

Abstract
The EMT-transcription factor ZEB1 is heterogeneously expressed in tumor cells and in cancer-associated fibroblasts (CAFs) in colorectal cancer (CRC). While ZEB1 in tumor cells regulates metastasis and therapy resistance, its role in CAFs is largely unknown. Combining fibroblast-specific Zeb1 deletion with immunocompetent mouse models of CRC, we observe that inflammation-driven tumorigenesis is accelerated, whereas invasion and metastasis in sporadic cancers are reduced. Single-cell transcriptomics, histological characterization, and in vitro modeling reveal a crucial role of ZEB1 in CAF polarization, promoting myofibroblastic features by restricting inflammatory activation. Zeb1 deficiency impairs collagen deposition and CAF barrier function but increases NFκB-mediated cytokine production, jointly promoting lymphocyte recruitment and immune checkpoint activation. Strikingly, the Zeb1-deficient CAF repertoire sensitizes to immune checkpoint inhibition, offering a therapeutic opportunity of targeting ZEB1 in CAFs and its usage as a prognostic biomarker. Collectively, we demonstrate that ZEB1-dependent plasticity of CAFs suppresses anti-tumor immunity and promotes metastasis.
Keywords: Colorectal Cancer, Tumor Microenvironment, Cancer-Associated Fibroblast Plasticity, Immune Checkpoint Blockade
Subject terms: Cancer, Immunology, Signal Transduction
Synopsis
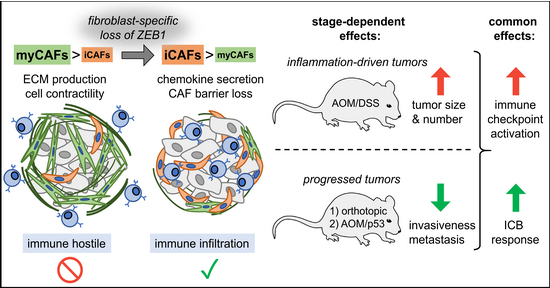
Fibroblast-specific Zeb1 knock-out in colorectal cancer models leads to a shift in myCAF and iCAF populations, promoting immune infiltration. This increases colitis-associated tumorigenesis, reduces progression and metastasis and renders tumors susceptible to immune checkpoint blockade.
Zeb1 loss in cancer-associated fibroblasts (CAFs) accelerates inflammation-driven colorectal tumorigenesis.
In sporadic tumors CAF-specific Zeb1-loss results in decreased progression and metastasis.
Impaired myofibroblast and increased inflammation result in increased immune cell infiltration and checkpoint activation.
Zeb1 deficiency in CAFs sensitizes colorectal cancers to immune checkpoint inhibition.
Fibroblast-specific Zeb1 knock-out in colorectal cancer models leads to a shift in myCAF and iCAF populations, promoting immune infiltration. This increases colitis-associated tumorigenesis, reduces progression and metastasis and renders tumors susceptible to immune checkpoint blockade.
Introduction
Colorectal cancer (CRC) is the third most frequent tumor type that accounts for the second highest cancer-related mortality worldwide (Morgan et al, 2023). Despite improved diagnosis and treatment options at an early stage, advanced CRC frequently leads to fatal metastatic relapse. Molecular classification has shown that the stroma-rich consensus molecular subtype 4 (CMS4) is linked with the worst prognosis in patients (Guinney et al, 2015). A refinement of this classification based on recent single-cell RNA sequencing (scRNA-seq) demonstrated that fibroblast enrichment together with tumor cell-intrinsic features contribute to poor prognosis (Joanito et al, 2022). These analyses identify cancer-associated fibroblasts (CAFs) as crucial players in the tumor microenvironment (TME) that drive disease progression, therapy resistance, and metastasis (Calon et al, 2015; Isella et al, 2015; Sahai et al, 2020; Schmitt and Greten, 2021).
Immune checkpoint blockade (ICB) therapy has shown impressive efficacy for patients with microsatellite instable (MSI) tumors (Andre et al, 2020; Le et al, 2015). These tumors display an elevated tumor mutation burden resulting in high immunogenicity. However, for most patients with microsatellite stable (MSS) tumors, no immunotherapies are available. In preclinical mouse models, inhibition of TGFβ signaling in CAFs promote lymphocyte infiltration and response to ICB (Tauriello et al, 2018). This example highlights the concept of a stroma-directed intervention to improve therapy of MSS tumors. However, the identification of specific targets to alter the fibroblast-rich stroma is needed.
Recent scRNA-seq analyses have shown that CAFs represent a heterogeneous cell population with several prototypic subtypes as initially identified in PDAC and later in other solid cancers including CRC (Bartoschek et al, 2018; Biffi et al, 2019; Elyada et al, 2019; Kieffer et al, 2020; Lee et al, 2020; Ohlund et al, 2017; Sebastian et al, 2020). Myofibroblast-like myCAFs localize closely to tumor cells and secrete extracellular matrix (ECM) encapsulating tumor cells, whereas iCAFs in the tumor periphery create an inflammatory milieu (Ohlund et al, 2017; Sahai et al, 2020). In addition, MHCII+ antigen-presenting CAFs (apCAFs) have been described (Elyada et al, 2019). The importance of CAF plasticity has been recently illustrated in rectal cancer, where IL1-induced iCAFs adopted a senescence phenotype, which mediates radioresistance and disease progression. Therapeutic inhibition of IL1 signaling allowed to restore radiosensitivity, indicating iCAFs as promising therapeutic target (Nicolas et al, 2022).
ZEB1 is a core EMT-transcription factor and is frequently upregulated at the invasive front of CRC and other cancer entities, where it orchestrates tumor stemness, metastasis, and therapy resistance (Caramel et al, 2018; Stemmler et al, 2019). Its tumor cell-specific loss leads to profound changes in gene expression, impairing cell plasticity (Krebs et al, 2017; Meidhof et al, 2015; Preca et al, 2015; Wellner et al, 2009). In pancreatic and breast cancer, ZEB1 was also found to be upregulated in the dysplastic fibroblast-rich stroma, which was correlated with poor survival (Bronsert et al, 2014; Fu et al, 2019). However, the functional relevance of ZEB1 in CAFs and its effect on tumor progression is yet unknown. Here, we have studied the role of ZEB1 in fibroblasts using mouse models for colitis-associated cancer and sporadic metastatic CRC demonstrating a key requirement for CAF diversification. Analysis of the immune TME identified a stage-specific effect on lymphocyte infiltration and CRC progression. Zeb1 loss reduced liver metastasis and augmented responsiveness to ICB, highlighting the potential of a CAF-directed therapy.
Results
ZEB1 in fibroblasts affects CRC tumorigenesis in a context-dependent manner
ZEB1 is expressed heterogeneously in the TME of both human and murine tumors (Fig. 1A,B), and high expression of ZEB1 in stromal cells of pancreatic cancer is a prognostic factor for poor survival in patients (Bronsert et al, 2014). To investigate a functional involvement, we generated fibroblast-specific Zeb1-deleted mice (FibΔZeb1), by combining the Zeb1fl/fl genotype (Brabletz et al, 2017) with the constitutive Col6a1-Cre transgene (Koliaraki et al, 2015) or with the tamoxifen-inducible Col1a2-CreERT2 knock-in allele (Zheng et al, 2002). Cre recombinase activity in both lines was restricted to fibroblasts as identified by Cre reporter alleles (Appendix Fig. S1A,B), and deletion of Zeb1 was confirmed at the genomic (Appendix Fig. S1C) and protein level in the colon of both models (Appendix Fig. S1D,E). Of note, loss of Zeb1 in fibroblasts did not compromise colon morphogenesis or homeostasis (Appendix Fig. S1F,G).
Figure 1. Context-dependent role of stromal ZEB1 during colorectal carcinogenesis.

(A, B) ZEB1 IHC on human (A) and ZEB1/E-cadherin IF stainings on mouse (B) CRC samples. Red and blue arrowheads depict cells with high and low/absent ZEB1 detection, respectively. (C–H) AOM/DSS model (C) showing representative endoscopic images (D) (dotted lines indicate unobstructed area) and quantification of colon obstruction in FibCtrl and FibΔZeb1 mice (n = 24/17 for FibCtrl/FibΔZeb1, Genotype: P = 0.0282, two-way ANOVA, day 75: P = 0.0382, Šídák’s multiple comparisons test), macroscopic images (E, colons opened longitudinally, arrowheads point to individual tumors) and quantification of tumor volume and number (F) (n = 23/16 for FibCtrl/FibΔZeb1, number: P = 0.0714, volume: P = 0.0180, Student’s t test). Quantitative analysis of ZEB1 depletion, proliferation (KI67) and apoptosis (cl. Caspase 3) (G) with representative KI67 IHC is given (H). For ZEB1, the fraction of positive stromal cells was quantified. For KI67 and cl. Caspase (CASP) 3, the fraction of all tumor cells was quantified (n = 17/11 for FibCtrl/FibΔZeb1, Zeb1: P = 0.0054, KI67: P = 0.0020, cl. CASP3: P = 0.1126, Mann–Whitney test). (I–L) Schematic overview of orthotopic transplantation of tumor organoids into the cecum of FibCtrl and FibΔZeb1 mice (I). AKPre organoids were generated after re-culturing cells retrieved from orthotopic AKP tumors. Analysis of tumor onset (detected by palpation) after orthotopic AKP organoid transplantation (J) (n = 12/10 for FibCtrl/FibΔZeb1, P = 0.6401, Mantel–Cox test) as well as of liver metastasis incidence (K) (AKP: P = 0.0427, AKPre: P = 0.0425, Fisher’s exact test) and numbers (L) after orthotopic transplantation of AKP or AKPre tumor organoids (n = 12/10 for FibCtrl/FibΔZeb1 with AKP and 12/14 for FibCtrl/FibΔZeb1 with AKPre, AKP: P = 0.8469, AKPre: P = 0.0007, two-way ANOVA). All mice with AKPre transplantation were treated with control IgG and are shown also in Fig. 5. Mice with AKP transplantation were treatment-naïve. Data information: Data are presented as mean ± SEM (D) or mean ± SD (F, G, L). Scale bars represent 50 µm (A, B), 5 mm (E) or 100 µm (H). Source data are available online for this figure.
FibΔZeb1 mice were subjected to the inflammation-driven AOM/DSS model (Neufert et al, 2007) (Fig. 1C), where deletion in fibroblasts did not affect overall or tumor-free survival of mice (Appendix Fig. S2A,B). Intriguingly, endoscopic scoring revealed increased colonic obstruction (Fig. 1D), which was confirmed by significantly increased individual and total adenoma volumes and a trend to increased tumor numbers in FibΔZeb1 mice at endpoints (Fig. 1E,F; Appendix Fig. S2C). Histologic differentiation was not affected by loss of stromal Zeb1 (Appendix Fig. S2D), but endoscopic analysis showed slightly increased colonic inflammation (Appendix Fig. S2E–G). By immunohistochemistry (IHC) fewer ZEB1-positive stromal cells and increased epithelial proliferation accompanied by slightly reduced cell death was observed in FibΔZeb1 mice (Fig. 1G,H). These data demonstrate that loss of Zeb1 in fibroblast promotes inflammation-driven adenoma growth.
Sporadic CRC progression was modeled by orthotopic transplantation of tumor organoids (Fumagalli et al, 2018) into FibΔZeb1 and FibCtrl mice (Fig. 1I). Tumor organoids were genetically engineered from normal colonic organoids to harbor mutations in Apc, Tp53 and Kras loci (ApcΔ/Δ, KrasG12D, Tp53Δ/Δ; AKP) and either transplanted directly into syngeneic mice, or re-cultured upon one round of orthotopic growth to promote further tumor progression in vivo (AKPre). AKPre tumors indeed displayed more aggressive tumor growth with earlier onset and less differentiated morphology. However, deletion of Zeb1 in fibroblasts did not affect the engraftment of organoids, overall survival, primary tumor size, and tumor morphology upon transplantation of AKP (Figs. 1J and EV1A–D) or AKPre organoids (Fig. EV1E–K). Strikingly, spontaneous metastasis to the liver was decreased in FibΔZeb1 mice, regardless of AKP or AKPre transplantation, as reflected in the fraction of metastasis-bearing mice and the number of metastases per mouse (Fig. 1K,L), indicating a pro-metastatic role of ZEB1 in CAFs of sporadic and progressed CRC. Overall, these findings suggest that ZEB1 in fibroblasts regulates colon cancer initiation and progression in a tumor context- and stage-dependent manner.
Figure EV1. Loss of Zeb1 in fibroblasts does not affect morphology of primary tumors in the orthotopic transplantation model.

