


Abstract
A library of three synthetic ribozymes with randomized arms, targeting NUX, GUX and NXG triplets, respectively, were used to identify ribozyme-accessible sites on the HIV-1 LTR transcript comprising positions –533 to 386. Three cleavable sites were identified at positions 109, 115 and 161. Ribozymes were designed against these sites, either unmodified or with 2′-modifications and phosphorothioate groups, and their cleavage activities of the transcript were determined. Their biological activities were assessed in cell culture, using a HIV-1 model assay system where the LTR is a promoter for the expression of the reporter gene luciferase in a transient expression system. Intracellular efficiency of the ribozymes were determined by cotransfection of ribozyme and plasmid DNA, expressing the target RNA. Modified ribozymes, directed against positions 115 and 161, lowered the level of LTR mRNA in the cell resulting in inhibition of expression of the LTR-driven reporter gene luciferase of 87 and 61%, respectively. In the presence of Tat the inhibitions were 43 and 25%. The inactive variants of these ribozymes exhibited a similar inhibitory effect. RNase protection revealed a reduction of RNA which was somewhat stronger for the active than the inactive ribozymes, particularly for ribozyme 115. Unmodified ribozymes showed no inhibition in the cell. The third ribozyme, targeting a GUG-triplet at position 109, possessed only low cleavage activity in vitro and no inhibitory effect in cell culture.
INTRODUCTION
Ribozymes are RNA-based enzymes that bind to and cleave RNA molecules in a sequence-specific manner. The development of ribozyme-mediated cleavage of mRNA in trans has resulted in an increasing number of reports using ribozymes for potentially therapeutic application. A wide variety of sequences in the HIV genome have also been the target for hammerhead and hairpin ribozyme-mediated interference with gene expression. Some of these examples have demonstrated the ability of ribozymes either to inhibit expression of reporter genes under control of HIV-1 genes or to suppress HIV (1–11). These results were all achieved by delivering the ribozymes endogenously, i.e. using plasmids or retroviral vectors containing the ribozyme sequence behind a suitable promoter. Exogenous delivery of synthetic ribozymes is an alternative for the inhibition of gene expression particularly with the hammerhead ribozyme (2–4). However, exogenous delivery has not yet been applied for the inhibition of the expression of HIV-1 genes, with the exception of mainly a cellular-uptake study with a kinetically not characterized DNA–RNA ribozyme chimera (12). We have therefore decided to explore this mode of application and have chosen the HIV-1 LTR as a target because of its high degree of conservation among the known HIV-1 isolates and because of its presence in both early and late viral gene products (13). Also, several cellular factors interact with regulatory elements of the LTR to regulate the expression of the HIV-1 genome (14). We have approached this question by studying the effect of hammerhead ribozymes on a model system where the expression of luciferase is driven by the HIV-1 LTR as promoter.
As degradation of ribozymes by nucleases can be a problem in the exogenous delivery mode, we not only used unmodified ribozymes but also derivatives which are known to stabilize the ribozymes without interference with catalytic activity (15–17).
RNA molecules exhibit extensive secondary structures of single- and double-stranded regions which renders many potential ribozyme-cleavable triplets inaccessible. This limited accessibility has been most convincingly demonstrated not only for oligodeoxynucleotides (18–20) but also for ribozymes (21). The success of ribozymes in interfering with gene expression thus depends critically on the identification of those regions of the chosen target RNA which are accessible for ribozyme annealing. One experimental strategy is the annealing of a completely randomized oligodeoxynucleotide to the target transcript and cleavage with RNase H which has been successful not only for the identification of oligodeoxynucleotide sites (22,23) but also for ribozyme sites on the human acetylcholinesterase RNA (24). This strategy has also been extended to native RNA as target in cell extracts which relates more closely to the intracellular environment (25). It has also been used in conjunction with the computer-predicted secondary structures of local sequence stretches (26,27). This method is well suited for the design of oligodeoxynucleotides for the antisense-oligodeoxynucleotide-mediated inhibition of gene expression but does not necessarily accurately identify ribozyme cleavage sites although this has been successful in two cases (24,28). More suitable presumably is the use of ribozymes with randomized binding arms. This has been described for the group I intron ribozyme (29), for the hairpin ribozyme (30,31) and a DNAzyme (32). We have decided to use three different hammerhead ribozyme libraries, directed against either NUX, GUX or NXG triplets, to probe the HIV-1 LTR transcript for cleavage. Although this RNA has been among the targets for endogenous ribozyme cleavage, no analysis for ribozyme accessibility has been performed so far.
MATERIALS AND METHODS
Phosphoramidites were purchased from Per Septive Biosystems. T7 Polymerase was purified from the overproducer Escherichia coli BL 21/pAR1219 kindly supplied by F. W. Studier (Brookhaven). [α-32P]UTP and [γ-32P]ATP were purchased from Amersham (Braunschweig). Ribonucleoside triphosphates, T4 Polynucleotidkinase, AMV Reverse Transcriptase, DNase I (RNase free), SP6 RNA Polymerase, restriction endonucleases HindIII, BamHI, EcoRI and its 10× buffer were obtained from Boehringer Mannheim. Dulbecco’s modified Eagle’s medium (DMEM), fetal bovine serum and coenzyme A were supplied by Sigma (München). BCA protein assay reagents A and B were obtained from Pierce (Rockford, IL). Penicillin/streptomycin were obtained from ICN Biomedicine. Tfx-50, fmol sequencing system and Beetle luciferin were purchased from Promega. Nitrocellulose transfer membrane (PROTRAN BA 86, 0.34 µm) was obtained from Schleicher & Schuell (Dassel). Ultraspec RNA was purchased from Biotecx (Houston, TX). RPA II Kit and pTRI-β-actin-human were purchased from Ambion (Austin, TX). Opti-MEM was obtained from Gibco BRL (Karlsruhe) and RNase inhibitor from Fermentas.
Plasmids and oligonucleotides
For the in vitro cleavage reaction and RNase protection assays plasmid pOTH33 was used that directs the in vitro transcription of the HIV-1 LTR from –533 to +386 under control of the bacteriophage T7 or SP6 promoter to produce a sense or antisense transcript, respectively (16). For the detection of ribozyme-mediated cleavage in cell culture experiments plasmid pLTRLuc was constructed, expressing the luciferase reporter gene under control of the HIV-1 LTR-promoter (Fig. 1). Thus, the HIV-1 LTR sequence from position –533 to +190 was excised from plasmid pOTH22 (16) by XbaI and StuI restriction and was cloned into pBHELuc (33), containing the luciferase gene of Photinus pyralis, to replace the SV40 promoter. The sequence of pLTRLuc was confirmed by DNA sequencing. Plasmid pRSVTat (34,35) was used for transactivation of LTR-driven luciferase expression in cell culture experiments. Both pBHELuc and pRSVTat were obtained from H. Hauser (GBF, Braunschweig, Germany). Plasmid pTRI-β-actin-human (Ambion) was used as a standard in RNase protection assays and northern blot.
