

Abstract
Key Clinical Message
Creutzfeldt‐Jakob disease (CJD) is a rapidly progressive, fatal neurodegenerative disorder. This case highlights parkinsonism as a rare initial manifestation of sporadic CJD (sCJD), emphasizing the need for heightened clinical awareness to prevent misdiagnosis. Early and accurate diagnosis of sCJD is crucial for preventing potential iatrogenic transmission and optimizing patient management.
Abstract
Creutzfeldt‐Jakob disease (CJD) is a fatal neurodegenerative illness. While movement disorders may be present at the onset of the disease in about half of those with sporadic CJD (sCJD), parkinsonism is a rare initial presentation. In this article, we report a case of CJD with parkinsonism as the initial presentation of the disease. We report a 69‐year‐old lady with initial symptoms of gait difficulty, tremor, and bradykinesia. Later, she developed cognitive impairment, ataxia, chin tremor, and myoclonic jerks. Her condition worsened to the point of akinetic mutism. She was diagnosed with probable sCJD after detecting protein 14‐3‐3 in her cerebrospinal fluid and observing typical imaging features.This case report illustrates important aspects of an inevitably fatal and rapidly progressing disease's early presentation and clinical features. The uncommon initial presentations of sCJD should be considered with the intent of preventing misdiagnosis in the future. Early diagnosis of sCJD can prevent possible iatrogenic disease transmission and improve patient care.
Keywords: Creutzfeldt‐Jakob disease (CJD), neurodegenerative disease, parkinsonism, prion disease, protein 14‐3‐3, sporadic CJD (sCJD)
1. INTRODUCTION
Creutzfeldt‐Jakob disease (CJD) is a deadly neurodegenerative illness that belongs to the family of transmissible spongiform encephalopathies (TSEs), sometimes referred to as “prion diseases”, and its annual incidence ranges from 1.0 to 1.4 per million in the United States. 1
CJD is characterized by rapidly progressive dementia in older adults, with death typically occurring within 1 year of illness onset. 2 In addition to rapidly progressive dementia, several characteristic features may aid in diagnosing CJD. They include focal or generalized myoclonus (may be stimulus responsive), progressive visual symptoms, cerebellar ataxia, or dysarthria. 3
Approximately half of the patients with sporadic CJD (sCJD) may have movement disorder at the onset of the disease presentation. 4 Several movement disorders have been described in many patients with CJD. In addition, the frequency of movement disorders will be increased with the disease duration. These movement disorders can also occur early and have been described as initial manifestations of the illness. 5 Myoclonus is the most frequent abnormal movement in sCJD and may be stimulus‐responsive. 6 Others include chorea, dystonia, tremor, rigidity, and parkinsonism. A recent systematic review reported parkinsonism and isolated rigidity in 7.3% and 43.9% of patients with sCJD, respectively. The median time from the onset of parkinsonism to death was 3 months, indicating a late presentation during the disease course. 4
In this article, we present a case of CJD initially manifesting with extrapyramidal symptoms. This case is notable for its unusual presentation of parkinsonism characterized by prominent gait disturbance, rigidity, and bradykinesia. It underscores that parkinsonism can be an initial feature of sCJD. This highlights the importance of considering CJD in the differential diagnosis of parkinsonism with atypical features and demonstrates the rapidly progressive and fatal nature of sCJD, which remains incurable despite immunotherapy.
2. CASE HISTORY
A 69‐year‐old woman was referred to our hospital with a history of gait disturbance that had begun 8 months earlier. Within the onset of gait difficulty, the family also noticed a symmetrical tremor in her hands and slowness of movements during daily activities accompanied by frequent falls.
About a month after the initiation of her symptoms, she reported a strange sensation inside her body without any objective phenomenon that was described as “chills inside her body.” Afterward, behavioral change became apparent in the form of delusion of persecution, changes in personality, and feelings of fear without reason. She became paranoid about others and had less confidence in close relatives.
A few months later, her gait worsened with the need for a walking aid and intensified tremors in both hands and her chin. Progressively she lost the ability to walk without help, and generalized myoclonic jerks appeared.
Notably, her past medical history included breast cancer diagnosed 2 years before this admission. She has undergone lumpectomy and chemoradiotherapy and, subsequently, oral tamoxifen. The follow‐up had not shown any evidence of the relapse of the tumor.
