link to the electronic supplementary material.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgements
We thank the patients and their families who participated in these studies and physicians and staff who conducted and/or managed these studies.
Declarations
Funding
This work was supported by Genentech Inc., a member of the Roche group.
Conflict of Interest
A.M., J.Y.J., M.W., and B.A. are Genentech employees and Roche shareholders. H.W. is Faculty of Pharmaceutical Sciences at the University of British Columbia working under contract with Genentech.
Data Availability Statement
Qualified researchers may request access to individual patient-level data through the clinical study data request platform (https://Vivli.org/).
Ethics Approval
Clinical trials (NCT00854152 and NCT00854126) were conducted in accordance with Good Clinical Practice guidelines and the Declaration of Helsinki. Prior to enrollment, written informed consent was acquired from all patients, in alignment with the approved protocols from the ethics committees at each study site. All clinical trial information can be accessed through the following website. https://clinicaltrials.gov/.
Consent to participate/Consent for publication
All patients provided written informed consent.
Code availability
Codes provided in Supplementary Information.
Author Contributions
Wrote manuscript: A.M., J.Y.J., M.R.W., B.A., and H.W. Designed research: A.M. and H.W. Performed research: A.M. Analyzed data: A.M.
References
- 1.Zhu AZ. Quantitative translational modeling to facilitate preclinical to clinical efficacy & toxicity translation in oncology. Future Sci OA. 2018;4(5):FSO306. 10.4155/fsoa-2017-0152. 10.4155/fsoa-2017-0152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Polivka J Jr, Janku F. Molecular targets for cancer therapy in the PI3K/AKT/mTOR pathway. Pharmacol Ther. 2014;142(2):164–75. 10.1016/j.pharmthera.2013.12.004. 10.1016/j.pharmthera.2013.12.004 [DOI] [PubMed] [Google Scholar]
- 3.Vanhaesebroeck B, Perry MWD, Brown JR, Andre F, Okkenhaug K. PI3K inhibitors are finally coming of age. Nat Rev Drug Discov. 2021;20(10):741–69. 10.1038/s41573-021-00209-1. 10.1038/s41573-021-00209-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dolgin E. Cancer drug approvals and setbacks in 2022. Nat Cancer. 2022;3(12):1406–8. 10.1038/s43018-022-00482-y. 10.1038/s43018-022-00482-y [DOI] [PubMed] [Google Scholar]
- 5.2023 Guidance document. Optimizing the Dosage of Human Prescription Drugs and Biological Products for the Treatment of Oncologic Diseases. US Food and Drug Administration. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/optimizing-dosage-human-prescription-drugs-and-biological-products-treatment-oncologic-diseases. Accessed 8 Jan 2023.
- 6.2023 Project Optimus. Reforming the dose optimization and dose selection paradigm in oncology. US Food and Drug Administration. https://www.fda.gov/about-fda/oncology-center-excellence/project-optimus. Accessed 8 Jan 2023.