(A) IF images and quantification of ZEB1 expression in tumor stroma after orthotopic transplantation of AKP tumor organoids (n = 10/9 independent mice for FibCtrl/FibΔZeb1, P < 0.0001, Student’s t test). (B) Quantification of primary tumor engraftment in treatment-naïve FibCtrl and FibΔZeb1 mice after orthotopic transplantation of AKP tumor organoids. Numbers of experimental mice are indicated. (C) Tumor volume after orthotopic transplantation of AKP tumor organoids (n = 8/5 independent mice for FibCtrl/FibΔZeb1, P = 0.7584, Student’s t test). (D, E) Representative H&E stainings of AKP (D) and AKPre (E) tumor sections. Top left corners show higher magnification of the indicated regions. (F–H) Analysis of tumors after orthotopic transplantation of AKPre tumor organoids in treatment-naïve FibCtrl and FibΔZeb1 mice. (F) Tumor onset (n = 4/5 for FibCtrl/FibΔZeb1; P = 0.6401, Mantel–Cox test). (G) Quantification of tumor engraftment. Numbers of experimental mice are indicated. (H) Tumor volume (n = 4/4 independent mice for FibCtrl/FibΔZeb1). (I–K) Analysis of tumors after orthotopic transplantation of AKPre tumor organoids in control IgG-treated FibCtrl and FibΔZeb1 mice. These mice are shown again as controls in Fig. 5. (I, J) Tumor onset (I) and quantification (J) after orthotopic transplantation of AKPre tumor organoids (n = 12/14 for FibCtrl/FibΔZeb1, P = 0.4564, Mantel–Cox test). (K) Tumor volumes (n = 8/10 independent mice for FibCtrl/FibΔZeb1). Only tumors collected after day 28 were included. Data information: Data are represented as mean ± SD (A, C, H, K). Scale bars represent 50 µm (A) or 1 mm (D, E). Source data are available online for this figure.
Fibroblast diversity is reduced upon deletion of Zeb1
We applied scRNA-seq to gain insights into the cellular heterogeneity of CAFs and the transcriptional changes upon Zeb1 loss. AOM/DSS tumors were enzymatically dissociated, and CAFs were enriched by fluorescence-activated cell sorting (FACS) of CD326 (EPCAM)−, CD45−, and CD31− cells before sequencing (SORT-seq, Fig. 2A, left). Integrated clustering of cells from both genotypes was performed, followed by annotation using previously reported CAF signatures (Bartoschek et al, 2018; Elyada et al, 2019) (Fig. EV2A). Differential abundance analysis (Zhao et al, 2021) was performed for unbiased identification of genotype-specific differences. Prominent abundance differences were observed in two regions that were enriched for myCAF and iCAF-specific genes (Fig. 2B–D). FibCtrl cells were overrepresented in the myCAF region and FibΔZeb1 cells in the iCAF region (Fig. EV2B–D). Genotypes were studied independently to avoid that FibΔZeb1 cells are influenced by FibCtrl cells. Here, walktrap clustering resulted in eight and six CAF clusters in FibCtrl and FibΔZeb1 tumors, respectively (Fig. 2E). Cross-correlation of transcriptomes indicated ambiguity in FibΔZeb1 clusters number 1 and 4 (Fig. 2F). Furthermore, no Zeb1-deficient CAFs were matched to FibCtrl cluster 8, altogether suggesting impaired diversification and subtype specification in FibΔZeb1 CAFs. The transcriptomes of FibCtrl CAFs correlated well with “iCAF” and “myCAF” archetypes described in pancreatic cancer (Elyada et al, 2019; Ohlund et al, 2017) (Fig. 2G). In contrast, FibΔZeb1 CAFs displayed less clusters with myCAF-like identity.
Figure 2. scRNA sequencing of CAFs reveals a key role of ZEB1 for fibroblast plasticity.

(A) Experimental scheme for isolation of CAFs and other cell types. AOM/DSS adenomas (left) and primary tumors from the orthotopic AKP model (right) of FibCtrl and FibΔZeb1 mice were enzymatically dissociated, and fibroblasts were enriched by depletion of CD31+, CD45+, and EPCAM+ cells by flow cytometry. Numbers of analyzed mice and sorted cells are shown. (B–G) Fibroblast analysis from the AOM/DSS model. (B) Integrated UMAP-(Leiden) clusters of CAFs from n = 3 mice per genotype were subjected to differential abundance (‘DA’) analysis. 3 significant DA regions between FibCtrl and FibΔZeb1 were found (Wilcoxon P values for regions 1, 2, and 3: 5.37 × 10−14; 1.35 × 10−12 and 5.62 × 10−06). Note that region 2 derived exclusively from one mouse and was therefore neglected. (C) Projections of iCAF and myCAF gene signature scores (Elyada et al, 2019). Note the highlighted DA regions from (B). (D) Log2 fold changes of representative DAseq marker genes in DA regions 1 and 3, designated as “iCAF” and “myCAF” regions, respectively (P < 0.01, as determined by STG (stochastic gates) within DAseq). (E) Independent t-SNE clustering in FibCtrl (left) and FibΔZeb1 mice (right). (F) Cluster similarities defined by cluster annotations based on “SingleR” scores (see “Methods” for details) (Aran et al, 2019). Grayscale shows the log2-transformed numbers of FibΔZeb1 cells assigned to the different FibCtrl clusters. (G) Heatmap showing the similarity (annotation scores) of gene expression in CAF clusters with published gene sets. (H–J) scRNA-seq of fibroblasts, immune cells and tumor cells after orthotopic transplantation of AKP organoids. (H) Data from n = 4 mice per genotype of the orthotopic model were subjected to scRNA sequencing and t-SNE clustering in FibCtrl (left) or FibΔZeb1 tumors (right). (I, J) Down- and upregulated gene sets in FibΔZeb1 CAFs in comparison to the two FibCtrl CAF clusters, as determined using Enrichr. Differential genes between FibΔZeb1 CAFs and each of the two FibCtrl CAF clusters were individually determined and pooled before enrichment analysis (FDR ≤ 0.1; P ≤ 0.05, as determined by default Welch t tests within the findMarkers function). (K, L) Tumor sections from mice after transplantation with AKP organoids were subjected to multiplexed immunostaining (n = 5/6 for FibCtrl/FibΔZeb1). (K) Representative images of fibroblast and CAF markers (VIM, αSMA, C3, MHCII). (L) Individual cells were UMAP embedded based on their CAF marker intensities and the density distribution of cells from FibCtrl or FibΔZeb1 mice is shown. Data information: Scale bars represent 100 µm (K). Source data are available online for this figure.
Figure EV2. scRNA-seq in AOM/DSS and orthotopic models show reduced CAF diversity and impaired subtype-specific gene expression in FibΔZeb1 tumors.

(A) Phenotypic annotation of AOM/DSS CAFs (n = 3 mice per genotype; corresponding to Fig. 2) upon integrated clustering by scoring of iCAF/myCAF/apCAF/mCAF/vCAF gene signatures (Bartoschek et al, 2018; Elyada et al, 2019) and displayed on UMAP Leiden clusters, pooled according to the determined scores. Note that ‘i/myCAFs’ share features of iCAFs and myCAFs and that the identities of 3 cell clusters could not be determined (ND) using these gene sets. (B) Genotype distribution of cells in AOM/DSS CAFs in DA regions (please refer to Fig. 2). The fraction of FibCtrl and FibΔZeb1 CAFs in each DA region or among all CAFs is shown. (C, D) Gene set enrichment analysis using Enrichr of marker genes from DAseq region 1 (C) and 3 (D) as compared to all other CAFs (Benjamini-Hochburg corrected Fisher’s exact test). (E) t-SNE sub-clustering of fibroblasts separately in FibCtrl (left) and FibΔZeb1 mice (right) of the orthotopic model showing less clusters in FibΔZeb1. (F) Cluster similarities defined by cluster annotations based on ‘SingleR’ scores (see "Methods" for details) (Aran et al, 2019). Grayscale shows the log2-transformed number of cells across clusters. Note the low similarity of FibΔZeb1 cells with FibCtrl clusters 2 and 5. (G) Heatmap showing the similarity (annotation scores) of gene expression in CAF clusters with published gene sets. Note, the high scores of FibCtrl clusters 2 and 5 when compared with ‘iCAF’ and ‘myCAF’ signatures, respectively, and absence in FibΔZeb1. Source data are available online for this figure.
We also examined non-inflammation-driven orthotopic tumors by scRNA-seq. Following FACS, CAFs were analyzed in parallel to isolated CD45+ immune and EPCAM+ tumor cells (Fig. 2A, right). A separate analysis of genotypes resulted in two CAF clusters for FibCtrl but only one for FibΔZeb1 tumors (Fig. 2H). Individual analysis of CAFs confirmed that loss of Zeb1 limits the CAF repertoire (Fig. EV2E) and cross-annotation confirmed increased ambiguity in FibΔZeb1 (Fig. EV2F). We observed a higher correlation of CAF clusters with “iCAF” and “myCAF” archetypes in FibCtrl compared to FibΔZeb1 (Fig. EV2G). Consistent with AOM/DSS CAFs, differential gene expression analysis between FibCtrl and FibΔZeb1 CAFs showed reduced signatures for ECM organization and increased inflammatory terms (Fig. 2I,J). In summary, our data show that independent of the CRC model, CAF diversification and specification is strongly impaired when ZEB1 is absent.
To study the differentiation defects in situ, we performed multiplex immunofluorescence staining. In orthotopic tumors, fibroblasts were defined as VIM+, CD45−, EPCAM− cells, and CAFs were further characterized by αSMA, C3, and MHCII staining (Figs. 2K and EV3A,B). Quantitative image analysis and dimensionality reduction allowed classification into myCAF, iCAF, and apCAF-enriched populations (Fig. EV3C). While the majority of cells in FibCtrl displayed a myofibroblast-like phenotype, CAFs in FibΔZeb1 tumors were predominated by the inflammatory subtype (Fig. 2L). Here, Zeb1 loss did not affect apCAFs or result in a mixed αSMA+ /C3+ iCAF/myCAF identity (Fig. EV3A–D).
Figure EV3. Multiplexed IF staining of tumor sections reveals an enrichment of iCAF-like cells in FibΔZeb1 tumors.

(A, B) Single marker images (VIM, αSMA, C3, MHCII, EPCAM, CD45, DAPI) and merge of all channels for the representative images of FibCtrl (A) and FibΔZeb1 (B) orthotopic tumors shown in Fig. 2K. (C) Normalized staining intensities of CAF markers (αSMA, C3, MHCII) in UMAP embedding of CAFs from FibCtrl and FibΔZeb1 tumors. (D) Quantification of the distribution of CAF subtypes based on thresholds for αSMA (myCAF-like), C3 (iCAF-like) or MHCII (apCAF) staining intensity (n = 5/6 independent mice for FibCtrl/FibΔZeb1, myCAF: P = 0.0469, iCAF: P = 0.0499, Student’s t test). Data information: Data are presented as mean ± SD (D). Scale bars represent 100 µm (A, B). Source data are available online for this figure.
ZEB1 is crucial for myofibroblast differentiation
We next studied the functional consequences of Zeb1 loss and CAF polarization. We established adherent colon fibroblast cultures from Zeb1fl/fl mice, which showed a typical myofibroblast-like morphology. Induction of Cre recombinase activity resulted in efficient Zeb1 deletion in vitro, which was confirmed by immunofluorescence staining (Fig. 3A). Of note, Zeb1 deficiency-induced loss of fibrillary αSMA staining, demonstrating reduced myofibroblast differentiation and CAF activation (Sahai et al, 2020). Consistently, Zeb1-deleted fibroblasts showed lower expression of myofibroblast markers by qRT-PCR and bulk RNA sequencing analyses (Fig. 3B,C) and strongly reduced contraction in collagen (Fig. 3D). In contrast, we observed no major changes in proliferation or senescence of Zeb1-deficient fibroblasts (Appendix Fig. S3A,B). To study myofibroblast-specific functions in vivo, skin wound healing assays revealed delayed wound closure in FibΔZeb1 compared to FibCtrl mice (Fig. 3E; Appendix Fig. S3C). Furthermore, the impaired myCAF polarization in AOM/DSS adenomas and orthotopic tumors was associated with reduced deposition of collagen (Fig. 3F,G). Since collagenous ECM can constitute a physical barrier for immune cell infiltration, we conducted an in vitro migration assay to test the ability of fibroblasts to block the passage of mouse splenocytes (Fig. 3H). Strikingly, FibΔZeb1 fibroblasts showed a pronounced barrier defect. Collectively, these data demonstrate the fundamental role of ZEB1 in myofibroblast specification and function.
Figure 3. ZEB1 is critically involved in myofibroblast differentiation and functionality.

(A) Representative IF staining of ZEB1 and αSMA in fibroblasts after in vitro recombination. (B) qRT-PCR analysis of myofibroblast markers Acta2 and Pdgfrb in fibroblasts after in vitro recombination, (n = 4/6; 2 independent fibroblast lines per genotype in 2/3 biological replicates for Acta2/Pdgfrb, Acta2: P = 0.0014, Pdgfrb: P = 0.0190, Student’s t test). (C) Gene set enrichment analysis using myCAF and extracellular matrix (GO: 0031012) signatures. (D) Representative images and quantification of collagen contraction assay (n = 5/4 independent FibCtrl/FibΔZeb1 lines; P < 0.0001, Student’s t test). (E) Quantification of relative wound area in FibCtrl and FibΔZeb1 mice during a skin wound healing model (n = 16/22), day 1: P = 0.0539, day 2: P = 0.0326, two-way ANOVA). (F, G) Representative images (F) and quantification (G) of Picrosirius red staining of tumor sections from FibCtrl and FibΔZeb1 mice after AOM/DSS and orthotopic transplantation (45/50 image sets derived from n = 13/9 FibCtrl/FibΔZeb1 mice for AOM/DSS and 84/76 image sets derived from n = 8/9 FibCtrl/FibΔZeb1 mice for the orthotopic tumor model, AOM/DSS: P = 0.0023, orthotopic: P = 0.0015, Mann–Whitney test). Areas from representative images are marked in green. (H) Quantification of T-cell migration through a transwell insert alone or with a layer of fibroblasts after in vitro recombination of Zeb1 (n = 7 independent lines, no Fib vs FibCtrl: P < 0.0001, no Fib vs FibΔZeb1: P = 0.0002, FibCtrl vs FibΔZeb1: P = 0.0012, Tukey’s multiple comparisons test). Data information: Data are presented as mean ± SD (B, D, G, H) or mean ± SEM (E). Scale bars represent 200 µm (A) or 80 µm (F). Source data are available online for this figure.