After 8 months of initial presentation, the neurological examination revealed an awake and alert lady but not completely oriented to time and place. The Mini‐Mental Status Examination (MMSE) score was 13 out of 30. Rigidity and bradykinesia were detected symmetrically on both sides. She had a resting tremor in both hands and her chin. The gait examination revealed a stooped posture with a short stepping gait and occasional freezing. Deep tendon reflexes were all symmetrically +1 to +2, and plantar reflexes were downward bilaterally. Although the sensory examination revealed no remarkable findings, the patient stated distal paresthesia of the upper limbs. Other positive findings in the examination were grasp reflex, utilization behavior, and the Myerson sign.
3. METHODS
After admission, a comprehensive workup was performed with the clinical features of rapidly progressive dementia, parkinsonism, behavioral change, and frontal release signs. Blood and urine tests were normal, including electrolytes, liver, and kidney function tests, and level of vitamin B12. Tests for systemic vasculitis were all negative. Thyroid function tests and Anti‐thyroid peroxidase antibody were in the normal range. According to the history of breast cancer, paraneoplastic and autoimmune antibodies in the blood and cerebrospinal fluid (CSF) were assessed, which showed no positive result. CSF analysis revealed no white blood cells and normal values for glucose and protein (39 mg/dL). The complementary tests revealed that the 14‐3‐3 protein was positive in CSF.
The brain MRI had typical features of sCJD. It showed T2 and FLAIR hyperintensities in bilateral caudate, putamina, and medical thalami with water restriction in Diffusion‐weighted imaging (DWI). In addition, there were typical cortical ribboning patterns in bilateral parieto‐occipital and bilateral medial frontal regions in DWI (Figure 1). The electroencephalography (EEG) revealed nonspecific generalized slowing without any periodic sharp waves (Figure 2).
FIGURE 1.
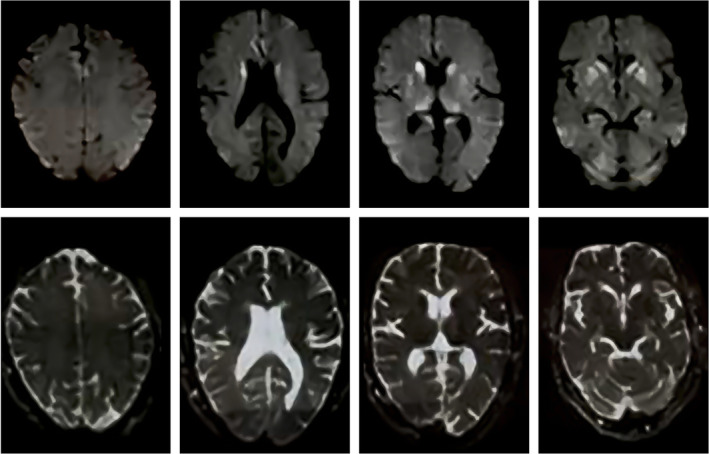
Axial brain MRI, DWI, and corresponding ADC sequences show water‐restriction appearance (DWI hyperintensity and ADC hypointensity) in bilateral caudate, putamina, and medial thalamic cortical ribboning at bilateral parieto‐occipital and medial frontal lobes.
FIGURE 2.
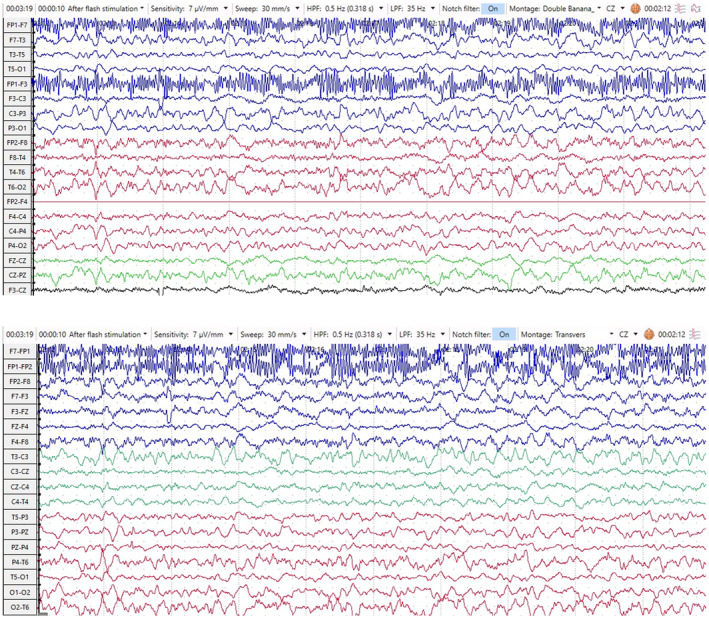
EEG Trace (10‐20 System) longitudinal (double banana) (above) and transverse (below) montages revealed a generalized slowing in theta range. Note that high‐frequency muscle artifact was seen in the FP1 lead.
