


Abstract
A new technique of PCR hot start using oligonucleotide primers with a stem–loop structure is developed here. The molecular beacon oligonucleotide structure without any chromophore addition to the ends was used. The 3′-end sequence of the primers was complementary to the target and five or six nucleotides complementary to the 3′-end were added to the 5′-end. During preparation of the reaction mixture and initial heating, the oligonucleotide has a stem–loop structure and cannot serve as an effective primer for DNA polymerase. After heating to the annealing temperature it acquires a linear structure and primer extension can begin.
INTRODUCTION
PCR hot start is used to minimize the yield of non-specific products and to increase reaction sensitivity. There are several different methods for carrying out PCR hot start. These include: (i) the manual addition of the critical reaction components at the annealing temperature (1); (ii) the preliminary separation of the reaction components with a high-melting substance (2,3); (iii) the use of DNA polymerase antibodies (4); (iv) the use of chemically-modified AmpliTaq Gold DNA polymerase (PE Biosystems); (v) the use of oligonucleotides that reversibly inhibit DNA polymerase (5); and (vi) the use of thermolabile DNA helicases (6).
In this report, we describe a new technique for PCR hot start that uses oligonucleotide primers with molecular beacon structures. Molecular beacons were introduced for real-time detection of PCR products (7). The oligonucleotide probe contains a fluorescence label at the 5′-end, a quencher chromophore at the 3′-end and has a hairpin-like structure. The loop sequence is complementary to the target sequence. The stem is formed by the annealing of complementary arm sequences 5–7 nt long on the ends of the oligonucleotide. The fluorescence label and the quencher chromophore are near to each other and the probe in this form (hairpin-like structure) has minimal fluorescence. During hybridization, a conformational change allows the formation of a linear structure that maximises fluorescence. If a mismatched nucleotide is present in the loop sequence, the hairpin-like structure becomes energetically more stable and fluorescence diminishes. Thus, the molecular beacons can be used for identifying point mutations (8). Bonnet et al. have previously discussed the use of molecular beacons as primers for enhancing specificity in PCR (9). We used the oligonucleotide primers with hairpin-like structure without any chromophore added to the ends. The 3′-end sequence was complementary to the target sequence and five or six nucleotides complementary to the 3′-end were added to the 5′-end. During preparation of the reaction mixture and initial heating the oligonucleotide maintains this hairpin-like structure and cannot serve as an effective primer for DNA polymerase. After heating to the annealing temperature it acquires a linear structure allowing hybridization with the target and only then can PCR begin.
MATERIALS AND METHODS
The oligonucleotides were synthesized via the phosphoramidite method and purified by electrophoresis in an acrylamide gel under denaturing conditions. PCR was carried out as described by Kaboev et al. (6). Two examples of PCR hot start are given.
One target was the IS6110 sequence of Mycobacterium tuberculosis (10) and the other was exon 4 of the human p53 gene (11). The forward primer (for M.tuberculosis) was complementary to the sequence between nucleotides 924 and 944 (IS6110 5′-TGGTCCTCGACGCGATCGAGC-3′) and the reverse primer to the sequence between nucleotides 1117 and 1096. Six random or complementary nucleotides to the 3′-end were added to the 5′-end (5′-GCTCCTGGCTAGTGCATTGTCATAGGAGC-3′). The sequences of primers to exon 4 of human p53 gene were as follows. The forward primer sequence (for exon 4 of the human p53 gene) was 5′-TTGCCGTCCCAAGCAATGGATG-3′ (7F) and the reverse primer sequences were (278R) 5′-GGCTGGTGCAAGTCACAGACTTGGCTG-3′ (linear) and (278RB) 5′-CCGACGTGCAAGTCACAGACTTGGCTG-3′ (hairpin-like). The template for PCR was prepared from a known number of M.tuberculosis cells or human lymphocytes by heating at 94°C in 0.2 M NaOH or ammonia for 10 min and subsequent neutralization with acetic acid.
RESULTS AND DISCUSSION
In this study we sought to establish a new method for PCR hot start. To diminish the unspecific DNA synthesis during preparation of the reaction mixture we tried to decrease the effective primer concentration by using primers with strongly pronounced secondary structure. The 5′ sequences of the oligonucleotides were complementary to the 3′ sequences. Such oligonucleotides were compared with normal, linear primers to estimate the yield and specificity of DNA fragment amplification from low copy M.tuberculosis and human genomes. Platinum Taq DNA polymerase (complexed with anti-Taq antibodies; Life Technologies) was used as a control. The results are shown in Figures 1 and 2. Primers with hairpin-like structures and platinum Taq DNA polymerase increased the yield and specifity of PCR. We obtained good results for several diagnostics for urogenital disease by this technique (data not shown). The rules for design of such primers are similar to the rules for molecular beacon design and general PCR primer design. A minimum requirement is that the stem must contain a GC-rich sequence of 5–6 nt. Thus, in this report we present the data for successful use of oligonucleotides with molecular beacon (hairpin-like) structures as primers to enhance PCR specificity and sensitivity.
Figure 1.