Loss of Zeb1 in fibroblasts induces a pro-inflammatory tumor microenvironment
To clarify whether the inflammatory polarization in combination with the reduced collagen deposition of Zeb1-deleted CAFs affects tumor immune cell infiltration, we performed IHC analyses. Strikingly, in inflammation-induced AOM/DSS tumors, we found increased infiltration of CD4+ T cells and FOXP3+ Tregs, but not of CD8+ cells or F4/80+ macrophages into FibΔZeb1 tumors (Fig. 4A), indicating modulation of adaptive anti-tumor immunity. Concomitantly, increased B-cell infiltration and an overall upregulation of the immune checkpoint molecule PD-L1 were observed, suggesting tolerance induction upon T-cell activation. Similar to AOM/DSS adenomas, increased T- and B-cell infiltration and expression of PD-L1 was observed in FibΔZeb1 orthotopic tumors, reflecting sporadic CRC (Fig. 4B). Yet, in contrast to AOM/DSS tumors, CD8+ T cells and macrophages were enriched. Since the primary tumor size was comparable in FibCtrl and FibΔZeb1 orthotopic tumors (Fig. EV1C,H,K), the observed T-cell infiltration apparently did not result in an effective anti-tumor response, presumably due to T-cell inactivity.
Figure 4. ZEB1 attenuates inflammatory signaling in fibroblasts and limits immune cell infiltration in multiple CRC models.

(A, B) IHC-based quantification of immune cell infiltration and PD-L1 expression in tumors from FibCtrl and FibΔZeb1 mice in the AOM/DSS model (A) and after orthotopic transplantation of AKP tumor organoids (B). Number of experimental mice per genotype are indicated (CD4, CD8, FOXP3, F4/80, B220, PD-L1: P = 0.0414, 0.2745, 0.0005, 0.3751, 0.0134, 0.0082 (AOM/DSS), P = 0.3383, 0.0068, 0.5852, 0.0401, 0.0122, 0.0545 (orthotopic), Student’s t test). (C) qRT-PCR analysis of Cxcl1 and Ccl2 mRNA expression in fibroblasts after in vitro recombination of Zeb1 and stimulation with IL1α (n = 4, two independent fibroblast lines per genotype in two biological replicates, 0, 5, 24 h: P = 0.9248, 0.0070, 0.1816 (Ccl2), P = 0.9998, 0.0337, 0.0890 (Cxcl1), Šídák’s multiple comparisons test). (D) Western blot of NFκB pathway activity in fibroblasts after 15 min of IL1α stimulation. β-ACTIN detection was used as a loading control. (E) IHC-based quantification of phospho-NFκB p65 (Ser536) in mice after AOM/DSS tumorigenesis or transplantation of AKPre organoids. Numbers of experimental mice per genotype are indicated (AOM/DSS: P = 0.0435, orthotopic: P = 0.0238, Student’s t test). (F) Quantification of T-cell attraction to fibroblasts after in vitro recombination of Zeb1 (n = 7 independent lines, no Fib vs FibCtrl: P = 0.0014, no Fib vs FibΔZeb1: P < 0.0001, FibCtrl vs FibΔZeb1: P = 0.0043, Tukey’s multiple comparisons test). (G–I) FibCtrl and FibΔZeb1 fibroblasts were seeded in 3D ECM and expression of fibroblast subtype markers was quantified by qRT-PCR. Baseline marker expression of FibCtrl and FibΔZeb1 fibroblasts without additional treatment (G) and after treatment with IL1α (H) or TGFβ (I) relative to the respective untreated condition (n = 9/6 for FibCtrl/FibΔZeb1 fibroblast lines, Ccl2, Cxcl1, Acta2, Tagln: P = 0.6830, 0.8861, 0.0184, 0.0084 (baseline), P = 0.9318, 0.5463, 0.3182, 0.7161 ( + IL1α), P = 0.0670, 0.0077, 0.1469, 0.0346 ( + TGFβ), Student’s t test). Data information: Data are presented as mean ± SD (A–C, E–I). Source data are available online for this figure.
To investigate if the differences between both models can be explained by the inflammatory environment in the AOM/DSS model, we employed another model of sporadic CRC, in which AOM-induced mutagenesis in combination with epithelial cell-specific Tp53 deletion results in invasive CRC (Neufert et al, 2021; Schwitalla et al, 2013) (Fig. EV4A). Here, deletion of Zeb1 in fibroblasts resulted in fewer, smaller and less invasive tumors, indicating delayed tumor progression in FibΔZeb1 mice in the AOM/p53 model (Fig. EV4B–D). We observed enhanced infiltration of CD4+ and CD8+ cells but no and only trending increase in FOXP3+ Tregs and stromal PD-L1 levels, respectively (Fig. EV4E). Collectively, our data show a context-dependent immunomodulatory role of ZEB1 expression in fibroblasts. This notion was supported by analysis of AOM/DSS tumors at day 50 during the acute phase of inflammation. Even before overt tumor outgrowth, significantly increased CD8+ and FOXP3+ cell infiltration was observed (Fig. EV4F–H), suggesting that the acute inflammation triggers a compensatory regulation of the immune microenvironment upon Zeb1 loss in fibroblasts.
Figure EV4. Loss of Zeb1 in fibroblasts impairs tumor progression and increases T-cell infiltration in the invasive non-inflammation-driven AOM/p53 model.

(A) Schematic representation of the AOM/p53 model. (B) Quantification of numbers and volumes of tumors in the colons of FibCtrl and FibΔZeb1 mice at the endpoint (n = 15/22 for FibCtrl/ FibΔZeb1, number: P = 0.0433, volume: P = 0.0202, Mann–Whitney test). (C) Macroscopic evaluation of the most advanced/progressed tumor per mouse categorizing ‘T4’ as fully invasive (penetrating the muscle) or not (‘T1-T3’) (fraction of mice is given, n = 15/22 for FibCtrl/FibΔZeb1, P = 0.0252, Fisher’s exact test). (D) Representative H&E stainings with a higher magnification of the indicated region to the right. (E) IHC-based quantification of immune cell infiltration and stromal PD-L1 expression of tumors from FibCtrl and FibΔZeb1 mice. Numbers of experimental mice per genotype are indicated (CD4, CD8, FOXP3, B220, PD-L1: P < 0.0001, P = 0.0010, 0.3618, 0.3748, 0.0745, Student’s t test). (F–H) AOM/DSS model until day 50 in FibCtrl and FibΔZeb1 mice. Macroscopic analysis of early adenomas (number: P = 0.8339, volume: P = 0.2088, Student’s t test) (F) and IHC quantification of epithelial proliferation and cell death (KI67: P = 0.5403, cl. CASP3: P = 0.2773, Student’s t test) (G), as well as immune cell infiltration (H). Numbers of experimental mice per genotype are indicated (CD4, CD8, FOXP3, B220: P = 0.0296, 0.9515, 0.0524, 0.3996, Student’s t test). Data information: Data are presented as mean ± SD (B, E–H). Scale bars represent 1.5 mm (D, left) or 100 µm (D, right). Source data are available online for this figure.
To explore the mechanism for the altered immune cell infiltration in Zeb1-deficient CAFs, we tested the response of cultured fibroblasts to the chemokine IL1α, a crucial stimulator of inflammatory polarization in CRC CAFs (Nicolas et al, 2022). We monitored basal and IL1α-induced NFκB activation in cultured FibCtrl and FibΔZeb1 fibroblasts by qRT-PCR and Western blot analyses. Upon stimulation, FibΔZeb1 fibroblasts showed increased expression of NFκB targets including Ccl2 and Cxcl1 that both have been described as iCAF markers (Biffi et al, 2019; Elyada et al, 2019; Nicolas et al, 2022) (Fig. 4C). Consistently, strongly increased levels of phosphorylated NFκB and IκBα were observed in lysates of FibΔZeb1 fibroblasts that were stimulated with IL1α for 15 min (Fig. 4D), when FibCtrl cells already partially resolved the signal, suggesting an overshooting pathway activation. Indeed, immunostaining of phospho-NFκB showed increased pathway activity in both FibΔZeb1 AOM/DSS and orthotopic tumors (Fig. 4E). Furthermore, secretome analysis in cultured CAFs from AOM/DSS tumors identified increased basal levels of CCL2 upon Zeb1 loss (Appendix Fig. S4A,B), which was confirmed by IHC (Appendix Fig. S4C). Notably, increased phospho-NFκB and CCL2 staining were also observed during the acute inflammation in FibΔZeb1 mice at day 50 of AOM/DSS treatment (Appendix Fig. S4D,E). Our data suggest that elevated cytokine production from Zeb1-deficient CAFs may act as a potent lymphocyte chemoattractant. Thus, the influence of FibCtrl or FibΔZeb1 fibroblasts on T-cell recruitment was tested in a transwell migration assay using stimulated T cells in co-culture with fibroblasts, where migration was significantly increased towards a FibΔZeb1 fibroblast monolayer (Fig. 4F).
As a plastic cell population, the observed changes could result from an intrinsic differentiation bias, or a failure to respond to external signals. To tackle this question, we seeded CAFs in a 3D ECM, representing a quiescent status, or applied IL1α or TGFβ treatment, to direct them into the myCAF and iCAF lineages, respectively (Ohlund et al, 2017). qRT-PCR showed that under baseline conditions FibΔZeb1 expressed unaffected levels of iCAF markers (Ccl2 and Cxcl1) but significantly reduced levels of myCAF markers (Acta2 and Tagln) (Fig. 4G). While the induction of inflammatory genes by IL1α was comparable in this setting (Fig. 4H), after TGFβ addition FibΔZeb1 CAFs showed a striking defect to downregulate inflammatory genes (Fig. 4I). In contrast, the TGFβ-mediated induction of myCAF markers was less prominently affected. Collectively, our data indicate that ZEB1 limits the sensitivity of fibroblasts to NFκB-activating signals, which causes an iCAF bias affecting tumor immune infiltration.
Loss of Zeb1 in CAFs enhances the response of CRC to immune checkpoint blockade
Given the refractoriness of non-hypermutated CRC to ICB therapy, the increased immune cell infiltration and induction of checkpoint molecules observed in FibΔZeb1 mice may point to a strategy to render MSS tumors more sensitive. We investigated this hypothesis in orthotopic and AOM/DSS tumors. Following transplantation of AKPre organoids, tumor-bearing mice were injected subcutaneously with anti-PD-L1 antibodies for 3 weeks. Strikingly, anti-PD-L1 therapy but not control IgG significantly delayed tumor growth of FibΔZeb1 in comparison with FibCtrl mice, eventually resulting in smaller FibΔZeb1 tumors at the endpoint (Fig. 5A,B). In addition, infiltration of CD4+ , CD8+ T cells, and B cells was further increased by ICB in tumors from treated FibΔZeb1 mice, whereas FOXP3+ cells were not affected. PD-L1 expression was reduced in FibΔZeb1 mice upon ICB, indicating successful reactivation of the adaptive immune response (Fig. EV5A). In a more clinically relevant setting, we explored whether ICB sensitivity is also induced by later Zeb1 deletion, when tumors have already formed. For this purpose, we inactivated Zeb1 in orthotopic tumors simultaneously with ICB administration (Fig. EV5B–D). Although not significant, a similar trend towards smaller tumors was observed in FibΔZeb1 mice, when Zeb1 was depleted from a preformatted immune TME. In autochthonous AOM/DSS tumors, we employed dual ICB targeting PD-L1 and CTLA-4, as we found a substantial increase in the number of Tregs in tumors of FibΔZeb1 mice in this model (Fig. 4A). Tumor growth was monitored longitudinally via endoscopy, and adenoma-bearing mice were subjected to ICB after recovery from cyclic DSS-induced inflammation at day 70. Strikingly, dual ICB abrogated tumor growth in FibΔZeb1 mice combined with a more robust increase in CD8+ T cells, ablation of FOXP3+ cells, and reduced PD-L1 expression (Figs. 5C,D and EV5E). Together, our analyses demonstrate that loss of Zeb1 in fibroblasts induces sensitivity to ICB in both colitis-induced and sporadic tumor models.
Figure 5. Loss of Zeb1 in fibroblasts enables response to ICB therapy.

