

Abstract
Current treatment outcome of patients with glioblastoma (GBM) remains poor. Following standard therapy, recurrence is universal with limited survival. Tumors from 173 GBM patients are analysed for somatic mutations to generate a personalized peptide vaccine targeting tumor-specific neoantigens. All patients were treated within the scope of an individual healing attempt. Among all vaccinated patients, including 70 treated prior to progression (primary) and 103 treated after progression (recurrent), the median overall survival from first diagnosis is 31.9 months (95% CI: 25.0–36.5). Adverse events are infrequent and are predominantly grade 1 or 2. A vaccine-induced immune response to at least one of the vaccinated peptides is detected in blood samples of 87 of 97 (90%) monitored patients. Vaccine-specific T-cell responses are durable in most patients. Significantly prolonged survival is observed for patients with multiple vaccine-induced T-cell responses (53 months) compared to those with no/low induced responses (27 months; P = 0.03). Altogether, our results highlight that the application of personalized neoantigen-targeting peptide vaccine is feasible and represents a promising potential treatment option for GBM patients.
Subject terms: CNS cancer, Peptide vaccines
Despite new treatment options, prognosis for patients with glioblastoma (GBM) remains poor. Here the authors report the clinical course of patients with GBM treated with a personalized neoantigen-derived peptide vaccine treated within the scope of an individual healing attempt.
Introduction
Glioblastomas (GBMs; for the designation GBM, please refer to the “Methods” section, Study design and participants) are incurable primary brain tumors and patients have a very poor prognosis. The current first-line standard of care treatment, established by the EORTC/NCIC in 2005 and consisting of radiation therapy with concurrent and adjuvant temozolomide (TMZ), leads to a median overall survival of about 15 months1. Since then, only two additional treatment approaches have been approved by the U.S. Food and Drug Administration (FDA) including bevacizumab for recurrent GBM and tumor-treating fields (TTF) for newly diagnosed and relapsed GBM2–4.
Bevacizumab increases progression-free survival (PFS) but does not increase overall survival (OS) in newly diagnosed GBM5,6. The addition of nivolumab, an anti-programmed cell death-1 (PD-1) antibody, generated no improvement in overall survival (OS) compared to standard therapy in phase 3 trials among patients with newly diagnosed GBM7,8, and no improvement in OS compared to addition of bevacizumab in patients with recurrent GBM9. Phase 3 trials evaluating vaccination against EGFRvIII with rindopepimut or against αvβ3/αvβ5 integrins with cilengitide also did not increase OS in patients with newly diagnosed GBM10,11. Despite efforts to improve treatment efficacy, GBM patients inevitably experience recurrence and OS has not improved.
Neoantigen-derived personalized peptide vaccines are uniquely tailored to individual tumors and are a promising approach for the treatment of cancer12,13. They are currently being tested in clinical trials14. A recent in-human treatment with a vaccine targeting the H3K27M neoantigen among eight patients with diffuse midline glioma showed safety and immunogenicity15. Neoantigen-derived peptide vaccines have already been applied to treat brain tumors in clinical trials16–18. The vaccines used for these clinical trials as well as our vaccines are designed based on the identification of tumor-specific somatic mutations and their translation into tumor-specific amino acid sequences (neoantigens). Computational methods are used to predict which neoantigens might be presented by HLA on the surface of tumor cells and could therefore be recognized by both cytotoxic CD8+ as well as CD4+ helper T-cells. Personalized neoantigen-derived peptide vaccines are then applied to (re)activate tumor-specific T-cell responses. The aim is to induce tumor regression and/or control by T-cell mediated targeted cytotoxicity and thereby reduce the risk of recurrence.
Here we describe the clinical course of 173 GBM patients treated with a personalized neoantigen-derived peptide vaccine within the scope of an individual healing attempt between 2015 and 2023. We report on feasibility and immunogenicity of this personalized peptide vaccine approach as well as clinical efficacy relative to historical, propensity-matched controls.
Results
Patient characteristics
One hundred and seventy-three GBM patients (IDH wildtype) received a personalized neoantigen-derived peptide vaccine between October 2015 and August 2023. Detailed patient characteristics are outlined in Table 1 and in Supplementary Table 1. Median age at the start of the peptide vaccine treatment was 54 years (range: 9–87) and 68% of patients were male. Thirty patients came from Germany, 42 patients came from other European countries, 77 from the United States, and 24 from other countries. Median time from GBM diagnosis to first vaccination was 10.3 months (range: 3–54). Seventy patients (40%) received vaccine prior to progression (primary) and 103 patients (60%) were treated after progression (recurrent). Participating patients were self-referred or were referred by their treating neuro-oncologist. There were no demographic, clinical, or treatment-related factors that excluded patients from treatment. However, all patients were required to travel to our treatment center in Germany for each vaccine dose.
Table 1.
Clinical information and diagnostic markers of the vaccinated patients
| Patients | % | |
|---|---|---|
| Total | 173 | 100% |
| Male | 117 | 68% |
| Female | 56 | 32% |
| Alive | 94 | 54% |
| Karnofsky performance status | ||
| 100 | 50 | 28% |
| 90 | 64 | 37% |
| 85 | 1 | 1% |
| 80 | 35 | 20% |
| 70 | 17 | 10% |
| 60 | 6 | 4% |
| Median age at diagnosis (years) | 53.1 | (range 9–87) |
| “7 gain combined with 10 loss” signature | 34 | 20% |
| EGFR gain of function (amplification and/or mutation) | 76 | 44% |
| TERT promoter status | ||
| Mutant | 143 | 83% |
| Wildtype | 30 | 17% |
| MGMT promoter status | ||
| Methylated | 75 | 43% |
| Unmethylated | 89 | 51% |
| Unknown | 9 | 5% |
| Therapy (during the course of the disease) | ||
| Radiation | 165 | 95% |
| Temozolomide | 167 | 97% |
| Glucocorticoids | 87 | 50% |
| Immune checkpoint inhibitor | 77 | 45% |
| Bevacizumab | 101 | 58% |
| Tumor status at personalized peptide treatment | ||
| Primary | 70 | 40% |
| Recurrent | 103 | 60% |
| Median time from diagnosis to personalized peptide vaccine treatment (months) | 10.3 | (range 3–54) |
| PD-L1 status (CP score >2) | ||
| Positive | 31 | 18% |
| Negative | 71 | 41% |
| Unknown | 70 | 40% |
| Unclear | 1 | 1% |
| Post-vaccination T-cell responses to in vitro stimulation with vaccinated peptides | ||
| Immunological responders | 77 | 45% |
| Immunological non-responders | 20 | 12% |
| Unknown (no immune monitoring) | 76 | 44% |
Source data are provided as a Source Data file.
