

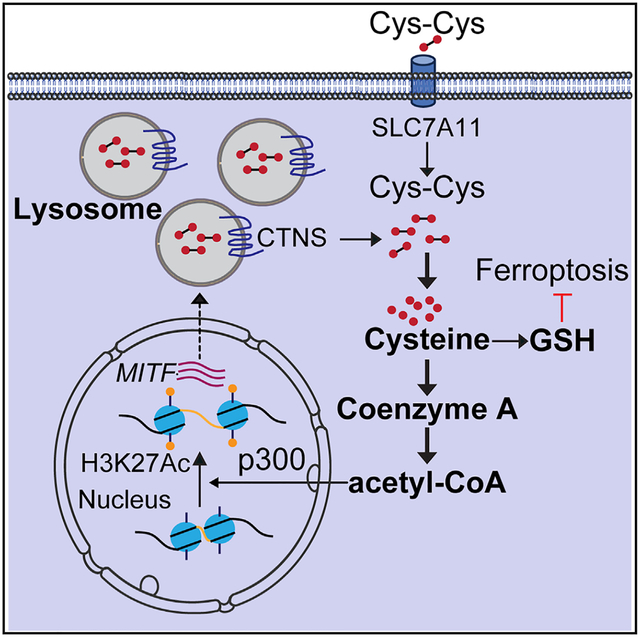
SUMMARY
The inherent ability of melanoma cells to alter the differentiation-associated transcriptional repertoire to evade treatment and facilitate metastatic spread is well accepted and has been termed phenotypic switching. However, how these facets of cellular behavior are controlled remains largely elusive. Here, we show that cysteine availability, whether from lysosomes (CTNS-dependent) or exogenously derived (SLC7A11-dependent or as N-acetylcysteine), controls melanoma differentiation-associated pathways by acting on the melanocyte master regulator MITF. Functional data indicate that low cysteine availability reduces MITF levels and impairs lysosome functions, which affects tumor ferroptosis sensitivity but improves metastatic spread in vivo. Mechanistically, cysteine-restrictive conditions reduce acetyl-CoA levels to decrease p300-mediated H3K27 acetylation at the melanocyte-restricted MITF promoter, thus forming a cysteine feedforward regulation that controls MITF levels and downstream lysosome functions. These findings collectively suggest that cysteine homeostasis governs melanoma differentiation by maintaining MITF levels and lysosome functions, which protect against ferroptosis and limit metastatic spread.
Graphical Abstract
In brief
Yu et al. show that cysteine availability is key for melanoma cells to maintain their melanocytic identity, lysosomal functions, defend against an iron-dependent cell death termed ferroptosis, and prevent metastatic spread. This study highlights how melanoma cells rely on cysteine to allow epigenetic control of the differentiated melanocytic fate.
INTRODUCTION
Melanoma arises from the malignant transformation of normal pigment-producing melanocytes in the skin and eye.1,2 During organismal development, melanocytes originate from the neural crest and follow conserved developmental pathways wherein they depend on expression and signaling cues acting on microphthalmia-associated transcription factor (MITF) that regulates critical genes involved in melanocyte differentiation and survival, functions that are largely maintained also in melanoma.3,4 It is somewhat paradoxical that MITF is a genuine melanoma oncogene activated by genomic amplification5 or point mutation,6,7 despite being a lineage master regulator that controls cell-fate choice and differentiation-associated pathways. However, melanoma tumors and even cultured melanoma cells display a significant degree of transcriptional plasticity, which has been attributed to functional switching between a proliferative differentiation-associated state and an invasive dedifferentiated (undifferentiated) state that promotes metastatic spread.8 Unlike genetic heterogeneity that increases with disease progression and drives acquired therapeutic resistance,9 accumulating evidence in melanoma indicates that reversible metabolic and epigenetic mechanisms associated with cancer cell differentiation states enable transcriptional and functional plasticity.10 Melanoma plasticity share features with epithelial-mesenchymal transition observed in epithelial tumors,11,12 which as a dichotomous concept is detailed as upregulated neural crest markers, increased extracellular matrix remodeling, and resistance to the growth inhibitory action of TGF-β, which occurs at the expense of downregulating MITF and downstream regulated genes.10 The use of more refined molecular modeling approaches has suggested that melanomas can be further classified into one of four states described as (1) differentiated, (2) neural crest-like, (3) transitory, or (4) dedifferentiated.13 Interestingly, melanoma cells exhibiting the dedifferentiated state were found resistant to multiple therapeutic drugs but exquisitely sensitive to induction of ferroptosis,13 which is a specific type of cell death caused by excessive iron-dependent lipid peroxidation.14 Ferroptosis specifically occurs when lipid peroxidation exceeds the detoxification ability of the glutathione system,15 whose biosynthesis depends on an adequate supply of the conditionally essential sulfur amino acid cysteine.16 Intracellular cysteines are derived from several sources, including endogenous synthesis from methionine via the transsulfuration pathway17 and extracellular oxidized cysteine imported by the cystine/glutamate antiporter solute carrier family 7 member 11 (SLC7A11).18 Recently, lysosomes, organelles required for general protein degradation and cell survival through autophagy, have emerged as an important source of cysteine.19–22 By recycling cysteine from protein degradation, lysosomes export oxidized cysteine using the lysosomal cystine transporter cystinosin (CTNS) and thus contribute to the intracellular cysteine pool.23 In addition to its requirement for glutathione synthesis, which is important for oxidative stress detoxification, the sulfur amino acid cysteine also ensures proteostasis and maintains adequate levels of thiol-containing biomolecules, including coenzyme A (CoA).19,23,24
Given that MITF directly regulates the mitochondrial biogenesis master regulator PGC-1α (encoded by PPARGC1A) to promote resistance to exogenous oxidative stress and bioenergetic loss induced by targeted BRAF inhibition in melanoma cells,25,26 and that specifically PGC-1α limits melanoma metastatic spread,27 we wanted to functionally explore whether MITF or PGC-1α controls sensitivity to ferroptosis and how this regulation mechanistically pertains to melanoma phenotype switching.
RESULTS
MITF maintains glutathione homeostasis and promotes resistance to ferroptosis
As our chosen experimental system for the functional interrogation of ferroptosis sensitivity, we used a panel of melanoma cell lines stratified by high expression vs. low expression of MITF (MITFhigh vs. MITFlow).13 Correspondingly, low vs. high AXL levels within this cohort also served as correlates of dedifferentiation and resistance to targeted kinase inhibitors (Figure 1A).8,28 We included the K029A-BRAF inhibitor-resistant melanoma cell line (K029A-Res) generated as described previously (cell line information provided in Table S1).29 Compared with MITFhigh melanoma cell lines, MITFlow cell lines showed comparable expression levels of SLC7A11 and glutathione peroxidase 4 (GPX4) (Figure 1A). However, MITFlow cell lines were more susceptible to cell death induced by cysteine restriction produced by limiting the cystine concentration in the medium (Figure 1B), and treatment using the GPX4 inhibitor RSL3 (Figure 1C), generated significantly more lipid peroxidation upon cysteine restriction and RSL3 treatment (Figure S1A). Supporting that cell death caused by cysteine restriction and RSL3 was indeed ferroptosis, treatment with ferroptosis-specific inhibitors ferrostatin 1 (Fer-1) and liproxstatin-1 (Lip-1), but not apoptosis inhibitor Z-VAD-FMK (Z-VAD) or necroptosis inhibitor necrostatin-1 (NEC-1), significantly attenuated cell death induced by cysteine restriction and RSL3 (Figures S1B and S1C). Importantly, the addition of N-acetylcysteine (NAC) or glutathione (GSH) also markedly protected against both cysteine restriction- and RSL3-induced cell death, indicating that disruption of cysteine and glutathione homeostasis under these conditions causes cell death through ferroptosis (Figure S1D). Furthermore, inhibition of glutathione production using the inhibitor of γ-glutamylcysteine synthetase, buthionine-sulfoximine, significantly aggravated cell death caused by cysteine restriction or RSL3 (Figure S1E). Taken together, these findings suggest that other, yet undefined, mechanisms of sensitivity to ferroptosis may be operational in melanoma cells.
Figure 1. MITF regulates sensitivity to cysteine restriction and ferroptosis induction.
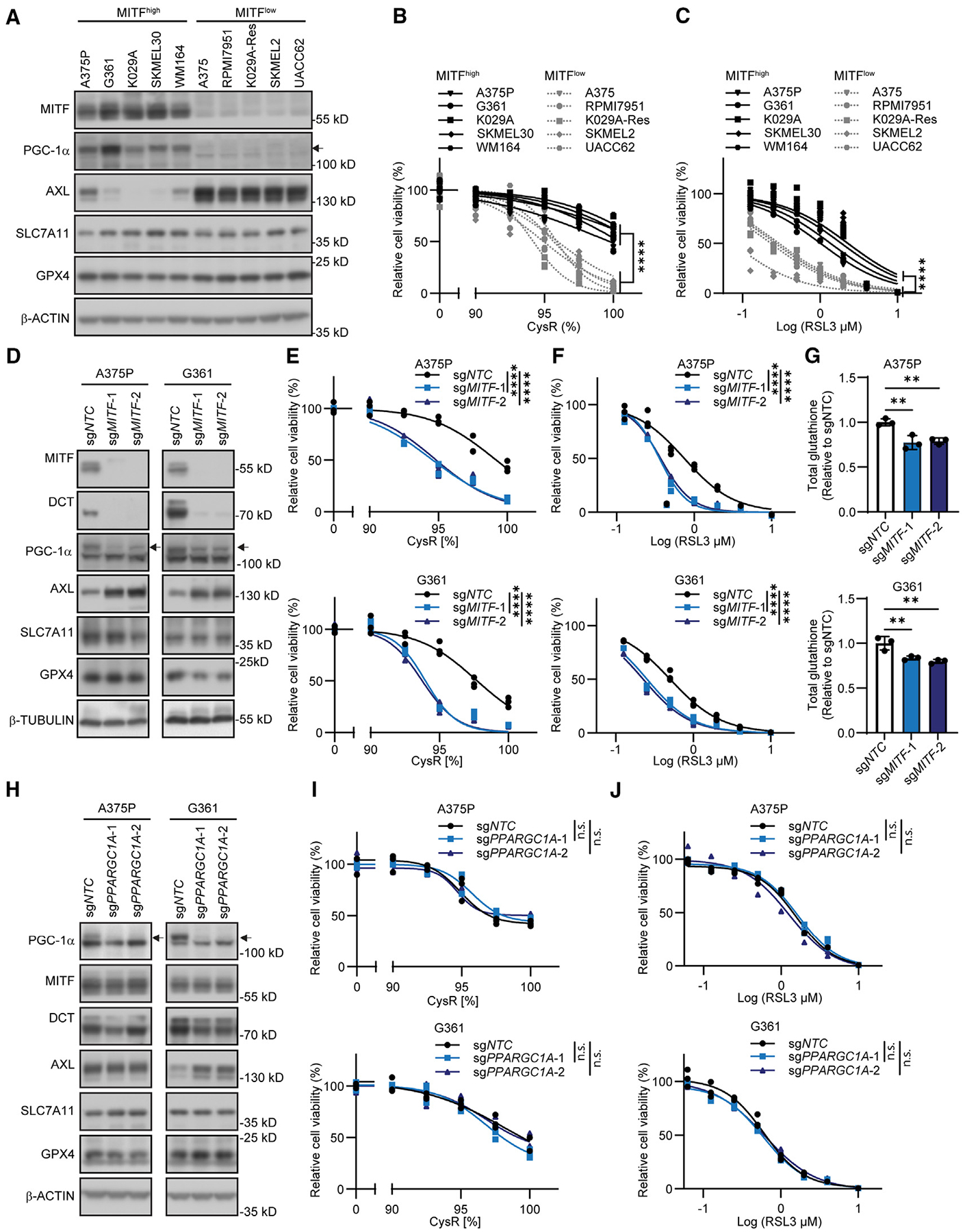
(A) Immunoblot analysis of the indicated proteins across a panel of MITFhigh and MITFlow melanoma cell lines.
(B and C) Differential sensitivity of MITFhigh and MITFlow melanoma cell lines to (B) cysteine restriction (CysR) and (C) RSL3 treatment as an inducer of ferroptosis.
(D) Immunoblot analysis of the indicated proteins in MITF-deleted A375P and G361 cells.
(E and F) MITF deletion induces sensitivity to (E) CysR and (F) ferroptosis induction using RSL3 treatment in A375P (top) and G361 (bottom) cells.
(G) MITF deletion reduces total glutathione levels across A375P (top) and G361 (bottom) cells.
(H) Immunoblot analysis of the indicated proteins in PPARGC1A-deleted A375P and G361 cells.
(I and J) PPARGC1A deletion does not alter sensitivity to (I) CysR or (J) ferroptosis induction using RSL3 in A375P (top) and G361 (bottom) cells.
Data shown as mean ± SD. Statistical significance was calculated using two-way ANOVA (B, C, and E–J) and one-way ANOVA (G). *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001; n.s., not significant.
See also Figures S1 and S2.
Because we found that levels of MITF were associated with sensitivity to cysteine depletion and ferroptosis induction, we used CRISPR-Cas9 genome editing to disrupt the MITF gene in each of the MITFhigh A375P and G361 melanoma cell lines. Deletion of MITF consistently led to decreased expression of downstream targets DCT and PGC-1α, and upregulation of AXL (Figure 1D). However, we could only detect negligible effects on SLC7A11 and GPX4, paralleling their comparable expression we had observed across MITFhigh and MITFlow cell lines (Figure 1A). Importantly, MITF deletion potentiated the effects of cysteine restriction and RSL3 treatment on reducing cell viability and generating lipid peroxides (Figures 1E, 1F, and S1F). Furthermore, MITF deletion reduced baseline total glutathione and the GSH/GSSG ratio and accentuated these effects following cysteine restriction (Figures 1G and S1G).
To assess whether overexpression of MITF was able to alter the sensitivity to cysteine restriction and RSL3 treatment, we used lentiviral transduction in A375 and K029A-Res melanoma cells. As expected, MITF overexpression upregulated DCT, PGC-1α, and reduced AXL levels, but did not alter the expression of SLC7A11 or GPX4 (Figure S1H). In these cells, increased MITF levels provided robust resistance to lipid peroxide production (Figure S2A), growth inhibition by cysteine restriction (Figure S2B), and RSL3 treatment (Figure S2C), and significantly increased the baseline and post-cysteine restriction total glutathione and GSH/GSSG ratios (Figures S2D and S2E).