(A) Kaplan–Meier analysis showing tumor-free survival of FibCtrl and FibΔZeb1 mice after orthotopic transplantation of AKPre organoids and intraperitoneal injection of anti-(a)-PD-L1 antibodies or control IgGs, as indicated by red arrows. Recombination of Zeb1 was induced by tamoxifen food starting from day (d) 3. Mice were considered tumor-free if no tumor was detected by palpation. Numbers of experimental mice per condition are indicated (FibCtrl-ICB vs FibΔZeb1-ICB: P = 0.0189, Mantel–Cox test). (B) Representative H&E images and quantification of tumor volumes after orthotopic transplantation of AKPre tumor organoids and ICB. Only tumors collected after d28 were included in this analysis. Number of experimental mice per condition are indicated. IgG-treated mice contributed to the initial AKPre analysis (FibCtrl-ICB vs FibΔZeb1-ICB: P = 0.0397, FibΔZeb1-IgG vs FibΔZeb1-ICB: P = 0.0792, Šídák’s multiple comparisons test). (C, D) ICB in the AOM/DSS model, applied by intraperitoneal injection of a-PD-L1 and a-CTLA-4 antibodies or control IgGs. Starting at d70 of AOM/DSS tumorigenesis antibodies were administered two times/week to all mice with at least 25–30% colon obstruction. This time point corresponds to d0 of ICB. Representative endoscopic images at d0 and d35 of ICB in the AOM/DSS model. Dotted line indicates unobstructed areas (C). Quantification of colon obstruction relative to d0 (D). Numbers of experimental mice per condition are indicated (FibCtrl-ICB vs FibΔZeb1-ICB: two-way ANOVA, day 35: P = 0.0211. Data information: Data are presented as mean ± SD (B) or mean ± SEM (D). Scale bars represent 1 mm (B). Source data are available online for this figure.
Figure EV5. Monitoring of immune cell infiltration after ICB and effect of late-stage deletion of Zeb1 on ICB.

(A) IHC-based quantification of immune cell infiltration and PD-L1 expression in tumors from FibCtrl and FibΔZeb1 mice after orthotopic transplantation of AKPre organoids and intraperitoneal injection of a-PD-L1 antibodies or control IgGs. Numbers of experimental mice per condition are indicated (CD4, CD8, FOXP3, B220, PD-L1: P = 0.0032, 0.0076, 0.6442, 0.0027, >0.9999 (FibCtrl-ICB vs FibΔZeb1-ICB), p = 0.1568, 0.0525, 0.0304, 0.2150, 0.0004 (FibΔZeb1-IgG vs FibΔZeb1-ICB), Šídák’s multiple comparisons test). IgG-treated mice contributed to initial AKPre analysis (Fig. 1). (B) Kaplan–Meier analysis showing tumor-free survival of FibCtrl and FibΔZeb1 mice after orthotopic transplantation of AKPre organoids and intraperitoneal injection of a-PD-L1 antibodies or control IgGs, as indicated by red arrows. Recombination of Zeb1 was induced by tamoxifen food starting from day (d) 14. Mice were considered tumor-free if no tumor was detected by palpation. Numbers of experimental mice per condition are indicated (P = 0.7583, Mantel–Cox test). (C) Quantification of tumor volumes after orthotopic transplantation of AKPre tumor organoids and ICB with late recombination of Zeb1. Only tumors collected after d28 were included in this analysis (n = 8 independent mice, P = 0.6276, Student’s t test). (D) IF-based quantification of stromal cells expressing ZEB1 (n = 8/8 independent mice for FibCtrl/FibΔZeb1, P = 0.0002, Student’s t test). (E) IHC-based quantification of immune cell infiltration and PD-L1 expression in AOM/DSS tumors from FibCtrl and FibΔZeb1 mice receiving ICB (2 intraperitoneal injections of a-PD-L1 and a-CTLA-4 antibodies or control IgGs per week starting at d70 of AOM/DSS tumorigenesis). Numbers of experimental mice per condition are indicated (CD4, CD8, FOXP3, B220, PD-L1: P = 0.9902, 0.0802, <0.0001, 0.0070, 0.0003 (FibΔZeb1-IgG vs FibΔZeb1-ICB), P = 0.6241, 0.7615, 0.0070, 0.3163, 0.0144 (FibCtrl-IgG vs FibΔZeb1-IgG), Šídák’s multiple comparisons test). Data information: Data are presented as mean ± SD (A, C–E). Source data are available online for this figure.
Discussion
Functional diversification of CAFs is a result of coevolution with tumor cells in the TME. Using autochthonous and organoid transplantation models of CRC, we have discovered a key role of ZEB1 in governing this plasticity. Zeb1 deletion severely impairs myofibroblastic and modulates inflammatory CAF functions, jointly affecting immune cell infiltration. Consequently, inflammation-driven adenoma formation is augmented, yet more progressed cancer models show reduced invasiveness and metastasis. Despite this stage-dependent outcome, the common immunomodulation leads to checkpoint activation in FibΔZeb1 tumors. We show that targeting ZEB1 sensitizes to ICB reinforcing a translational rationale to target specific CAF subtypes for therapy of MSS tumors.
Our data identify that ZEB1 in CAFs acts as an immunosuppressor that promotes malignant progression. As both archetypes, i.e., iCAFs and foremost myCAFs, are strongly dysregulated in the absence of ZEB1, we conclude that ZEB1 controls general CAF plasticity. This is supported by our single-cell analysis in both models, the impaired myofibroblastic functionality and the augmented response to inflammatory stimuli in vitro. While previous reports have established that EMT(-related-) TFs ZEB1, SNAIL, TWIST, and PRRX1 can regulate the classical mechanoinvasive features of CAFs directly affecting tumor malignancy (Feldmann et al, 2021; Stanisavljevic et al, 2015; Yeo et al, 2018), our data highlight a critical impact of CAF plasticity on modulation of anti-tumor immunity that may contribute to the poor prognosis reported in breast and pancreatic cancer patients with elevated ZEB1+ stromal cells (Bronsert et al, 2014; Ouled Dhaou et al, 2020).
As mechanistic basis for enhanced inflammation and immune cell infiltration, we show reduced collagen deposition, increased inflammatory signaling and chemoattraction in Zeb1-deficient CAFs collectively modulating the immune TME. The stage-dependent consequences can be explained by the known tumor-promoting role of inflammation in the AOM/DSS model (Greten et al, 2004; Koliaraki et al, 2015; Neufert et al, 2007; Neufert et al, 2021; Schmitt and Greten, 2021). Consistently, phospho-NFκB and CCL2 levels were upregulated already during the acute phase of inflammation, and we found strongly increased infiltration of B cells, CD4+ T cells and FOXP3+ Tregs in FibΔZeb1 tumors accompanied by enhanced PD-L1. CD8+ T cells were unaffected, yet strongly induced upon dual ICB, indicating an immune checkpoint involving Tregs. This agrees with previous reports on tolerance induction in AOM/DSS and colitis-associated CRC (Olguin et al, 2018; Pastille et al, 2014; Yassin et al, 2019). In contrast, increased CD8+ T cells and delayed tumor progression in the sporadic FibΔZeb1 tumors showed an unaffected FOXP3+ infiltration, pointing towards Tregs as part of a compensatory immunomodulatory mechanism in the AOM/DSS model. Taken together, we conclude that loss of Zeb1 in CAFs facilitates inflammation, immune infiltration, and co-activated immunosuppression.
We furthermore identified ZEB1 as an important regulator balancing myofibroblastic and inflammatory functions of CAFs. Of note, TGFβ signaling has been shown essential for the acquisition of classical myofibroblast phenotypes (Biffi et al, 2019; Elyada et al, 2019; Sahai et al, 2020; Tauriello et al, 2018; Tauriello et al, 2022) and in tumor cells, ZEB1 is a key mediator of TGFβ signaling (Krebs et al, 2017; Schuhwerk et al, 2022; Stemmler et al, 2019). Our data indicates that ZEB1 acts downstream of TGFβ signaling and facilitates myofibroblast polarization at least in part by downregulation of inflammatory gene expression. Consistently, Zeb1-deleted fibroblasts displayed increased NFκB activation in vitro and in vivo. In this regard, it is important to mention that ZEB1 has been shown as a direct transcriptional (co-)inducer of inflammatory gene expression, like IL6, IL8 and others in several cell types, such as breast cancer cells and fibroblasts (Fu et al, 2019; Katsura et al, 2017), corneal fibroblasts, hematopoietic and myeloid cells (Cortes et al, 2017; Liang et al, 2022; Qian et al, 2021; Scott and Omilusik, 2019; Wang et al, 2009). Together these data suggest that ZEB1 elicits context-dependent effects on inflammation.
The sensitization of unresponsive tumors to ICB that was observed in two independent mouse CRC models points towards a more general CAF-engaging immune checkpoint in CRC, in line with the refractoriness of non-hypermutated tumors (Le et al, 2015; Tauriello et al, 2022). Thus, ZEB1 expression in CAFs may serve as a negative predictive marker for ICB efficacy and agents that interfere with ZEB1 function may be beneficial to enhance immune infiltration and ICB sensitivity. Yet, before clinical translation is possible several limitations need to be considered: Because pharmacologic targeting of ZEB1 as a TF is challenging, new strategies will be required such as the development of PROTACs. Alternatively, interference with ZEB1 downstream programs could allow to modulate CAF identities. For instance, inhibiting the DNA damage response (DDR) kinase ATM has recently been shown to inhibit myofibroblastic features, increase immune infiltration and sensitize to immune checkpoint therapy in subcutaneous tumor models (Mellone et al, 2022). In this regard, we recently discovered an actionable vulnerability in ZEB1high cancer cell sub-populations by inhibiting the DDR nuclease MRE11 (Schuhwerk et al, 2022). In addition, a more detailed understanding will be essential, how CAF polarization affects the tumor immune environment in patients. Here in particular, the impact of standard therapies should be considered, because radiotherapy-induced senescence in iCAFs favors therapy resistance and a poor outcome in rectal cancer (Nicolas et al, 2022). Likewise, ablation of αSMA-high myCAFs, or deletion of type 1 collagen in myofibroblasts was shown to aggravate disease course in PDAC and experimental liver metastasis (Bhattacharjee et al, 2021; Chen et al, 2021; Ozdemir et al, 2014). Hence, targeting regulators of CAF plasticity, rather than fully depleting integral components of the ECM or CAFs in general, may be beneficial. Our study suggests that reduction of ZEB1 in CAFs may turn immunologically “cold” into “hot” CRCs and thereby sensitize patients to ICB. Given the known role of ZEB1 in tumor cells to induce EMT, stemness, and chemoresistance, combined targeting of ZEB1 in fibroblast and tumor cells might act synergistically to improve CRC therapy. However, correct timing may be crucial to allow reformatting of the immune TME for efficient sensitization to ICB.
Methods
Ethics statement
Animal husbandry and all experiments were performed according to the European Animal Welfare laws and guidelines. The protocols were approved by the committee on ethics of animal experiments of Bavaria (Regierung von Unterfranken, Würzburg; TS-18/14, 55.2-DMS-2532-2-270, -2-952, and -2-1133) and of Hessen, Germany (Regierungspräsidium Darmstadt F123/1031, F123/1040, and F123/2001). Power analysis was used to calculate the sample size required for animal experiments. Animals were kept on a 12:12 h light-dark cycle and provided with food and water ad libitum in the animal facilities of the Friedrich-Alexander University of Erlangen-Nürnberg and the Georg-Speyer-Haus Frankfurt. Col1a2-CreERT2Tg/+;Zeb1fl/fl and Col6a1-CreTg/+;Zeb1fl/fl mice were kept on FvB and C57BL/6 backgrounds, respectively. Col6a1-CreTg/+;Zeb1fl/fl;Vil-FlpTg/+;Tp53FRT/FRT FibΔZeb1 mice were kept on a mixed FvB/C57BL/6 background.
Mouse models of CRC
Rosa26-tdTomato (Madisen et al, 2010); RRID:IMSR_JAX:007914) and Rosa26-mTmG (Muzumdar et al, 2007); RRID:IMSR_JAX:007576) mice have been described previously. Conditional Zeb1 knockout mice (Brabletz et al, 2017); MGI:5901939) were crossed with mice expressing Col6a1-Cre (Armaka et al, 2008); GI:3775430) or Col1a2-CreERT2 (Zheng et al, 2002); RRID:IMSR_JAX:029567) to generate Cre-positive FibΔZeb1 mice or Cre-negative FibCtrl mice. Age-matched littermates of both sexes were used for experiments and randomly assigned to groups within genotypes. Genotyping primer sequences are listed in Appendix Table S1.