EGFR epidermal growth factor receptor, MGMT methylguanine methyltransferase, PD-L1 programmed death ligand 1, TERT telomerase, CP combined positive score.
At data cut-off (01.08.2023), 94 of 173 patients (54%) are still alive, median observation time from initial diagnosis to date of last follow-up or death is 21.3 months (range 6–65 months, mean 23.9 months). At the time of first vaccination, 159 patients (92%) had received standard-of-care treatment with radiation therapy and TMZ chemotherapy. Three patients had received radiotherapy only, five patients had received TMZ only, and one patient was treated with combined radiotherapy and lomustine chemotherapy. Eighty-seven patients were treated with glucocorticoids including dexamethasone for which timing and dosing were not comprehensively recorded. Other therapeutic strategies were applied at the discretion of the patients’ primary treating physicians including standard-of-care agents as well as agents available on a compassionate-use, non-approved basis.
Interestingly, 31/173 (18%) patients had one or more germline variants associated with a hereditary tumor predisposition. All identified germline variants were classified as inactivating or likely inactivating in the following genes: ATM, APC, BARD1, BRCA1, BRIP1, CDKN2A/B, CHEK2, DDB2, EPCAM, ERCC2, ERCC5, FANCC, FANCE, FANCI, FANCM, MLH1, MTAP, MSH2, MSH6, MUTYH, NBN, PMS2, POLE, TP53, RECQL4, UBE2T. Characteristics of patients with germline variants are featured in Supplementary Data 1. Additionally, 7/173 (4%) patients showed a tumor mutation burden (TMB) of at least 10 Mut/Mb associated with DNA damage repair gene mutations. Of those seven patients, three were associated with a TMZ-induced hypermutation signature.
Vaccine
The median time from tumor tissue acquisition to initiation of vaccine administration was 16 weeks. Vaccine administration could commence within 12 weeks after completion of tumor genomic sequencing. In total, 2955 peptides were synthesized, formulated and vaccinated. The median number of peptides included per vaccine was 19 (mean 16.4, range 6–24). At the cut-off date, patients had received between 1 and 31 vaccinations (median 8). 115 patients (66%) received at least 7 vaccinations including 170 patients (98%) who completed all of the planned four vaccinations of the priming phase.
Immune monitoring
At least one post-vaccination blood sample for evaluation of vaccine-induced neoantigen-specific T-cell response was available for 97 of 173 patients (56%). The median time after vaccine initiation and initial immune monitoring was 97 days and it was most commonly conducted 2–3 months after the first vaccine administration. The median time between diagnosis to the first immune monitoring (at 7th vaccination) was 13.6 months.
Overall, 87 of 97 patients (88%) with available immune monitoring data exhibited post-vaccination T-cell responses against at least one vaccinated neoantigen-derived peptide, while 10 of 97 patients (10%) exhibited no T-cell response against any vaccinated neoantigen-derived peptide. The median time from vaccine initiation to first detection of vaccine-induced immune response was 116 days (range: 53–606) (Table 1).
Safety
Adverse events (AEs) attributable to vaccine therapy were nearly all grade 1 or 2 (Supplementary Table 2). Four patients experienced grade 3 AE including allergic reaction, anaphylaxis, and skin reaction. These reactions rapidly resolved with appropriate medical therapy and did not require hospitalization. One patient chose to discontinue vaccine therapy while the remaining three continued vaccine administrations. No grade 4 AEs were observed.
Treatment outcomes
The median OS in our cohort was 31.9 months (95% CI: 25.0–36.5). At the end of the observation period, 94 of 173 patients (54%) were still alive. As expected, patients starting a personalized peptide vaccine prior to progression (in the following labeled as “primary”) had a longer OS than patients with progressive disease (referred to as “recurrent”). Median OS has not been reached for primary patients as less than 50% had died at data cut-off. Median OS was 23.8 months for recurrent patients with progressive tumor (p < 0.0001, Fig. 1A). Likewise, on-treatment survival measured from initiation of vaccine was longer for primary patients compared to recurrent (median 28.9 months vs 9.8 months p < 0.0001, Fig. 1B). The overall survival rates of our cohort compare favorably to recently reported datasets19–22 and OS noted among our primary patients compares favorably to that reported among high-grade glioma patients undergoing alternative tumor-specific antigen vaccines15–18. To compare the overall survival of our cohort with public data we used propensity score matching23–25, to select closely matched GBM patients from four public datasets19–22.
Fig. 1. Survival of patients with stable or progressive disease.

OS (A) and on-treatment survival (B) of patients with either stable (primary; black) or progressive (recurrent; red) disease at the time of personalized peptide vaccine administration (P < 0.0001). Statistical significance of survival differences was computed using the log-rank test. Source data are provided as a Source Data file.
We acknowledge that our collective might be biased towards patients with longer survival due to the production time required for the personalized peptide vaccines. To mitigate this possible bias, we only included patients from the public datasets who survived longer than the median time between diagnosis and first vaccination observed in our cohort. The difference in OS between the matching patients and our cohort is highly significant (median overall survival 22.7 months vs 31.1 months, P = 0.0032, Fig. 2). We also performed multivariate Cox regression analysis on the matching cohort to estimate the effect of the peptide vaccine on survival while controlling for other variables (age at diagnosis, gender, MGMT status, TMZ, and concurrent chemoradiotherapy). In this analysis, receiving neoantigen-derived peptide vaccine was significantly associated with a lower risk of death (estimated hazard ratio of 0.65, 95% confidence interval [0.48, 0.87], P = 4.0e-03, Supplementary Table 5).
Fig. 2. OS of patients who received a personalized peptide vaccine (red) in comparison to the matching cohort (publication data, propensity score-matched, black) (P = 0.0032).
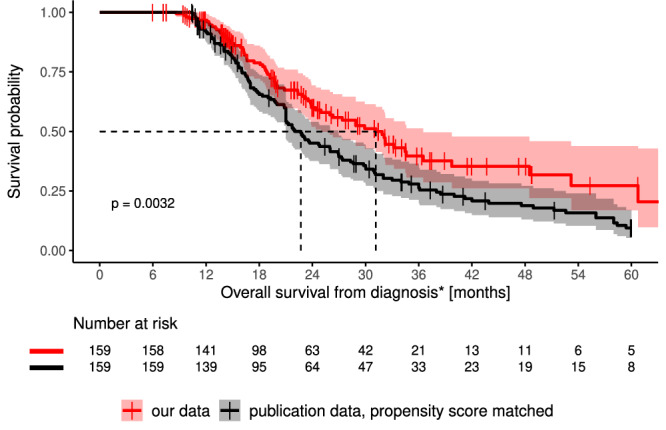
Statistical significance of survival differences was computed using the log-rank test. 95% confidence interval of cumulative hazard (log survival) is shown as shaded areas. Source data are provided as a Source Data file.