Figure 1.
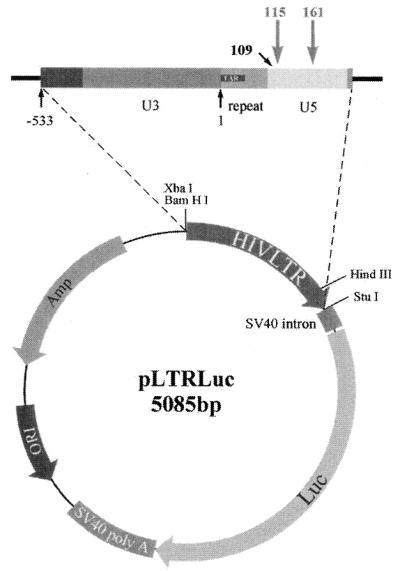
Plasmid pLTRLuc, expressing the reporter gene luciferase under control of the HIV-1 LTR promoter for cell culture experiments.
Ribozymes and randomized ribozyme libraries were chemically synthesized and purified as described previously (36,37). Mass spectral analysis (Dalton): Rz115M [M+H]+ calc. 11 718.1; found for active and inactive, 11 710.0; Rz161M [M+H]+ calc. 11 660.1; found for active, 11 641.5; for inactive, 11 651.4.
DNA-primer LTR250 (5′-dAGCCGAGTCCTGCGTCGAGA-3′) was used for the primer extension assay and sequencing. DNA oligonucleotides AS-115 (5′-dTGCCCGTTTCGGCCTAACGGCCTCATCAGTGTTGTG-3′) and AS-161 (5′-dCTTTAGTTTCGGCCTAACGGCCTCATCAGAGTGTGG-3′) were used for northern blot analysis.
In vitro transcription
For in vitro transcription a plasmid containing the appropriate sequence, pOTH33, was linearized and either gel purified or phenol–chloroform extracted and ethanol precipitated. Two micrograms of linearized plasmid DNA, 20 U of RNase inhibitor, 2500 U T7-polymerase and 50 µCi [α-32P]UTP were incubated in a volume of 50 µl for 1 h at 37°C in a buffer containing 10 mM DTT, 500 µM of each NTP, 50 mM Tris–HCl, pH 7.5, 2 mM spermidine and 6 mM MgCl2. Thereafter 50 U of DNase I (RNase-free) were added for 15 min. Non-incorporated NTPs were removed by centrifugation with microconcentrators Micron-50 (Amicon) according to the protocol applied by the manufacturer. The concentration of the aqueous RNA solution was determined by UV absorption at 260 nm to range from 0.1 to 7.0 µM. RNA was stored at –20°C.
Mapping of accessible sites
The EcoRI-linearized plasmid pOTH33 containing the HIV-1 LTR was in vitro transcribed and used as substrate for the mapping procedure. The transcript and randomized pool of ribozymes were preincubated at 75°C for 1 min in the presence of 50 mM Tris–HCl, pH 7.5, followed by 10 min of incubation at 37°C. MgCl2 was added to a final concentration of 10 mM. Cleavage reactions were performed in a total volume of 20 µl with 5 nM LTR substrate and 100 µM of the randomized ribozyme pool. After 4 h incubation at 37°C RNA was ethanol precipitated, the RNA pellet was resuspended in 5 µl of water and used for primer extension assay. As a control the pOTH33 transcript was incubated without ribozyme libraries.
Cleavage sites were identified by primer extension analysis mediated by AMV reverse transcriptase and DNA primer LTR250. The RNA pellet from the cleavage reaction (5 µl) was first denatured in the presence of [γ-32P]ATP-labeled DNA primer by heating to 80°C for 10 min in 5 µl hybridization buffer (60 mM NaCl, 50 mM Tris–HCl, pH 7.5, 5 mM DTT) and then slowly cooled to 30°C. Annealed samples were then added to 5 µl of primer extension buffer (0.5 mM dNTPs, 4 mM MgCl2, 1× hybridization buffer).
The reverse transcriptase reaction was initiated by adding 12.5 U of AMV reverse transcriptase. After 30 min incubation at 50°C the reaction was stopped by addition of an equal volume of gel-loading buffer (95% formamide, 0.025% xylene cyanol, 0.025% bromophenol blue, 18 mM EDTA, 0.025% SDS) and analyzed on an 8% polyacrylamide–7 M urea gel in the presence of a DNA sequencing ladder of the analyzed LTR-region generated by sequencing pOTH33 (0.6 µg) with the fmol sequencing system (Promega) using the [γ-33P]ATP-labeled DNA primer LTR250 according to the protocol described by the manufacturer.
In vitro ribozyme cleavage kinetics
For cleavage kinetics the EcoRI linearized plasmid pOTH33 containing the HIV-1 LTR was in vitro transcribed as described. Cleavage reactions were performed under single turnover conditions with a range of ribozyme concentrations (between 250 and 2000 nM). The ribozymes were incubated at 75°C for 1 min in the presence of 50 mM Tris–HCl, pH 7.5. After cooling to 37°C MgCl2 was added to a final concentration of 10 mM. Cleavage reactions with 100 nM substrate were performed in a total volume of 25 µl, containing 50 mM Tris–Cl, pH 7.5, and 10 mM MgCl2. A reaction was initiated by addition of the ribozyme and incubated at 37°C for 1 h. Reactions were stopped by the addition of 10 µl stop-mix (7 M urea, 50 mM EDTA, 0.05% bromophenol blue, 0.05% xylene cyanol). The resulting cleavage products were analyzed on a 6% polyacrylamide–8 M urea gel (40 cm long for 1 h at 50 W) and the remaining fraction of the substrate was determined by laser scanning densitometry on a Fuji Bas-2000 Phosphorimager. Kinetic constants for the cleavage were obtained by plotting the observed cleavage rate kobs against the quotient of kobs over the ribozyme concentration [E]g according to the following equation:
–ln(Frac S)/t = kobs = –KM (kobs/[E]g) + kreact
The reaction rate kobs equals the negative logarithm of the remaining substrate (Frac S) divided by the reaction time t. The negative slope represents the KM value, and the intercept of the regression line with the ordinate gives the maximal reaction rate kreact under single turnover conditions (17).