Given that MITF is a known upstream regulator of the master regulator of mitochondrial biogenesis PGC-1α (encoded by PPARGC1A), which promotes resistance to exogenous oxidative stress and bioenergetic loss induced by targeted BRAF inhibition in melanoma cells,25,26 we explored whether PGC-1α was an effector downstream of MITF on these functional endpoints. CRISPR-Cas9-mediated knockout of the PPARGC1A gene in A375P and G361 did not reduce MITF levels, nor did it consistently affect DCT, AXL, SLC7A11, or GPX4 (Figure 1H). Furthermore, PPARGC1A deletion did not affect sensitivity to cysteine restriction or RSL3 treatment (Figures 1I and 1J). We complemented these analyses further using overexpression of PGC-1α in A375 and K029A-Res cells, where we could not discern any effect on DCT, SLC7A11, GPX4 levels, or any differential functional effects from cysteine restriction or RSL3 treatment (Figures S2F–S2H). Taken together, these data indicate that MITF, independent of PGC-1α, is a causal regulator of glutathione homeostasis and ferroptosis sensitivity.
Attenuated lysosome biogenesis and ferroptosis sensitivity are signatures of reduced MITF
In melanocytes and melanoma, MITF controls cellular identity, survival, and differentiation-associated functions.4 Because of the dichotomous association between high MITF levels and resistance to cysteine restriction and ferroptosis induction, we sought to identify biological processes that could be relevant mechanisms explaining these functional endpoints. Using publicly available quantitative proteomic data for melanoma cell lines within the Cancer Cell Line Encyclopedia (CCLE),30 and classifying the profiled cell lines based on high vs. low MITF levels (Table S2), we could discern a significant enrichment (adj. p < 0.05) of lysosome-associated processes (Figures S3A and S3B). Based on this observation, we found the expression of the lysosomal proteins LAMP1, LAMP2, and cathepsin B (CTSB) was downregulated in our panel of MITFlow cells (Figure 2A). Correspondingly, MITF deletion resulted in a strong downregulation of these lysosomal proteins in A375P and G361 cells (Figure 2B), accompanied by decreased fluorescence from the acidotropic LysoTracker dye (Figure S3C). Conversely, overexpression of MITF in A375 and K029A-Res cells upregulated the expression of LAMP1, LAMP2, and CTSB (Figure S3D), suggesting a role for MITF in regulating lysosomal abundance and function.
Figure 2. Low levels of MITF are associated with reduced lysosome abundance and increased sensitivity to ferroptosis in vitro and in vivo.
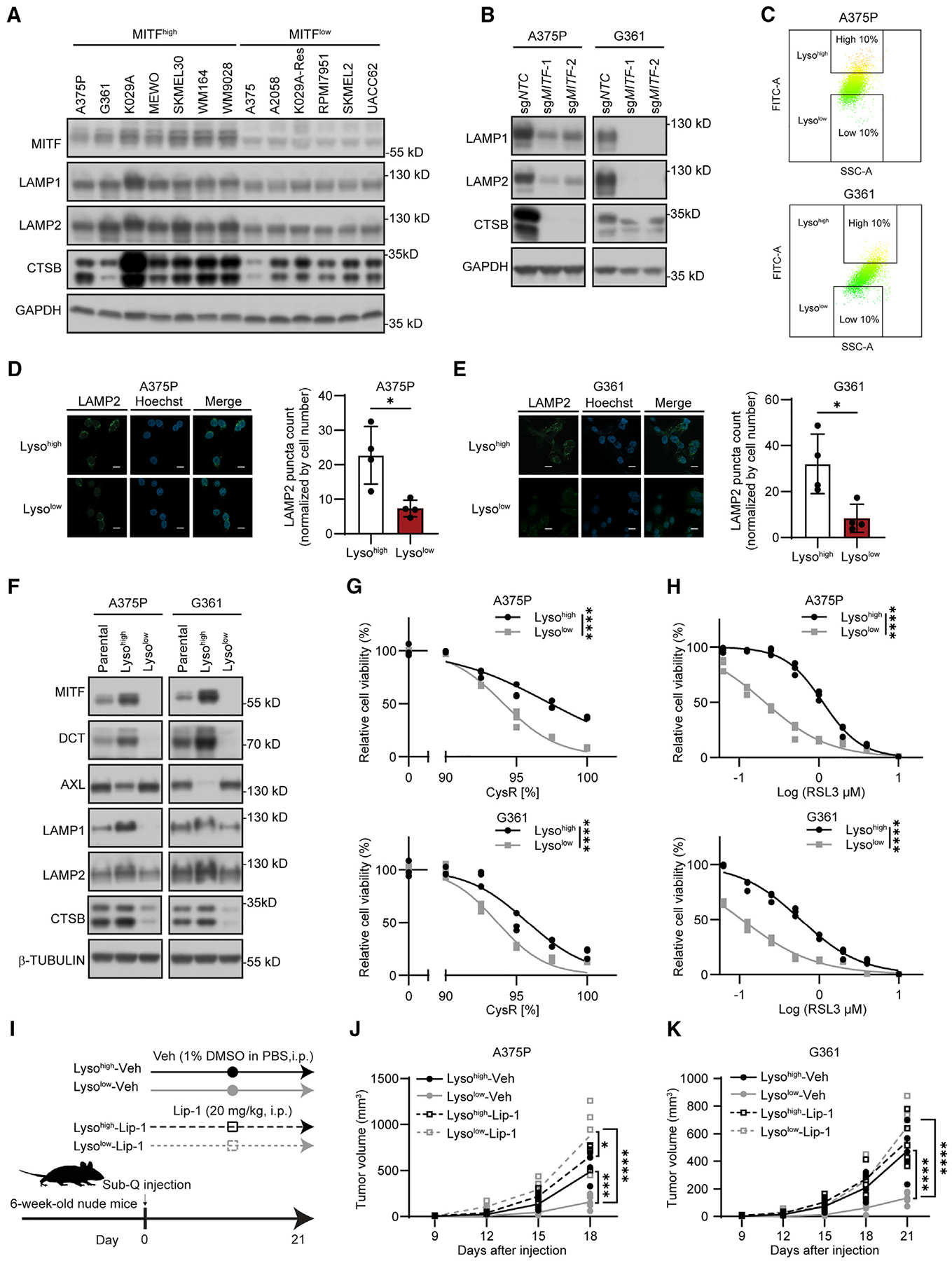
(A and B) Immunoblot analysis of the indicated proteins across (A) a panel of MITFhigh and MITFlow melanoma cell lines, and (B) MITF-deleted A375P and G361 cells.
(C) Scatterplots (SSC-A vs. FITC-A) of LysoTracker-green stained cells by FACS sorting to separate A375P (top) and G361 (bottom) cells into Lysohigh and Lysolow populations.
(D and E) Representative fluorescent images and quantifications of immuno-stained LAMP2 in A375P (D) and G361 (E) Lysohigh and Lysolow cells. Scale bars, 20 μm.
(F) Immunoblot analysis of the indicated proteins across Lysohigh and Lysolow populations of A375P and G361 cells, respectively.
(G and H) Differential sensitivity of Lysohigh and Lysolow populations of A375P and G361 to (G) CysR and (H) ferroptosis induction using RSL3.
(I) Schematic of xenograft tumor treatment regimen (vehicle, veh), 1% DMSO in PBS, compared with liproxstatin1 (Lip-1, 20 mg/kg) to evaluate effects on Lysohigh and Lysolow on tumor growth in nude (FoxN1nu) mice (n = 5).
(J and K) Resulting tumor growth measures (DxDxL/2) for Lysohigh and Lysolow populations of (J) A375P and (K) G361 for 3 weeks daily i.p. treatment with vehicle (Veh) vs. Lip-1.
Data shown as mean ± SD. Statistical significance was calculated using unpaired two-tailed t test (D and E) or two-way ANOVA (G, H, J, and K). *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
See also Figures S3 and S4.
The MITF family member TFEB is considered the master regulator of lysosomal biogenesis, whose nuclear localization is regulated by phosphorylation by mTORC1.31 Therefore, we used CRISPR-Cas9 to delete TFEB in MITFhigh A375P and G361 cells. Ablation of TFEB did not consistently affect the expression levels of LAMP2, MITF, DCT, or AXL (Figure S3E), nor did it change the sensitivity to cysteine restriction or RSL3 treatment (Figures S3F and S3G). These data support a dominant role for melanocyte-restricted MITF in maintaining lysosomal function, which is in consistent with previous reports that MITF drives endolysosomal biogenesis.32,33
Given the insight that MITF overexpression was sufficient to increase levels of lysosomal proteins, and that melanoma phenotype switching involves MITF downregulation,8 we wanted to assess whether there is evidence of intracellular lysosomal heterogeneity within populations of melanoma cells. To this end, we used the LysoTracker dye to label each of the MITFhigh A375P and G361 cell lines and used fluorescence-activated cell sorting to segregate them based on the high vs. low fluorescence intensity (Figure 2C). We denoted cells with high and low LysoTracker intensity cells as Lysohigh and Lysolow cells, respectively. Immunostaining using a LAMP2 as a specific lysosome marker validated significantly higher lysosome abundance in Lysohigh cells (Figures 2D and 2E). Interestingly, Lysohigh cells exhibited higher than average levels of MITF, DCT, LAMP1, LAMP2, and CTSB, and lower AXL, whereas Lysolow cells had markedly lower levels of these melanocytic and lysosomal proteins (Figure 2F). The expression levels of organelle markers for nucleus and ER were comparable between Lysohigh and Lysolow cells, while mitochondrial pyruvate carboxylase showed a slightly lower expression in Lysolow cells, in accordance with lower PGC-1α level in Lysolow cells (Figure S3H). Given this observation, we compared the sensitivity with cysteine restriction, RSL3 treatment, and resulting lipid peroxidation and found that the Lysolow cells had gained vulnerability to each of these endpoints (Figures 2G, 2H, and S3I). Supporting MITF’s key role in differentiating this phenotype, low MITF cells sorted with LysoTracker did not show any difference in expression of lysosome markers or sensitivity to cysteine restriction or RSL3 treatment (Figures S4A–S4D). Since ferroptosis is considered a barrier to the growth of tumors,34 we implanted each of these Lysohigh and Lysolow A375P and G361 cell lines subcutaneously in immunocompromised (nude) mice and treated with the ferroptosis inhibitor (Lipoxstatin-1, Lip-1) or vehicle (Veh) (Figure 2I). Ferroptosis inhibitor treatment (Lip-1) rescued subcutaneous tumor growth of Lysolow cells in both cell lines, while Lysohigh cells that readily formed tumors were largely unaffected (Figures 2J, 2K, S4E, and S4F). These results collectively indicate that melanoma cells display intracellular lysosome heterogeneity that associates with MITF expression levels, and that Lysolow cells are compromised for subcutaneous tumor growth in a manner that can be rescued by ferroptosis inhibition.
Overexpression of MITF and TFEB restores lysosomal functions to maintain glutathione homeostasis and ferroptosis resistance in Lysolow cells
To understand how lysosomal function maintains glutathione sufficiency and ferroptosis resistance, we performed metabolomic profiling of Lysohigh and Lysolow cells cultured in normal medium or in cystine-restricted medium (Figure S5A). In response to 16 h of cysteine restriction, cysteine and glutathione metabolism was found to be significantly affected processes across Lysohigh cells and Lysolow cells (Figure S5B). While cysteine-related metabolites and glutathione were generally downregulated by cysteine restriction, they were lower at baseline in Lysolow cells (Figure S5B). Given these results, we assessed total glutathione, as well as the ratio of reduced glutathione to oxidized glutathione (GSH/GSSG), in response to cysteine restriction. Not surprisingly, we found that total glutathione levels along with GSH/GSSG ratios were significantly decreased in Lysolow cells at baseline, and cysteine restriction reduced these measures across both Lysohigh and Lysolow states (Figures S5C and S5D).
To determine the role of lysosomes in this context, we overexpressed a Lyso-tag construct (3×HA-tagged Tmem192)35 in G361 Lysohigh and Lysolow cells and subsequently proceeded to isolate the now-tagged lysosomes for metabolite profiling, either from cells grown in normal or cystine-restrictive media (Figure S5E). Organelle analyses by western blot showed a predominant enrichment of lysosome proteins and absence of nuclear, ER, and mitochondrial protein markers, highlighting the specificity of this approach toward lysosomes (Figure S5E). At the metabolite level, lysosomal glutathione levels were significantly lower in Lysolow cells under both conditions compared with Lysohigh cells (Figure 3A). Furthermore, while cysteine restriction significantly altered the levels of many metabolites including amino acids (likely derived from lysosomal proteolysis) in Lysohigh cells, these alterations were noticeably attenuated in Lysolow cells following cysteine restriction (Figure 3A), indicating an impairment in lysosomal function in response to cysteine restriction in Lysolow cells. Next, given that proteins are proteolytically degraded in lysosomes via the autophagy-lysosome and/or the endosome-lysosome pathways, we assessed if Lysolow cells displayed markers of impaired lysosomal function during cysteine restriction and how the lysomotrophic and autophagy-inhibitory agent chloroquine (CQ)36 affected these measures. We observed that cysteine restriction moderately increased the autophagy cargo markers p62/SQSTM1 and lipidated LC3B levels in Lysohigh cells and CQ treatment augmented this effect (Figure 3B). In Lysolow cells, however, a negligible increase in p62/SQSTM1 and lipidated LC3B was detected during cysteine-restrictive growth, although it could be induced by CQ treatment (Figure 3B). Given that the autophagy lysosomal pathway is closely related to ferroptosis,37 these observations indicate that Lysohigh cells’ higher lysosomal activity could be a key mechanism to sustain survival in response to ferroptosis. To test this, we used CQ and bafilomycin A1 (Baf-A1) to inhibit the autophagy-lysosomal pathway in both Lysohigh and Lysolow cells. We subjected the cells to cysteine restriction and RSL3 treatment and found that both CQ and Baf-A1 further exacerbated cell death in Lysohigh cells (Figure S5F). Thus, Lysohigh cells are more competent to increase lysosome function to sustain viability in response to ferroptotic stresses. Because Lysolow cells exhibited lower lysosomal proteins LAMP1 and LAMP2, decreased levels of lysosome-derived amino acids, we surmised that these Lysolow cells with reduced MITF expression exhibited compromised lysosomal functions, leading to an inability to circumvent ferroptotic stress.
Figure 3. MITF or TFEB overexpression restores lysosome functions and hallmarks of autophagy in response to cysteine restriction and rescues glutathione levels.