To further investigate clinical metadata, we performed Kaplan–Meier analysis and compared on-treatment survival between different subgroups of vaccinated patients by univariate analysis. The investigated parameters, the observed hazard ratios, as well as the results of the log-rank tests, can be found in Table 2. As expected, longer overall and on-treatment survival was observed for patients with methylated MGMT promoter (OS: HR 0.38 (0.23–0.62), P = 6.5e-05; on-treatment survival: HR 0.41, (0.25–0.66), P = 2e-04) and related to the Karnofsky index (OS: 0.97 (0.95–0.99), P = 0.003; on-treatment survival: HR 0.95, (0.93–0.97), P = 3.1e-07).
Table 2.
Univariate analysis of covariates with on-treatment survival
| Covariate | Hazard Ratio | 95% CI for HR | log-rank test | p value |
|---|---|---|---|---|
| Patient characteristics | ||||
| Age at first diagnosis | 1 | (1–1) | 0.21 | 0.65 |
| Sex (male) | 1 | (0.63–1.6) | 0.00059 | 0.98 |
| Karnofsky Index | 0.95 | (0.93–0.97) | 26 | **3.1e-07 |
| PD-L1 status | 1.1 | (0.56–2.1) | 0.049 | 0.82 |
| MGMT methylated | 0.41 | (0.25–0.66) | 14 | **2e-04 |
| HRD score | 1 | (0.99–1) | 0.3 | 0.58 |
| HRD high (≥30) | 1 | (0.56–1.8) | 0.00017 | 0.99 |
| TMB mut/Mb | 0.99 | (0.99–1) | 2.6 | 0.11 |
| TMB high (≥10) | 0.47 | (0.17–1.3) | 2.2 | 0.14 |
| MSI vs MSS | 0.44 | (0.11–1.8) | 1.3 | 0.25 |
| Germline variantsa | 0.8 | (0.44–1.5) | 0.51 | 0.47 |
| Treatment (during the course of the disease) | ||||
| Checkpoint Inhibitor | 0.88 | (0.57–1.4) | 0.29 | 0.59 |
| Radiation | 0.82 | (0.33–2) | 0.19 | 0.66 |
| Temozolomide | 0.53 | (0.21–1.3) | 2 | 0.16 |
| Bevacizumab | 1.9 | (1.2–3.2) | 7.2 | **0.0073 |
| Tumor treating fields | 0.61 | (0.36–1) | 3.5 | 0.06 |
| Steroids | 1.3 | (0.82–2) | 1.2 | 0.27 |
| Positive T-cell response after 7th vaccination | 0.47 | (0.24–0.94) | 4.7 | *0.03 |
Source data are provided as a Source Data file.
HRD homologous recombination deficiency, TMB tumor mutational burden, MSI microsatellite instability, MSS microsatellite stability.
P values from likelihood ratio tests. Significance: *P < 0.05, **P < 0.01.
aAssociated with cancer risk and predisposition.
No significant on-treatment survival differences were noted after stratifying patients for age at first diagnosis (95% CI, 1 to 1; P = 0.65), sex (95% CI, 0.63 to 1.6; P = 0.98), time from initial diagnosis to vaccination (95% CI, 1 to 1; P = 0.65), homologous recombination deficiency score (95% CI, 0.99–1; P = 0.58), TMB (95% CI, 0.17–1.3; P = 0.14), MSI status (95% CI, 0.11–1.8; P = 0.25), presence of a germline mutation (95% CI, 0.44–1.5; P = 0.47), application of TMZ (95% CI, 0.21–1.3; P = 0.16), tumor treating fields (TTF; 95% CI, 0.36–1; P = 0.06) or checkpoint inhibitor treatment (95% CI, 0.57–1.4; P = 0.59).
Moreover, stratifying patients by bevacizumab treatment resulted in significant differences in survival (bevacizumab therapy yes versus no; HR 1.8 (1.1–2.9), P = 0.0022). Bevacizumab treatment was associated with reduced OS (median 26.3 months [95% CI: 24–33] with bevacizumab vs. 40 months [95% CI: 32, no upper limit available] without bevacizumab) which likely reflects poorer clinical condition (Fisher test for bevacizumab treatment yes/no versus disease state primary/recurrent; p < 0.0001).
T-cell response and outcome
To deepen the analysis of the impact of neoantigen-specific T-cells, we grouped all patients with available immune monitoring data (n = 97) into immunological responders (iR; n = 77) and immunological non-responders (iNR; n = 20; for details, see “Method” section). We found that iR had a significantly higher median on-treatment survival than iNR (28.7 months versus 17.4 months; HR 0.47 (0.24–0.94); P = 0.03; Fig. 3A).
Fig. 3. Survival of immunological responders and non-responders and the matching cohort.
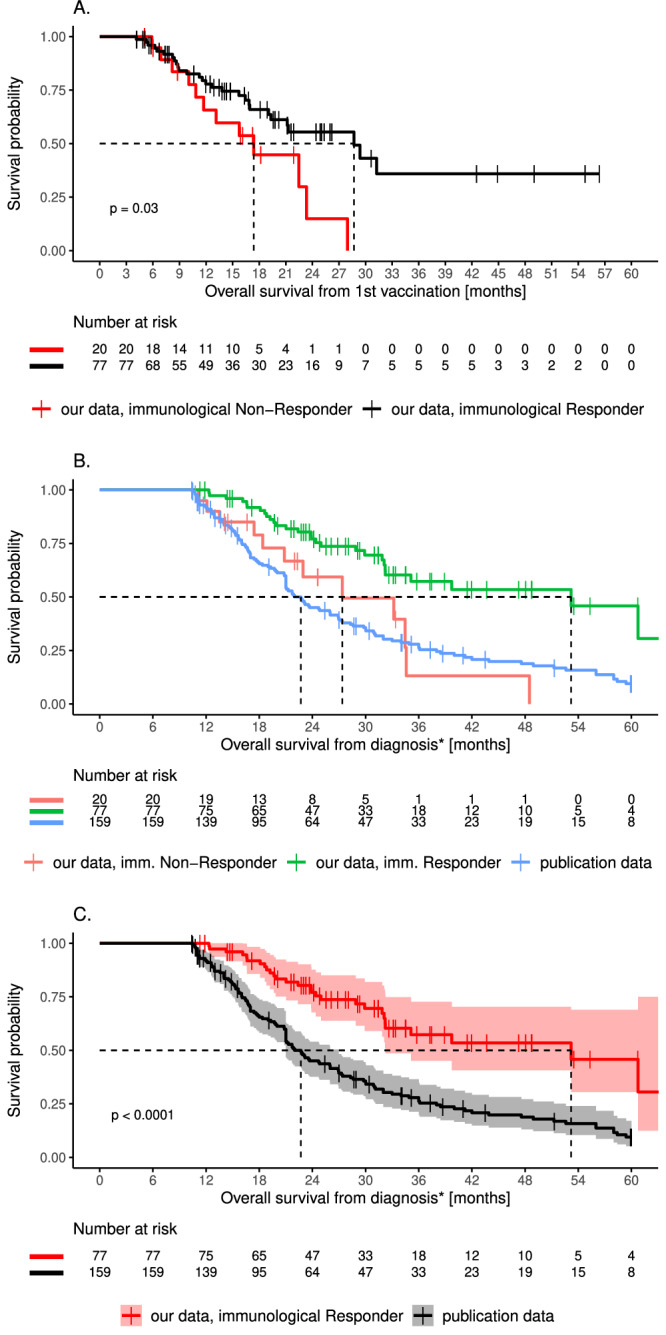
A On-treatment survival of immunological responders (iR; black) and non-responders (iNR; red). Significance is given for the comparison between patients with and without positive T-cell response (P = 0.03). B OS of our iR (green, 53.2 months, P < 0.0001 vs publication data) and iNR (red, 27.4 months, P = 0.74 vs publication data) and the matching cohort (publication data) (blue, 22.7 months). C OS of iR (red) and the matching cohort (publication data; black) (P < 0.0001). Statistical significance of survival differences was computed using the log-rank test. 95% confidence interval of cumulative hazard (log survival) is shown as shaded areas. Source data are provided as a Source Data file.