Cell culture experiments
HeLa cells were cultured in DMEM supplemented with 10% heat-inactivated fetal calf serum (FCS), 100 U/ml penicillin and 10 µg/ml streptomycin. Cells were kept in a moist atmosphere with 10% CO2 at 37°C. The day before transfection exponentially growing cells were seeded onto 6-well tissue culture plates according to standard protocols at a density of 2.5 × 105 cells/well. Cells were grown overnight (at least 18 h) to yield ~50–70% confluency for transfection. Prior to transfection ribozymes and Tfx-50 were complexed in 100 µl Opti-MEM for 20 min (500 pmol ribozyme/4 µl Tfx-50). At the same time the target plasmid pLTRLuc together with plasmid pRSVTat were complexed with Tfx-50 in 100 µl Opti-MEM (0.25 µg pLTRLuc and 0.002 µg pRSVTat/1 µl Tfx-50). Cells were incubated at 37°C in 800 µl DMEM with FCS, supplemented with the Tfx-50–ribozyme and Tfx-50–plasmid complexes (final concentration of ribozyme 0.5 µM). After 2.5 h cells were washed twice with DMEM (10% FCS). Transfected cells were cultured for 18 h.
Inhibition of gene expression by ribozymes was determined by measuring the activity of luciferase. At defined time points after transfection cells were washed twice with phosphate buffered saline (PBS) (0.14 M NaCl, 2.7 mM KCl, 16 mM Na2HPO4, 1.5 mM KH2PO4, pH 7.4) and cell extracts were made using 130 µl of lysis buffer (0.1 M potassium phosphate, pH 7.8, 0.2% Triton X-100, 1 mM DTT).
Luciferase expression was quantified on 20 µl of centrifuged lysate supernatant using a buffer containing 20 mM Tricine, 1.07 mM (MgCO3)4.Mg(OH)2.5H2O, 2.67 mM MgSO4, 0.1 mM EDTA, 33.3 mM DTT, 270 µM CoA, 470 µM Luciferin and 530 µM ATP. Light emission was measured by a Lumat LB9501 luminometer (Berthold) and relative light units (RLU) were calculated versus background activity. Luciferase activity was normalized to the protein concentration, determined using the Pierce BCA assay according to the protocol supplied by the vendor. Results present an average of at least three different experiments, each done in duplicate.
RNase protection
For the quantitation of LTR-RNA levels HeLa cells were plated and transfected as described previously. After 18 h cells were washed with PBS and RNA was extracted using an Ultraspec RNA kit. To measure the level of LTR-luciferase RNA the RNase protection kit RPA II was used. An antisense RNA probe targeting portions of the LTR region or the β-actin-human gene were prepared by in vitro transcription in the presence of [α-32P]UTP. The antisense probe for HIV-1 LTR contained 341 nt and was generated by in vitro transcription of plasmid pOTH33, linearized with HindIII and transcribed by SP6 polymerase. After hybridization with LTR mRNA the protected fragment contained 112 nt including the LTR region from position 78 to 190. The RNase protection assay was carried out with 20 µg of total RNA according to the standard protocol as described by the manufacturer. Hybridization of cellular RNA with both transcripts, 32P-labeled β-actin-human- and LTR-probe was carried out at 42°C overnight. The protected fragments were precipitated, analyzed with 6% polyacrylamide–urea gel electrophoresis and quantitated using the Fuji Bas-2000 Phosphorimager. The β-actin mRNA level served as an internal RNA quantitation reference.
Northern blot
For the detection of intracellular ribozymes, cells were grown and transfected with ribozymes as described. Following growth for 18 h cells were washed with PBS and RNA was isolated with Ultraspec RNA as described by the manufacturer. RNA was separated by a 1.5% (w/v) agarose–formaldehyde gel. Northern transfer and hybridization were performed according to standard procedures (Maniatis). The same blot was probed with the Rz161 and Rz115 specific oligonucleotides (AS-161, AS-115) which were 32P-end labeled and an internally 32P-labeled β-actin antisense transcript. Ribozyme and β-actin-levels were quantified on a Fuji Bas-2000 Phosphorimager. The β-actin mRNA served as an internal RNA quantitation reference.
RESULTS
Identification of susceptible target sites
Three different ribozyme libraries were generated by chemical synthesis (Fig. 2). GUX and NUX libraries represent conventional hammerhead ribozymes while the NXG library is specific for a purine nucleotide-specific hammerhead-like ribozyme previously selected in vitro (38). The binding arms are randomized except for the 3′-terminal nucleoside.
Figure 2.

Sequences of hammerhead and hammerhead-like ribozymes with randomized substrate binding arms.
To identify cleavage sites on the HIV-1 LTR promoter a 940 nt long LTR transcript of pOTH33 was incubated with one of the three ribozyme libraries. Locations of the resulting cleavage sites were identified by reverse transcriptase-mediated primer extension analysis of the 3′-fragments aligned with a dideoxy sequencing ladder of the corresponding LTR region (Fig. 3). As a control the LTR substrate was incubated without libraries to ensure that sites do not arise from reverse transcription termination. Two prominent bands at positions 115 and 161 were observed using the GUX and NUX libraries. The NXG library identified one weak cleavage band at position 109. According to the sequences flanking the cleavage sites, three different ribozymes, containing 7 nt for each binding arm, were designed (Fig. 4). Modified ribozymes were used to protect against nuclease degradation by modifications at the 2′-position of pyrimidine nucleotides and by incorporation of phosphorothioates at the termini. Ribozymes with inverted arms served as a control for the sequence specific effect of the active ribozyme in cell culture experiments. Catalytically inactive ribozymes were identical to the active ribozyme except for two mutations in the catalytic core, G5A and A14G, leading to complete abolishment of cleavage capacity. These constructs were used as a control for the effect of cleavage on the inhibition of luciferase expression.
Figure 3.
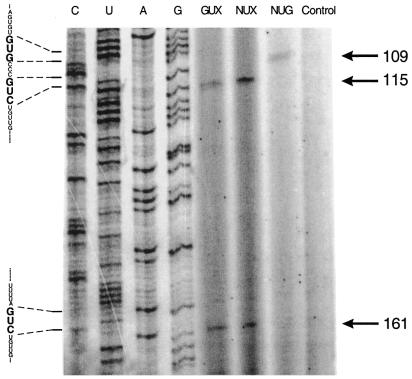
Detection of cleavage sites of the HIV-1 LTR mRNA by primer extension analysis. The in vitro transcript pOTH33, containing the HIV-1 LTR region, was incubated with three different synthetic ribozyme pools, containing randomized binding arms. Cleavage sites were detected by primer extension of the 3′-cleavage product. As a control LTR-transcript was incubated under the same conditions without ribozyme pool. Products were analyzed with 8% polyacrylamide–urea gel electrophoresis, aligned to the DNA sequencing ladder of pOTH33 and generated by using the same primer as described in Materials and Methods. Cleavage products are indicated by an arrow. Numbers are according to the cleavage positions in the LTR region.