AOM/DSS driven adenoma were induced as described (Neufert et al, 2007). Briefly, mice were injected intraperitoneally (i.p.) with 10 mg/kg of azoxymethane (AOM, Sigma, A5486) in 0.9% NaCl), prior to administration of three cycles of 1.75% dextran sulfate sodium (DSS, MP Biomedicals, SKU-0216011080) in the drinking water ad libitum, each interrupted by 2 weeks of regular water and finally sacrificed at day 84 or at ethical endpoints. A separate cohort of mice was sacrificed at day 50 at the peak of inflammation. Colonic inflammation and tumor growth was monitored by endoscopy under isoflurane anesthesia employing the Colorview endoscopic system (Karl Storz), as described before (Becker et al, 2006), expressed as numbers of adenoma and/or the maximal colon obstruction by the adenoma in percent of the full colonic diameter at the most blocked site. Colonic inflammation was visually scored from ‘0’ (zero) to ‘4’ (four), with ‘0’ representing normal, ‘1’ signs of thickening, increased vascularity, and/or fibrin visible, moderate granularity and/or stool being still shaped, ‘2’ stronger thickening, vascularization, granularity and unshaped stool (no blood), ‘3’ loss of transparency, extreme granularity, rectal blood (clots) and/or traces of blood in the mucosa and/or in the unshaped or spread stool, and ‘4’ (partially) liquid stool, several sites of bloody mucosal damage or non-rectal liquid blood in the colon. Stool consistency was scored individually from ‘1’ to ‘4’ as well, with ‘0’ being normal and solid, ‘1’ softer than usual, ‘2’ unshaped, ‘3’ almost liquid but containing solid pieces of stool and/or containing traces of (clotted) blood, ‘4’ being liquid and/or mostly bloody. For immune checkpoint inhibition, mice were injected i.p. with anti-PD-L1 (BXC-BE0101) and anti-CTLA-4 (BXC-BE0131) antibodies or isotype controls (BXC-BE0094 and BXC-BP0087) twice per week (10 mg/kg each, Bio X cell). Therapies started at day 70 for all mice, unless colon obstruction by adenomas did not reach 25–30%, as determined by endoscopy. Adenoma growth was then monitored weekly via endoscopy, and mice were sacrificed at day 105.
The AOM/p53 model was based on the previously reported Tp53ΔIEC model (Neufert et al, 2021; Schwitalla et al, 2013), using Villin-Flp mediated recombination and intestinal epithelium-specific inactivation of biallelic FRT-flanked Tp53 (intron 1 and 10). Experimental animals were generated by sequential crossings to generate experimental Col6a1-CreTg/+;Zeb1fl/fl;Vil-FlpTg/+;Tp53FRT/FRT FibΔZeb1;Δp53 and Col6a1-Cre+/+;Zeb1fl/fl;Vil-FlpTg/+;Tp53FRT/FRT FibCtrl;Δp53 littermates. AOM/p53 tumors were induced by six weekly doses of AOM (10 mg/kg, i.p.) in 0.9% NaCl, as described previously (Neufert et al, 2021; Schwitalla et al, 2013). Tumor growth was monitored by endoscopy as in the AOM/DSS model and animals were sacrificed after 161 days or at any other ethical endpoint.
Orthotopic transplantation of tumor organoids was performed as described (Fumagalli et al, 2018). In brief, AKP or AKPre organoids were mechanically dissociated, seeded in high-concentration collagen pads (6.5 mg/mL) and allowed to recover for 48 h. Collagen pads were transplanted below the muscularis externa of the mouse cecum, and serosal wounds were covered by an anti-adhesion barrier. Recombination of Zeb1 was induced at day 14 for AKP organoids and at day 4 for AKPre organoids by 400 mg/kg tamoxifen diet (custom order based on Altromin 1824P). Tumor growth was monitored three times a week by palpation, and mice were sacrificed when tumors exceeded 1 cm in diameter. For immune checkpoint inhibition, mice were injected i.p. with anti-PD-L1 (Bio X cell, BE0101) or matched control antibodies (Bio X cell, BE0090) at days 14, 21, and 28. To ensure equal treatment of mice, only tumors collected after day 28 were considered for size comparison. For AKPre transplantation, only a small batch of untreated mice was used, and most analyses were performed in mice treated with control antibodies as mentioned in the figure legends for the reduction of mouse numbers.
Skin wound healing
Wound closure of a skin excision wound in mice was determined for 10 days (Lin et al, 2011). Briefly, circular wounds were applied under sterile conditions in anaesthetized mice into the dorsum after depilation. The dorsal skin was lifted at the midline and punched through two layers of skin using a disposable biopsy punch (6 mm in diameter, Kai medical, Solingen #BP-60F). Wound size was determined at indicated time points with a digital calliper and by transferring wound outline to a transparent film for area calculation using ImageJ.
Organoid culture and genetic engineering
Primary mouse intestinal organoid cultures were established as reported (Fan et al, 2019). Briefly, mouse colons were cut into small fragments, washed with PBS, and epithelial crypts were dissociated by incubation with 10 mM EDTA, passed through a 100 µm mesh and collected by centrifugation. Colon crypts were seeded at high density in BME (R&D Systems, 3533-010-02) and cultured in Advanced DMEM/F12 (Thermo Fisher Scientific, 12634028) with 20% Wnt3a conditioned medium, 10% Noggin conditioned medium, 10% R-spondin1 conditioned medium, B27 supplement (Thermo Fisher Scientific, 17504-044), 500 mM N-Acetylcysteine (Merck, A9165), 500 µg/mL human EGF (Peprotech, AF-100-15) and 500 µM A83-01 (Tocris, 2939/10). Established organoids were passaged twice a week by mechanical dissociation and reseeding.
For generation of tumor organoids, colons from Lox-Stop-Lox-KrasG12D mice were used (Jackson et al, 2001). Both Apc and Tp53 alleles were mutated by co-transfection of Cas9 with the respective sgRNA plasmids. The oncogenic KrasLSL.G12D allele was recombined by pAC1-Cre (ATCC, 39532) transfection. Modified lines were clonally expanded, and successful modification confirmed by Sanger sequencing. The hCas9 and gRNA_GFP-T2 plasmids were a kind gift from George Church (Addgene plasmids #30205 and #41820) and the Apc sgRNA plasmid kindly provided by Hans Clevers (Schwank et al, 2013). sgRNA oligonucleotides targeting Tp53 (sense: 5’-GTTTTAGAGCTAGAAATAGCAAG-3’ antisense: 5’-AGTGAAGCCCTCCGAGTGTCGGTGTTTCGTCCTTTCCACAAGAT-3’) were inserted into gRNA_GFP-T2 plasmid by inverse cloning as published (Schwank et al, 2013).
Tumor organoids from established tumors were obtained by enzymatic digestion of primary tumors with 0.1% Collagenase I (Thermo Fisher Scientific, 17018029), 0.2% Dispase II (Merck, D4693) and 2 U/mL DNase I (New England Biolabs, M0303L). Cells were passed through a 40 µm mesh and seeded at high density in BME.
Primary colon fibroblast isolation and culture experiments
For fibroblast cultures, normal colon fragments after epithelial crypt isolation or minced AOM/DSS tumors were washed in PBS and further incubated with 10 mM EDTA. After 1 h, tissue fragments were seeded into 10-cm culture plates coated with 0.1% gelatine (Sigma, G2500) in DMEM (Thermo Fisher Scientific, 31966-047) with 10% FBS (Sigma, F7524) and Pen/Strep (Thermo Fisher Scientific, 15140-122). After several days, fibroblasts expanded from tissue fragments attached to the plates, which were collected by trypsinization and propagated as regular two-dimensional cell cultures on gelatine-coated plates. Rates of proliferation were analyzed by plating 200 fibroblasts in hexaplicates in 96-wells grown for 10 days, respectively, and cell confluence was determined by live-cell microscopy using an IncuCyte S3 instrument (Sartorius). Cell senescence was determined by the Senescence β-Galactosidase Staining Kit (Cell Signaling, 9860) 3 days after plating 1 × 104 fibroblasts in six-well plates according to the manufacturer’s instructions. SA-β-Gal positive and total cell numbers were counted from 15 random images each (>700 cells per line).
For the collagen contraction assay, 1 × 105 fibroblasts were seeded in 500 µL Collagen I solution (Thermo Fisher Scientific, A1048301) adjusted to 1 mg/mL according to the manufacturer’s instructions into a 24-well suspension plate. After polymerization of collagen, the collagen disks were detached from the plate and contraction was determined by monitoring the area of the discs over time.
For the barrier function and T-cell attraction assays, splenocytes were isolated from OT1 mice (Hogquist et al, 1994). T cells were stimulated by the addition of SIINFEKL peptide (AnaSpec, AS-60193-1) for 4 h and expanded for 3 days prior to an experiment. For the barrier function assay, 12-well transwells with 8-µm pore size (Greiner, 665638) were coated with 0.1% gelatine, and 2 × 105 fibroblasts were seeded on top of the transwell. After 48 h, 3 × 104 T cells were added on top of the confluent fibroblast layer and cells in the lower compartment were monitored microscopically. For the T-cell attraction assay, 1 × 105 fibroblasts were seeded into a 12-well adhesion culture plate. After 24 h, a transwell with 3-µm pore size (Corning, CLS3462-48EA) containing 3 × 104 T cells was added to the well, and attracted cells in the lower compartment were monitored microscopically.
For inflammatory activation, colon fibroblasts were plated in a six-well plate and after 24 h treated with 1 ng/mL murine recombinant IL1α (Biolegend, 575002) in PBS for the indicated periods. The medium was replaced by 1 mL of fresh medium 2 h before treatment and supplemented with 1 mL 2 ng/mL IL1α containing medium at the starting point. At indicated time points, cells were harvested and processed for RNA and protein isolation.
For analysis in 3D, 2 × 105 fibroblasts were seeded in 8 × 25 µL of BME and cultured in Advanced DMEM/F12. Cells were analyzed either untreated or treated with 10 ng/mL recombinant hTGFβ (Peprotech, 100-21) or 1 ng/mL recombinant mIL1α. Medium was replaced by fresh medium every 24 h and cells were harvested and processed for RNA isolation after 96 h.
Histology, immunohistochemistry (IHC), and immunofluorescence (IF)
Tumors of orthotopic transplantation were dissected and fixed in 4% PFA/PBS overnight at 4 °C. For histological and IHC analyses, paraffin-embedded tumors were sectioned at 3 µm and stained using the Bond-Max device (Leica) and the Bond Polymer Refine Detection system (Leica, DS9800). Sections were imaged using an Aperio CS2 digital pathology slide scanner (Leica) and marker expression was quantified with macros in Aperio ImageScope (v12.4).
AOM/DSS and AOM/p53 colons were collected, flushed with PBS and longitudinally opened for imaging and determining tumor sizes and numbers using a caliper. Colons were mounted as Swiss Rolls, fixed in 4% PFA/PBS overnight at 4 °C, paraffin-embedded, sectioned at 3–4 µm and subjected to hematoxylin/eosin (H&E), IHC or IF staining as described previously (Krebs et al, 2017). After antibody incubations, washing steps, and DAB reactions, the slides were counterstained with Mayer’s hematoxylin before dehydration and mounting (Roti®-Histokitt, 6638.2). Analysis and image acquisition was performed using a Leica DM5500B microscope. Scoring of infiltration into AOM/DSS tumors was estimated from the cellularities of infiltrated cell-type marker-positive cells on IHC slides, with none (0), rare (1), few (2), several (3), and many/abundant (4) classification. For anti-ZEB1 IF of FibCtrl/FibΔZeb1 (Col6a1-Cre) mice, cryosections from fresh frozen colon specimens were fixed in 4% PFA for 10 min, permeabilized for 10 min in 0.25% Triton X-100/PBS, blocked in 3% BSA/PBS and incubated with anti-ZEB1 antibodies. After washing and incubation with Alexa594-conjugated secondary antibodies, DAPI-stained sections were mounted (Antifadent AF1, Citifluor). Images were acquired using a Leica DM5500B microscope.
For immunofluorescence staining of tumors from orthotopic transplantation, citrate-based antigen retrieval was applied with deparaffinized sections. Slides were blocked with 20% goat serum (Merck, G9023) in PBST and stained for 1 h with primary antibodies and for 30 min with secondary Alexa488/647-conjugated antibodies. Sections were imaged using an Evos FL microscope and marker expression was quantified using CellProfiler (McQuin et al, 2018). Antibodies and dilutions are listed in Appendix Table S2.
Multiplex immunofluorescence analysis
FFPE sections (3 μm) from orthotopic tumors (AKP) were stained with Opal 7‐Color Automation IHC Kits (Akoya Bioscience) in the BOND‐RX Multiplex IHC Stainer (Leica) following established protocols (Strack et al, 2020). Each section was put through 6 sequential rounds of staining, which included blocking in 5% BSA followed by incubation with primary antibodies; for staining conditions see Appendix Table S2. Nuclei were counterstained with 4′,6‐diamidino‐2‐phenylindole (DAPI) contained in the Opal 7‐Color Automation IHC Kits and slides were mounted with Fluoromount‐G (SouthernBiotech). Imaging was performed with the PhenoImager HT imaging system (Akoya Bioscience). Tumor regions of interest were defined manually, and out-of-focus images or high background signals were excluded from further analysis. Images were analyzed using the phenotyping application of the ‘inForm’ software V2.54.10 (Akoya Bioscience) and the fluorescence intensities for individual cells were exported. Epithelial cells (33 ± 6%) and immune cells (12 ± 5%) were excluded by EPCAM or CD45 intensity thresholds, respectively. CAFs (7 ± 2%) were separated from other cells by thresholds of αSMA, C3 or MHCII. Assigned phenotypes were myCAFs (αSMA+ , C3−, MHCII−), iCAFs (C3+ , αSMA−, MHCII−), apCAFs (MHCII+ , αSMA−, C3−) and mixed myCAF/iCAF identity (αSMA+ , C3+ , MHCII−). Cell numbers were then exported and statistically analyzed by Student’s t test. Normalized intensity values of αSMA, C3 and MHCII were used to plot CAFs by UMAP (umap V0.2.10.0) and to display the staining intensities and the cell density distribution (ndensity, ggplot2 V3.4.2).