Interestingly, OS of iNR was not significantly different from OS of the propensity-matched historical control patients (median 27.4 months vs. 22.7 months, P = 0.74, Fig. 3B). Furthermore, iR showed remarkably long OS (53.2 months). This difference in median OS was highly significant when compared to the propensity-matching cohort (P < 0.001; Fig. 3C). MGMT status, recurrent status, and immunosuppressant intake (for example Dexamethasone) were not significantly associated with developing higher (>0.1) immune responses (Fisher’s exact test, P = 0.12, 0.32 and 0.62 respectively; Supplementary table 6).
This real-world dataset also includes 22 GBM patients (13%) with OS longer than 3 years including 11 who received vaccine prior to progression and 11 who received vaccine after progression. Of those, 19 (86%) showed T-cell responses after the priming phase, one showed no response after priming and immune monitoring was not available for two patients.
Discussion
Personalized therapy is a rapidly growing and promising approach for cancer treatment that conventionally involves biologically based therapies against molecular targets driving dysregulated cell signaling pathways. In this work, we present the largest real-world observation involving GBM patients treated with a personalized peptide vaccine to date. This retrospective analysis shows that our personalized vaccine is immunogenic, well tolerated and that production and application are feasible within a reasonable time frame. One hundred and seventy-three GBM patients received a neoantigen-derived peptide vaccine as an additional treatment to standard-of-care for newly diagnosed disease or as salvage therapy after recurrence. The results of our observation will also serve as groundwork for the design of a future prospective clinical trial.
The peptide vaccine was well tolerated even when integrated with other treatments typically administered among newly diagnosed and recurrent patients, respectively. Most treatment-related adverse events were grade 1 or 2 with only four observed grade 3 AE which were readily treated, and no observed grade 4 events.
We have shown that it is technically feasible to produce a fully personalized neoantigen-derived peptide vaccine within a reasonable time frame (within 12 weeks after completion of next-generation sequencing (NGS) analysis). Furthermore, the vaccine was able to elicit robust T-cell responses in a high proportion of patients.
Most importantly, vaccine-induced (re)activation of neoantigen-specific T-cells throughout treatment correlated strongly with a significantly prolonged on-treatment survival. The patients who developed multiple T-cell responses against vaccinated antigens (iR) showed a significantly longer overall survival than those patients with no/low induced T-cell responses (iNR). In line with these findings, Walter et al. observed in a phase 2 study that among peptide-vaccinated renal cell cancer patients, T-cell responses against multiple peptides were associated with longer overall survival26.
As expected, stratification of our patients according to molecular and clinical parameters showed a longer OS associated with methylated MGMT promoter status and Karnofsky index. Interestingly, the addition of bevacizumab to the treatment regimen is associated with a significantly reduced overall survival but this likely reflects its preferential use among more heavily pre-treated patients with diminished clinical status.
Our results align with those of other studies showing that peptide vaccines may elicit stable T-cell responses and that these immune responses may be associated with promising clinical results. For instance, the clinical phase 1 NOA16 trial (NCT02454634) showed safety, immunogenicity, and efficacy of an IDH1 peptide vaccine in newly diagnosed IDH1 mutated grade 3 and grade 4 astrocytoma18. Immune responses were observed in 93% of the patients together with three-year progression-free and death-free rates of 0.63 and 0.84, respectively, whereas patients with no immune responses showed progression within 2 years of first diagnosis. Personalized peptide vaccines together with adjuvant TMZ also showed feasibility and immunogenicity in newly diagnosed GBM in the phase 1 GAPVAC-101 trial (NCT02149225)16. Targeting unmutated antigens and neoantigens induced T-cell responses with a median PFS and OS of 14.2 and 29 months from diagnosis, respectively. Additionally, Keskin et al17. showed in a phase 1/1b trial the induction of circulating polyfunctional neoantigen-specific T-cell responses as well as an increase in the number of tumor-infiltrating T-cells in newly diagnosed patients with MGMT promoter unmethylated glioblastoma following surgical resection, conventional radiotherapy, and application of personalized neoantigen-targeting vaccines. In that study, vaccine-specific neoantigen reactive T-cells were also detected in resected tumors after progression confirming that systemic, vaccine-specific T-cells can traffic to the CNS and into GBM tumors. In line with our findings but on a much smaller scale, Grassl et al. recently demonstrated the feasibility and efficacy of an H3K27M-targeted vaccine in diffuse midline glioma patients on a compassionate used basis, with tumor-specific T-cell responses and clinical benefit15.
Very interestingly, 31/173 (18%) of the patients in our cohort had one or more germline variants associated with a hereditary tumor predisposition. As this finding may be relevant not only for therapy guidance but also for genetic counseling of family members, routine testing for germline variants associated with a tumor predisposition should be considered for GBM patients.