Figure 4.

Sequences and modifications of ribozymes targeted against the HIV-1 LTR mRNA at position 109, 115 and 161. 2′-Fluoro-2′-deoxypyrimidine nucleotides, circled; 2′-amino-2′-deoxyuridine nucleotides, boxed. S, phosphorothioate linkages.
Cleavage activity in vitro
Active ribozymes Rz115 and Rz161, modified and unmodified, and unmodified ribozyme Rz109, were incubated with the pOTH33 in vitro transcript as described in Materials and Methods. Cleavage activities were determined under single turnover conditions (Table 1). Active ribozyme variants cleaved the transcribed LTR mRNA substrate in two fragments of predicted size. Neither the inverted ribozyme nor the non-functional ribozyme produced any cleavage products. The activities of ribozymes Rz115 and Rz161, modified or unmodified, were almost the same. A lower degree of cleavage was observed with ribozyme Rz109.
Table 1. Cleavage constants of ribozymes.
| Ribozyme | kreact (10–5 s–1) | KM (nM) | kreact/KM (M–1 s–1) |
|---|---|---|---|
| Rz115 | 24 | 398 | 606 |
| Rz115M | 30 | 398 | 760 |
| Rz161 | 22 | 289 | 787 |
| Rz161M | 22 | 252 | 896 |
| Rz109 | 10 | 437 | 243 |
Ribozymes were incubated with the in vitro transcript of HIV-1 LTR for 1 h at 37°C. Cleavage kinetics were performed under single turnover conditions as described in Materials and Methods. M, modified ribozymes (17).
Activities of ribozymes in cell culture
The activity of ribozymes against the LTR target mRNA in cell culture was examined with the plasmid construct pLTRLuc containing the ribozyme substrate LTR as a promoter to direct the expression of the reporter gene of luciferase (Fig. 1). HeLa cells were transiently cotransfected simultaneously with pLTRLuc and ribozymes using Tfx-50 as the carrier as described in Materials and Methods. The efficiency of ribozyme activity against the target LTR-RNA was determined by a decrease in luciferase activity after 18 h of growth as RLU/mg protein. Assays were normalized to protein concentration of the cell lysate. Activity in cells transfected with 0.25 µg pLTRLuc varied between 0.4 and 1.5 × 105 RLU/mg protein. Additionally in some experiments, cotransfection of the plasmid pRSVTat was performed. Transfection of 2 ng pRSVTat resulted in an increase of the luciferase activity by a factor of up to 100. The results, shown in Figure 5, present the average of at least three experiments, each in duplicate, of the luciferase activity in cells, transfected with the active or inactive ribozymes compared to cells transfected with the inverted form. Double determinations deviated by <5%.
Figure 5.

Inhibition of HIV-1 LTR dependent luciferase expression by ribozymes. HeLa cells were cotransfected with plasmid and active ribozymes or their corresponding controls. After 18 h of growth luciferase activity was determined. Luciferase activity of cells, transfected with the inverted form of the ribozyme, was defined as 100% control. Results present an average of at least three experiments each done in duplicate. (A and B) Luciferase activity of cells transfected with ribozymes 161M and 115M, respectively, in the absence of Tat. (C and D) Activity in the presence of Tat.
The inhibition levels of luciferase activity reached 87 and 61% after 18 h for 0.5 µM of active ribozymes Rz115M and Rz161M, respectively, when cotransfected only with plasmid pLTRLuc. In the presence of pRSVTat, leading to a transactivation of the LTR-dependent luciferase activity, the inhibition levels were 43% for Rz115M and 25% for Rz161M. Inhibition was essentially identical for the inactive Rz115M with 85% in the absence of Tat, and 39% in its presence. There was a bigger difference for the two forms with R161M where the inhibition was only 53% with the inactive form without Tat, and 39% in its presence.
The active and inactive forms of Rz115M were also tested in single experiments, in the absence of Tat, at 1 µM concentration with essentially the same results. Lowering the concentrations to 0.1 µM gave approximately half the degree of inhibitions, and at even lower concentration of 0.025 µM the active ribozyme achieved only 11% inhibition and the inactive none.
In contrast to the in vitro results the unmodified ribozyme variants of Rz115 and Rz161 were inactive in the cellular environment. Also Rz109, demonstrating only a low cleavage activity in vitro, as well as the modified form, Rz109M, did not show any inhibitory effect in the cell (data not shown).
To confirm that inhibition of ribozymes was sequence specific for LTR-RNA, cells were cotransfected with ribozymes and pBHELuc, a control-plasmid expressing luciferase under control of the SV40 early promoter. By the substitution of the promoter region LTR in this plasmid the corresponding transcript should no longer be a target for the ribozymes and no inhibitory effect on luciferase activity was to be expected. Transfection of ribozyme and plasmid DNA was performed as described above. No inhibition of luciferase activity was observed (data not shown). This result indicates that HIV-1 LTR-directed ribozymes inhibit sequence-specifically gene expression of the appropriate target sequence.
Decrease of LTR mRNA
An RNase protection assay with total RNA of cells, cotransfected with pLTRLuc, pRSVTat and ribozyme under the same conditions as previously described, was carried out in order to see whether the decrease of luciferase activity correlated with a decrease in the level of the target LTR transcript in the cells. The amount of LTR mRNA was correlated with the amount of β-actin mRNA for the same sample (Fig. 6). In cells transfected with the active ribozyme Rz115M, uncleaved LTR-RNA was reduced to 31% compared to cells transfected with the inverted control. The inactive ribozyme reduced the amount of the substrate RNA to 53%. In cells transfected with ribozyme Rz161M the amount of substrate RNA decreased to 64% with the active ribozyme and to 76% with the inactive variant. No products of RNA cleavage could be observed, only the full-length RNA, presumably because of degradation of the cleaved RNA.
Figure 6.

RNase protection analysis of the LTR mRNA in cells transfected with ribozymes, their corresponding controls and plasmid pLTRLuc and pRSVTat. Total RNA of transfected cells was purified after 18 h of growth and probed with the antisense transcript of the LTR. LTR mRNA levels were normalized to the level of β-actin mRNA. As a marker dephosphorylated pUC19 Sau3AI digested DNA, labeled with [γ-32P]ATP, was used.