- 7.Bueno L, de Alwis DP, Pitou C, Yingling J, Lahn M, Glatt S, et al. Semi-mechanistic modelling of the tumour growth inhibitory effects of LY2157299, a new type I receptor TGF-beta kinase antagonist, in mice. Eur J Cancer. 2008;44(1):142–50. 10.1016/j.ejca.2007.10.008. 10.1016/j.ejca.2007.10.008 [DOI] [PubMed] [Google Scholar]
- 8.Wong H, Belvin M, Herter S, Hoeflich KP, Murray LJ, Wong L, et al. Pharmacodynamics of 2-[4-[(1E)-1-(hydroxyimino)-2,3-dihydro-1H-inden-5-yl]-3-(pyridine-4-yl)-1H-pyrazol-1-yl]ethan-1-ol (GDC-0879), a potent and selective B-Raf kinase inhibitor: understanding relationships between systemic concentrations, phosphorylated mitogen-activated protein kinase kinase 1 inhibition, and efficacy. J Pharmacol Exp Ther. 2009;329(1):360–7. 10.1124/jpet.108.148189. 10.1124/jpet.108.148189 [DOI] [PubMed] [Google Scholar]
- 9.Yang J, Nie J, Ma X, Wei Y, Peng Y, Wei X. Targeting PI3K in cancer: mechanisms and advances in clinical trials. Mol Cancer. 2019;18(1):26. 10.1186/s12943-019-0954-x. 10.1186/s12943-019-0954-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Josephs DH, Sarker D. Pharmacodynamic biomarker development for PI3K pathway therapeutics. Transl Oncogenom. 2015;7(Suppl 1):33–49. 10.4137/TOG.S30529. 10.4137/TOG.S30529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Owonikoko TK, Khuri FR. Targeting the PI3K/AKT/mTOR pathway: biomarkers of success and tribulation. Am Soc Clin Oncol Educ. 2013. 10.14694/EdBook_AM.2013.33.e395. 10.14694/EdBook_AM.2013.33.e395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wallin JJ, Edgar KA, Guan J, Berry M, Prior WW, Lee L, et al. GDC-0980 is a novel class I PI3K/mTOR kinase inhibitor with robust activity in cancer models driven by the PI3K pathway. Mol Cancer Ther. 2011;10(12):2426–36. 10.1158/1535-7163.MCT-11-0446. 10.1158/1535-7163.MCT-11-0446 [DOI] [PubMed] [Google Scholar]
- 13.Dolly SO, Wagner AJ, Bendell JC, Kindler HL, Krug LM, Seiwert TY, et al. Phase I study of apitolisib (GDC-0980), dual phosphatidylinositol-3-kinase and mammalian target of rapamycin kinase inhibitor, in patients with advanced solid tumors. Clin Cancer Res. 2016;22(12):2874–84. 10.1158/1078-0432.CCR-15-2225. 10.1158/1078-0432.CCR-15-2225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Salphati L, Pang J, Plise EG, Lee LB, Olivero AG, Prior WW, et al. Preclinical assessment of the absorption and disposition of the phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor GDC-0980 and prediction of its pharmacokinetics and efficacy in human. Drug Metab Dispos. 2012;40(9):1785–96. 10.1124/dmd.112.046052. 10.1124/dmd.112.046052 [DOI] [PubMed] [Google Scholar]
- 15.Therasse P, Arbuck SG, Eisenhauer EA, Wanders J, Kaplan RS, Rubinstein L, et al. New guidelines to evaluate the response to treatment in solid tumors. European Organization for Research and Treatment of Cancer, National Cancer Institute of the United States, National Cancer Institute of Canada. J Natl Cancer Inst. 2000;92(3):205–16. 10.1093/jnci/92.3.205. [DOI] [PubMed]
- 16.Byrne MJ, Nowak AK. Modified RECIST criteria for assessment of response in malignant pleural mesothelioma. Ann Oncol. 2004;15(2):257–60. 10.1093/annonc/mdh059. 10.1093/annonc/mdh059 [DOI] [PubMed] [Google Scholar]
- 17.Beal S, Boeckmann L, Bauer R, Sheiner L. NONMEM User’s Guides. (1989–2009). NONMEM User’s Guides (1989–2009). Elliot City: Icon Development Solutions; 2009.
- 18.R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing. Vienna: R Core Team; 2019.