The following limitations of the presented analysis must be considered. Our results reflect a highly heterogeneous patient population including primary patients as well as recurrent patients. Importantly, our data suggest a possible association of T-cell responses with survival among both primary and recurrent patients relative to historical propensity-matched controls. Nonetheless, further investigation of a potential benefit of vaccine therapy will require a prospective, randomized trial among a homogeneous patient population. The heterogeneity of our patient population reflects “real-world” experience including that there were no exclusion factors specified that prevented patients from being considered for neoantigen vaccine therapy. Another potential bias is related to the fact that our vaccine requires time to be produced, and improved survival is to be expected because outcome measures may also represent patients who were able to wait for vaccine preparation. To address this, our propensity-matched controls were limited to only those patients who survived long enough to reach our patients’ median time from diagnosis to first vaccination (10.3 months). Another potential source of bias was the patients’ socio-economic status, as they were able to afford the cost of vaccine production and travel to Germany for its administration. However, this bias is not relevant to the subgroup analysis (Fig. 3A) demonstrating improved outcomes associated with vaccine-induced T-cell responses. Importantly, our retrospective analysis can show correlation only, but no causal links between positive T-cell response to vaccination and survival. It could be that anergic patients who lacked response to the vaccine had decreased overall immune system performance which itself contributed to their earlier deaths. Another limitation to consider is that data on post-vaccination T-cell responses have not yet been available for all our patients. Finally, each patient received additional treatment to the personalized vaccine based on recommendations of their primary treating team. Such treatments were highly variable and may likely have impacted outcome parameters. Nevertheless, the established benefit on survival for these therapies is in general quite limited. The extent of resection might also have influenced the course of the disease. However, only limited information was available for our cohort as well as for the patients from the public data set. A clinical trial setting would allow the integration of such parameters for further analysis. To address the limitations mentioned above a randomized and controlled clinical trial is planned.
In summary, this retrospective analysis provides further evidence that it is technically feasible to produce a fully individualized neoantigen-derived peptide vaccine under real-world conditions. Further investigation in the framework of randomized, controlled clinical trials is necessary to assess survival of GBM patients treated with neoantigen-based peptide vaccines and to analyse the specific contributions of the vaccination as well as of other immune therapies.
Methods
Study design and participants
One hundred seventy-three GBM patients were treated with a personalized peptide vaccine within the scope of an individual healing attempt [statement WD 9 – 3000 – 083/23 of the German Parliament, guidelines 2001/20/EG and 2005/28/EG, Declaration of Helsinki of the World Medical Association (Article 37)]. An approval by the Institutional Review Board and ethics committees is not required. Our report includes patients with histologically defined and molecularly confirmed IDH-wildtype GBM, as well as patients with previously diagnosed GBM, recently reclassified as diffuse pediatric-type high-grade glioma, H3-wildtype, and IDH-wildtype according to the 5th edition of the CNS WHO classification27. Our findings apply to both sexes. Sex was determined based on patient documents, reflecting genetic (chromosomal) sex. All patients provided informed consent for our personalized neoantigen vaccine therapy in an individual healing attempt and for the use of results for scientific research. The patients did not receive any compensation.
Neoantigen prediction and peptide selection
For the prediction of neoantigens, somatic variants were phased and the resulting coding sequences were translated into the local amino acid sequence context to generate epitope candidates. The patient’s HLA type was identified using OptiType algorithm28. HLA-epitope binding affinity was predicted as described before29.
Selection of epitopes was performed using an in-house developed and proprietary neoantigen selection algorithm as previously described29. Briefly, potential HLA class I epitopes with high predicted binding affinity, high allele frequency, and high potential expression were selected. When possible, predicted neoantigens for all patient HLA class I molecules were selected. Furthermore, peptides that potentially bind to multiple HLA class I molecules of the patient were preferred. In addition, possible HLA class II epitopes with high allele frequency and expected high expression were selected. Expression of tumor mutations was confirmed in the patient’s tumor transcriptome data or, if such data was lacking, expression of respective proteins in human gliomas was manually checked in the Human Protein Atlas database (https://www.proteinatlas.org/) and integrated into the peptide selection process.
We aimed to vaccinate 20 peptides. However, for some patients fewer peptides could be selected due to reduced presence of mutations, HLA binders, synthesizable peptides, etc. Finally, the median number of peptides selected was 19 (mean 16.4).
Vaccination
Vaccine formulation and administration were performed as previously reported30. Peptides were synthesized by solid-phase peptide synthesis (SPPS) and purified to at least 95% (synthesized by Intavis Peptide Services GmbH, Tübingen, Germany). Lyophilized peptides (HCl salt) were dissolved in water (Aqua ad iniectabilia; BBraun, Melsungen, Germany) + 33% dimethylsulfoxide (Miltenyi, Bergisch Gladbach, Germany). Peptides were mixed and sterile-filtered through a PTFE-membrane filter (Millex-LG sterile filter; Millipore) and bottled in glass vials (Thermo Scientific). The final concentration of the multi-peptide solution was 0.8 mg/mL per peptide. The resulting peptide vaccine was controlled for identity and purity of contained peptides as well as sterility and absence of endotoxins followed by QC/QA release. Vaccine vials were stored at −80 °C.
Per vaccination, 0.5 ml multi-peptide solution (0.8 mg/mL per peptide) was injected intracutaneously in the left or right lower abdomen followed by subcutaneous injection of 83 µg sargramostim and/or superficial application of imiquimod in the same area. Patients were vaccinated four times in the first 2 weeks of the vaccination process with subsequent boosting vaccinations every four to six weeks (Supplementary Fig. 5). For every vaccination, the following patient data were recorded: known allergies, current state of health, current medications, vaccine tolerability, complete blood count, vital parameters, information about the vaccine (dose, lot number, injection site, adjuvant). The patients were observed for at least 30 minutes after each vaccine dose.
Additional details are provided in the supplementary information file.
Detection of vaccine-induced T-cell responses
To monitor neoantigen-specific T-cells, 80 ml whole blood was routinely drawn before the first vaccination and usually before the seventh vaccination. Detection of neoantigen-specific T-cells was performed as previously described30,31 using an assay which is primarily detecting functional memory T-cells32. Briefly, peripheral blood mononuclear cells (PBMCs) were isolated from whole blood using density gradient centrifugation and were cryopreserved in freezing medium containing 10% DMSO until further usage. After thawing, PBMCs were stimulated with peptides (1 µg/ml for MHC class I peptides and 5 µg/ml for MHC class II peptides) either separately or in pools of 2–5 peptides (MHC class I and II peptides were not combined) if cell numbers were low. Cells were cultivated in presence of IL-2 (10 U/ml; Miltenyi Biotec, Bergisch Gladbach, Germany) and IL-7 (10 ng/ml; Miltenyi Biotec) for 12 days. After 12 days of cultivation, expanded cells were restimulated with corresponding peptides at the same concentration and additionally incubated for 14 h in presence of Golgi inhibitors (1 µl/ml; Golgi Plug, BD Biosciences, Franklin Lakes, NJ, USA).