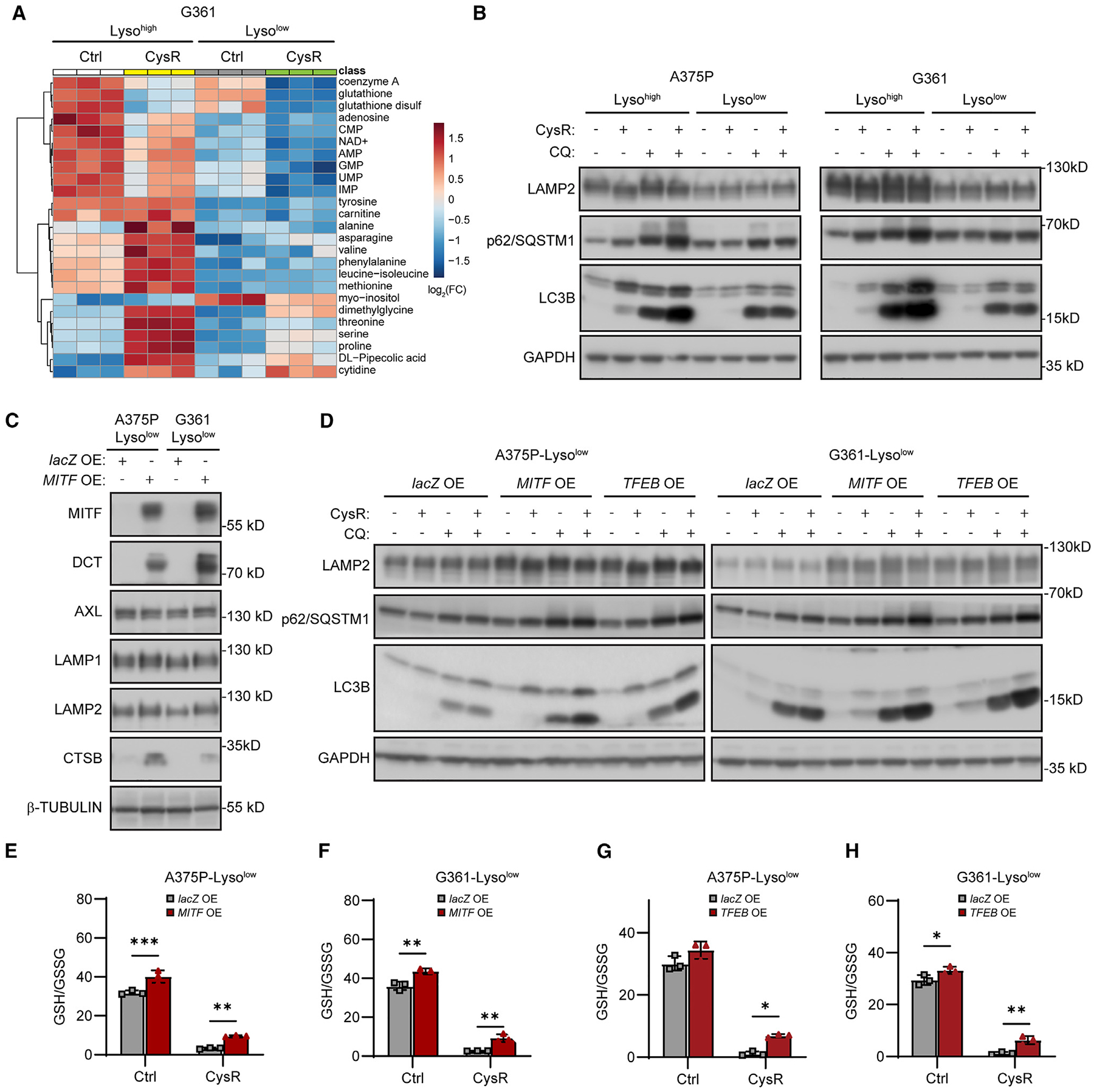
(A) Heatmap of the top 25 altered lysosomal metabolites between G361 Lysohigh and Lysolow in response to 16 h cysteine restriction (CysR). Lyso-IP experiment performed in triplicate followed by metabolite extraction, and metabolite level changes compared in pairs Lysohigh Ctrl vs. Lysohigh CysR, and Lysolow Ctrl vs. Lysolow CysR.
(B) Immunoblot analysis of LAMP2, p62/SQSTM1, and LC3B in the indicated Lysohigh and Lysolow cells in response to 16 h CysR and 100 μM chloroquine (CQ) treatment.
(C) Immunoblot analysis of the indicated proteins in Lysolow A375P and G361 cells overexpressing lacZ or MITF.
(D) Immunoblot analysis of LAMP2, p62/SQSTM1 and LC3B levels in Lysolow cells overexpressing lacZ, MITF, or TFEB in response to 16 h CysR or treatment with 100 μM CQ.
(E–H) Effects of CysR for 16 h on GSH/GSSG ratios within (E) A375P and (F) G361 Lysolow cells overexpressing lacZ or MITF, and (G) A375P and (H) G361 overexpressing lacZ or TFEB.
Data shown as mean ± SD with statistical significance based on unpaired two-tailed t test. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
See also Figures S5 and S6.
Because Lysolow cells displayed these features of compromised lysosomal functions, we examined whether MITF and/or TFEB could rescue these processes, given their largely overlapping roles as regulators of lysosomal biogenesis and functions.33 While only overexpression of MITF induced DCT levels as expected (Figures 3C and S5G), overexpression of either MITF or TFEB in Lysolow cells increased the lysosomal proteins LAMP1 and LAMP2 and upregulated lysosome abundance measured by LysoTracker intensity (Figures 3C and S5G–S5I). Importantly, each of the MITF and TFEB overexpressed Lysolow cell line could restore the induction of lipidated LC3B after cysteine restriction and augment the effects of CQ treatment on p62/SQSTM1 levels (Figure 3D). In addition, there was a significant increase in total glutathione levels (Figures S5J and S5K) and an increase in GSH/GSSG ratios at baseline and robust improvement during cysteine restriction (Figures 3E–3H). Furthermore, Lysolow cells overexpressing MITF or TFEB were markedly less susceptible to lipid peroxidation and reduced viability in response to cysteine restriction or treatment with RSL3 (Figures S6A–S6F). Collectively, these findings indicate that each of MITF and TFEB could increase lysosomal functions in Lysolow melanoma cells that served to maintain glutathione homeostasis, oxidative stress scavenging ability, and to protect against ferroptosis.
Cysteine restriction downregulates MITF by decreasing acetyl-CoA for p300-mediated H3K27 acetylation at the melanocyte-restricted MITF promoter region
Since cells with higher MITF levels were less sensitive to cysteine restriction, we decided to investigate whether cysteine could affect MITF levels by acting as a feedforward regulatory loop. In MITFhigh A375P and G361 melanoma cells, cysteine restriction progressively decreased MITF and DCT levels, while levels of AXL increased (Figure 4A), suggesting that adequate cysteine availability is required to maintain MITF expression. Next, we decided to explore whether this effect was due to altered protein stability. To this end, we examined whether proteasome inhibition (MG132), or autophagy inhibition (CQ), could prevent the downregulation of MITF, and observed that these inhibitors did not affect MITF levels (Figure S7A). However, medium supplementation with NAC or GSH did rescue the effects on MITF following cysteine restriction (Figure S7B), indicating that cysteine availability, and its bioproduct glutathione, suffice to rescue the effects of cysteine restriction on MITF levels. To determine whether there were specific transcriptional effects of cysteine restriction that could explain the protein level alterations, we performed quantitative PCR (qPCR) of MITF and DCT, as well as AXL. While MITF and DCT were reduced in RNA levels, and AXL increased (Figure S7C), none of the genuine upstream regulators of MITF, including LEF1, SOX10, or PAX3,4 was consistently decreased (Figure S7D), suggesting that the effects of cysteine restriction could be affecting MITF transcription directly. Importantly, ectopically expressed MITF in low-MITF cells did not significantly decrease upon cysteine restriction (Figure S7E), further supporting that cysteine restriction-induced MITF decrease is more likely due to transcriptional suppression.
Figure 4. Cysteine limits CoA and available Ac-CoA that affects p300-mediated H3K27ac marks and resulting MITF levels.
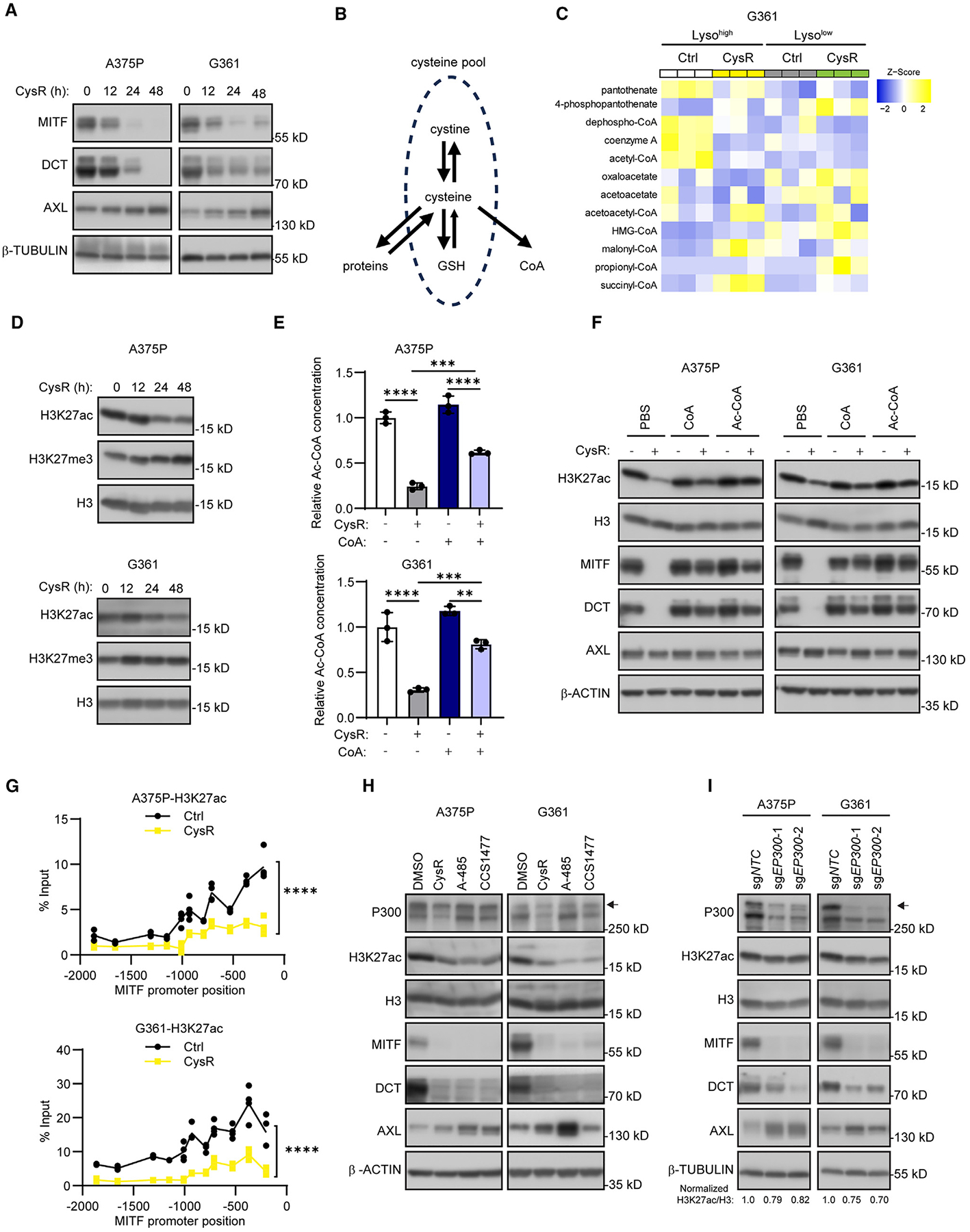
(A) Immunoblot analysis of indicated differentiation markers in A375P and G361 cells as a time course of cysteine restriction (CysR).
(B) Schematic view of the cellular sources and uses of cysteine, including protein, GSH, and CoA biosynthesis.
(C) Heatmap of CoA and acetyl-CoA metabolite level changes in Lysohigh and Lysolow G361 cells after 16 h CysR for experiment performed in triplicate with resulting changes as Z scores.
(D) Immunoblot analysis of alterations in histone H3 lysine K27 (H3K27) histone marks in A375P and G361 cells as a time course of CysR.
(E) Relative acetyl-CoA levels in A375P (top) and G361 (bottom) cells subjected to 24 h CysR and cotreated with 1 mM CoA.
(F) Immunoblot analysis of indicated proteins in A375P (top) and G361 (bottom) in A375P and G361 cells in response to 48 h CysR and co-treatment with 1 mM coenzyme A (CoA) and 1 mM of acetyl-CoA (Ac-CoA).
(G) ChIP-qPCR analysis of H3K27ac occupancy across the proximal melanocyte-restricted MITF promoter region (0 position indicates transcription start) in A375P (top) and G361 (bottom) following 48 h CysR.
(H) Immunoblot analysis of the indicated proteins in A375P and G361 cells in response to 48 h CysR, A-485 at 5 μM, or CCS1477 at 5 μM treatment.
(I) Immunoblot analysis of the indicated proteins and quantification of H3K27ac/H3 ratio in EP300-deleted A375P and G361 cells.
Data shown as mean ± SD. Statistical significance was calculated based on one-way ANOVA (E) and two-way ANOVA (G). *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
See also Figures S7 and S8.
Because cysteine is a conditionally essential amino acid that is involved in many biosynthetic pathways, including proteins and the biomolecules GSH and CoA (Figure 4B),38 we analyzed metabolites related to CoA metabolism from whole-cell metabolite profiling of Lysohigh and Lysolow cells during cysteine restriction and found that levels of CoA and acetyl-CoA (Ac-CoA) in Lysolow cells were significantly decreased compared with Lysohigh cells (Figure 4C). Cellular cysteine is indispensable for CoA synthesis starting from pantothenate,22,39 and as a central energy carrier, Ac-CoA fuels both anabolic and catabolic pathways in cells, as well as modifying transcription factor functions and chromatin structure directly to facilitate transcription.40,41 Given that we observed that MITF RNA levels were reduced upon cysteine restriction (Figure S7C), and that Ac-CoA enables histone acetylation, where histone H3 lysine 27 acetylation (H3K27ac) is a histone mark associated with an open transcriptionally active chromatin structure,42 we hypothesized that reduced Ac-CoA levels may explain MITF downregulation. To this end, we used cysteine restriction in a time course to monitor its effects on H3K27ac, compared with the competing H3 lysine 27 tri-methylation (H3K27me3) mark in A375P and G361 cells. Importantly, while H3K27me3 marks were seemingly upregulated by cysteine restriction (Figure 4D), the gradual decrease in global H3K27ac marks during cysteine restriction largely paralleled a decrease in MITF and DCT levels with that of a corresponding increase in AXL (Figure 4A). This observation prompted us to explore whether supplementation of CoA and Ac-CoA could rescue the effects of cysteine restriction on the differentiation markers MITF and DCT. To this end, we supplemented the medium with exogenous CoA, which, as 4′-phosphopantetheine is taken up by cells and is used by the CoA biosynthesis pathway.43 Remarkably, while cysteine restriction markedly attenuated Ac-CoA levels, CoA supplementation significantly increased Ac-CoA production in cysteine-restricted cells (Figure 4E). Furthermore, supplementation of CoA or Ac-CoA robustly rescued the effects on H3K27ac, MITF, and DCT in both A375P and G361 cells (Figure 4F). These results clearly indicate that reduction of Ac-CoA levels mediates the effects of cysteine restriction through altering MITF levels in melanoma cells.