Picrosirius red staining
Slides were deparaffinized, rehydrated, and then incubated in Picrosirius Red solution (abcam, ab246832) for 1 h at RT before washing in 0.5% acidified water, dehydration, and mounting (Roti®-Histokitt, 6638.2). Polarized light imaging was done by using a Leica DM5500B microscope equipped with a polarization filter to monitor green thinner collagen fibers and yellow-orange bundled fibers by refraction and birefringence. Percentages of total stained areas were analyzed on polarized light images using CellProfiler by RGB conversion, global background-based thresholding for pixel identification, followed by binary image generation of each channel for measuring area occupied by pixels.
Western blot analysis
Protein extraction and western blotting were carried out as described (Krebs et al, 2017; Schuhwerk et al, 2022). Briefly, cells grown in 6-well plates were lysed in 150 mM NaCl, 50 mM Tris-HCl pH 8.0, 0.5% Na-Desoxycholate (w/v), 0.1% SDS (w/v), 1% NP40 (v/v), 1× complete protease inhibitor (Roche, 4693132001), 1 mM PMSF, 1× PhosSTOP (Roche, 4906837001) for 20 min at 4 °C. Protein concentrations were determined by using the BCA Protein Assay (Thermo Fisher Scientific, 23225) according to manufacturer’s instructions. Up to 30 µg of protein lysate was separated by SDS-PAGE and transferred to nitrocellulose membranes before antibody incubation. Detection was carried out using Western Lightning Plus-ECL (Perkin Elmer, NEL103001EA) and a ChemiDocTM Imaging System (BioRad). Antibodies and dilutions are listed in Appendix Table S2.
Secretome analysis
For secretome analysis, a commercially available secretome array kit was used (R&D systems, ARY028) according to the manufacturer’s instructions. Samples from AOM/DSS CAF supernatants were prepared by surgically detaching adenomas and mincing using scalpels and incubating the tissue in digestion buffer containing 0.05% Collagenase D (w/v), 0.3% Dispase II (w/v), 0.05% DNase I (w/v), 4% FBS (v/v), in DMEM/F12 medium (Thermo Fisher Scientific, 31331028) for 30-45 min at 37 °C with constant agitation. In total, 10 mL of cold washing buffer (sterile PBS containing 2% FBS) was added, and the suspension passed through a 70 µm strainer. After centrifugation and erythrocyte lysis in ACK buffer (150 mM NH4Cl, 10 mM KHCO3, 0.1 mM EDTA; pH 7.2-7.4) for 2 min at RT, washed and collected cells were resuspended in DMEM/F12/10% FBS and plated in 12-well plates. Adherent cells were passaged in 1:1 ratios when reaching confluence. After 2–3 passages, the supernatants of individual confluent 6-well vessels were collected and kept on ice. After centrifugation at 1000 × g for 5 min at 4 °C, supernatants were aliquoted and frozen at −80 °C. For the secretome array, samples were added to the equilibrated and blocked membranes for incubation overnight at 4 °C. After incubation with the detection antibody cocktail for 1 h, with the Streptavidin-HRP mix for 30 min and the detection mix for 1 min at RT, secretomes were detected by imaging like for western blot. Intensities were normalized to the background of each respective membrane.
RNA isolation and quantitative reverse transcriptase (qRT-)PCR
Total RNA of cultured cells was isolated using the RNeasy Plus Mini Kit (Qiagen, 74136) or the NucleoSpin RNA kit (Macherey-Nagel, 740955.250) and 200–500 ng of total RNA was used to synthesize cDNA using the RevertAid First Strand cDNA Synthesis Kit (Thermo Fisher Scientific, K1622) or M-MLV Reverse Transcriptase, RNase H Minus, Point Mutant (Promega, M3682) according to manufacturers’ instructions. Subsequent qRT-PCR was performed in triplicates in 384-well plates using primers and Roche universal probe library (UPL) with TaqManTM Universal MasterMix II (Thermo Fisher, 4440044) and LightCycler® 480 II (Roche) or in 96-well plates using primers with PowerUpTM SYBRTM Green MasterMix (Thermo Fisher, A25778) and CFX Opus 96 Real-Time PCR System (Biorad). Primer sequences are listed in Appendix Table S3.
Single-cell RNA sequencing (scRNA-seq) and analysis
Single-cell transcriptomes were generated using a commercially available 384-well plate approach (SORT-Seq2, Single Cell Discoveries, SCD (Muraro et al, 2016)). To this end, primary tumors were dissected and dissociated into single cells as for establishment of organoid lines from tumors. After erythrocyte lysis in ACK buffer, cells were incubated with Fc Block and antibodies (concentrations listed in Appendix Table S2) in PBS/2% FCS/2 mM EDTA. Single viable cells (eFluor-) were gated for EPCAM+ or CD45+ or double negative cells. EPCAM−, CD45− cells were further gated for CD31 expression and triple-negative cells were considered fibroblasts for sequencing. Cells were index-sorted into 384-well capture plates containing 50 µL lysis buffer and barcoded primers covered by 10 µL of mineral oil by flow cytometry using a BD FACSAria Fusion sorter (BD biosciences) and capture plates were sent for paired-end sequencing at SCD (Illumina Nextseq™ 500). Sequences from read 1 were used for assigning reads to cells and libraries, whereas read 2 was aligned to the ensemble transcriptome (genome assembly GRCm38) using bwa version 0.7.10 (Li and Durbin, 2010). The transcript count table was generated by SCD using a custom-written script (https://github.com/anna-alemany/transcriptomics/tree/master/mapandgo). Transcript counts and metadata for all samples were imported and stored as a single-cell experiment object (SingleCell-Experiment (Amezquita et al, 2020) version 1.12.0). During quality control, cells with high mitochondrial content (isOutlier, scater (McCarthy et al, 2017) version 1.18.5) were discarded to remove low-quality cells that may have been damaged during processing or may not have been fully captured by the sequencing protocol. For analysis of CAFs, cells with high expression of Ptprc and Epcam genes were excluded to avoid contamination by residual immune/epithelial cells. The samples were normalized by computing the log2-transformed normalized expression values across all genes for each cell (logNormCounts, scater). Next, all batches were subset to the common features across all samples to enable downstream analysis. To account for the differences in samples due to plates (“batch effect”), we used the fastMNN algorithm (correctExperiments, batchelor (Haghverdi et al, 2018) version 1.6.2). Subsequently, clusters were identified using a graph-based approach (buildSNNGraph with k set to 15, scran (Lun et al, 2016) version 1.18.5) and the walktrap algorithm (igraph version 1.2.6). Cluster-specific marker genes were identified using the findMarkers function (scran) that uniquely define one cluster against the rest. Functional enrichment analysis in Metascape (https://metascape.org/) and Enrichr (https://maayanlab.cloud/Enrichr/) was applied to identify pathways and processes that were enriched in each cluster based on differentially expressed genes (FDR ≤ 0.1; P ≤ 0.05). Furthermore, expression profiles of identified clusters were compared with those previously published for CAF subtypes using the AddModuleScore function (Tirosh et al, 2016) from Seurat (Hao et al, 2021) version 4.0.0. To this end, each cell was assigned a score using the module of genes associated with the “published clusters”. A positive score suggested that this module of genes is expressed in a particular cell more than would be expected, given the average expression of this module across the population. A mean score for cluster-specific cells was calculated to obtain scores per cluster for each module of genes associated with “published clusters”. Using a reference dataset with known labels, the SingleR (Aran et al, 2019) approach (version 1.4.1) labels new cells from a test dataset based on similarity to the reference. Thereby, we assessed the similarity between clusters from FibΔZeb1 and FibCtrl, where FibCtrl was set as a reference. For the scRNA-seq dataset from non-inflammation-driven orthotopic tumors, lower quality clusters with indistinct or ambiguous cell-type identities were excluded by further sub-clustering.
The DAseq tool (Zhao et al, 2021) was used to study the differential abundance of cells from AOM/DSS scRNA FibCtrl and FibΔZeb1, using read counts after basic quality control filtering implemented in the standard Seurat (Hao et al, 2021) workflow. Cells with 100–6000 detected genes and less than 10% mitochondrial counts were included. The entire dataset which includes FibCtrl and FibΔZeb1 samples, was used to normalize the data, identify the highly variable genes, scale the data, compute the PCA, use nearest neighbor embedding, integrate the batches using harmony and recluster the data. Cell phenotypes were assigned by using Seurat’s AddModuleScore and published gene sets (Bartoschek et al, 2018; Elyada et al, 2019). DAseq was applied to identify differential abundance (Zhao et al, 2021) and three differentially abundant cell populations were identified. One contained almost exclusively cells from one SORT-Seq plate and was therefore neglected. Statistical significance is reported in the respective figures or figure legends. Characteristic genes for the two other DA populations were identified using the marker gene function included in the DAseq package, which uses stochastic gates.
Bulk RNA-Seq and GSEA
The integrity of total RNA samples was assessed by Bioanalyzer2100 before sequencing and processed at Novogene UK, Cambridge. RNA-Seq raw reads files were analyzed using nfcore RNA-seq pipeline version 3.11.0 (Ewels et al, 2020). Briefly, raw sequencing reads were aligned to the mouse genome GRCm38 using STAR (Dobin et al, 2013), the reads aligning to each annotated gene were quantified by the Salmon software (Patro et al, 2017), and differential gene expression was performed using DESeq2 R package (Love et al, 2014). Gene set enrichment analysis (GSEA) was performed on all (filtered) genes across the different experimental conditions using the R packages “clusterProfiler” (v4.10.0) and “enrichPlot” (v1.22.0) with MSigDB and custom gene sets (Elyada et al, 2019; Ohlund et al, 2017; Wu et al, 2021). To account for multiple comparisons, P values were adjusted using the Benjamini–Hochberg method.
Statistics and reproducibility
Information on the number of biologically independent samples analyzed and the number of times experiments were performed is included in the figure legends. All the representative experiments were repeated at least three times unless otherwise stated. Statistical analyses were performed using GraphPad Prism, within the provided R packages or online tools for gene set enrichment analyses, and details on statistical methods are included in the figure legends. All error bars represent SD or SEM as indicated. Where possible animals were assigned to experimental groups using simple randomization and the investigators were blinded for initial animal data collection on tumor/metastasis parameters. No blinding was applied for other experiments since investigators needed information about the groups to correctly perform and analyze the experiments.
Supplementary information
Figure EV1-EV5, Appendix Fig. S1-S4 Source Data
Acknowledgements
The authors thank Britta Schlund, Eva Bauer, Friederike Gräbner, Tahmineh Darvishi, Margarete Mijatovic, and Petra Dinse for excellent technical assistance. The authors are grateful to Stefan Stein and Annette Trzmiel (Core Facility Flow Cytometry GSH Frankfurt) for support with all fluorescence-activated cell sorting (FACS) and the animal facilities at FAU Erlangen and GSH Frankfurt for excellent animal care and welfare. The authors thank all members from the Farin, Stemmler, and Brabletz laboratories for critical input and fruitful discussions. This work was supported by the German Research Foundation (FOR2438/P04, TRR305 TP A03, A04, B01 and B07, and BR1399/9-1, BR1399/10-1, BR1399/15-1, BR1399/17-1, BR4145/1-1, BR4145/2-1 and BR4145/3-1), the Wilhelm Sander-Stiftung (2020.039.1), IZKF-Erlangen (IZKF P34, P133 and D39) and the European Union’s Horizon 2020 research and innovation program under the Marie Skłodowska-Curie grant agreement (No. 861196, PRECODE). AH and DBB were supported by the German Federal Ministry of Education and Research (BMBF) within the framework of the CompLS funding concept [031L0309A (NetMap)].
Expanded view
Author contributions
Constantin Menche: Formal analysis; Validation; Investigation; Visualization; Methodology; Writing—original draft; Writing—review and editing. Harald Schuhwerk: Conceptualization; Formal analysis; Funding acquisition; Validation; Investigation; Visualization; Methodology; Writing—original draft; Writing—review and editing. Isabell Armstark: Investigation. Pooja Gupta: Data curation; Software; Formal analysis; Investigation; Visualization; Methodology. Kathrin Fuchs: Investigation. Ruthger van Roey: Data curation; Investigation. Mohammed H Mosa: Data curation; Investigation. Anne Hartebrodt: Software; Formal analysis; Investigation; Visualization; Methodology. Yussuf Hajjaj: Data curation; Software; Investigation. Ana Clavel Ezquerra: Investigation. Manoj K Selvaraju: Data curation; Software; Investigation; Visualization. Carol I Geppert: Resources. Stefanie Bärthel: Resources. Dieter Saur: Resources. Florian R Greten: Resources; Supervision. Simone Brabletz: Funding acquisition; Investigation. David B Blumenthal: Software; Formal analysis; Supervision; Funding acquisition; Investigation; Visualization. Andreas Weigert: Investigation; Visualization; Methodology. Thomas Brabletz: Conceptualization; Supervision; Funding acquisition; Investigation; Writing—original draft; Writing—review and editing. Henner F Farin: Conceptualization; Formal analysis; Supervision; Funding acquisition; Investigation; Visualization; Writing—original draft; Writing—review and editing. Marc P Stemmler: Conceptualization; Formal analysis; Supervision; Funding acquisition; Investigation; Visualization; Writing—original draft; Project administration; Writing—review and editing.