The readout was Flow Cytometric Analysis after Intracellular Cytokine Staining (ICS). After cultivation, cells were washed and stained extra- and intracellularly using fluorochrome-conjugated antibodies titrated to their optimal concentrations. Finally, cells were measured on a Novocyte 3005R cytometer (Agilent, Santa Clara, CA, USA).
Data were analysed using FlowJo version 10.5.3 (FlowJo LLC, Ashland, AZ, USA). Briefly, CD4+ and CD8+ T-cells were gated within viable CD3+ lymphocytes and analysed separately for each functional marker (CD154, IFN-γ, TNF, and IL-2). Peptide-specific responses were evaluated using the stimulation index (SI). The stimulation index is the calculated ratio of polyfunctional activated CD4+ or CD8+ T-cells (positive for at least 2 markers of CD154, IFN-γ, TNF, and/or IL-2) in the peptide-stimulated sample to the mock-stimulated sample (NC). Cells stimulated with an antibody-based nonspecific stimulus (10 µl/mL; human CytoStimTM, Miltenyi Biotec) served as positive control (PC). Neoantigen-specific T-cells were defined as being present if SI was ≥2. Additionally, a minimum frequency of 0.1% of reactive T-cells positive for at least one activation marker including CD154, IFN-γ, TNF, and/or IL-2 had to be reached among a minimum of 10,000 measured CD4+ or CD8+ events.
Immunological responders vs. non-responders
We calculated the ratio between vaccine-induced T-cell responses (either tested individually or pooled) and the number of vaccinated neoantigen-derived peptides for each patient. Patients were defined as immunological non-responders (iNR) for ratios <0.1 (e.g., one detected vaccine-induced T-cell response among 20 vaccinated neoantigens). Patients were defined as immunological responders (iR) for ratios ≥ 0.1 (e.g., two detected vaccine-induced T-cell responses among 20 vaccinated neoantigens). A detailed gating strategy can be found in Supplementary Fig. 1.
Propensity score matching
Variable selection
According to literature and guidelines of propensity score matching24,25, all variables should be included that affect both the treatment assignment and the dependent variable. The variables should be unaffected by the treatment assignment. Additionally, too many variables should also be avoided in order not to have a high variance. In the literature of GBM patient prognosis analysis, the following factors were often shown to be significantly associated with survival1,33: age at diagnosis, sex, MGMT promoter methylation status, concurrent TMZ chemoradiotherapy, and TMZ adjuvant therapy, extent of resection, performance scores.
Based on the completeness of patient variables of the publication data and our data, five variables were selected for the matching: age at diagnosis, sex, MGMT promoter methylation status, concurrent TMZ chemoradiotherapy, and TMZ adjuvant therapy. As a result, 159 treated patients with all five variables and 507 patients from the public data have been defined.
Other relevant variables such as performance scores and extent of resection were only available for a much smaller fraction of public and/or our patients. By including these variables, the number of patients for matching would have been reduced, and matching with replacement should have been performed to have one matched control patient for each vaccine patient.
Public GBM clinical data for constructing a matching cohort
Clinical data of GBM patients (IDH-wildtype), diagnosed according to the WHO 2016 Classification of Tumors of the Central Nervous System, from four publications were collected. 324 out of the 507 patients were selected from the public datasets for matching. The 324 patients all survived longer than 10.3 months, which is the median time from diagnosis to first vaccination in our cohort.
The median follow-up time of the four publications ranges from 34.8 months to infinite. Taking all 324 patients together, the median follow-up time is 61.2 months. Only 5% of public patients and 3% of treated patients survived longer than 60 months. Thus, 60 months was used as follow-up cut-off and patients were censored from both groups who survived longer than 60 months. The numbers of patients from each publication19–22 and their median survival months, median follow-up months are given in Supplementary Table 4.
Matching implementation
The matching of our vaccine patients (treatment group) to patients from four public datasets (control group) was implemented using the MatchIt R package23. Propensity scores were estimated with logistic regression. The nearest neighbor matching method was used to select one closest matching control patient for each vaccine patient. The ordering of vaccine patients for matching was set as random. Additionally, the constraint was set that each pair of treatment and control patients must have the same MGMT status. Unmatched patients from both the treatment and the control groups to be discarded was also allowed. The following parameters were given to the matchit function to fulfill these criteria: method = “nearest”, distance = “glm”, discard = “both”, m.order = “random”, exact = “MGMT”, ratio = 1.
Assessment of matching quality
The balance of the propensity scores before and after matching was visually assessed using jitter plot and histograms. The balance of individual variables was evaluated using the standardized mean difference before and after the matching (variable balance plot). By using the common threshold of 0.1, all variables became balanced after the matching. The results are shown in Supplementary Fig. 4.
Multivariate analyses using matched treated and control patients
The matching has balanced the variable distributions between our cohort and the control cohort. However, it could not remove all biases because the matching is not exact for every variable. Fitting multivariate regression models on balanced datasets is suggested to have less biases and less model dependence25. Multivariate Cox regression model was fitted with the 5 variables mentioned above together with the treatment group variable. This allows us to estimate the effect of receiving vaccine on survival while controlling for other variables (Supplementary Table 5).
Statistical analysis
Patient data were collected and curated in tabular form and imported into R Version 4.0.434. On-treatment survival refers to the time between first vaccination and date of death/cut-off date. Overall survival (OS) indicates the time between first diagnosis and date of death/cut-off date. The term ‘censored’ used in the context of Kaplan–Meier statistics refers to patients still alive at the time of data cut-off. All survival statistics were computed using the “survival” package35,36. Statistical significance of survival differences was computed using the log-rank test (function survfit). Kaplan–Meier curves were generated using the package “survminer”37. The median follow-up time was calculated with reverse Kaplan–Meier. All univariate statistics were computed using the Cox Proportional Hazards model implemented in function coxph. Continuous variables (HRD score, TMB) were tested both as-is and as binary variables using thresholds (Table 2). R codes for the propensity score matching and the survival analysis (Supplementary Codes 1 and 2, respectively) are provided as Supplementary Data.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Supplementary information
Description of Additional Supplementary Files
Source data
Acknowledgements
We would like to thank RNDr. Bc. Iveta Selingerová, Ph.D. for sharing the data of the patients treated with standard chemo-radiation regimen in 2014–2017 in real-world care outside of clinical trials (data published in July 2020 by Lakomý et al.19). Most importantly, we thank all patients for donating their data to help us publish this work.