Since both decreased H3K27ac and increased H3K27me3 reduce chromatin accessibility and cause epigenetic suppression of gene transcription,42,44 we performed chromatin immunoprecipitation for H3K27ac and H3K27me3 occupancy during normal and cystine restrictive conditions. We did not detect any changes in H3K27me3 occupancy, but cysteine restriction robustly diminished H3K27ac occupancy within the MITF promoter region (Figures 4G and S7F). To further validate this observation, we inhibited H3K27me3 through depletion of EZH2, the major enzyme responsible for H3K27me3, and found that loss of H3K27me3 did not affect differentiation markers (Figure S7G). Because p300/CBP are enzymes catalyzing acetylation of H3K27, we chose to examine the effects of the p300/CBP enzymatic inhibitors A-48545 and CCS147746 on expression of MITF, DCT, and AXL. Importantly, A-485 and CCS1477 treatment phenocopied the effects of cysteine restriction (Figure 4H), and while CRISPR-Cas9-mediated deletion of CREBBP (encoding CBP) did not recapitulate these effects (Figure S7H), deletion of EP300 (encoding p300) led to strong downregulation of MITF and DCT, accompanied by upregulation of AXL at both protein and RNA levels (Figures 4I and S8A). As control for these experiments, NAC supplementation did not affect the A-485 treatment effects on MITF, DCT, and AXL, suggesting that cysteine availability is upstream of p300 activity (Figures S8B–S8D). Prompted by the observation that thiol availability is key to cells’ antioxidant defenses, we furthermore tested if other oxidative stressors could also cause downregulation of MITF and DCT, with upregulation of AXL. Similar to the effects of cysteine restriction, RSL3, iron salophene complex, non-ferroptotic stressor piperlongumine, and tert-butyl hydroperoxide, all led to downregulation of MITF, DCT, and H3K27ac levels, as well as upregulation of AXL, effects that could be reversed by NAC supplementation (Figure S8E). Taken together, these findings clearly suggest that the sulfur amino acid cysteine is conditionally essential to ensure adequate levels of acetyl-CoA for p300-dependent transcriptional activation of the MITF gene, thereby maintaining the melanoma differentiation state.
Lysosomal cysteine maintains MITF to protect against ferroptosis
Since cysteine availability modulates MITF levels, which in turn control lysosome functions33 (Figures 3C–3F and S4A–S4D), an organelle responsible for providing reduced cysteine from degradation of proteins20 through the activity of the lysosomal cystine transporter (CTNS),19,24 we wanted to examine the relationship between these endpoints. To this end, we subjected A375P and G361 melanoma cells to cysteine restriction in a time course experiment and found a concomitant increase in NRF2 levels (Figure 5A) in parallel with LC3B lipidation, indicating an oxidative stress response. Furthermore, both SLC7A11 and CTNS were upregulated by cysteine restriction. Expression correlation analysis using a CCLE melanoma dataset showed a clear correlation between CTNS and MITF expression (Figure S9A), which was further corroborated by lower CTNS protein level in MITFlow melanoma cells (Figure S9B). Furthermore, modulation of MITF also led to a corresponding change of CTNS (Figures S9C and S9D). Similarly, CTNS expression levels were lower in Lysolow cells compared with Lysohigh cells, and overexpression of either MITF or TFEB in these Lysolow cells upregulated CTNS expression (Figures 5B, 5C, and S9E). Because CTNS mediates lysosomal cystine export,47 this prompted us to investigate if CTNS mediates ferroptosis sensitivity. Surprisingly, CRISPR-Cas9-mediated knockout of CTNS in A375P and G361 consistently reduced expression of MITF and DCT, and concomitantly increased AXL levels (Figure 5D). Accordingly, these CTNS-deleted melanoma cells had significantly lower total glutathione levels, and a substantial decrease in the GSH/GSSG ratio at baseline and in response to cysteine restriction (Figures 5E and S9F). Furthermore, and similar to MITF-deleted cells, CTNS deletion increased lipid peroxidation and ferroptosis sensitivity (Figures S9G–S9I). Collectively, these results suggest that lysosome-derived cysteine controls MITF levels and sensitivity to ferroptosis.
Figure 5. Compromised cysteine homeostasis suppresses melanoma differentiation and attenuates oxidative stress scavenging.
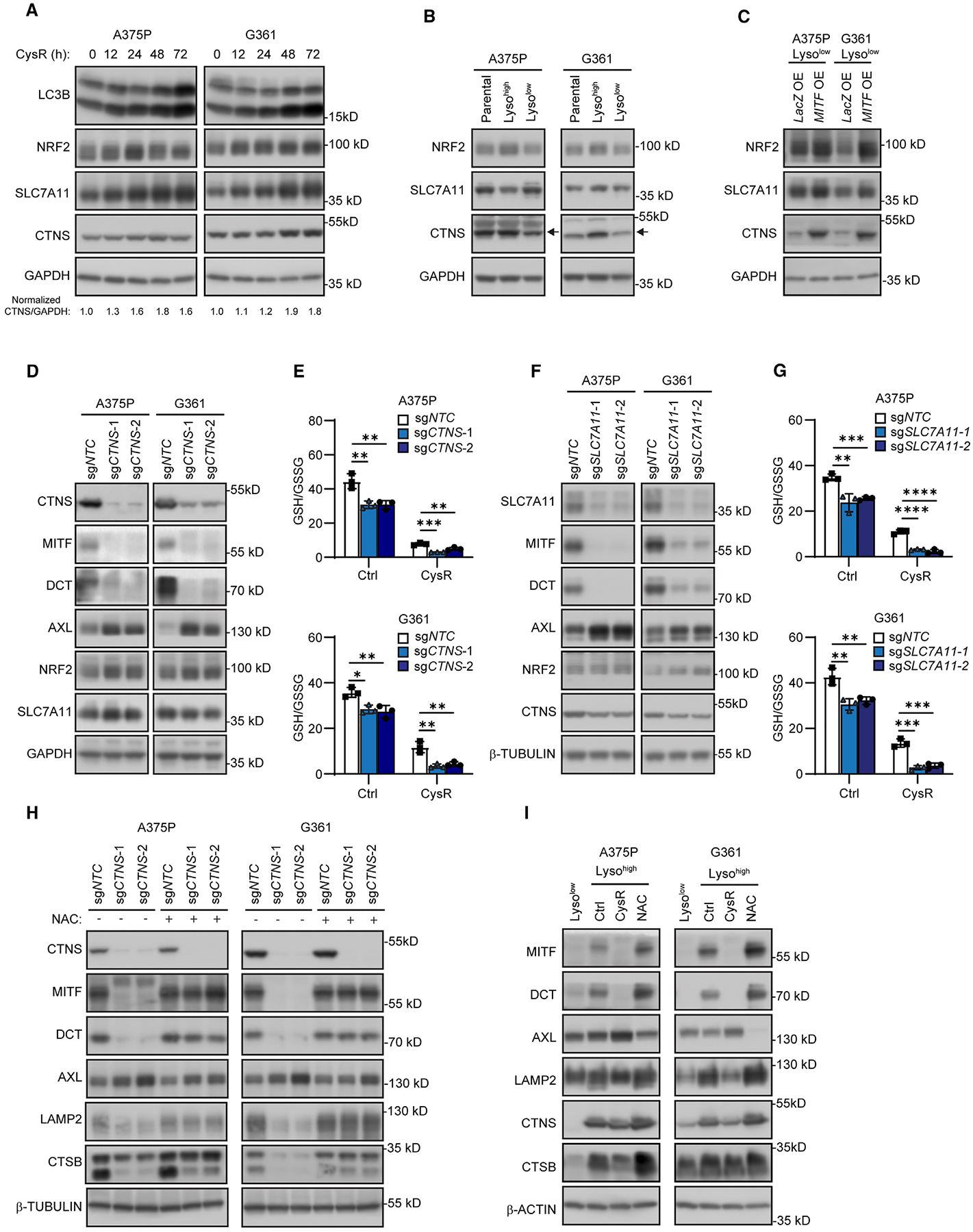
(A) Immunoblot analysis of the indicated proteins and quantification of CTNS/GAPDH ratio in A375P and G361 cells in response to a time course of cysteine restriction (CysR).
(B) Immunoblot analysis of the indicated proteins in Lysohigh and Lysolow A375P and G361 cells.
(C) Immunoblot analysis of the indicated proteins in Lysolow A375P and G361 cells overexpressing lacZ or MITF.
(D) Immunoblot analysis of the indicated proteins in CTNS-deleted A375P and G361 cells.
(E) Effects on GSH/GSSG ratios in CTNS-deleted A375P (top) and G361 (bottom) cells after 16 h CysR.
(F) Immunoblot analysis of the indicated proteins in SLC7A11-deleted A375P and G361 cells.
(G) Effects on GSH/GSSG ratios in SLC7A11-deleted A375P (top) and G361 (bottom) cells after 16 h CysR.
(H) Immunoblot analysis on the effects on the indicated proteins in A375P and G361 cells when N-acetylcysteine (NAC) (1 mM) was present during selection for genetic deletion of CTNS.
(I) Immunoblot analysis of the indicated proteins in Lysolow cells and Lysohigh A375P and G361 cells after 2 weeks culture in cystine (10 μM)-supplemented DMEM or with NAC (1 mM).
Data shown as mean ± SD with statistical significance based on one-way ANOVA. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
See also Figures S9–S11.
As aforementioned, cysteine is also derived from extracellular oxidized cystine through the plasma cystine transporter SLC7A11,48 although seemingly not regulated by MITF or TFEB, so we examined whether deletion of SLC7A11 would parallel the results. Accordingly, SLC7A11 deletion similarly caused downregulation of MITF and DCT and upregulation of AXL in A375P and G361 cells (Figure 5F). Furthermore, SLC7A11 deleted cells displayed impaired glutathione production and exhibited higher levels of lipid peroxidation and sensitivity to ferroptosis (Figures 5G and S10A–S10D).
Since NRF2 (encoded by NFE2L2) is a known upstream regulator of SLC7A11,49 we assessed whether CRISPR-Cas9 mediated NFE2L2 deletion would cause downregulation of MITF levels and downstream genes. Surprisingly, NFE2L2 deletion did not alter baseline levels of SLC7A11, yet caused MITF, DCT, and CTNS downregulation, with comparable effects on glutathione, lipid peroxides, and ferroptosis sensitivity as CTNS or SLC7A11 deletion (Figures S10E–S10G and S11A–S11C).
Given that disruption of CTNS effectively blocks lysosome-derived cysteine, we assessed whether exogenous NAC during the CRISPR-Cas9 genome-editing would affect the phenotypic outcome of the cells. Remarkably, NAC supplementation caused the CTNS-deleted cells to retain differentiation of MITF and DCT, and lysosome markers LAMP2 and CTSB, accompanied by an attenuated AXL upregulation (Figure 5H). In addition, we compared the culture of Lysohigh A375P and G361 cells during cysteine restriction (10 μM cystine; 95% reduction from 200 μM cystine in DMEM) or in the presence of 1 mM NAC for 2 weeks. Culture of Lysohigh cells in restrictive cystine medium consistently resulted in a Lysolow phenotype with decreased MITF and DCT, and elevated AXL expression, while culture in NAC even increased MITF levels within Lysohigh cells (Figure 5I). Furthermore, LysoTracker indicated that cystine-restricted Lysohigh cells displayed reduced staining, while NAC supplementation conversely increased staining intensities (Figures S11D and S11E). Consequently, these results support the conclusion that cysteine availability maintains MITF levels and markers of melanocyte/melanoma differentiation.
Low lysosomal melanoma cell population exhibits increased invasion and metastasis
Given that our data indicated that cells selected based on low LysoTracker staining consistently downregulated MITF levels, which is suggestive of phenotypic switching,8 we decided to compare global transcriptional changes between Lysohigh and Lysolow cells. To this end, we performed bulk RNA sequencing of G361 Lysohigh and Lysolow cells as quadruplet replicates. After mapping the reads and quantifying gene expression levels, we found that the differential changes were substantial and nearly affected a third of the global transcriptional repertoire. Because MITF, and a number of canonical downstream targets (DCT, TYRP1, TYR) were changed more than 2-fold, while most members of the CLEAR (coordinated lysosomal expression and regulation) gene network regulated by TFEB50 were not, we chose to focus on genes (L2F > 2, p < 0.05) and of those that were known to be bound by MITF or TFEB from chromatin enrichment analysis (ChEA database). Enrichr/GSEA51 analyses revealed that Lysohigh cells harbored signatures (adj. p < 0.05) related to melanoma and MITF (Figure 6A). Interestingly, Lysolow cells were correspondingly enriched for signatures of invadopodia formation of cancer cells and glioma/astrocytoma proteins, suggestive of migration and dedifferentiation, respectively. Because increased migration and invasion closely associate with poor clinical outcome, we used the identified invadopodia signature genes to retrospectively analyze primary melanoma patient outcomes within a publicly available high-quality dataset (GSE57715).52 Using ssGSEA53 analyses, we found that this signature correlates with heightened clinical risk (worse overall survival; Mantel-Cox log rank, *p < 0.015; HR = 1.80) (Figure 6B). Among the genes within this invadopodia signature, TGFB1 and MMP2 are known drivers and effectors of cancer cell migration and invasion. To this end, we analyzed their expression and modulation by cysteine restriction (CysR) to find that both A375P and G361 Lysolow cells had consistently higher baseline expression of TGFB1 and MMP2. However, while TGFB1 and MMP2 became upregulated in Lysohigh cells following cysteine restriction, they became downregulated in Lysolow cells (Figures 6C–6F). Although reduced expression TGFB1 and MMP2 in Lysolow cells by restricting cysteine could suggest a modality to blunt invasion driven by these genes, their induction by cysteine restriction in Lysohigh cells challenges this.
Figure 6. Melanoma dedifferentiation is associated with upregulation of TGF-β and invadopodia signatures and increased capacity for invasion and metastasis.