Source data underlying figure panels in this paper may have individual authorship assigned. Where available, figure panel/source data authorship is listed in the following database record: biostudies:S-SCDT-10_1038-S44319-024-00186-7.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Data availability
scRNA-seq and bulk RNA-seq data have been deposited to Gene Expression Omnibus (GEO) database with the accession codes GSE253368, GSE253639 and GSE253546. All original codes have been deposited at GitHub (bulk RNA-Seq: https://github.com/AG-Stemmler/Menche-Schuhwerk-et-al.-Zeb1_CRC_Manuscript; scRNA-Seq: https://github.com/CUBiDA/zeb1_crc_manuscript).
The source data of this paper are collected in the following database record: biostudies:S-SCDT-10_1038-S44319-024-00186-7.
Disclosure and competing interests statement
The authors declare no competing interests. Thomas Brabletz is a member of the Advisory Editorial Board of EMBO reports. This has no bearing on the editorial consideration of this article for publication.
Footnotes
These authors contributed equally to this work as first authors: Constantin Menche, Harald Schuhwerk.
These authors contributed equally to this work as senior authors: Henner F Farin, Marc P Stemmler.
Contributor Information
Thomas Brabletz, Email: thomas.brabletz@fau.de.
Henner F Farin, Email: h.farin@georg-speyer-haus.de.
Marc P Stemmler, Email: marc.stemmler@fau.de.
Supplementary information
Expanded view data, supplementary information, appendices are available for this paper at 10.1038/s44319-024-00186-7.
References
- Amezquita RA, Lun ATL, Becht E, Carey VJ, Carpp LN, Geistlinger L, Marini F, Rue-Albrecht K, Risso D, Soneson C et al (2020) Orchestrating single-cell analysis with Bioconductor. Nat Methods 17:137–145 10.1038/s41592-019-0654-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andre T, Shiu KK, Kim TW, Jensen BV, Jensen LH, Punt C, Smith D, Garcia-Carbonero R, Benavides M, Gibbs P et al (2020) Pembrolizumab in microsatellite-instability-high advanced colorectal cancer. New Engl J Med 383:2207–2218 10.1056/NEJMoa2017699 [DOI] [PubMed] [Google Scholar]
- Aran D, Looney AP, Liu L, Wu E, Fong V, Hsu A, Chak S, Naikawadi RP, Wolters PJ, Abate AR et al (2019) Reference-based analysis of lung single-cell sequencing reveals a transitional profibrotic macrophage. Nat Immunol 20:163–172 10.1038/s41590-018-0276-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armaka M, Apostolaki M, Jacques P, Kontoyiannis DL, Elewaut D, Kollias G (2008) Mesenchymal cell targeting by TNF as a common pathogenic principle in chronic inflammatory joint and intestinal diseases. J Exp Med 205:331–337 10.1084/jem.20070906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartoschek M, Oskolkov N, Bocci M, Lövrot J, Larsson C, Sommarin M, Madsen CD, Lindgren D, Pekar G, Karlsson G et al (2018) Spatially and functionally distinct subclasses of breast cancer-associated fibroblasts revealed by single cell RNA sequencing. Nat Commun 9:5150 10.1038/s41467-018-07582-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker C, Fantini MC, Neurath MF (2006) High resolution colonoscopy in live mice. Nat Protoc 1:2900–2904 10.1038/nprot.2006.446 [DOI] [PubMed] [Google Scholar]
- Bhattacharjee S, Hamberger F, Ravichandra A, Miller M, Nair A, Affo S, Filliol A, Chin L, Savage TM, Yin D et al (2021) Tumor restriction by type I collagen opposes tumor-promoting effects of cancer-associated fibroblasts. J Clin Invest 131:e146987 10.1172/JCI146987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biffi G, Oni TE, Spielman B, Hao Y, Elyada E, Park Y, Preall J, Tuveson DA (2019) IL1-induced JAK/STAT signaling is antagonized by TGFβ to shape CAF heterogeneity in pancreatic ductal adenocarcinoma. Cancer Discov 9:282–301 10.1158/2159-8290.CD-18-0710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brabletz S, Lasierra Losada M, Schmalhofer O, Mitschke J, Krebs A, Brabletz T, Stemmler MP (2017) Generation and characterization of mice for conditional inactivation of Zeb1. Genesis 55:e23024 [DOI] [PubMed]
- Bronsert P, Kohler I, Timme S, Kiefer S, Werner M, Schilling O, Vashist Y, Makowiec F, Brabletz T, Hopt UT et al (2014) Prognostic significance of Zinc finger E-box binding homeobox 1 (ZEB1) expression in cancer cells and cancer-associated fibroblasts in pancreatic head cancer. Surgery 156:97–108 10.1016/j.surg.2014.02.018 [DOI] [PubMed] [Google Scholar]
- Calon A, Lonardo E, Berenguer-Llergo A, Espinet E, Hernando-Momblona X, Iglesias M, Sevillano M, Palomo-Ponce S, Tauriello DV, Byrom D et al (2015) Stromal gene expression defines poor-prognosis subtypes in colorectal cancer. Nat Genet 47:320–329 10.1038/ng.3225 [DOI] [PubMed] [Google Scholar]
- Caramel J, Ligier M, Puisieux A (2018) Pleiotropic roles for ZEB1 in cancer. Cancer Res 78:30–35 10.1158/0008-5472.CAN-17-2476 [DOI] [PubMed] [Google Scholar]
- Chen Y, Kim J, Yang S, Wang H, Wu CJ, Sugimoto H, LeBleu VS, Kalluri R (2021) Type I collagen deletion in alphaSMA(+) myofibroblasts augments immune suppression and accelerates progression of pancreatic cancer. Cancer Cell 39:548–565.e546 10.1016/j.ccell.2021.02.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortes M, Sanchez-Moral L, de Barrios O, Fernandez-Acenero MJ, Martinez-Campanario MC, Esteve-Codina A, Darling DS, Gyorffy B, Lawrence T, Dean DC et al (2017) Tumor-associated macrophages (TAMs) depend on ZEB1 for their cancer-promoting roles. EMBO J 36:3336–3355 10.15252/embj.201797345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR (2013) STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29:15–21 10.1093/bioinformatics/bts635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elyada E, Bolisetty M, Laise P, Flynn WF, Courtois ET, Burkhart RA, Teinor JA, Belleau P, Biffi G, Lucito MS et al (2019) Cross-species single-cell analysis of pancreatic ductal adenocarcinoma reveals antigen-presenting cancer-associated fibroblasts. Cancer Discov 9:1102–1123 10.1158/2159-8290.CD-19-0094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewels PA, Peltzer A, Fillinger S, Patel H, Alneberg J, Wilm A, Garcia MU, Di Tommaso P, Nahnsen S (2020) The nf-core framework for community-curated bioinformatics pipelines. Nat Biotechnol 38:276–278 10.1038/s41587-020-0439-x [DOI] [PubMed] [Google Scholar]
- Fan YY, Davidson LA, Chapkin RS (2019) Murine colonic organoid culture system and downstream assay applications. Methods Mol Biol 1576:171–181 10.1007/7651_2016_8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldmann K, Maurer C, Peschke K, Teller S, Schuck K, Steiger K, Engleitner T, Ollinger R, Nomura A, Wirges N et al (2021) Mesenchymal plasticity regulated by Prrx1 drives aggressive pancreatic cancer biology. Gastroenterology 160:346–361.e324 10.1053/j.gastro.2020.09.010 [DOI] [PubMed] [Google Scholar]
- Fu R, Han CF, Ni T, Di L, Liu LJ, Lv WC, Bi YR, Jiang N, He Y, Li HM et al (2019) A ZEB1/p53 signaling axis in stromal fibroblasts promotes mammary epithelial tumours. Nat Commun 10:3210 10.1038/s41467-019-11278-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fumagalli A, Suijkerbuijk SJE, Begthel H, Beerling E, Oost KC, Snippert HJ, van Rheenen J, Drost J (2018) A surgical orthotopic organoid transplantation approach in mice to visualize and study colorectal cancer progression. Nat Protoc 13:235–247 10.1038/nprot.2017.137 [DOI] [PubMed] [Google Scholar]
- Greten FR, Eckmann L, Greten TF, Park JM, Li ZW, Egan LJ, Kagnoff MF, Karin M (2004) IKKbeta links inflammation and tumorigenesis in a mouse model of colitis-associated cancer. Cell 118:285–296 10.1016/j.cell.2004.07.013 [DOI] [PubMed] [Google Scholar]
- Guinney J, Dienstmann R, Wang X, de Reynies A, Schlicker A, Soneson C, Marisa L, Roepman P, Nyamundanda G, Angelino P et al (2015) The consensus molecular subtypes of colorectal cancer. Nat Med 21:1350–1356 10.1038/nm.3967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haghverdi L, Lun ATL, Morgan MD, Marioni JC (2018) Batch effects in single-cell RNA-sequencing data are corrected by matching mutual nearest neighbors. Nat Biotechnol 36:421–427 10.1038/nbt.4091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao Y, Hao S, Andersen-Nissen E, Mauck 3rd WM, Zheng S, Butler A, Lee MJ, Wilk AJ, Darby C, Zager M et al (2021) Integrated analysis of multimodal single-cell data. Cell 184:3573–3587.e3529 10.1016/j.cell.2021.04.048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogquist KA, Jameson SC, Heath WR, Howard JL, Bevan MJ, Carbone FR (1994) T cell receptor antagonist peptides induce positive selection. Cell 76:17–27 10.1016/0092-8674(94)90169-4 [DOI] [PubMed] [Google Scholar]
- Isella C, Terrasi A, Bellomo SE, Petti C, Galatola G, Muratore A, Mellano A, Senetta R, Cassenti A, Sonetto C et al (2015) Stromal contribution to the colorectal cancer transcriptome. Nat Genet 47:312–319 10.1038/ng.3224 [DOI] [PubMed] [Google Scholar]
- Jackson EL, Willis N, Mercer K, Bronson RT, Crowley D, Montoya R, Jacks T, Tuveson DA (2001) Analysis of lung tumor initiation and progression using conditional expression of oncogenic K-ras. Genes Dev 15:3243–3248 10.1101/gad.943001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joanito I, Wirapati P, Zhao N, Nawaz Z, Yeo G, Lee F, Eng CLP, Macalinao DC, Kahraman M, Srinivasan H et al (2022) Single-cell and bulk transcriptome sequencing identifies two epithelial tumor cell states and refines the consensus molecular classification of colorectal cancer. Nat Genet 54:963–975 10.1038/s41588-022-01100-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katsura A, Tamura Y, Hokari S, Harada M, Morikawa M, Sakurai T, Takahashi K, Mizutani A, Nishida J, Yokoyama Y et al (2017) ZEB1-regulated inflammatory phenotype in breast cancer cells. Mol Oncol 11:1241–1262 10.1002/1878-0261.12098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kieffer Y, Hocine HR, Gentric G, Pelon F, Bernard C, Bourachot B, Lameiras S, Albergante L, Bonneau C, Guyard A et al (2020) Single-cell analysis reveals fibroblast clusters linked to immunotherapy resistance in cancer. Cancer Discov 10:1330–1351 10.1158/2159-8290.CD-19-1384 [DOI] [PubMed] [Google Scholar]
- Koliaraki V, Pasparakis M, Kollias G (2015) IKKbeta in intestinal mesenchymal cells promotes initiation of colitis-associated cancer. J Exp Med 212:2235–2251 10.1084/jem.20150542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krebs AM, Mitschke J, Lasierra Losada M, Schmalhofer O, Boerries M, Busch H, Boettcher M, Mougiakakos D, Reichardt W, Bronsert P et al (2017) The EMT-activator Zeb1 is a key factor for cell plasticity and promotes metastasis in pancreatic cancer. Nat Cell Biol 19:518–529 10.1038/ncb3513 [DOI] [PubMed] [Google Scholar]
- Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, Skora AD, Luber BS, Azad NS, Laheru D et al (2015) PD-1 blockade in tumors with mismatch-repair deficiency. New Engl J Med 372:2509–2520 10.1056/NEJMoa1500596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HO, Hong Y, Etlioglu HE, Cho YB, Pomella V, Van den Bosch B, Vanhecke J, Verbandt S, Hong H, Min JW et al (2020) Lineage-dependent gene expression programs influence the immune landscape of colorectal cancer. Nat Genet 52:594–603 10.1038/s41588-020-0636-z [DOI] [PubMed] [Google Scholar]