Author contributions
P.L. wrote the first draft of the report with input from A. Reinhardt, F.B., J.H., H.Z., B. Shao, and S.B. F.B. and B. Shao did the statistical analysis. A. Rabsteyn, M.S., T.O., A.G., C.K.F., S. Kayser, N.P., M.F., J.W., C.S., D.H., C.G., T.K.H., C.l.F., D. Biskup, D. Brooke, D.P., U.M.M., G.I., D.T.B., R.M., L.S.L., M.H., PdR, S. Kebir, W.W.L., V.W.L., M.W., A.M.M., S. Kesari, M.C., A.D., D.M.A., H.S.F., P.Y.W., E.C.N., F.M.I., B. Sipos, K.G., L.Z., M.G. and D.A.R. provided critical review and revision of the text. All authors approved the final version of the manuscript. All authors had full access to all the data in the study and had final responsibility for the decision to submit for publication. P.L., A. Reinhardt, F.B., J.H., H.Z., O.B., and S.B. have directly accessed and verified the underlying data reported in the manuscript.
Peer review
Peer review information
Nature Communications thanks Annette Molinaro and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Data availability
With publication, the data collected, generated, and evaluated for this article can be provided by the corresponding author, specifically exome, transcriptome, and flow cytometry data. Individual participant data are not publicly available because they contain information that compromises the privacy of the research participants. Information can be made available as de-identified data after both parties have signed a data access agreement. Sequencing data are available on the EGA repository (Study ID: EGAS50000000449, Dataset ID: EGAD50000000650).
The publicly available data used in this analysis are available in the supplementary tables of the corresponding publications20–22.
The data published by Lakomý et al19. were provided by the authors of the study upon request. The following data were then provided by Dr. Selingerová: sex, age, ECOG, deep brain location, resection, IDH, MGMT, time to RT initiation, RT dose, contouring, Stupp yes/no, chemoradiotherapy duration, corticosteroid use, adjuvant chemotherapy, number of adjuvant chemotherapy cycles, treatment after progression, type of treatment after progression, OS, censOS, PFS, and censPFS.
The remaining data are available within the Article, Supplementary Information, or Source Data file. Source data are provided with this paper.
Code availability
Supplementary Code 1 refers to the R code used for the propensity score matching and the multivariate analysis. Supplementary Code 2 refers to the R code used for the survival analysis, the univariate analysis, and the figures.
Competing interests
Disclosures for Patrick Y. Wen as research support: Astra Zeneca, Black Diamond, Bristol Meyers Squibb, Celgene, Chimerix, Eli Lily, Erasca, Genentech/Roche, Kazia, MediciNova, Merck, Novartis, Nuvation Bio, Servier, Vascular Biogenics, VBI Vaccines. Disclosures for Patrick Y. Wen as advisory board/consultant: Astra Zeneca, Black Diamond, Celularity, Chimerix, Day One Bio, Genenta, Glaxo Smith Kline, Insightec, Kintara, Merck, Mundipharma, Novartis, Novocure, Prelude Therapeutics, Sapience, Servier, Sagimet, Vascular Biogenics, VBI Vaccines. Conflict of interest for Uwe M. Martens as advisory role: BMS, MSD, Roche, Guardant Health, Sanofi-Aventis, GSK, Novartis. Conflict of interest for Uwe M. Martens as scientific support: Dieter Schwarz Foundation. Saskia Biskup and Dirk Biskup have ownership interests in CeCaVa GmbH and CeGaT GmbH. Dirk Hadaschik is an employee of CeCaVa GmbH. Florian Battke, Johannes Harter, and Magdalena Feldhahn are employed by CeGaT GmbH. Alexander Golf, Julian Wünsche, and Thomas Okech are employed by MVZ Zentrum für ambulante Onkologie GmbH. The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest. No competing interests were disclosed by the other authors.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
The online version contains supplementary material available at 10.1038/s41467-024-51315-8.
References
- 1.Stupp, R. et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 352, 987–996 (2005). 10.1056/NEJMoa043330 [DOI] [PubMed] [Google Scholar]
- 2.Stupp, R. et al. NovoTTF-100A versus physician’s choice chemotherapy in recurrent glioblastoma: a randomised phase III trial of a novel treatment modality. Eur. J. Cancer48, 2192–2202 (2012). 10.1016/j.ejca.2012.04.011 [DOI] [PubMed] [Google Scholar]
- 3.Stupp, R. et al. Maintenance therapy with tumor-treating fields plus temozolomide vs temozolomide alone for glioblastoma: a randomized clinical trial. JAMA314, 2535–2543 (2015). 10.1001/jama.2015.16669 [DOI] [PubMed] [Google Scholar]
- 4.Stupp, R. et al. Effect of tumor-treating fields plus maintenance temozolomide vs maintenance temozolomide alone on survival in patients with glioblastoma: a randomized clinical trial. JAMA318, 2306–2316 (2017). 10.1001/jama.2017.18718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chinot, O. L. et al. Bevacizumab plus radiotherapy-temozolomide for newly diagnosed glioblastoma. N. Engl. J. Med.370, 709–722 (2014). 10.1056/NEJMoa1308345 [DOI] [PubMed] [Google Scholar]
- 6.Gilbert, M. R. et al. A randomized trial of bevacizumab for newly diagnosed glioblastoma. N. Engl. J. Med.370, 699–708 (2014). 10.1056/NEJMoa1308573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Omuro, A. et al. Radiotherapy combined with nivolumab or temozolomide for newly diagnosed glioblastoma with unmethylated MGMT promoter: an international randomized phase III trial. Neuro Oncol.25, 123–134 (2023). 10.1093/neuonc/noac099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lim, M. et al. Phase III trial of chemoradiotherapy with temozolomide plus nivolumab or placebo for newly diagnosed glioblastoma with methylated MGMT promoter. Neuro Oncol.24, 1935–1949 (2022). 10.1093/neuonc/noac116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Reardon, D. A. et al. Effect of nivolumab vs bevacizumab in patients with recurrent glioblastoma: the CheckMate 143 phase 3 randomized clinical trial. JAMA Oncol.6, 1003–1010 (2020). 10.1001/jamaoncol.2020.1024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Weller, M. et al. Rindopepimut with temozolomide for patients with newly diagnosed, EGFRvIII-expressing glioblastoma (ACT IV): a randomised, double-blind, international phase 3 trial. Lancet Oncol.18, 1373–1385 (2017). 10.1016/S1470-2045(17)30517-X [DOI] [PubMed] [Google Scholar]
- 11.Stupp, R. et al. Cilengitide combined with standard treatment for patients with newly diagnosed glioblastoma with methylated MGMT promoter (CENTRIC EORTC 26071-22072 study): a multicentre, randomised, open-label, phase 3 trial. Lancet Oncol.15, 1100–1108 (2014). 10.1016/S1470-2045(14)70379-1 [DOI] [PubMed] [Google Scholar]
- 12.Saxena, M. et al. Therapeutic cancer vaccines. Nat. Rev. Cancer21, 360–378 (2021). 10.1038/s41568-021-00346-0 [DOI] [PubMed] [Google Scholar]