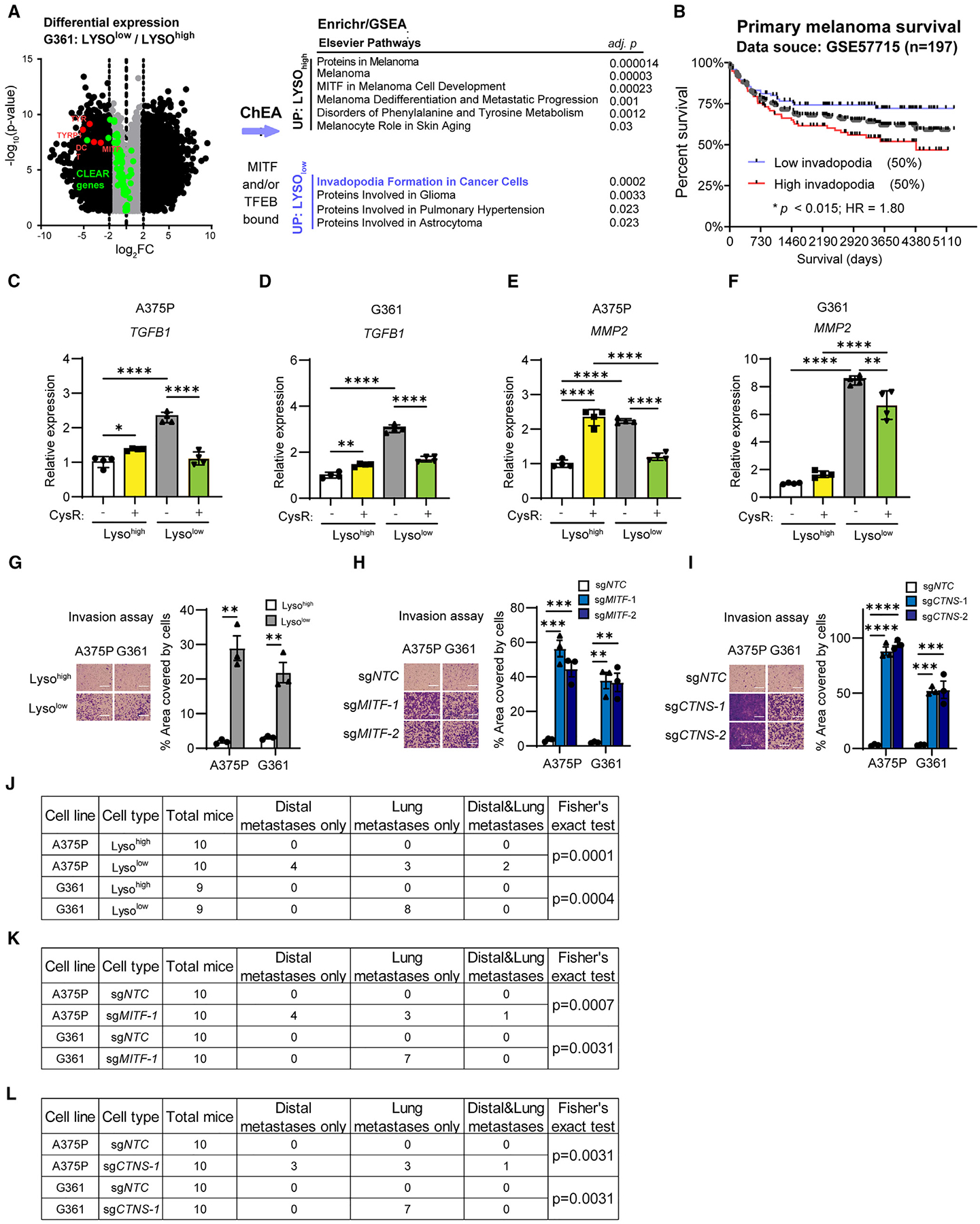
(A) Volcano plot (Log2 fold changes vs. −Log(p)) of transcripts significantly different (p < 0.05) across Lysohigh and Lysolow G361 cells and gene set enrichment analysis (GSEA) by Enrichr to compare genes between Lysohigh and Lysolow that are bound by MITF or TFEB (ChEA database) and 4-fold or more (L2F ≥ 2) differentially expressed identifies a collection of MITF signatures being increased while invadopodia formation in cancer cells and glioma/astrocytoma signatures decreased.
(B) Overall survival analysis using the identified and differentially expressed invadopodia signature genes (SH3PXD2A, MMP2, FSCN1, AFAP1, PDGFA, PRKCA, KCNN3, and EGFR) across a cohort of primary cutaneous melanoma patients (GSE57715; AJCC stages I and II) segregated by ssGSEA to define the 50% highest compared with the 50% lowest reveals statistically significant segregation of clinical risk (Mantel-Cox log rank; p < 0.013).
(C–F) Quantitative PCR assessment of TGFB1 expression in Lysohigh and Lysolow (C) A375P and (D) G361, and MMP2 expression in (E) A375P and (F) G361, each following 16 h cysteine restriction (CysR) compared with control DMEM medium.
(G–I) Representative images and quantification of transwell invasion assay of A375P and G361 Lysohigh and Lysolow (G), MITF knockout (H), and CTNS knockout
(I) cells. Experiments performed as triplicates.
(J–L) Table of metastasis incidence and statistics for A375P and G361 Lysohigh and Lysolow (J), MITF knockout (K), and CTNS knockout (L) cells.
Scale bars, 150 μm. Data shown as mean ± SD. Statistical significance was calculated based on one-way ANOVA (C–F, H, and I), unpaired two-tailed t test (G), and Fisher’s exact test (J–L). *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
See also Figure S12.
To functionally ascertain whether Lysolow cells could satisfy endpoints of metastatic spread, which could contrast their compromised subcutaneous tumor growth hindered by ferroptosis, we chose to use transwell invasion and tail vein implantation assays. Using each of A375P and G361, comparing Lysohigh to Lysolow cells, MITF-deleted (sgMITF) cells, CTNS-deleted (sgCTNS) to their respective non-targeting control (sgNTC), we consistently found that Lysolow, sgMITF, and sgCTNS cells were more migratory and invasive compared with Lysohigh or control counterparts (Figures 6G–6I and S12A–S12C). Strikingly, tail vein injection of these selected or modified melanoma cells in immunocompromised (nude) mice revealed that Lysolow, sgMITF, and sgCTNS cells displayed an increased ability for metastatic spread in vivo, which in the A375P cell model even proceeded beyond lung colonization (Figures 6J–6L and S12D–S12F).
These data, taken together, clearly suggest that compromised lysosome function, impaired cysteine homeostasis, or MITF suppression directly, promotes invasive and migratory features associated with poor clinical outcome. These processes have to be balanced to enable local tumor growth and to protect against ferroptotic cues in the microenvironment.
DISCUSSION
Melanoma is a complex and heterogeneous disease, and its inherent ability to undergo phenotypic switching has been associated with resistance to oncogene-targeted and immune checkpoint treatments, as well as facilitating metastatic spread.10 MITF is the master transcriptional regulator of differentiation- and proliferation-associated pathways and its downregulation implicitly drives this plasticity, yet the underlying mechanisms involved have been incompletely understood. Here, we provide data demonstrating that the sulfur amino acid cysteine is at the center of controlling the phenotype switching of melanomas through limiting CoA availability, which in turn supplies Ac-CoA as a substrate for p300-catalyzed H3K27ac histone marks that serve to maintain MITF transcription. MITF also coordinately regulates lysosome functions that provide proteolysis-derived cysteine, specifically by regulating the lysosomal cysteine transporter CTNS, indicating a functional cysteine feedforward regulation. It is noteworthy that the cellular cysteine pool serves to satisfy the demands for protein synthesis, glutathione, and CoA, and other thiol-containing biomolecules. Hence, a feedforward regulation of cysteine, by providing Ac-CoA to sustain a transcriptionally active H3K27ac chromatin state on gene promoters that maintain the differentiated cell state, becomes self-fulfilling for the cell. Furthermore, pigment synthesis, to which normal differentiated melanocytes are dedicated, is an oxidative process,54 and in this regard, thiol-containing antioxidants are essential for ensuring the proper operation of melanogenesis while also sustaining cell survival. Because most antioxidants primarily depend on reduced cysteine, this suggests a fundamental dependency of the differentiated melanocyte cell state on adequate cysteine availability. Disruption of cysteine homeostasis by limiting the cellular availability of cysteine (experimentally as cystine-restrictive medium, cysteine restriction, or disrupting SLC7A11), disabling lysosome derived from proteolysis (disrupting CTNS function), or increasing oxidative stress, depletes the total available pool of reduced cysteine, with metabolic and viability consequences for the cell.
Lysosomes are cellular organelles specialized in proteolytic degradation of proteins and have emerged as a key source for cysteine.19,23,24 The MiT family member TFEB is the established master regulator of lysosome biogenesis,31 whose nuclear localization is regulated by mTORC1 phosphorylation, which unlike the melanocyte and melanoma restricted M-MITF isoform that are constitutively nuclear.55 To this end, our data suggest that TFEB is not required to maintain lysosome functions in melanoma cells. However, TFEB or MITF overexpression was able to rescue compromised lysosomal functions in cells selected based on low LysoTracker staining and low MITF expression. These data are consistent with the notion that MITF, as part of driving the differentiated melanoma phenotype, regulates lysosomes, and with reports of partial overlapping functions between MITF and TFEB in maintaining lysosome functions32,33,56
To manage oxidative stress cells must be able to generate reducing equivalents, i.e., NADPH, which is produced by the oxidative branch of the pentose phosphate pathway and is used by cellular reductases such as glutathione reductase as well as thioredoxin reductase, generating oxidized NADP+ and reduced thiol.57 In the absence of adequate thiols, such as cysteine, the oxidative stress threshold is lower; similarly, excessive oxidative stress demands increased levels of cysteine. To this end, the fact that dedifferentiated melanoma cells are susceptible to ferroptosis13 is not surprising, given the antioxidant properties of cysteine and lower MITF expression associated with features of compromised lysosome functions.
While we found that cells with compromised lysosomal function were specifically limited in their ability to grow as primary (subcutaneous) tumors in vivo due to ferroptosis, these cells and intracellular cysteine-restricted variants exhibited increased avidity for metastatic spread, a feature closely associated with the phenotype switching concept of differentiated melanomas. Experimental analyses of metastatic behavior have previously indicated a promoting role for cysteine as an antioxidant that increases metastatic frequency,58 and that blood vessel but not lymph vessel distant site dissemination is limited by ferroptosis,59 suggesting that microenvironmental cysteine availability in particular may facilitate or restrict metastatic spread. Taken together with our results here and prior published work,27,29 this suggests an overarching role for oxidative stress resistance to promote cellular survival and metastasis, while switching the inherent melanoma state toward a dedifferentiated phenotype associated with reduced MITF levels, lysosomal functions, and cysteine homeostasis causes improved metastatic spread, albeit with collateral dependencies, i.e., ferroptosis.
In conclusion, we find that the melanocyte-lineage master regulator MITF drives resistance to ferroptosis by promoting lysosome functions that supply cysteine for oxidative stress scavenging, which protects against ferroptosis. When cellular cysteine becomes limited, CoA availability is reduced, and Ac-CoA becomes restricted as a substrate for p300-mediated H3K27 acetylation, which ensures MITF transcription, and thus disrupts a normally operating feedforward mechanism that sustains the differentiated melanoma phenotype and limits metastasis.
Limitations of the study
While studies have demonstrated that ferroptosis occurs naturally during tumor progression and hampers tumor growth,60,61 our data suggest that ferroptosis sensitivity constitutes a barrier to melanoma progression, but further in vivo characterization is needed. We present data showing that a tumor cell invadopodia signature that predicts poor primary melanoma outcome includes the known metastasis effectors TGF-β and MMP2, which were upregulated individually in Lysolow cells but consistently curtailed by cysteine restriction (CysR). Because these two factors were downregulated during ferroptotic stress (i.e., CysR), it certainly indicates that this may constitute a barrier to metastatic spread. However, a direct functional requirement for these factors as metastatic drivers is not provided herein, nor could we ascertain whether the molecular signature derived from the transcriptomic comparison between Lysohigh and Lysolow cells could reliably predict patient outcomes across different cancer stages. Furthermore, our focused experimental approach to assess metastatic spread was entirely based on evaluating lung and distal metastases following tail vein implantation. To this end, metastatic spread following orthotopic injection would possibly represent a more appropriate metric of disseminating disease and better recapitulate the multiple steps required for local and distant metastasis.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Pere Puigserver (pere_puigserver@dfci.harvard.edu).
Materials availability
Plasmids and cell lines generated in this study are available from the lead contact upon request.
Data and code availability
RNA seq data generated in this study was deposited in the Gene Expression Omnibus (GEO) under the accession numbers GEO: GSE269922.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this work paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND STUDY PARTICIPANT DETAILS
Animal experiments and human cancer cell lines
All animal procedures were performed in conformance with Institutional Animal Care and Use Committee (IACUC) protocol approved by the Beth Israel Deaconess Medical Center Animal Facility. Except as noted otherwise, all studies described here used 4/6-week-old male outbred homozygous nude (Foxn1nu/Foxn1nu) mice purchased from The Jackson Laboratory (Bar Harbor, ME, USA). All mice were acclimated to the Beth Israel Deaconess Medical Center Animal Facility for at least one week before beginning the studies. Mice were housed in a specific pathogen free (SPF) mouse facility with a 12:12 h light/dark cycle and with free access to food and water. Mice were euthanized with CO2 according to the Institutional Animal Care and Use Committee protocol when their body weight declined by more than 20% of the maximum or if their tumor volume exceeded 2000 mm3.
Human melanoma cell lines were obtained from ATCC and the Broad Institute of Harvard and MIT.62K029X-Braf inhibitor-resistant melanoma cell line (abbreviated as K029A-Res) was generated as previously described.29 Cell lines were authenticated by either DNA fingerprinting with small tandem repeat profiling or in-house PCR testing of melanoma marker genes and BRAF mutation status (information provided in Table S1). Cells were maintained, if not otherwise indicated, in DMEM with 10% FBS, 100 U/ml penicillin, and 100 mg/mL streptomycin. To restrict cysteine/cysteine (cysteine restriction) in vitro, cystine-deficient DMEM was prepared by adding back missing components, except cystine, to the deficient DMEM media (High Glucose DMEM w/o L-Glutamine, Methionine, Cystine, Magnesium Sulfate, D9812–06B) purchased from United States Biological (Swampscott, Massachusetts, USA). All cells were cultured in a humidified incubator at 37°C with 5% CO2.
METHOD DETAILS
Plasmid construction, lentiviral generation, and transduction
CRISPR/Cas9-mediated gene knockout was performed using the GeCKO system. Briefly. The pLentiCRISPRv2 plasmid with puromycin resistance (Addgene, 98290) was digested with the BbsI enzyme. The guide RNA oligos (provided in Table S3) were phosphorylated with T4 PNK (NEB) and then inserted into the digested pLentiCRISPRv2 plasmid. Ligation was performed using Quick Ligase (NEB). The resulting ligation reaction was used to transform Stabl3 Competent E. coli. Plasmids from each clone were sequenced by GENEWIZ. The Gateway Recombination Cloning Technology was used to generate plasmids for overexpressing MITF and TFEB. The full-length MITF or TFEB sequence was transferred from the pDONR221 vector to the pLX304 destination vector to generate lentiviral plasmids that express MITF or TFEB.
Lentiviral plasmids were transfected into HEK293T cells in six-well plates using Lipofectamine 3000 reagents following the manufacturer’s instructions. The medium was changed 24 h after transfection. The next day, medium containing the lentivirus was collected, filtered through a 0.22-μm filter, and added to the targeted cells in the presence of 8 μg/mL polybrene. At 24 h after the infection, the medium was replaced with fresh medium. The infected cells were then selected with 2 μg/mL of puromycin or 10 μg/mL blasticidin for 4 days and cultured in normal growth medium for another week prior to experiments.