Cellular uptake of ribozymes and stability in cells
The intracellular concentration of ribozymes after 18 h of growth was determined by northern blot. In order to compare cellular uptake and stability of ribozymes HeLa cells were transfected with Rz115M, Rz161M and Rz161 as described above. Total RNA purified from cells transfected with 0.5 µM ribozyme was first probed with ribozyme-specific oligonucleotides and then with a human β-actin specific probe to normalize ribozyme values for the amount of cellular RNA (Fig. 7). The levels of both variants of the modified ribozymes, Rz115M and Rz161M, were found to be nearly identical. In contrast, the level of the unmodified ribozyme Rz161 was much lower than that of the modified ribozymes.
Figure 7.
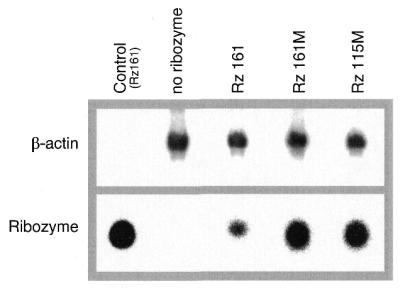
Northern blot analysis of intracellular ribozymes. HeLa cells were transfected with ribozymes. After 18 h of growth total RNA was isolated and subjected to northern blot analysis as described. Upper panel, β-actin; lower panel, ribozymes.
DISCUSSION
Synthetic ribozymes have been used for the inhibition of gene expression less often than the plasmid- or retroviral vector-derived ribozymes (2,3). This is probably due to the considerable synthetic effort which is involved in the preparation of the chemically modified ribozymes which are required to prevent degradation by nucleases (15,17,39,40). Targets for chemically modified ribozymes in cell culture have been c-myb (41), N-ras (42), luciferase (43), multiple drug resistance gene (44), VEGF receptor (45), Ki-ras (46), dopamine D2 receptor (47) and Hepatitis C virus 5′ LTR (48). In one study anti-Tat ribozymes were microinjected in human cells and shown to be effective when placed in the cytoplasm (49). In vivo model studies have been reported for inhibition of amelogenin (50), protein kinase Cα (51) and for reduction of stromelysin (52).
The HIV-1 LTR has also been previously described as a target for ribozymes in several publications but only for endogenous delivery. (5,11,53–56). In none of these studies was the RNA or transcripts checked for ribozyme accessibility. This is important as the HIV-LTR, like all RNAs, is highly structured as shown by chemical probing (57). The three ribozyme libraries we used for the identification of such sites in a transcript of nucleotides produced strong cleavage bands at positions 115 and 161 by the NUX and GUX libraries, containing the core sequence of the conventional ribozyme, and a weak band at position 109 for the NXG library with the core of the purine-specific hammerhead-like ribozyme (38). Interestingly, positions 115 and 161 had been earlier selected by us for ribozyme cleavage, simply by checking the sequence of this LTR region for GUC sites (16). Other groups had directed their endogenously produced ribozymes also against position 115, either as hammerhead or hairpin ribozymes with good results (9,11,53,56). Both the RNA at positions 115 and 161 are followed downstream by either a bulge or a loop according to the structural model by Baudin et al. (57) which might facilitate the nucleation of the ribozymes which subsequently invade the double-stranded regions. This model is consistent with the findings for the annealing of oligodeoxynucleotides to tRNA (58,59).
Ribozymes 109, 115 and 161 were kinetically characterized for cleavage of a transcript in the unmodified form (Table 1). As expected from the weaker band in transcript cleavage with the randomized form (Fig. 3) ribozyme 109 was less effective in this cleavage than ribozymes 115 and 161. Only these two were further characterized with the chemically modified forms which showed comparable activity to the unmodified (Table 1). HeLa cell culture experiments were then initiated using the plasmid pLTRLuc for the HIV-1 LTR-driven expression of the luciferase and the chemically modified ribozymes (Figs 1 and 4). The inverted form of the ribozymes were used to neutralize the positive charge of the carrier and served as a control for the sequence specificity of the inhibitory effect. Eighteen hours after transfection the chemically modified form of the active ribozyme 161M showed 67% inhibition of expression (Fig. 5). The catalytically inactive form produced 53% of inhibition. Ribozyme 115M had a more pronounced effect, giving 87% inhibition for the active and approximately the same with the inactive form. Ribozyme 109M did not show an effect on luciferase expression. It was of interest to test whether the ribozymes could still cope with the larger amount of RNA stimulated by the presence of Tat which is known to increase the expression of chimeric LTR-genes by stimulating transcription (60,61). When cotransfection was performed with pRSVTat inhibition was reduced to 22% for active ribozyme 161M and similarly for the inactive form, and to 40% for ribozyme 115M. Thus, the larger amount of mRNA induced by Tat results in a lower degree of inhibition. This indicates that a certain ratio of ribozyme to RNA is required for optimal inhibition. In summary, these results also show that exogenously applied ribozymes can suppress the expression of an LTR-driven gene by interacting with the HIV-1 LTR promoter RNA.
The similar degree of inhibition of expression by the catalytically active and inactive ribozymes could be ascribed to an essentially antisense effect by both, indicating a lack of turnover for the active ribozyme. This mechanism would explain the effect by a physical block of the ribosome in translation. If this was the correct interpretation, the level of mRNA should not be reduced in the ribozyme-treated cells. However, the RNase protection analysis (Fig. 6) shows a reduction of the LTR RNA to 31% by the active ribozyme 115M and to 53% for the inactive. Similarly, these values are 53 and 64%, respectively, for the two forms of ribozyme 161M. Rz115M, which exhibited a more pronounced inhibition of protein expression than Rz161M, also reduced the level of RNA more strongly. One also observes a higher degree in reduction of the RNA for the active than the inactive forms, particularly for Rz115M. This difference of 22% could possibly be ascribed to ribozyme-dependent cleavage indicating that this pathway makes a certain contribution in degradation of the RNA at least for this ribozyme. However, inhibition of expression does not reflect the contribution by the two mechanisms of RNA degradation where it might be obscured by the life time of the protein.
The reduction of the target RNA by the inactive forms of the ribozymes would suggest that they did not act by a simple antisense effect but that a degradation of the target RNA must have occurred. The mechanism for such a degradation can only be hypothesized on at present. One possibility is that a double-stranded RNase is responsible for this effect. The presence of such an enzyme in human cellular extracts and its partial purification from rat liver has been described (62).