- 13.Sellars, M. C., Wu, C. J. & Fritsch, E. F. Cancer vaccines: building a bridge over troubled waters. Cell185, 2770–2788 (2022). 10.1016/j.cell.2022.06.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dunn, G. P., Sherpa, N., Manyanga, J. & Johanns, T. M. Considerations for personalized neoantigen vaccination in malignant glioma. Adv. Drug Deliv. Rev.186, 114312 (2022). 10.1016/j.addr.2022.114312 [DOI] [PubMed] [Google Scholar]
- 15.Grassl, N. et al. A H3K27M-targeted vaccine in adults with diffuse midline glioma. Nat. Med.29, 2586–2592 (2023). 10.1038/s41591-023-02555-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hilf, N. et al. Actively personalized vaccination trial for newly diagnosed glioblastoma. Nature565, 240–245 (2019). 10.1038/s41586-018-0810-y [DOI] [PubMed] [Google Scholar]
- 17.Keskin, D. B. et al. Neoantigen vaccine generates intratumoral T cell responses in phase Ib glioblastoma trial. Nature565, 234–239 (2019). 10.1038/s41586-018-0792-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Platten, M. et al. A vaccine targeting mutant IDH1 in newly diagnosed glioma. Nature592, 463–468 (2021). 10.1038/s41586-021-03363-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lakomy, R. et al. Real-world evidence in glioblastoma: Stupp’s regimen after a decade. Front Oncol.10, 840 (2020). 10.3389/fonc.2020.00840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Barthel, F. P. et al. Longitudinal molecular trajectories of diffuse glioma in adults. Nature576, 112–120 (2019). 10.1038/s41586-019-1775-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brennan et al. The somatic genomic landscape of glioblastoma. Cell155, 462–477 (2013). 10.1016/j.cell.2013.09.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jonsson, P. et al. Genomic correlates of disease progression and treatment response in prospectively characterized gliomas. Clin. Cancer Res.25, 5537–5547 (2019). 10.1158/1078-0432.CCR-19-0032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ho, D. et al. MatchIt: nonparametric preprocessing for parametric causal inference. J. Stat. Softw.42, 1–28 (2011). 10.18637/jss.v042.i08 [DOI] [Google Scholar]
- 24.Caliendo, M. & Kopeinig, S. Some practical guidance for the implementation of propensity score matching. J. Economic Surv.22, 31–72 (2008). 10.1111/j.1467-6419.2007.00527.x [DOI] [Google Scholar]
- 25.Ho, D. E. et al. Matching as nonparametric preprocessing for reducing model dependence in parametric causal inference. Polit. anal.15, 199–236 (2007). 10.1093/pan/mpl013 [DOI] [Google Scholar]
- 26.Walter, S. et al. Multipeptide immune response to cancer vaccine IMA901 after single-dose cyclophosphamide associates with longer patient survival. Nat. Med.18, 1254–1261 (2012). 10.1038/nm.2883 [DOI] [PubMed] [Google Scholar]
- 27.WHO Classification of Tumours Editorial Board. Central nervous system tumours. WHO Classification of Tumours Series, 5th edn, Vol. 6 (Lyon (France), International Agency for Research on Cancer, 2021).
- 28.Szolek, A. et al. OptiType: precision HLA typing from next-generation sequencing data. Bioinformatics30, 3310–3316 (2014). 10.1093/bioinformatics/btu548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Blumendeller, C. et al. Use of plasma ctDNA as a potential biomarker for longitudinal monitoring of a patient with metastatic high-risk upper tract urothelial carcinoma receiving pembrolizumab and personalized neoepitope-derived multipeptide vaccinations: a case report. J. Immunother. Cancer9, e001406 (2021). 10.1136/jitc-2020-001406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zelba, H. et al. Adjuvant treatment for breast cancer patients using individualized neoantigen peptide vaccination—a retrospective observation. Vaccines10, 1882 (2022). 10.3390/vaccines10111882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zelba H., et al. Case report: targeting of individual somatic tumor mutations by multipeptide vaccination tailored for HLA class I and II presentation induces strong CD4 and CD8 T-cell responses in a patient with metastatic castration sensitive prostate cancer. Front. Immunol. 14, 1271449 (2023). [DOI] [PMC free article] [PubMed]
- 32.Zelba, H. et al. A highly specific assay for the detection of SARS-CoV-2-reactive CD4+ and CD8+ T cells in COVID-19 patients. J. Immunol.206, 580–587 (2021). 10.4049/jimmunol.2000811 [DOI] [PubMed] [Google Scholar]
- 33.Michaelsen, S. R. et al. Clinical variables serve as prognostic factors in a model for survival from glioblastoma multiforme: an observational study of a cohort of consecutive non-selected patients from a single institution. BMC cancer13, 402 (2013). 10.1186/1471-2407-13-402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.R Core Team. R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. https://www.R-project.org/ (2020).
- 35.Therneau T. _A Package for Survival Analysis in S_. version 2.38, https://CRAN.R-project.org/package=survival (2015).
- 36.T. M. Therneau, P. M. Grambsch. _Modeling Survival Data: Extending the Cox Model_. Springer, New York. ISBN 0-387-98784-3. (2000).
- 37.A. Kassambara, M. Kosinski and P. Biecek. survminer: Drawing Survival Curves using ‘ggplot2’. R package version 0.4.9. https://CRAN.R-project.org/package=survminer (2021).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Description of Additional Supplementary Files
Data Availability Statement
With publication, the data collected, generated, and evaluated for this article can be provided by the corresponding author, specifically exome, transcriptome, and flow cytometry data. Individual participant data are not publicly available because they contain information that compromises the privacy of the research participants. Information can be made available as de-identified data after both parties have signed a data access agreement. Sequencing data are available on the EGA repository (Study ID: EGAS50000000449, Dataset ID: EGAD50000000650).
The publicly available data used in this analysis are available in the supplementary tables of the corresponding publications20–22.
The data published by Lakomý et al19. were provided by the authors of the study upon request. The following data were then provided by Dr. Selingerová: sex, age, ECOG, deep brain location, resection, IDH, MGMT, time to RT initiation, RT dose, contouring, Stupp yes/no, chemoradiotherapy duration, corticosteroid use, adjuvant chemotherapy, number of adjuvant chemotherapy cycles, treatment after progression, type of treatment after progression, OS, censOS, PFS, and censPFS.
The remaining data are available within the Article, Supplementary Information, or Source Data file. Source data are provided with this paper.
Supplementary Code 1 refers to the R code used for the propensity score matching and the multivariate analysis. Supplementary Code 2 refers to the R code used for the survival analysis, the univariate analysis, and the figures.