Cell viability assay
Cell viability was measured using a CellTiter-Glo Luminescent Cell Viability Assay (G7572, Promega) according to the manufacturer’s protocol. In brief, cells in 100 μL growth medium were seeded onto 96-well plates at a density of 1 × 104 cells per well and compounds were added at the indicated concentrations. After the indicated treatment time, CellTiter-Glo Reagent (100 μL per well) was added and incubated at room temperature for 10 min before measuring luminescence using a FLUOstar Omega microplate reader (BMG Labtech). Viability was normalized to respective controls.
Western blotting
For immunoblotting, cells were lysed in RIPA buffer and protein concentration was quantified using a BCA Protein Assay Kit. 20–30 μg of proteins per sample were loaded to precast polyacrylamide gels (Thermo Fisher Scientific) and separated by electrophoresis. Following electrophoresis, proteins were transferred to PVDF membranes and incubated with primary antibodies at 4° overnight and then respective secondary antibodies for 1 h at room temperature. Imaging was performed using an Amersham Imager 680.
RT-qPCR
RNA was isolated using Trizol reagent. 1 μg of RNA was used to generate cDNA using the iScript Advanced cDNA Synthesis Kit following the manufacturer’s protocol. For gene expression analysis, cDNA samples were mixed with SYBR Green quantitative PCR master mix and run on an Applied Biosystems QuantStudio 6 Flex Real-Time PCR System. All qPCR primer sequences used in this study are provided in Table S4.
Total glutathione and GSH/GSSG measurements
Total glutathione levels were measured using a glutathione colorimetric detection kit (EIAGSHC; Thermo Fisher Scientific) according to the manufacturer’s instructions. GSH/GSSG ratios were determined using the GSH/GSSG-Glo Assay kit by Promega according to the manufacturer’s instructions. Briefly, following 16 h of culturing in normal media or cystine-free media, about 6 × 105 cells were detached, centrifuged, and resuspended in 1 mL of pre-warmed Hank’s Buffered Salt Solution (HBSS). Total glutathione and oxidized glutathione were then measured using the GSSG-Glo assay.
Immunostaining and confocal microscopy
Immunostaining of LAMP2 was performed as previously described.63 Briefly, cells grown on coverslips were fixed with 4% PFA in PBS at room temperature for 10 min and permeabilized with 0.1% Triton-100 in PBS at room temperature for another 10 min. Cells were then blocked with 5% BSA in PBS at room temperature for 30 min and followed by incubation with anti-LAMP2 antibody (1:200) in the blocking buffer at 4°C overnight. The next morning, cells were washed three times with PBS and incubated with Alexa Fluor 488 anti-mouse antibody for 1 h at room temperature. After washing by PBS three times, the cells were then stained with 5 μg/mL Hoechst 33342 in PBS at room temperature for 5 min and mounted with Shandon-Mount permanent mounting medium. All samples were visualized using a Zeiss LSM 880 inverted Confocal Laser Scanning Microscope and image quantification was conducted using Fiji. The LAMP2 positive puncta count was measured using the “find maxima” function built in Fiji. Quantification of puncta count was conducted by normalizing the total puncta count to the cell number in the image.
Acetyl-Coenzyme A measurements
Acetyl-Coenzyme A levels were measured according to the manufacturer’s instructions. Briefly, 1×106 cells were resuspended in 500 μL of the Assay Buffer and homogenized with a Dounce homogenizer prior to centrifugation at 10,000 g for 10 min at 4°C. The supernatant was collected and subjected to deproteination with a final concentration of 1 M perchloric acid (PCA). Following incubation on ice for 5 min and centrifugation at 13,000 g for 2 min, the subsequent supernatant was then subjected to neutralization with ice-cold 2M KOH and a further centrifugation at 13,000 g for 15 min at 4°C. The supernatant was then collected for Acetyl-CoA measurement following the manufacturer’s protocol. The final Acetyl-CoA concentration was normalized to the control sample.
Flow cytometry
To quantify lipid peroxides, 2–3 ×105 cells were seeded in 6-well plates one day prior to 16-h cystine-free media culturing or 4-h RSL3 treatment. Following treatment, the cells were incubated with 2 mM BODIPY 581/591 C11 dye for 30 min in a humidified incubator (at 37°C, 5% CO2) in the dark before trypsinization. To measure lysosome abundance, specified cells were stained with 100 nM Lysotracker green at 37°C for 30 min before trypsinization. Cells were then washed with PBS twice and subjected to flow cytometry analyses using a BD LSR Fortessa cell analyzer.
Cell sorting
A375P and G361 melanoma cells were sorted based on the labeling of LysoTracker Green (Invitrogen). Briefly, 1 ×107 of A375P or G361 cells were stained with 100 nM LysoTracker Green in growth media at 37°C for 30 min before trypsinization. Following detachment, cells were washed with PBS twice and subjected to FACS sorting at DFCI Flow Cytometry Core. The 10% of cells with the highest Lysotracker Green signal intensity and the 10% of cells with the lowest were collected as Lysohigh and Lysolow cells, respectively, and propagated for subsequent in vitro and in vivo analyses.
ChIP-qPCR
Chromatin immunoprecipitation assay was performed using Simplechip Plus Enzymatic Chromatin IP Kit (Cell Signaling Technology) as previously described.64 Briefly, 5–10 million cells treated with DMSO or 10 μM Ibrutinib for 4 h were fixed with formaldehyde for 10 min at room temperature. The cross-linking reaction was stopped by addition of 1×glycine solution for 5 min. Cells were washed twice with ice-cold PBS. Pellets were lysed and digested with micrococcal nuclease for 20 min at 37°C, and then sonicated 20 s on, 30 s off for nine cycles at 4°C. Equal amounts of precleared lysates were incubated with IgG or specific antibodies (H3K27ac and H3K27me3 from Cell Signaling Technology) overnight, followed by precipitation with protein A/G-Dynabeads for 2 h qPCR with SYBR green was performed to quantify the promoter occupancy. The ChIP-qPCR primers were provided in Table S5.
LC/MS-based whole cell metabolite profiling
LC/MS was employed to profile and quantify the polar metabolite content of both whole cell and IP samples, as previously described.65 Briefly, following 16-h cysteine restriction treatment, 10 million cells were washed with ice-cold PBS and then incubated with 4 mL of 80% methanol (cooled to −80°C) at −80°C for 20 min. Cells were then scraped on dry ice with cell scraper and transferred the cell lysate/methanol mixture to a 15-mL conical tube on dry ice. Cell lysate/methanol mixture was then centrifuged at 14,000g for 5 min at 4°C–8°C to pellet the cell debris and the metabolite-containing supernatant was then centrifuged to a new 15-mL conical tube on dry ice. The remaining cell pellet was then extracted with another 500 μL 80% (v/v) methanol (−80°C) and vortexed for 1 min at 4°C before centrifugation at 14,000g for 5 min at 4°C. The supernatant from the second extraction was transferred to that from the first extraction and the combined extract dried using a SpeedVac. Subsequent metabolomics profiling was conducted using the AB/SCIEX 5500 QTRAP triple quadrupole instrument at as described previously.65 Enrichment, pathway, and statistical analyses were carried out using the online tool MetaboAnalyst.66 Processed whole-cell metabolomics data is provided in Table S6.
Lyso-tag IP and lysosome metabolomics
Lyso-tag plasmid (pLJC5-Tmem192–3xHA) was a gift from David Sabatini (Addgene plasmid # 102930; RRID: Addgene_102930). G361 Lysohigh and Lysolow cell lines expressing Lyso-Tag were generated as described above. Following 16-h cysteine restriction, Lyso-Tag IP was conducted as previously described.35 Briefly, approximately 40 million cells were utilized for each LysoIP. Cells were washed twice with ice-cold PBS, then scraped in KPBS (136 mM KCl, 10 mM KH2PO4, pH 7.25 adjusted with KOH), and centrifuged at 1000 × g for 2 min at 4°C. 2.5% of total cells was reserved for further processing of the whole-cell fraction. The remaining cells were gently homogenized, and the homogenate was centrifuged at 1000 × g for 2 min at 4°C. The supernatant, containing cellular organelles including lysosomes, was incubated with 150 μL of prewashed anti-HA magnetic beads on a gentle rotator shaker for 3 min. Immunoprecipitates underwent three gentle washes with KPBS on a DynaMag Spin Magnet. For metabolite extraction from lysosomes, beads with bound lysosomes were resuspended in 50 μL ice-chilled metabolite extraction buffer (80% methanol, 20% water with internal standards). After a 5-min incubation on ice, beads were removed, and the metabolite extract (liquid fraction) was centrifuged at 1000 × g for 2 min at 4°C. The supernatant was collected and analyzed by LC/MS to determine the relative abundance of each metabolite. Processed lysosome metabolomics data is provided in Table S7.
Bulk RNA-seq
Samples in quadruplet for each of G361 Lysohigh and Lysolow cells were isolated using TRIzol reagent. Purified RNA was further treated with DNase I at 37°C for 10 min, followed by cleanup with phenol/chloroform extraction. RNA quality assessment (Agilent Bioanalyzer 2100), library preparation (purification and fragmentation of mRNA using oligo (dT)-attached magnetic beads, cDNA synthesis and processing, followed by amplification and further purification for PE100 libraries), and RNA sequencing (BGISEQ-500) followed by sample deconvolution were performed by the Beijing Genomics Institute (BGI, ShenZhen, China). Raw sequencing reads were mapped to GRCh38.90 using HiSat2.1and quantitated based on genes as counts per million reads (CPM) using SeqMonk v48.2.67 Raw read data were brokered through SRA and quantitated gene level matrix data is available through GEO (GSE269922). Differential gene expression across each of the two states Lysohigh and Lysolow were calculated as average Log2fold (L2F) changes with significance (p) based on unpaired two-sided students’ t-test (Table S8). Genes that satisfied at least 4-fold difference (log2F = 2) up or down, were cross referenced with ChEA data on genes bound by MITF and/or TFEB. The resulting joint genes; 167 increased in Lysolow and 366 decreased, were analyzed by Enrichr/GSEA for signature enrichment using an adjusted p-value cut-off of 0.05.
Gene expression correlation analysis
Gene expression correlation analysis in melanoma cell lines was performed using the Broad Institute DepMap website (https://depmap.org/). The expression data (Expression 23Q3) were accessed in Nov. 2023 for the analyses of expression correlation.
In vitro migration and invasion assay
Transwell chambers (29442–120) were purchased from Corning Life Science. For migration assay, 1× 105 cells in 0.1 mL serum-free medium were seeded directly into the transwell insert. For invasion assay, 50 μL of Matrigel is added to a 24-well transwell insert and solidified in a 37°C incubator for 15–30 min before adding1× 105 cells in 0.1 mL of FBS-free medium. 600 μL growth medium containing 10% FBS was used in the lower chamber as chemoattractant. Following 16-h incubation, the remaining cells on the top of the membrane were removed by a cotton swab. The migrated cells were then fixed and stained with 0.2% crystal violet solution. After drying, the membrane with migrated cells was removed from the transwell insert and placed on a glass slide for imaging using an EVOS M5000 microscope imaging system.
Cell line-derived xenograft model
Cells were detached by trypsin treatment and suspended in cold serum-free DMEM. 1 × 106 cells were injected into the right flank of mice subcutaneously. Liproxstatin-1 (Lip-1) treatment was conducted as previously described.60 Briefly, Lip-1 was first dissolved in DMSO then diluted with PBS and injected into mice at 20 mg/kg body weight daily. Liproxstatin-1 and vehicle control (1% DMSO in PBS) were initiated following cell engraftment and administered once daily by i.p. injection for 3 weeks. Tumor volumes were quantified by measuring the length (L) and width (W) of the tumor using a caliper and calculated according to V = (L*W*W)/2. Mice were sacrificed after 3 weeks. No mice exhibited severe loss of body weight (>15%) or evidence of infections or wounds during our experiments.
In vivo metastasis assay
To assess the metastatic capacity, cells grown at around 60–80% confluency were detached by trypsin and resuspended in serum-free DMEM. 1 X 106 cells in 0.1 mL DMEM were injected into 6-week-old male nude mice via the tail vein. 5–10 nude mice were used for each cell type. Mice were monitored weekly and euthanized upon observing signs of large surface tumor, labored breath, or rapid weight loss (20%) or upon resident veterinarian’s request. Upon euthanasia or notified death, mice were necropsied, and lung tissue was dissected for imaging.