The difference in degrees of inhibition by active and inactive forms by exogenously delivered chemically modified ribozymes differs when comparing data provided in other publications. Scherr et al. (42) report 54% reduction of luciferase activity for the active and 20% for the inactive ribozyme in their N-ras chimeric construct. The values reported for inhibition of PKCα (51) are 70 and 50%, respectively, and mRNA was reduced by 97% for the active and by 50% for the inactive form. Inhibition of proliferation by ribozymes directed against the VEGF receptor was between 56 and 77% for the active and no change observed for the inactive ribozyme; levels of mRNA were reduced to 50% by the active and to 13% by the inactive (45). Our own work on expression of luciferase showed essentially no difference in expression for the two forms (43). This comparison shows a considerable variation in results for the expression as well as for the mRNA levels. Most authors observe a certain degree of inhibition of protein expression by the inactive ribozyme and also reduction in the mRNA level. Although the reasons for these different results are not clear at present, the type of chemical modification of the ribozyme might play a role, for example in the stability of the ribozyme–target RNA complex. The more stable this complex is, the more difficult the dissociation of the cleavage products will be. Conversely, the stability of the complex with the inactive ribozyme might facilitate degradation by nucleases.
It is worthy of note that only the chemically modified ribozymes showed any inhibitory effect but not the unmodified. Analysis of ribozyme cellular uptake indicated a considerably weaker cellular concentration of the unmodified form of ribozyme 161 measured after 18 h (Fig. 7). The low cellular concentration of the unmodifed form apparently is not sufficient to have an effect on translation. It is not clear whether the low concentration is due to poorer uptake or to a more pronounced degradation by cellular nucleases. The difference in inhibitory power for the modified and the unmodified ribozyme is in contrast to our finding with ribozymes directed against the luciferase directly where the unmodified ribozyme also had an inhibitory effect (43). However, a different cell line was used there which also stably expressed the reporter protein. Thus a direct comparison is not possible.
In conclusion, this report shows the successful identification of ribozyme cleavage sites by a hammerhead ribozyme library and the usefulness of exogenously applied chemically stabilized hammerhead ribozymes for the inhibition of HIV-1 LTR driven gene expression. It also suggests that in the system investigated here, the inactive forms of the ribozymes, and possibly the active, might exert their effect by stimulating a double-stranded RNase.
Acknowledgments
ACKNOWLEDGEMENTS
We thank H. Hauser (GBF, Braunschweig) for the gift of pBHELuc and pRSVTat, U. Kutzke for the synthesis of oligonucleotides, F. Hillenkamp and B. Spottke (University of Münster) for the mass spectra and the Fonds der chemischen Industrie for financial support.
REFERENCES
- 1.Birikh K.R., Heaton,P.A. and Eckstein,F. (1997) The structure, function and application of the hammerhead ribozyme. Eur. J. Biochem., 245, 1–16. [DOI] [PubMed] [Google Scholar]
- 2.Bramlage B., Luzi,E. and Eckstein,F. (1998) Designing ribozymes for the inhibition of gene expression. Trends Biotechnol., 16, 434–438. [DOI] [PubMed] [Google Scholar]
- 3.James H.A. and Gibson,I. (1998) The therapeutic potential of ribozymes. Blood, 91, 371–382. [PubMed] [Google Scholar]
- 4.Muotri A.R., da Veiga Pereira,L., dos Reis Vasques,L. and Menck,C.F. (1999) Ribozymes and the anti-gene therapy: how a catalytic RNA can be used to inhibit gene function. Gene, 37, 303–310. [DOI] [PubMed] [Google Scholar]
- 5.Lee C.G.L., Jeang,K.-T., Martin,M.A., Pastan,I. and Gotteman,M.M. (1997) Efficient long-term coexpression of a hammerhead ribozyme targeted to the U5 region of HIV-1 LTR by linkage to the multidrug-resistance gene. Antisense Nucleic Acid Drug Devel., 7, 511–522. [DOI] [PubMed] [Google Scholar]
- 6.Bertrand E., Castanatto,D., Zhou,C., Carbonnelle,C., Lee,N.S., Good,P., Chatterjee,S., Grange,T., Pictet,R., Kohn,D., Engelke,D. and Rossi,J.J. (1997) The expression cassette determines the functional activity of ribozymes in mammalian cells by controlling their intracellular concentration. RNA, 3, 75–88. [PMC free article] [PubMed] [Google Scholar]
- 7.Wang L., Witherington,C., King,A., Gerlach,W.L., Carr,A., Penny,R., Cooper,D., Symonds,G. and Sun,L.-Q. (1998) Preclinical characterization of an anti-tat ribozyme for therapeutic application. Human Gene Ther., 9, 1283–1291. [DOI] [PubMed] [Google Scholar]
- 8.Jackson W.H., Moscoso,H., Nechtman,J.F., Galileo,D.S., Garver,F.A. and Lanclos,K.D. (1998) Inhibition of HIV-1 replication by an anti-tat hammerhead ribozyme. Biochem. Biophys. Res. Commun., 245, 81–84. [DOI] [PubMed] [Google Scholar]
- 9.Koizumi M., Ozawa,Y., Yagi,R., Nishigaki,T., Kaneko,M., Oka,S.-I., Kimura,S., Iwamoto,A., Komatsu,Y. and Ohtsuka,E. (1998) Design and anti-HIV-1 activity of hammerhead and hairpin ribozymes containing a stable loop. Nucl. Nucl., 17, 207–218. [DOI] [PubMed] [Google Scholar]
- 10.Kuwabara T., Warashina,M., Nakayama,A., Ohkawa,J. and Taira,K. (1999) tRNAVal -heterodimeric maxizymes with high potential as gene-inactivating agents: simultaneous cleavage at two sites in HIV-1 tat mRNA in cultured cells. Proc. Natl Acad. Sci. USA, 81, 1886–1891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Klebba C., Ottmann,O.G., Scherr,M., Pape,M., Engels,J.W., Grez,M., Hoelzer,D. and Klein,S.A. (2000) Retrovirally expressed anti-HIV ribozymes confer a selective survival advantage on CD4+ T cells in vitro. Gene Ther., 7, 408–416. [DOI] [PubMed] [Google Scholar]