Before completing the diagnostic workup and based on the significant history of old malignancy, a therapeutic trial of corticosteroid with 1000 mg methylprednisolone in 5 successive days was initiated. Still, no change was detected in the patient's condition. The patient was discharged for palliative care because there is no cure for CJD.
4. OUTCOME
In the aftermath, urinary incontinence appeared, and both ataxia and parkinsonism deteriorated. Throughout 4 months, she gradually lost the ability to talk and motor functions, becoming akinetic‐mutic. Finally, she was ventilated, and after 1 month, she passed away due to cardiorespiratory failure. The overall duration between the initial presentation and death lasts approximately 13 months.
5. DISCUSSION
According to Revised International Creutzfeldt‐Jakob Disease Surveillance Network Diagnostic Criteria, the patient's clinical presentation and laboratory findings were consistent with the probable sCJD. She had typical clinical features (i.e., dementia, extrapyramidal features, myoclonus, akinetic mutism) with a disease duration of fewer than 2 years, positive 14‐3‐3 assay, and typical MRI features. 7
Previous studies reported that apart from cognitive decline, the most common symptom of CJD is gait ataxia. The initial presentation of extrapyramidal signs may be seen in a minority of the patients which may be hypokinetic or hyperkinetic. 8 , 9 It should be noted that due to its low frequency and clinical heterogeneity, sCJD is likely under‐recognized, especially in low‐ and middle‐income nations. Several difficulties relating to the diagnosis of CJD have been reported, including financial limits, lack of testing facilities, underdeveloped healthcare infrastructure, scarcity of specialists, and regional disparities between rural and urban centers. 10
Moreover, several neurodegenerative diseases can mimic CJD. Alzheimer's disease (AD) and dementia with Lewy bodies (DLB) can both present with rapidly progressive dementia, but AD typically progresses more slowly and lacks the prominent extrapyramidal signs, myoclonus, and visual disturbances seen in CJD. DLB, while presenting with dementia, parkinsonism, and visual hallucinations like CJD, usually has a more gradual onset. Frontotemporal dementia (FTD) may resemble CJD in terms of behavioral changes and cognitive impairment, but it progresses more slowly and lacks prominent extrapyramidal features. Corticobasal degeneration (CBD) shares similarities with CJD, such as cognitive impairment and movement disorders, but it also progresses more gradually. 11 , 12 Additionally, the diversity of initial symptoms is one of the clinical difficulties that may lead to misdiagnosis. Several cases of CJD have been reported that atypical initial symptoms lead to misdiagnosis. 13 In one retrospective study, only 18% of patients were diagnosed correctly at their first assessment. 12
Cases of CJD with initial presentation of extrapyramidal symptoms have been reported before. 14 Amongst the movement disorders presented with CJD, myoclonus is by far the most seen movement disorder associated with CJD. Typically, myoclonus is generalized and distributed in the limbs but may be focal or unilateral, as reported by Razme and his colleagues. 6 , 15 Dystonia, choreoathetosis, tremor, and parkinsonism have also been reported. Generalized chorea accompanied by dystonia of upper limbs was reported in a 71‐year‐old lady by Tan and colleagues. 15 Extrapyramidal syndrome (typically akinetic mutism) is characteristic of the final stages of the disease; however, atypical parkinsonism may occur as an initial presentation. Cases have been reported with other features resembling CBD, Progressive supranuclear palsy, FTD, and Parkinson's disease.
In most cases, gait difficulty and rigidity were often associated with other signs. The subacute course of the symptoms with rapid cognitive decline and generalized myoclonus suggested CJD. 5
A post‐mortem brain pathology of spongiform degeneration was reported in a patient with progressive dementia and parkinsonism, and the disease course lasted 4 years. However, Lewy bodies were detected in the brain stem. The authors noted that the patient had both sCJD and Parkinson's disease simultaneously. 16 Interestingly, a case of rapidly progressive Parkinsonism, in which he could not perform his daily activities within 2 months, was reported in a 71‐year‐old Korean man. 14
By reporting this case, we aim to emphasize one of the uncommon initial presentations of sCJD to prevent future misdiagnosis. Early diagnosis of sCJD prevents possible iatrogenic transmission of the disease and is essential for the patient and caregivers to provide proper care.
AUTHOR CONTRIBUTIONS
Sahar Nikkhah Bahrami: Conceptualization; investigation; methodology; project administration; visualization; writing – original draft; writing – review and editing. Asal Sadat Karimi: Investigation; visualization; writing – original draft. Sepehr Khosravi: Supervision; writing – review and editing. Mostafa Almasi‐Dooghaee: Supervision; validation.