- 19.Goldmacher GV, Conklin J. The use of tumour volumetrics to assess response to therapy in anticancer clinical trials. Br J Clin Pharmacol. 2012;73(6):846–54. 10.1111/j.1365-2125.2012.04179.x. 10.1111/j.1365-2125.2012.04179.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tanaka C, O’Reilly T, Kovarik JM, Shand N, Hazell K, Judson I, et al. Identifying optimal biologic doses of everolimus (RAD001) in patients with cancer based on the modeling of preclinical and clinical pharmacokinetic and pharmacodynamic data. J Clin Oncol. 2008;26(10):1596–602. 10.1200/JCO.2007.14.1127. 10.1200/JCO.2007.14.1127 [DOI] [PubMed] [Google Scholar]
- 21.Yamazaki S, Skaptason J, Romero D, Lee JH, Zou HY, Christensen JG, et al. Pharmacokinetic–pharmacodynamic modeling of biomarker response and tumor growth inhibition to an orally available cMet kinase inhibitor in human tumor xenograft mouse models. Drug Metab Dispos. 2008;36(7):1267–74. 10.1124/dmd.107.019711. 10.1124/dmd.107.019711 [DOI] [PubMed] [Google Scholar]
- 22.Yates JW, Dudley P, Cheng J, D’Cruz C, Davies BR. Validation of a predictive modeling approach to demonstrate the relative efficacy of three different schedules of the AKT inhibitor AZD5363. Cancer Chemother Pharmacol. 2015;76(2):343–56. 10.1007/s00280-015-2795-7. 10.1007/s00280-015-2795-7 [DOI] [PubMed] [Google Scholar]
- 23.Dayneka NL, Garg V, Jusko WJ. Comparison of four basic models of indirect pharmacodynamic responses. J Pharmacokinet Biopharm. 1993;21(4):457–78. 10.1007/BF01061691. 10.1007/BF01061691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mager DE, Wyska E, Jusko WJ. Diversity of mechanism-based pharmacodynamic models. Drug Metab Dispos. 2003;31(5):510–8. 10.1124/dmd.31.5.510. 10.1124/dmd.31.5.510 [DOI] [PubMed] [Google Scholar]
- 25.Sutherlin DP, Bao L, Berry M, Castanedo G, Chuckowree I, Dotson J, et al. Discovery of a potent, selective, and orally available class I phosphatidylinositol 3-kinase (PI3K)/mammalian target of rapamycin (mTOR) kinase inhibitor (GDC-0980) for the treatment of cancer. J Med Chem. 2011;54(21):7579–87. 10.1021/jm2009327. 10.1021/jm2009327 [DOI] [PubMed] [Google Scholar]
- 26.Sarker D, Ang JE, Baird R, Kristeleit R, Shah K, Moreno V, et al. First-in-human phase I study of pictilisib (GDC-0941), a potent pan-class I phosphatidylinositol-3-kinase (PI3K) inhibitor, in patients with advanced solid tumors. Clin Cancer Res. 2015;21(1):77–86. 10.1158/1078-0432.CCR-14-0947. 10.1158/1078-0432.CCR-14-0947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tarantelli C, Lupia A, Stathis A, Bertoni F. Is there a role for dual PI3K/mTOR inhibitors for patients affected with lymphoma? Int J Mol Sci. 2020. 10.3390/ijms21031060. 10.3390/ijms21031060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wong H, Alicke B, West KA, Pacheco P, La H, Januario T, et al. Pharmacokinetic–pharmacodynamic analysis of vismodegib in preclinical models of mutational and ligand-dependent hedgehog pathway activation. Clin Cancer Res. 2011;17(14):4682–92. 10.1158/1078-0432.CCR-11-0975. 10.1158/1078-0432.CCR-11-0975 [DOI] [PubMed] [Google Scholar]
- 29.Claret L, Gupta M, Han K, Joshi A, Sarapa N, He J, et al. Evaluation of tumor-size response metrics to predict overall survival in Western and Chinese patients with first-line metastatic colorectal cancer. J Clin Oncol. 2013;31(17):2110–4. 10.1200/JCO.2012.45.0973. 10.1200/JCO.2012.45.0973 [DOI] [PubMed] [Google Scholar]
- 30.Wong H, Choo EF, Alicke B, Ding X, La H, et al. Antitumor activity of targeted and cytotoxic agents in murine subcutaneous tumor models correlates with clinical response. Clin Cancer Res. 2012;18(14):3846–55. 10.1158/1078-0432.CCR-12-0738. 10.1158/1078-0432.CCR-12-0738 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Qualified researchers may request access to individual patient-level data through the clinical study data request platform (https://Vivli.org/).