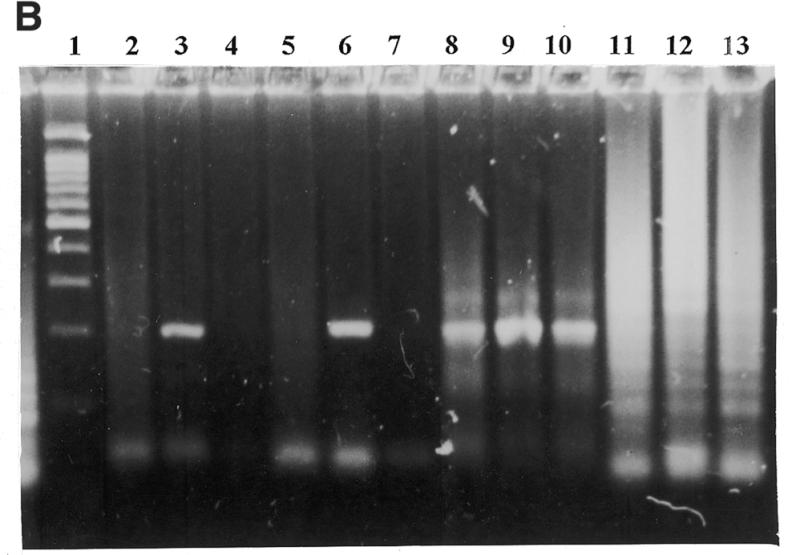
Effect of the molecular beacon primer on PCR specificity and sensitivity. (A) The target was M.tuberculosis DNA. All reaction mixtures contained 0.5 µg of human placental DNA. Gel electrophoresis in 2% agarose of the amplification products. Lane 1, DNA size marker (100 bp ladder), the prominent band is at 500 bp; lanes 2–4 and 8–10, reverse primer as molecular beacon; lanes 5–7 and 11–13, linear reverse primer; lanes 2, 5, 8 and 11, ∼1000 genome copies; lanes 3, 6, 9 and 12, 100 copies; lanes 4, 7, 10 and 13, 10 copies; lanes 2–7, platinum Taq DNA polymerase; lanes 8–13, normal Taq DNA polymerase. (B) Detection of M.tuberculosis DNA in clinical sample by the described technique. Gel electrophoresis in 2% agarose of the amplification products. Lane 1, DNA size marker (100 bp ladder); lanes 2–4 and 8–10, reverse primer as molecular beacon; lanes 5–7 and 11–13, linear reverse primer; lanes 2, 5, 8 and 11, DNA sample from sputum; lanes 3, 6, 9 and 12, ∼1000 genome copies; lanes 4, 7, 10 and 13, ∼100 genome copies; lanes 2–7, platinum Taq DNA polymerase; lanes 8–13, normal Taq DNA polymerase.
Figure 2.

Effect of the molecular beacon primer on the PCR specificity and sensitivity. The target was human DNA (exon 4 of p53 gene). Gel electrophoresis in 2% agarose of the amplification products. Lane 1, DNA size marker (100 bp ladder), the prominent band is 500 bp; lanes 2, 3 and 6, 7, reverse primer as molecular beacon; lanes 4, 5 and 8, 9, linear reverse primer; lanes 2, 4, 6 and 8, ∼1000 genome copies; lanes 3, 5, 7 and 9, ∼100 genome copies; lanes 2–5, platinum Taq DNA polymerase; lanes 8 and 9, usual Taq DNA polymerase. The platinum Taq DNA polymerase gave good amplification from 100 000+ genome copies (not shown).
REFERENCES
- 1.D’Aquila R.T., Bechtel,L.J., Videler,J.A., Eron,J.J., Gorczyca,P. and Kaplan,J.C. (1991) Nucleic Acids Res., 19, 3749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Horton R.M., Hoppe,B.L. and Conti-Tronconi,B.M. (1994) Biotechniques, 16, 42–43. [PubMed] [Google Scholar]
- 3.Bassam B.J. and Caetano-Anolles,G. (1993) Biotechniques, 14, 30–34. [PubMed] [Google Scholar]
- 4.Kellogg D.E., Rybalkin,I., Chen,S., Mukhamedova,N., Vlasik,T., Siebert,P.D. and Chenchik,A. (1994) Biotechniques, 16, 1134–1137. [PubMed] [Google Scholar]
- 5.Dang C. and Jayasena S.D., (1997) J. Mol. Biol., 264, 268–278. [DOI] [PubMed] [Google Scholar]
- 6.Kaboev O.K., Shevelev,I.V., Luchkina,L.A., Tret’iakov,A.N. and Shcherbakova,O.G. (1999) Bioorg. Khim., 25, 398–400. [PubMed] [Google Scholar]
- 7.Tyagi S. and Kramer,F.R. (1996) Nat. Biotechnol., 14, 303–306. [DOI] [PubMed] [Google Scholar]
- 8.Tyagi S., Bratu,D.P. and Kramer,F.R. (1998) Nat. Biotechnol., 16, 359–363. [DOI] [PubMed] [Google Scholar]
- 9.Bonnet G., Tyagi,S., Libchaber,A. and Kramer,F.R. (1999) Proc. Natl Acad. Sci. USA, 25, 6171–6176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Thierry D., Cave,M.D., Eisenach,K.D., Crawford,J.T., Bates,J.H., Gicquel,B. and Guesdon,J.L. (1990) Nucleic Acids Res., 18, 188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lamb P. and Crawford,L. (1986) Mol. Cell. Biol., 6, 1379–1385. [DOI] [PMC free article] [PubMed] [Google Scholar]