QUANTIFICATION AND STATISTICAL ANALYSIS
All measurements were taken from distinct biological samples. Unpaired two-tailed Student’s t-test was used for two-group comparisons, one-way ANOVA was used for comparisons of three (3) or more groups, and two-way repeated measures ANOVA was used to compare the effects of two independent variables. Statistical significance is represented by asterisks corresponding to *p < 0.05, **p < 0.01, and ***p < 0.001. GraphPad Prism software was used to generate graphs and perform statistical analyses.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Cathepsin B | Cell Signaling Technology | Cat#31718; AB_2687580 |
| xCT/SLC7A11 | Cell Signaling Technology | Cat#12691; AB_2687474 |
| AXL | Cell Signaling Technology | Cat#8661; AB_11217435 |
| Acetyl-Histone H3 Lys27 | Cell Signaling Technology | Cat#8173; AB_10949503 |
| Tri-Methyl-Histone H3 Lys27 | Cell Signaling Technology | Cat#9733; AB_2616029 |
| Histone H3 | Cell Signaling Technology | Cat#4499; AB_10544537 |
| TFEB | Cell Signaling Technology | Cat#4240; AB_11220225 |
| EZH2 | Cell Signaling Technology | Cat#5246; AB_10694683 |
| P300 | Cell Signaling Technology | Cat#86377; AB_2800077 |
| CBP | Cell Signaling Technology | Cat#7389; AB_2616020 |
| GAPDH | Cell Signaling Technology | Cat#2118; AB_561053 |
| beta-Actin | Cell Signaling Technology | Cat#4967; AB_330288 |
| HA-Tag | Cell Signaling Technology | Cat#2367; AB_10691311 |
| Calnexin | Cell Signaling Technology | Cat#2679; AB_2228381 |
| SQSTM1/p62 | Cell Signaling Technology | Cat#5114; AB_10624872 |
| LC3B | Cell Signaling Technology | Cat#3868; AB_2137707 |
| Vinculin | Cell Signaling Technology | Cat#4650; AB_10559207 |
| beta-Tubulin | Cell Signaling Technology | Cat#2146; AB_2210545 |
| Anti-rabbit IgG, HRP-linked Antibody | Cell Signaling Technology | Cat#7074; AB_2099233 |
| Anti-mouse IgG, HRP-linked Antibody | Cell Signaling Technology | Cat#7076; AB_330924 |
| Lamin B1 | Abcam | Cat# ab133741; AB_2616597 |
| MITF antibody | Abcam | Cat# ab12039; AB_298801 |
| LAMP1 | Santa Cruz Biotechnology | Cat# SC-20011; AB_626853 |
| LAMP2 | Santa Cruz Biotechnology | Cat# SC-18822; AB_626858 |
| DCT | Santa Cruz Biotechnology | Cat# SC-74439; AB_1130818 |
| PGC-1α | Santa Cruz Biotechnology | Cat# SC-518025; AB_2890187 |
| GPX4 | Thermo Fisher Scientific | Cat# MAB5457; AB_2232542 |
| CTNS | Thermo Fisher Scientific | Cat# 13085-1-AP; AB_2230084 |
| Pyruvate Carboxylase | Thermo Fisher Scientific | Cat# PA5-50101; AB_2635554 |
| NRF2 | Thermo Fisher Scientific | Cat# PA5-27882; AB_2545358 |
| Alexa Fluor488-conjugated goat anti-mouse IgG | Thermo Fisher Scientific | Cat# A32723; AB_2633275 |
| Bacterial and virus strains | ||
| One Shot Stbl3 chemically competent E. coli | Thermo Fisher Scientific | Cat# C737303 |
| Biological samples | ||
| RNase-free DNase I | New England Biolabs | Cat# M0303 |
| T4 DNA Ligase | New England Biolabs | Cat# M0202 |
| BbsI-HF | New England Biolabs | Cat# R3539 |
| T4 Polynucleotide Kinase | New England Biolabs | Cat# M0201 |
| Gateway BP clonase II enzyme mix | Thermo Fisher Scientific | Cat# 11789020 |
| LR clonase II enzyme mix | Thermo Fisher Scientific | Cat# 11791020 |
| Chemicals, peptides, and recombinant proteins | ||
| DMEM | Thermo Fisher Scientific | Cat# 11965126 |
| Sodium pyruvate | Thermo Fisher Scientific | Cat# 11360070 |
| Fetal Bovine Serum | Germini Bio | Cat# 11360070 |
| Penicillin-Streptomycin | Thermo Fisher Scientific | Cat# 15140122 |
| Trypsin-EDTA | Thermo Fisher Scientific | Cat# 25200056 |
| Blasticidin | Thermo Fisher Scientific | Cat# R21001 |
| Puromycin Dihydrochloride | Thermo Fisher Scientific | Cat# A1113803 |
| DIMETHYL SULFOXIDE | VWR | Cat# 80058-040 |
| RSL3 | Selleckchem | Cat# S8155 |
| Liproxstatin-1 | Selleckchem | Cat# S7699 |
| MG-132 | Selleckchem | Cat# S2619 |
| A-485 | Selleckchem | Cat# S8740 |
| CCS-1477 | Selleckchem | Cat# S9667 |
| Piperlongumine (PL) | Selleckchem | Cat# S7551 |
| Chloroquine diphosphate salt | Sigma Aldrich | Cat# C6628 |
| L-Buthionine-sulfoximine (BSO) | Sigma Aldrich | Cat# B2515 |
| tret-Butyl hydroperoxide solution | Sigma Aldrich | Cat# 416665 |
| High Glucose DMEM w/o L-Glutamine, Methionine, Cystine, Magnesium Sulfate | United States Biological | Cat# D9812-06B |
| Polybrene | Santa Cruz Biotechnology | Cat# SC-134220 |
| Lipofectamine 3000 | Thermo Fisher Scientific | Cat# L3000015 |
| LysoTracker™ Green DND-26 | Thermo Fisher Scientific | Cat# L7526 |
| BODIPY 581/591 C11 | Thermo Fisher Scientific | Cat# D3861 |
| Restore PLUS Western Blot Stripping Buffer | Thermo Fisher Scientific | Cat# PI46430 |
| Pierce anti-HA magnetic bead | Thermo Fisher Scientific | Cat# 88836 |
| tret-Butyl hydroperoxide solution | Sigma Aldrich | Cat# 416665 |
| Reduced glutathione | Thermo Fisher Scientific | Cat# G4251 |
| TRIzol reagent | Thermo Fisher Scientific | Cat# 15596018 |
| RIPA buffer | Thermo Fisher Scientific | Cat# AAJ62524AE |
| N-acetyl cysteine | Thermo Fisher Scientific | Cat# A1540914 |
| Hoechst 33342 | Thermo Fisher Scientific | Cat# H3570 |
| Epredia Shandon-Moun permanent mounting medium | Thermo Fisher Scientific | Cat# 1900331 |
| Iron salophene complex (ISC) | Cayman Chemical | Cat# 28788 |
| Critical commercial assays | ||
| CellTiter-Glo® Luminescent cell viability assay | Promega | Cat# G7572 |
| GSH/GSSG-Glo™ Assay(V6611) | Promega | Cat# V6611 |
| Quick Ligation Kit | New England Biolabs | Cat# M2200 |
| MycoAlert™ Plus Mycoplasma Detection Kits | VWR | Cat# 75860-358 |
| Pierce BCA protein assay Kit | Thermo Fisher Scientific | Cat# 23225 |
| Glutathione colorimetric detection kit | Thermo Fisher Scientific | Cat# EIAGSHC |
| QIAprep Spin Miniprep Kit | Qiagen | Cat# 27106 |
| Acetyl-Coenzyme A assay kit | Sigma Aldrich | Cat# MAK039 |
| iScript Advanced cDNA Synthesis Kit | Bio-Rad Laboratories | Cat# 1708891 |
| iTaq Universal SYBR Green Supermix | Bio-Rad Laboratories | Cat# 1725122 |
| Deposited data | ||
| RNA-Seq data | This study | GEO: GSE269922 |
| Experimental models: Cell lines | ||
| A375P | ATCC | N/A |
| G361 | ATCC | N/A |
| K029A | Broad Institute | N/A |
| MEWO | ATCC | N/A |
| SKMEL30 | Broad Institute | N/A |
| WM-164 | Broad Institute | N/A |
| WM902B | Broad Institute | N/A |
| A375 | ATCC | N/A |
| A2058 | Broad Institute | N/A |
| K029A-Res | Generated in house | N/A |
| SKMEL2 | ATCC | N/A |
| RPMI7951 | ATCC | N/A |
| UACC62 | Broad Institute | N/A |
| Experimental models: Organisms/strains | ||
| outbred homozygous nude (Foxn1nu/Foxn1nu) mice | The Jackson Laboratory | 007850 |
| Oligonucleotides | ||
| Oligonucleotides for sgRNA | Table S3 | N/A |
| Oligonucleotides for qPCR | Table S4 | N/A |
| Oligonucleotides for Chip-qPCR | Table S5 | N/A |
| Recombinant DNA | ||
| pLENTI-CRISPRv2-sgNTC | This study | N/A |
| pLENTI-CRISPRv2-sgMITF-1 | This study | N/A |
| pLENTI-CRISPRv2-sgMITF-2 | This study | N/A |
| pLENTI-CRISPRv2-sgPPARGC1A-1 | This study | N/A |
| pLENTI-CRISPRv2-sgPPARGC1A-2 | This study | N/A |
| pLENTI-CRISPRv2-sgTFEB-1 | This study | N/A |
| pLENTI-CRISPRv2-sgTFEB-2 | This study | N/A |
| pLENTI-CRISPRv2-sgCTNS-1 | This study | N/A |
| pLENTI-CRISPRv2-sgCTNS-2 | This study | N/A |
| pLENTI-CRISPRv2-sgSLC7A11-1 | This study | N/A |
| pLENTI-CRISPRv2-sgSLC7A11-2 | This study | N/A |
| pLENTI-CRISPRv2-sgNFE2L2-1 | This study | N/A |
| pLENTI-CRISPRv2-sgNFE2L2-2 | This study | N/A |
| pLENTI-CRISPRv2-sgEZH2-1 | This study | N/A |
| pLENTI-CRISPRv2-sgEZH2-2 | This study | N/A |
| pLENTI-CRISPRv2-sgEP300-1 | This study | N/A |
| pLENTI-CRISPRv2-sgEP300-2 | This study | N/A |
| pLENTI-CRISPRv2-sgCBP-1 | This study | N/A |
| pLENTI-CRISPRv2-sgCBP-2 | This study | N/A |
| pLX304-MITF | This study | N/A |
| pLX304-TFEB | This study | N/A |
| Software and algorithms | ||
| Prism (v8.0e) | GraphPad | https://www.graphpad.com/ |
| FlowJo | Treestar Inc. | https://www.flowjo.com/ |
| FIJI | ImageJ NIH | https://imagej.net/software/fiji/ |
Highlights.
Melanoma cells display heterogeneity in lysosome abundance and function dependent on MITF
MITF and lysosomes regulate cysteine homeostasis and ferroptosis sensitivity in melanoma
Cysteine sustains acetyl-CoA for p300-dependent epigenetic control of MITF expression
Low lysosome melanoma cells are local tumor growth challenged but metastasis efficient
ACKNOWLEDGMENTS
We thank all members of the Puigserver Laboratory for helpful discussions and insights regarding this project. This work was supported in part by the Claudia Adams Barr Program in Cancer Research (to P.P.), Dana-Farber Cancer Institute internal funds (to P.P.), Beyond the Sun Drenched Skies philanthropic fund (to H.R.W.), NIH R01CA181217 (to P.P.), Friends of Dana-Farber award (to J.L.), CRI Fellowship CRI4166 (to J.L.), and an AACR-Merck Immunooncology Research Fellowship 22-40-68-YU (to D.Y.).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2024.114484.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- 1.Long GV, Swetter SM, Menzies AM, Gershenwald JE, and Scolyer RA (2023). Cutaneous melanoma. Lancet Lond. Engl 402, 485–502. 10.1016/S0140-6736(23)00821-8. [DOI] [PubMed] [Google Scholar]
- 2.Massi D, Mihic-Probst D, Schadendorf D, Dummer R, and Mandalà M (2020). Dedifferentiated melanomas: Morpho-phenotypic profile, genetic reprogramming and clinical implications. Cancer Treat Rev. 88, 102060. 10.1016/j.ctrv.2020.102060. [DOI] [PubMed] [Google Scholar]
- 3.Kawakami A, and Fisher DE (2017). The master role of microphthalmia-associated transcription factor in melanocyte and melanoma biology. Lab. Invest 97, 649–656. 10.1038/labinvest.2017.9. [DOI] [PubMed] [Google Scholar]
- 4.Goding CR, and Arnheiter H (2019). MITF—the first 25 years. Genes Dev. 33, 983–1007. 10.1101/gad.324657.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Garraway LA, Widlund HR, Rubin MA, Getz G, Berger AJ, Ramaswamy S, Beroukhim R, Milner DA, Granter SR, Du J, et al. (2005). Integrative genomic analyses identify MITF as a lineage survival oncogene amplified in malignant melanoma. Nature 436, 117–122. 10.1038/nature03664. [DOI] [PubMed] [Google Scholar]
- 6.Yokoyama S, Woods SL, Boyle GM, Aoude LG, MacGregor S, Zismann V, Gartside M, Cust AE, Haq R, Harland M, et al. (2011). A novel recurrent mutation in MITF predisposes to familial and sporadic melanoma. Nature 480, 99–103. 10.1038/nature10630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bertolotto C, Lesueur F, Giuliano S, Strub T, de Lichy M, Bille K, Dessen P, d’Hayer B, Mohamdi H, Remenieras A, et al. (2011). A SUMOylation-defective MITF germline mutation predisposes to melanoma and renal carcinoma. Nature 480, 94–98. 10.1038/nature10539. [DOI] [PubMed] [Google Scholar]
- 8.Hoek KS, Eichhoff OM, Schlegel NC, Döbbeling U, Kobert N, Schaerer L, Hemmi S, and Dummer R (2008). In vivo switching of human melanoma cells between proliferative and invasive states. Cancer Res. 68, 650–656. 10.1158/0008-5472.CAN-07-2491. [DOI] [PubMed] [Google Scholar]
- 9.Marusyk A, Janiszewska M, and Polyak K (2020). Intratumor Heterogeneity: The Rosetta Stone of Therapy Resistance. Cancer Cell 37, 471–484. 10.1016/j.ccell.2020.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rambow F, Marine J-C, and Goding CR (2019). Melanoma plasticity and phenotypic diversity: therapeutic barriers and opportunities. Genes Dev. 33, 1295–1318. 10.1101/gad.329771.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sensi M, Catani M, Castellano G, Nicolini G, Alciato F, Tragni G, De Santis G, Bersani I, Avanzi G, Tomassetti A, et al. (2011). Human cutaneous melanomas lacking MITF and melanocyte differentiation antigens express a functional Axl receptor kinase. J. Invest. Dermatol 131, 2448–2457. 10.1038/jid.2011.218. [DOI] [PubMed] [Google Scholar]
- 12.Tirosh I, Izar B, Prakadan SM, Wadsworth MH, Treacy D, Trombetta JJ, Rotem A, Rodman C, Lian C, Murphy G, et al. (2016). Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science 352, 189–196. 10.1126/science.aad0501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tsoi J, Robert L, Paraiso K, Galvan C, Sheu KM, Lay J, Wong DJL, Atefi M, Shirazi R, Wang X, et al. (2018). Multi-stage Differentiation Defines Melanoma Subtypes with Differential Vulnerability to Drug-Induced Iron-Dependent Oxidative Stress. Cancer Cell 33, 890–904.e5. 10.1016/j.ccell.2018.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jiang X, Stockwell BR, and Conrad M (2021). Ferroptosis: mechanisms, biology and role in disease. Nat. Rev. Mol. Cell Biol 22, 266–282. 10.1038/s41580-020-00324-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Conrad M, Kagan VE, Bayir H, Pagnussat GC, Head B, Traber MG, and Stockwell BR (2018). Regulation of lipid peroxidation and ferroptosis in diverse species. Genes Dev. 32, 602–619. 10.1101/gad.314674.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Badgley MA, Kremer DM, Maurer HC, DelGiorno KE, Lee H-J, Purohit V, Sagalovskiy IR, Ma A, Kapilian J, Firl CEM, et al. (2020). Cysteine depletion induces pancreatic tumor ferroptosis in mice. Science 368, 85–89. 10.1126/science.aaw9872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhu J, Berisa M, Schwörer S, Qin W, Cross JR, and Thompson CB (2019). Transsulfuration Activity Can Support Cell Growth upon Extracellular Cysteine Limitation. Cell Metabol. 30, 865–876.e5. 10.1016/j.cmet.2019.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dixon SJ, Patel DN, Welsch M, Skouta R, Lee ED, Hayano M, Thomas AG, Gleason CE, Tatonetti NP, Slusher BS, and Stockwell BR (2014). Pharmacological inhibition of cystine-glutamate exchange induces endoplasmic reticulum stress and ferroptosis. Elife 3, e02523. 10.7554/eLife.02523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.He L, Chen J, Deng P, Huang S, Liu P, Wang C, Huang X, Li Y, Chen B, Shi D, et al. (2023). Lysosomal cyst(e)ine storage potentiates tolerance to oxidative stress in cancer cells. Mol. Cell 83, 3502–3519.e11, 0. 10.1016/j.molcel.2023.08.032. [DOI] [PubMed] [Google Scholar]