- 12.Konopka K., Rossi,J.J., Swiderski,P., Slepushki,V.A. and Düzgünes,N. (1998) Delivery of an anti-HIV-1 ribozyme into HIV-infected cells via cationic liposomes. Biochim. Biophys. Acta, 1372, 55–68. [DOI] [PubMed] [Google Scholar]
- 13.Turner M.F. and Summers,M.F. (1999) Structural Biology of HIV. J. Mol. Biol., 285, 1–32. [DOI] [PubMed] [Google Scholar]
- 14.Pereira L.A., Bentley,K., Peeters,A., Churchill,M.J. and Deacon,N.J. (2000) A compilation of cellular transcription factor interactions with the HIV-1 LTR promoter. Nucleic Acids Res., 28, 663–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pieken W., Olsen,D.B., Benseler,F., Aurup,H. and Eckstein,F. (1991) Kinetic characterization of ribonuclease-resistant 2′-modified hammerhead ribozymes. Science, 253, 314–317. [DOI] [PubMed] [Google Scholar]
- 16.Heidenreich O. and Eckstein,F. (1992) Hammerhead ribozyme-mediated cleavage of the long terminal repeat RNA of human immunodeficiency virus type 1. J. Biol. Chem., 267, 1904–1909. [PubMed] [Google Scholar]
- 17.Heidenreich O., Benseler,F., Fahrenholz,A. and Eckstein,F. (1994) High activity and stability of hammerhead ribozymes containing 2′-modified pyrimidine nucleosides and phosphorothioates. J. Biol. Chem., 269, 2131–2138. [PubMed] [Google Scholar]
- 18.Peyman A., Helsberg,M., Kretzschmar,G., Mag,M., Grabley,S. and Uhlmann,E. (1995) Biol. Chem. Hoppe-Seyler, 376, 195–198. [DOI] [PubMed] [Google Scholar]
- 19.Monia B.P., Johnston,J.F., Geiger,T., Muller,M. and Fabbro,D. (1996) Antitumor activity of a phoshorothioate antisense oligodeoxynucleotide targeted against C-raf kinase. Nature Med., 2, 669–675. [DOI] [PubMed] [Google Scholar]
- 20.Milner N., Mir,K.U. and Southern,E.M. (1997) Selecting effective antisense reagents on combinatorial oligonucleotide arrays. Nat. Biotechnol., 15, 537–541. [DOI] [PubMed] [Google Scholar]
- 21.Campbell T.B., McDonald,C.K. and Hagen,M. (1997) The effect of structure in a long target RNA on ribozyme cleavage efficiency. Nucleic Acids Res., 25, 4985–4993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lima W.F., Brown-Driver,V., Fox,M., Hanecak,R. and Bruice,T.W. (1997) Combinatorial screening and rational optimization for hybridization to folded hepatitis C virus RNA of oligonucleotides with biological antisense activity. J. Biol. Chem., 272, 626–638. [PubMed] [Google Scholar]
- 23.Ho S.P., Bao,Y., Lesher,T., Malhota,R., Ma,L.Y., Fluharty,S.J. and Sakai,R.R. (1998) Mapping of RNA accessible sites for antisense experiments with oligonucleotide libraries. Nat. Biotechnol., 16, 59–63. [DOI] [PubMed] [Google Scholar]
- 24.Birikh K.R., Berlin,Y.A., Soreq,H. and Eckstein,F. (1997) Probing accessible sites for ribozymes on human acetylcholinesterase RNA. RNA, 3, 429–437. [PMC free article] [PubMed] [Google Scholar]
- 25.Scherr M. and Rossi,J.J. (1998) Rapid determination and quantitation of the accessibility to native RNAs by antisense oligodeoxynucleotides in murine cell extracts. Nucleic Acids Res., 22, 5079–5085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Scherr M., Rossi,J.J., Sczakiel,G. and Patzel,V. (2000) RNA accessibility prediction: a theoretical approach is consistent with experimental studies in cell extracts. Nucleic Acids Res., 28, 2455–2461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Patzel V., Steidl,U., Kronenwett,R., Haas,R. and Sczakiel,G. (1999) A theoretical approach to select effective antisense oligodeoxyribonucleotides at high statistical probability. Nucleic Acids Res., 27, 4328–4334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Scherr M., Reed,M., Huang,C.-F., Riggs,A.D. and Rossi,J.J. (2000) Oligonucleotide scanning of native mRNAs in extracts predicts intracellular ribozyme efficiency: ribozyme-mediated reduction of the murine DNA methyltransferase. Mol. Ther., 2, 26–38. [DOI] [PubMed] [Google Scholar]
- 29.Lan N., Howrey,R.P., Lee,S., Snith,C.A. and Sullenger,B.A. (1998) Ribozyme-mediated repair of sickle β-globin mRNAs in erythrocyte precursors. Science, 280, 1593–1596. [DOI] [PubMed] [Google Scholar]
- 30.Yu Q., Pecchia,D.B., Kingslay,S.L., Heckman,J.E. and Burke,J.M. (1998) Cleavage of highly structured viral RNA molecules by combinatorial libraries of hairpin ribozymes. J. Biol. Chem., 273, 23524–23533. [DOI] [PubMed] [Google Scholar]
- 31.zur Pulitz J., Yu,Q., Burke,J.M. and Wands,J.R. (1999) Combinatorial screening and intracellular antiviral activity of hairpin ribozymes directed against hepatitis B virus. J. Virol., 73, 5381–5387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cairns M.J., Hopkins,T.M., Witherington,C., Wang,L. and Sun,L.-Q. (1999) Target site selection for an RNA-cleaving catalytic DNA. Nat. Biotechnol., 17, 480–486. [DOI] [PubMed] [Google Scholar]
- 33.Artelt P., Morelle,C., Ausmeier,M., Fitzek,M. and Hauser,H. (1988) Vectors for efficient expression in mammalian fibroblastoid, myeloid and lymphoid cells via tranfection or infection. Gene, 68, 213–219. [DOI] [PubMed] [Google Scholar]
- 34.Peterlin B.M., Luciw,P.A., Barr,P.J. and Walker,M.D. (1986) Elevated levels of mRNA can account for the transactivation of human immunodeficiency virus. Proc. Natl Acad. Sci. USA, 83, 9734–9738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gorman C. (1985) In Glover,D.M. (ed.), DNA Cloning II, IRL Press, Oxford, UK, pp. 143–190.