FUNDING INFORMATION
There is no funding or grant to disclose for this study.
CONFLICT OF INTEREST STATEMENT
The authors report there are no competing interests to declare.
CONSENT
Written informed consent was obtained from the patient's next of kin to publish this report in accordance with the journal's patient consent policy.
ACKNOWLEDGMENTS
We kindly respect and thank the patient's family for providing the information and well‐corporation.
Bahrami SN, Karimi AS, Khosravi S, Almasi‐Dooghaee M. Parkinsonism as an initial presentation of Creutzfeldt‐Jakob disease: A case report and review of literature. Clin Case Rep. 2024;12:e9278. doi: 10.1002/ccr3.9278
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available on request from the corresponding author. The data are not publicly available due to privacy or ethical restrictions.
REFERENCES
- 1. Maddox RA, Person MK, Blevins JE, et al. Prion disease incidence in the United States: 2003–2015. Neurology. 2020;94(2):e153‐e157. [DOI] [PubMed] [Google Scholar]
- 2. Holman RC, Belay ED, Christensen KY, et al. Human prion diseases in the United States. PLoS One. 2010;5(1):e8521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Moreno M, Romero J. Sporadic Creutzfeldt‐Jakob disease: phenotypic variability. Neurologia (Barcelona, Spain). 2002;17(7):366‐377. [PubMed] [Google Scholar]
- 4. Rodriguez‐Porcel F, Ciarlariello VB, Dwivedi AK, et al. Movement disorders in Prionopathies: a systematic review. Tremor Other Hyperkinet Mov (N Y). 2019;9:1‐9. doi: 10.7916/tohm.v0.712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Maltête D, Guyant‐Maréchal L, Mihout B, Hannequin D. Movement disorders and Creutzfeldt‐Jakob disease: a review. Parkinsonism Relat Disord. 2006;12(2):65‐71. [DOI] [PubMed] [Google Scholar]
- 6. Razmeh S, Almasi M, Habibi AH, Alizadeh E. Rapidly progressive dementia and unilateral jerky movements: a case of Creutzfeldt‐Jakob disease. Postgrad Med J. 2016;92(1093):688‐690. [DOI] [PubMed] [Google Scholar]
- 7. Watson N, Hermann P, Ladogana A, et al. Validation of revised international Creutzfeldt‐Jakob disease surveillance network diagnostic criteria for sporadic Creutzfeldt‐Jakob disease. JAMA Netw Open. 2022;5(1):e2146319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Krasnianski A, Kaune J, Jung K, Kretzschmar HA, Zerr I. First symptom and initial diagnosis in sporadic CJD patients in Germany. J Neurol. 2014;261(9):1811‐1817. [DOI] [PubMed] [Google Scholar]
- 9. Rabinovici G, Wang P, Levin J, et al. First symptom in sporadic Creutzfeldt–Jakob disease. Neurology. 2006;66(2):286‐287. [DOI] [PubMed] [Google Scholar]
- 10. Watson N, Brandel J‐P, Green A, et al. The importance of ongoing international surveillance for Creutzfeldt–Jakob disease. Nat Rev Neurol. 2021;17(6):362‐379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kojima G, Tatsuno BK, Inaba M, Velligas S, Masaki K, Liow KK. Creutzfeldt‐Jakob disease: a case report and differential diagnoses. Hawaii J Med Public Health. 2013;72(4):136‐139. [PMC free article] [PubMed] [Google Scholar]
- 12. Paterson RW, Torres‐Chae CC, Kuo AL, et al. Differential diagnosis of Jakob‐Creutzfeldt disease. Arch Neurol. 2012;69(12):1578‐1582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Xu Z, Zhao Y. A Creutzfeldt‐Jakob disease case misdiagnosed with acute cerebral infarction and review of the literature. Clin Case Reports. 2020;8(12):3310‐3314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Park Y, Lee CN. A case of Creutzfeldt‐Jakob disease presented as rapid progressive parkinsonism. Dement Neurocogn Disord. 2019;18(4):152‐156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Tan AH, Toh TH, Low SC, et al. Chorea in sporadic Creutzfeldt‐Jakob disease. J Mov Disord. 2018;11(3):149‐151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Iida T, Doh‐ura K, Kawashima T, Abe H, Iwaki T. An atypical case of sporadic Creutzfeldt‐Jakob disease with Parkinson's disease. Neuropathology. 2001;21(4):294‐297. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that support the findings of this study are available on request from the corresponding author. The data are not publicly available due to privacy or ethical restrictions.