- 20.Hecht F, and Harris IS (2023). Lysosomal cystine: an unexpected alarm bell for cysteine scarcity. Trends Cell Biol. 33, 1007–1009, 0. 10.1016/j.tcb.2023.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gao H, Bai Y, Jia Y, Zhao Y, Kang R, Tang D, and Dai E (2018). Ferroptosis is a lysosomal cell death process. Biochem. Biophys. Res. Commun 503, 1550–1556. 10.1016/j.bbrc.2018.07.078. [DOI] [PubMed] [Google Scholar]
- 22.Jouandin P, Marelja Z, Shih Y-H, Parkhitko AA, Dambowsky M, Asara JM, Nemazanyy I, Dibble CC, Simons M, and Perrimon N (2022). Lysosomal cystine mobilization shapes the response of TORC1 and tissue growth to fasting. Science 375, eabc4203. 10.1126/science.abc4203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Armenta DA, Laqtom NN, Alchemy G, Dong W, Morrow D, Poltorack CD, Nathanson DA, Abu-Remalieh M, and Dixon SJ (2022). Ferroptosis inhibition by lysosome-dependent catabolism of extracellular protein. Cell Chem. Biol 29, 1588–1600.e7. 10.1016/j.chembiol.2022.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Swanda RV, Ji Q, Wu X, Yan J, Dong L, Mao Y, Uematsu S, Dong Y, and Qian S-B (2023). Lysosomal cystine governs ferroptosis sensitivity in cancer via cysteine stress response. Mol. Cell 83, 3347–3359.e9. 10.1016/j.molcel.2023.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vazquez F, Lim J-H, Chim H, Bhalla K, Girnun G, Pierce K, Clish CB, Granter SR, Widlund HR, Spiegelman BM, and Puigserver P (2013). PGC1α expression defines a subset of human melanoma tumors with increased mitochondrial capacity and resistance to oxidative stress. Cancer Cell 23, 287–301. 10.1016/j.ccr.2012.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Haq R, Shoag J, Andreu-Perez P, Yokoyama S, Edelman H, Rowe GC, Frederick DT, Hurley AD, Nellore A, Kung AL, et al. (2013). Oncogenic BRAF regulates oxidative metabolism via PGC1α and MITF. Cancer Cell 23, 302–315. 10.1016/j.ccr.2013.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Luo C, Lim J-H, Lee Y, Granter SR, Thomas A, Vazquez F, Widlund HR, and Puigserver P (2016). A PGC1α-mediated transcriptional axis suppresses melanoma metastasis. Nature 537, 422–426. 10.1038/nature19347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Müller J, Krijgsman O, Tsoi J, Robert L, Hugo W, Song C, Kong X, Possik PA, Cornelissen-Steijger PDM, Geukes Foppen MH, et al. (2014). Low MITF/AXL ratio predicts early resistance to multiple targeted drugs in melanoma. Nat. Commun 5, 5712. 10.1038/ncomms6712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liang J, Yu D, Luo C, Bennett C, Jedrychowski M, Gygi SP, Widlund HR, and Puigserver P (2023). Epigenetic suppression of PGC1α (PPARGC1A) causes collateral sensitivity to HMGCR-inhibitors within BRAF-treatment resistant melanomas. Nat. Commun 14, 3251. 10.1038/s41467-023-38968-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nusinow DP, Szpyt J, Ghandi M, Rose CM, McDonald ER, Kalocsay M, Jané-Valbuena J, Gelfand E, Schweppe DK, Jedrychowski M, et al. (2020). Quantitative Proteomics of the Cancer Cell Line Encyclopedia. Cell 180, 387–402.e16. 10.1016/j.cell.2019.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Settembre C, Di Malta C, Polito VA, Garcia Arencibia M, Vetrini F, Erdin S, Erdin SU, Huynh T, Medina D, Colella P, et al. (2011). TFEB links autophagy to lysosomal biogenesis. Science 332, 1429–1433. 10.1126/science.1204592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ploper D, Taelman VF, Robert L, Perez BS, Titz B, Chen H-W, Graeber TG, von Euw E, Ribas A, and De Robertis EM (2015). MITF drives endolysosomal biogenesis and potentiates Wnt signaling in melanoma cells. Proc. Natl. Acad. Sci. USA 112, E420–E429. 10.1073/pnas.1424576112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Möller K, Sigurbjornsdottir S, Arnthorsson AO, Pogenberg V, Dilshat R, Fock V, Brynjolfsdottir SH, Bindesboll C, Bessadottir M, Ogmundsdottir HM, et al. (2019). MITF has a central role in regulating starvation-induced autophagy in melanoma. Sci. Rep 9, 1055. 10.1038/s41598-018-37522-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hassannia B, Vandenabeele P, and Vanden Berghe T (2019). Targeting Ferroptosis to Iron Out Cancer. Cancer Cell 35, 830–849. 10.1016/j.ccell.2019.04.002. [DOI] [PubMed] [Google Scholar]
- 35.Abu-Remaileh M, Wyant GA, Kim C, Laqtom NN, Abbasi M, Chan SH, Freinkman E, and Sabatini DM (2017). Lysosomal metabolomics reveals V-ATPase- and mTOR-dependent regulation of amino acid efflux from lysosomes. Science 358, 807–813. 10.1126/science.aan6298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mauthe M, Orhon I, Rocchi C, Zhou X, Luhr M, Hijlkema K-J, Coppes RP, Engedal N, Mari M, and Reggiori F (2018). Chloroquine inhibits autophagic flux by decreasing autophagosome-lysosome fusion. Autophagy 14, 1435–1455. 10.1080/15548627.2018.1474314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dai E, Chen X, Linkermann A, Jiang X, Kang R, Kagan VE, Bayir H, Yang WS, Garcia-Saez AJ, Ioannou MS, et al. (2024). A guideline on the molecular ecosystem regulating ferroptosis. Nat. Cell Biol 10.1038/s41556-024-01360-8. [DOI] [PubMed] [Google Scholar]
- 38.Bonifácio VDB, Pereira SA, Serpa J, and Vicente JB (2021). Cysteine metabolic circuitries: druggable targets in cancer. Br. J. Cancer 124, 862–879. 10.1038/s41416-020-01156-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dibble CC, Barritt SA, Perry GE, Lien EC, Geck RC, DuBois-Coyne SE, Bartee D, Zengeya TT, Cohen EB, Yuan M, et al. (2022). PI3K drives the de novo synthesis of coenzyme A from vitamin B5. Nature 608, 192–198. 10.1038/s41586-022-04984-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gout I (2018). Coenzyme A, protein CoAlation and redox regulation in mammalian cells. Biochem. Soc. Trans 46, 721–728. 10.1042/BST20170506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pietrocola F, Galluzzi L, Bravo-San Pedro JM, Madeo F, and Kroemer G (2015). Acetyl coenzyme A: a central metabolite and second messenger. Cell Metabol. 21, 805–821. 10.1016/j.cmet.2015.05.014. [DOI] [PubMed] [Google Scholar]
- 42.Creyghton MP, Cheng AW, Welstead GG, Kooistra T, Carey BW, Steine EJ, Hanna J, Lodato MA, Frampton GM, Sharp PA, et al. (2010). Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc. Natl. Acad. Sci. USA 107, 21931–21936. 10.1073/pnas.1016071107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Srinivasan B, Baratashvili M, van der Zwaag M, Kanon B, Colombelli C, Lambrechts RA, Schaap O, Nollen EA, Podgor sek A, Kosec G, et al. (2015). Extracellular 4’-phosphopantetheine is a source for intracellular coenzyme A synthesis. Nat. Chem. Biol 11, 784–792. 10.1038/nchembio.1906. [DOI] [PubMed] [Google Scholar]
- 44.Wiles ET, and Selker EU (2017). H3K27 methylation: a promiscuous repressive chromatin mark. Curr. Opin. Genet. Dev 43, 31–37. 10.1016/j.gde.2016.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lasko LM, Jakob CG, Edalji RP, Qiu W, Montgomery D, Digiammarino EL, Hansen TM, Risi RM, Frey R, Manaves V, et al. (2017). Discovery of a selective catalytic p300/CBP inhibitor that targets lineage-specific tumours. Nature 550, 128–132. 10.1038/nature24028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Welti J, Sharp A, Brooks N, Yuan W, McNair C, Chand SN, Pal A, Figueiredo I, Riisnaes R, Gurel B, et al. (2021). Targeting the p300/CBP Axis in Lethal Prostate Cancer. Cancer Discov. 11, 1118–1137. 10.1158/2159-8290.CD-20-0751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Adelmann CH, Traunbauer AK, Chen B, Condon KJ, Chan SH, Kunchok T, Lewis CA, and Sabatini DM (2020). MFSD12 mediates the import of cysteine into melanosomes and lysosomes. Nature 588, 699–704. 10.1038/s41586-020-2937-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Koppula P, Zhuang L, and Gan B (2021). Cystine transporter SLC7A11/xCT in cancer: ferroptosis, nutrient dependency, and cancer therapy. Protein Cell 12, 599–620. 10.1007/s13238-020-00789-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lim JKM, Delaidelli A, Minaker SW, Zhang H-F, Colovic M, Yang H, Negri GL, von Karstedt S, Lockwood WW, Schaffer P, et al. (2019). Cystine/glutamate antiporter xCT (SLC7A11) facilitates oncogenic RAS transformation by preserving intracellular redox balance. Proc. Natl. Acad. Sci. USA 116, 9433–9442. 10.1073/pnas.1821323116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Palmieri M, Impey S, Kang H, di Ronza A, Pelz C, Sardiello M, and Ballabio A (2011). Characterization of the CLEAR network reveals an integrated control of cellular clearance pathways. Hum. Mol. Genet 20, 3852–3866. 10.1093/hmg/ddr306. [DOI] [PubMed] [Google Scholar]
- 51.Chen EY, Tan CM, Kou Y, Duan Q, Wang Z, Meirelles GV, Clark NR, and Ma’ayan A (2013). Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinf. 14, 128. 10.1186/1471-2105-14-128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Staaf J, Harbst K, Lauss M, Ringnér M, Måsbäck A, Howlin J, Jirström K, Harland M, Zebary A, Palmer JM, et al. (2014). Primary melanoma tumors from CDKN2A mutation carriers do not belong to a distinct molecular subclass. J. Invest. Dermatol 134, 3000–3003. 10.1038/jid.2014.272. [DOI] [PubMed] [Google Scholar]
- 53.Barbie DA, Tamayo P, Boehm JS, Kim SY, Moody SE, Dunn IF, Schinzel AC, Sandy P, Meylan E, Scholl C, et al. (2009). Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1. Nature 462, 108–112. 10.1038/nature08460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Denat L, Kadekaro AL, Marrot L, Leachman SA, and Abdel-Malek ZA (2014). Melanocytes as instigators and victims of oxidative stress. J. Invest. Dermatol 134, 1512–1518. 10.1038/jid.2014.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Fock V, Gudmundsson SR, Gunnlaugsson HO, Stefansson JA, Ionasz V, Schepsky A, Viarigi J, Reynisson IE, Pogenberg V, Wilmanns M, et al. (2019). Subcellular localization and stability of MITF are modulated by the bHLH-Zip domain. Pigment Cell Melanoma Res. 32, 41–54. 10.1111/pcmr.12721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Perera RM, Stoykova S, Nicolay BN, Ross KN, Fitamant J, Boukhali M, Lengrand J, Deshpande V, Selig MK, Ferrone CR, et al. (2015). Transcriptional control of autophagy-lysosome function drives pancreatic cancer metabolism. Nature 524, 361–365. 10.1038/nature14587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Balsa E, Perry EA, Bennett CF, Jedrychowski M, Gygi SP, Doench JG, and Puigserver P (2020). Defective NADPH production in mitochondrial disease complex I causes inflammation and cell death. Nat. Commun 11, 2714. 10.1038/s41467-020-16423-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Piskounova E, Agathocleous M, Murphy MM, Hu Z, Huddlestun SE, Zhao Z, Leitch AM, Johnson TM, DeBerardinis RJ, and Morrison SJ (2015). Oxidative stress inhibits distant metastasis by human melanoma cells. Nature 527, 186–191. 10.1038/nature15726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ubellacker JM, Tasdogan A, Ramesh V, Shen B, Mitchell EC, Martin-Sandoval MS, Gu Z, McCormick ML, Durham AB, Spitz DR, et al. (2020). Lymph protects metastasizing melanoma cells from ferroptosis. Nature 585, 113–118. 10.1038/s41586-020-2623-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zou Y, Palte MJ, Deik AA, Li H, Eaton JK, Wang W, Tseng Y-Y, Deasy R, Kost-Alimova M, Dančík V, et al. (2019). A GPX4-dependent cancer cell state underlies the clear-cell morphology and confers sensitivity to ferroptosis. Nat. Commun 10, 1617. 10.1038/s41467-019-09277-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Li J, Liu J, Zhou Z, Wu R, Chen X, Yu C, Stockwell B, Kroemer G, Kang R, and Tang D (2023). Tumor-specific GPX4 degradation enhances ferroptosis-initiated antitumor immune response in mouse models of pancreatic cancer. Sci. Transl. Med 15, eadg3049. 10.1126/scitranslmed.adg3049. [DOI] [PubMed] [Google Scholar]
- 62.Jiang X, Zhou J, Yuen NK, Corless CL, Heinrich MC, Fletcher JA, Demetri GD, Widlund HR, Fisher DE, and Hodi FS (2008). Imatinib targeting of KIT-mutant oncoprotein in melanoma. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res 14, 7726–7732. 10.1158/1078-0432.CCR-08-1144. [DOI] [PubMed] [Google Scholar]
- 63.Yin Q, Jian Y, Xu M, Huang X, Wang N, Liu Z, Li Q, Li J, Zhou H, Xu L, et al. (2020). CDK4/6 regulate lysosome biogenesis through TFEB/TFE3. J. Cell Biol 219, e201911036. 10.1083/jcb.201911036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Li Y, Bouchlaka MN, Wolff J, Grindle KM, Lu L, Qian S, Zhong X, Pflum N, Jobin P, Kahl BS, et al. (2016). FBXO10 deficiency and BTK activation upregulate BCL2 expression in mantle cell lymphoma. Oncogene 35, 6223–6234. 10.1038/onc.2016.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yuan M, Breitkopf SB, Yang X, and Asara JM (2012). A positive/negative ion-switching, targeted mass spectrometry-based metabolomics platform for bodily fluids, cells, and fresh and fixed tissue. Nat. Protoc 7, 872–881. 10.1038/nprot.2012.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Pang Z, Zhou G, Ewald J, Chang L, Hacariz O, Basu N, and Xia J (2022). Using MetaboAnalyst 5.0 for LC-HRMS spectra processing, multiomics integration and covariate adjustment of global metabolomics data. Nat. Protoc 17, 1735–1761. 10.1038/s41596-022-00710-w. [DOI] [PubMed] [Google Scholar]
- 67.Kim D, Paggi JM, Park C, Bennett C, and Salzberg SL (2019). Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol 37, 907–915. 10.1038/s41587-019-0201-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNA seq data generated in this study was deposited in the Gene Expression Omnibus (GEO) under the accession numbers GEO: GSE269922.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this work paper is available from the lead contact upon request.