- 36.Nakamaye K.L. and Eckstein,F. (1994) AUA-cleaving hammerhead ribozymes: attempted selection for improved cleavage. Biochemistry, 33, 1271–1277. [DOI] [PubMed] [Google Scholar]
- 37.Tuschl T. and Eckstein,F. (1993) Hammerhead ribozymes: importance of stem-loop II for activity. Proc. Natl Acad. Sci. USA, 90, 6991–6994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vaish N.K., Heaton,P.A., Fedorowa,O. and Eckstein,F. (1998) In vitro selection of a purine nucleotide-specific hammerhead-like ribozyme. Proc. Natl Acad. Sci. USA, 95, 2158–2162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Paolella G., Sproat,B.S. and Lamond,A.I. (1992) Nuclease resistant ribozymes with high catalytic activity. EMBO J., 11, 1913–1919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Beigelman L., McSwiggen,J.A., Draper,K.G., Gonzalez,C., Jensen,K., Karpeisky,A., Modak,A.S., Matulic-Adamic,J., Direnzo,A.B., Haeberli,P., Sweedler,D., Tracz,D., Grimm,S., Wincott,F.E., Thackray,V.G. and Usman,N. (1995) Chemical modification of hammerhead ribozymes. J. Biol. Chem., 270, 25702–25708. [DOI] [PubMed] [Google Scholar]
- 41.Jarvis T.C., Alby,L.J., Beaudry,A.A., Wincott,F.E., Beigelman,L., McSwiggen,J.A., Usman,N. and Stinchcomb,D.T. (1996) Inhibition of vascular smooth muscle cell proliferation by ribozymes that cleave c-myb mRNA. RNA, 2, 419–428. [PMC free article] [PubMed] [Google Scholar]
- 42.Scherr M., Grez,M., Ganser,A. and Engels,J.W. (1997) Specific hammerhead ribozyme-mediated cleavage of mutant N-ras mRNA in vitro and ex vivo. J. Biol. Chem., 272, 14304–14313. [DOI] [PubMed] [Google Scholar]
- 43.Bramlage B., Alefelder,S., Marschall,P. and Eckstein,F. (1999) Inhibition of luciferase expression by synthetic hammerhead ribozymes and their cellular uptake. Nucleic Acids Res., 27, 3159–3167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mariani L., Citti,L., Nevischi,S., Eckstein,F. and Rainaldi,G. (2000) Ribozyme and free alkylated base: a dual approach for sensitizing Mex+ cells to the alkylating antineoplastic drug. Cancer Gene Ther., 7, 905–909. [DOI] [PubMed] [Google Scholar]
- 45.Parry T.J., Cushman,C., Gallegos,A.M., Agrawal,A.B., Richardson,M., Andrews,L.E., Maloney,L., Mokler,V.R., Wincott,F.E. and Pavco,P.A. (1999) Bioactivity of anti-angiogenic ribozymes targeting Flt-1 and KDR mRNA. Nucleic Acids Res., 27, 2569–2577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Giannini C.D., Roth,W.K., Piiper,A. and Zeuzem,S. (1999) Enzymatic and antisense effects of a specific anti-Ki-ras ribozyme in vitro and in cell culture. Nucleic Acids Res., 27, 2737–2744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Salmi P., Sproat,B.S., Ludwig,J., Hale,R., Avery,N., Kela,J. and Wahlestedt,C. (2000) Dopamine D2 receptor ribozyme inhibits quinpirole-induced stereotypy in rats. Eur. J. Pharmacol., 388, R1–R2. [DOI] [PubMed] [Google Scholar]
- 48.Macejak D.G., Jensen,K.L., Jamison,S.F., Domenico,K., Roberts,E.C., Chaudhary,N., von Carlowitz,I., Bellon,L., Tong,M.J., Conrad,A., Pavco,P.A. and Blatt,L.M. (2000) Inhibition of hepatitis C virus (HCV)-RNA-dependent translation and replication of a chimeric HCV poliovirus using synthetic stabilized ribozymes. Hepatology, 31, 769–776. [DOI] [PubMed] [Google Scholar]
- 49.Hormes R., Homann,M., Oelze,I., Marschall,P., Tabler,M., Eckstein,F. and Sczakiel,G. (1997) The subcellular localization and length of hammerhead ribozymes determine efficacy in human cells. Nucleic Acids Res., 25, 769–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lyngstadaas S.P., Risnes,S., Sproat,B.S., Thrane,P.S. and Prydz,H.P. (1995) A synthetic, chemically modified ribozyme eliminates amelogenin, the major translation product in developing mouse enamel in vivo. EMBO J., 14, 5224–5229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sioud M. and Sørensen,D.R. (1998) A nuclease-resistant protein kinase Cα ribozyme blocks glioma cell growth. Nat. Biotechnol. ,16, 556–561. [DOI] [PubMed] [Google Scholar]
- 52.Flory C.M., Pavco,P.A., Jarvis,T.C., Lesch,M.E., Wincott,F.E., Beigelman,L., Hunt,S.W. and Schrier,D.J. (1996) Nuclease-resistant ribozymes decrease stromelysin mRNA levels in rabbit synovium following exogenous delivery to the knee joint. Proc. Natl Acad. Sci. USA, 93, 754–758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dropulic B., Lin,N.H., Martin,M.A. and Jeang,K.-T. (1992) Functional characterization of a U5 ribozyme: intracellular suppression of human immunodeficiency virus type 1 expression. J. Virology, 66, 1432–1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Priesley S., Buonomo,S.B.C., Michienzi,A. and Bozzoni,I. (1997) Use of adenoviral VAI small RNA as carrier for cytoplasmic delivery of ribozymes. RNA, 3, 677–687. [PMC free article] [PubMed] [Google Scholar]
- 55.Ventura M., Wang,P., Franck,N. and Saragosti,S. (1994) Ribozyme targeting of HIV-1 LTR. Biochem. Biophys. Res. Commun., 203, 889–898. [DOI] [PubMed] [Google Scholar]
- 56.Andäng M., Hinkula,J., Hotchkiss,G., Larsson,S., Britton,S., Wong-Staal,F., Wahren,B. and Ährlund-Richter,L. (1999) Dose-response resistance to HIV-1/MuLV pseudotype virus ex vivo in a hairpin ribozyme transgenic mouse model. Proc. Natl Acad. Sci. USA, 96, 12749–12753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Baudin F., Marquet,R., Isel,C., Darlix,J.-L., Ehresmann,B. and Ehresmann,C. (1993) Functional sites in the 5′ region of human immunodeficiency virus type 1 RNA form defined structural domains. J. Mol. Biol., 229, 382–397. [DOI] [PubMed] [Google Scholar]
- 58.Mir K.U. and Southern,E.M. (1999) Determining the influence of structure on hybridization using oligonucleotide arrays. Nat. Biotechnol., 17, 788–792. [DOI] [PubMed] [Google Scholar]
- 59.Stein C.A. (1999) Hybridization predicition gets to first base. Nat. Biotechnol., 17, 751–752. [DOI] [PubMed] [Google Scholar]
- 60.Berkhout B., Silverman,R.H. and Jeang,K. (1989) Tat trans-activates the human immunodeficiency virus through a nascent RNA target. Cell, 59, 273–282. [DOI] [PubMed] [Google Scholar]
- 61.Laspia M.F., Rice,A.P. and Mathews,M.B. (1989) HIV-1 tat protein increases transcriptional initiation and stabilizes elongation. Cell, 59, 283–292. [DOI] [PubMed] [Google Scholar]
- 62.Wu H., MacLeod,R., Lima,W.F. and Crooke,S.T. (1998) Identification and partial purification of human double strand RNase activity. J. Biol. Chem., 273, 2532–2542. [DOI] [PubMed] [Google Scholar]