- Li H, Durbin R (2010) Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 26:589–595 10.1093/bioinformatics/btp698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang W, Zhang Y, Zhou L, Lu X, Finn ME, Wang W, Shao H, Dean DC, Zhang L, Liu Y (2022) Zeb1 regulation of wound-healing-induced inflammation in alkali-damaged corneas. iScience 25:104038 10.1016/j.isci.2022.104038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Q, Fang D, Fang J, Ren X, Yang X, Wen F, Su SB (2011) Impaired wound healing with defective expression of chemokines and recruitment of myeloid cells in TLR3-deficient mice. J Immunol 186:3710–3717 10.4049/jimmunol.1003007 [DOI] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S (2014) Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15:550 10.1186/s13059-014-0550-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lun AT, McCarthy DJ, Marioni JC (2016) A step-by-step workflow for low-level analysis of single-cell RNA-seq data with Bioconductor. F1000Res 5:2122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madisen L, Zwingman TA, Sunkin SM, Oh SW, Zariwala HA, Gu H, Ng LL, Palmiter RD, Hawrylycz MJ, Jones AR et al (2010) A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat Neurosci 13:133–140 10.1038/nn.2467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy DJ, Campbell KR, Lun AT, Wills QF (2017) Scater: pre-processing, quality control, normalization and visualization of single-cell RNA-seq data in R. Bioinformatics 33:1179–1186 10.1093/bioinformatics/btw777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McQuin C, Goodman A, Chernyshev V, Kamentsky L, Cimini BA, Karhohs KW, Doan M, Ding L, Rafelski SM, Thirstrup D et al (2018) CellProfiler 3.0: next-generation image processing for biology. PLoS Biol 16:e2005970 10.1371/journal.pbio.2005970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meidhof S, Brabletz S, Lehmann W, Preca BT, Mock K, Ruh M, Schüler J, Berthold M, Weber A, Burk U et al (2015) ZEB1-associated drug resistance in cancer cells is reversed by the class I HDAC inhibitor mocetinostat. EMBO Mol Med 7:831–847 10.15252/emmm.201404396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mellone M, Piotrowska K, Venturi G, James L, Bzura A, Lopez MA, James S, Wang C, Ellis MJ, Hanley CJ et al (2022) ATM regulates differentiation of myofibroblastic cancer-associated fibroblasts and can be targeted to overcome immunotherapy resistance. Cancer Res 82:4571–4585 10.1158/0008-5472.CAN-22-0435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan E, Arnold M, Gini A, Lorenzoni V, Cabasag CJ, Laversanne M, Vignat J, Ferlay J, Murphy N, Bray F (2023) Global burden of colorectal cancer in 2020 and 2040: incidence and mortality estimates from GLOBOCAN. Gut 72:338–344 10.1136/gutjnl-2022-327736 [DOI] [PubMed] [Google Scholar]
- Muraro MJ, Dharmadhikari G, Grun D, Groen N, Dielen T, Jansen E, van Gurp L, Engelse MA, Carlotti F, de Koning EJ et al (2016) A single-cell transcriptome atlas of the human pancreas. Cell Syst 3:385–394.e383 10.1016/j.cels.2016.09.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muzumdar MD, Tasic B, Miyamichi K, Li L, Luo L (2007) A global double-fluorescent Cre reporter mouse. Genesis 45:593–605 10.1002/dvg.20335 [DOI] [PubMed] [Google Scholar]
- Neufert C, Becker C, Neurath MF (2007) An inducible mouse model of colon carcinogenesis for the analysis of sporadic and inflammation-driven tumor progression. Nat Protoc 2:1998–2004 10.1038/nprot.2007.279 [DOI] [PubMed] [Google Scholar]
- Neufert C, Heichler C, Brabletz T, Scheibe K, Boonsanay V, Greten FR, Neurath MF (2021) Inducible mouse models of colon cancer for the analysis of sporadic and inflammation-driven tumor progression and lymph node metastasis. Nat Protoc 16:61–85 10.1038/s41596-020-00412-1 [DOI] [PubMed] [Google Scholar]
- Nicolas AM, Pesic M, Engel E, Ziegler PK, Diefenhardt M, Kennel KB, Buettner F, Conche C, Petrocelli V, Elwakeel E et al (2022) Inflammatory fibroblasts mediate resistance to neoadjuvant therapy in rectal cancer. Cancer Cell 40:168–184.e113 10.1016/j.ccell.2022.01.004 [DOI] [PubMed] [Google Scholar]
- Ohlund D, Handly-Santana A, Biffi G, Elyada E, Almeida AS, Ponz-Sarvise M, Corbo V, Oni TE, Hearn SA, Lee EJ et al (2017) Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J Exp Med 214:579–596 10.1084/jem.20162024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olguin JE, Medina-Andrade I, Molina E, Vazquez A, Pacheco-Fernandez T, Saavedra R, Perez-Plasencia C, Chirino YI, Vaca-Paniagua F, Arias-Romero LE et al (2018) Early and partial reduction in CD4(+)Foxp3(+) regulatory T cells during colitis-associated colon cancer induces CD4(+) and CD8(+) T cell activation inhibiting tumorigenesis. J Cancer 9:239–249 10.7150/jca.21336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouled Dhaou M, Kossai M, Morel AP, Devouassoux-Shisheboran M, Puisieux A, Penault-Llorca F, Radosevic-Robin N (2020) Zeb1 expression by tumor or stromal cells is associated with spatial distribution patterns of CD8+ tumor-infiltrating lymphocytes: a hypothesis-generating study on 113 triple negative breast cancers. Am J Cancer Res 10:3370–3381 [PMC free article] [PubMed] [Google Scholar]
- Ozdemir BC, Pentcheva-Hoang T, Carstens JL, Zheng X, Wu CC, Simpson TR, Laklai H, Sugimoto H, Kahlert C, Novitskiy SV et al (2014) Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell 25:719–734 10.1016/j.ccr.2014.04.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastille E, Bardini K, Fleissner D, Adamczyk A, Frede A, Wadwa M, von Smolinski D, Kasper S, Sparwasser T, Gruber AD et al (2014) Transient ablation of regulatory T cells improves antitumor immunity in colitis-associated colon cancer. Cancer Res 74:4258–4269 10.1158/0008-5472.CAN-13-3065 [DOI] [PubMed] [Google Scholar]
- Patro R, Duggal G, Love MI, Irizarry RA, Kingsford C (2017) Salmon provides fast and bias-aware quantification of transcript expression. Nat Methods 14:417–419 10.1038/nmeth.4197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preca BT, Bajdak K, Mock K, Sundararajan V, Pfannstiel J, Maurer J, Wellner U, Hopt UT, Brummer T, Brabletz S et al (2015) A self-enforcing CD44s/ZEB1 feedback loop maintains EMT and stemness properties in cancer cells. Int J Cancer 137:2566–2577 10.1002/ijc.29642 [DOI] [PubMed] [Google Scholar]
- Qian Y, Arellano G, Ifergan I, Lin J, Snowden C, Kim T, Thomas JJ, Law C, Guan T, Balabanov RD et al (2021) ZEB1 promotes pathogenic Th1 and Th17 cell differentiation in multiple sclerosis. Cell Rep 36:109602 10.1016/j.celrep.2021.109602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahai E, Astsaturov I, Cukierman E, DeNardo DG, Egeblad M, Evans RM, Fearon D, Greten FR, Hingorani SR, Hunter T et al (2020) A framework for advancing our understanding of cancer-associated fibroblasts. Nat Rev Cancer 20:174–186 10.1038/s41568-019-0238-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt M, Greten FR (2021) The inflammatory pathogenesis of colorectal cancer. Nat Rev Immunol 21:653–667 10.1038/s41577-021-00534-x [DOI] [PubMed] [Google Scholar]
- Schuhwerk H, Kleemann J, Gupta P, van Roey R, Armstark I, Kreileder M, Feldker N, Ramesh V, Hajjaj Y, Fuchs K et al (2022) The EMT transcription factor ZEB1 governs a fitness-promoting but vulnerable DNA replication stress response. Cell Rep 41:111819 10.1016/j.celrep.2022.111819 [DOI] [PubMed] [Google Scholar]
- Schwank G, Koo BK, Sasselli V, Dekkers JF, Heo I, Demircan T, Sasaki N, Boymans S, Cuppen E, van der Ent CK et al (2013) Functional repair of CFTR by CRISPR/Cas9 in intestinal stem cell organoids of cystic fibrosis patients. Cell Stem Cell 13:653–658 10.1016/j.stem.2013.11.002 [DOI] [PubMed] [Google Scholar]
- Schwitalla S, Ziegler PK, Horst D, Becker V, Kerle I, Begus-Nahrmann Y, Lechel A, Rudolph KL, Langer R, Slotta-Huspenina J et al (2013) Loss of p53 in enterocytes generates an inflammatory microenvironment enabling invasion and lymph node metastasis of carcinogen-induced colorectal tumors. Cancer Cell 23:93–106 10.1016/j.ccr.2012.11.014 [DOI] [PubMed] [Google Scholar]
- Scott CL, Omilusik KD (2019) ZEBs: novel players in immune cell development and function. Trends Immunol 40:431–446 10.1016/j.it.2019.03.001 [DOI] [PubMed] [Google Scholar]
- Sebastian A, Hum NR, Martin KA, Gilmore SF, Peran I, Byers SW, Wheeler EK, Coleman MA, Loots GG (2020) Single-cell transcriptomic analysis of tumor-derived fibroblasts and normal tissue-resident fibroblasts reveals fibroblast heterogeneity in breast cancer. Cancers 12:1307 10.3390/cancers12051307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanisavljevic J, Loubat-Casanovas J, Herrera M, Luque T, Pena R, Lluch A, Albanell J, Bonilla F, Rovira A, Pena C et al (2015) Snail1-expressing fibroblasts in the tumor microenvironment display mechanical properties that support metastasis. Cancer Res 75:284–295 10.1158/0008-5472.CAN-14-1903 [DOI] [PubMed] [Google Scholar]
- Stemmler MP, Eccles RL, Brabletz S, Brabletz T (2019) Non-redundant functions of EMT transcription factors. Nat Cell Biol 21:102–112 10.1038/s41556-018-0196-y [DOI] [PubMed] [Google Scholar]
- Strack E, Rolfe PA, Fink AF, Bankov K, Schmid T, Solbach C, Savai R, Sha W, Pradel L, Hartmann S et al (2020) Identification of tumor-associated macrophage subsets that are associated with breast cancer prognosis. Clin Transl Med 10:e239 10.1002/ctm2.239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tauriello DVF, Palomo-Ponce S, Stork D, Berenguer-Llergo A, Badia-Ramentol J, Iglesias M, Sevillano M, Ibiza S, Canellas A, Hernando-Momblona X et al (2018) TGFbeta drives immune evasion in genetically reconstituted colon cancer metastasis. Nature 554:538–543 10.1038/nature25492 [DOI] [PubMed] [Google Scholar]
- Tauriello DVF, Sancho E, Batlle E (2022) Overcoming TGFβ-mediated immune evasion in cancer. Nat Rev Cancer 22:25–44 10.1038/s41568-021-00413-6 [DOI] [PubMed] [Google Scholar]
- Tirosh I, Izar B, Prakadan SM, Wadsworth 2nd MH, Treacy D, Trombetta JJ, Rotem A, Rodman C, Lian C, Murphy G et al (2016) Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science 352:189–196 10.1126/science.aad0501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Lee S, Teh CE, Bunting K, Ma L, Shannon MF (2009) The transcription repressor, ZEB1, cooperates with CtBP2 and HDAC1 to suppress IL-2 gene activation in T cells. Int Immunol 21:227–235 10.1093/intimm/dxn143 [DOI] [PubMed] [Google Scholar]
- Wellner U, Schubert J, Burk UC, Schmalhofer O, Zhu F, Sonntag A, Waldvogel B, Vannier C, Darling D, zur Hausen A et al (2009) The EMT-activator ZEB1 promotes tumorigenicity by repressing stemness-inhibiting microRNAs. Nat Cell Biol 11:1487–1495 10.1038/ncb1998 [DOI] [PubMed] [Google Scholar]
- Wu T, Hu E, Xu S, Chen M, Guo P, Dai Z, Feng T, Zhou L, Tang W, Zhan L et al (2021) clusterProfiler 4.0: a universal enrichment tool for interpreting omics data. Innovation 2:100141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yassin M, Sadowska Z, Djurhuus D, Nielsen B, Tougaard P, Olsen J, Pedersen AE (2019) Upregulation of PD-1 follows tumour development in the AOM/DSS model of inflammation-induced colorectal cancer in mice. Immunology 158:35–46 10.1111/imm.13093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeo SY, Lee KW, Shin D, An S, Cho KH, Kim SH (2018) A positive feedback loop bi-stably activates fibroblasts. Nat Commun 9:3016 10.1038/s41467-018-05274-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Jaffe A, Li H, Lindenbaum O, Sefik E, Jackson R, Cheng X, Flavell RA, Kluger Y (2021) Detection of differentially abundant cell subpopulations in scRNA-seq data. Proc Natl Acad Sci USA 118:e2100293118 10.1073/pnas.2100293118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng B, Zhang Z, Black CM, de Crombrugghe B, Denton CP (2002) Ligand-dependent genetic recombination in fibroblasts: a potentially powerful technique for investigating gene function in fibrosis. Am J Pathol 160:1609–1617 10.1016/S0002-9440(10)61108-X [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure EV1-EV5, Appendix Fig. S1-S4 Source Data
Data Availability Statement
scRNA-seq and bulk RNA-seq data have been deposited to Gene Expression Omnibus (GEO) database with the accession codes GSE253368, GSE253639 and GSE253546. All original codes have been deposited at GitHub (bulk RNA-Seq: https://github.com/AG-Stemmler/Menche-Schuhwerk-et-al.-Zeb1_CRC_Manuscript; scRNA-Seq: https://github.com/CUBiDA/zeb1_crc_manuscript).
The source data of this paper are collected in the following database record: biostudies:S-SCDT-10_1038-S44319-024-00186-7.