


Abstract
i-Motifs (iMs) are non-canonical, four-stranded secondary structures formed by stacking of hemi-protonated CH+·C base pairs in cytosine-rich DNA sequences, predominantly at pH < 7. The presence of iM structures in cells was a matter of debate until the recent development of iM-specific antibody, iMab, which was instrumental for several studies that suggested the existence of iMs in live cells and their putative biological roles. We assessed the interaction of iMab with cytosine-rich oligonucleotides by biolayer interferometry (BLI), pull-down assay and bulk-FRET experiments. Our results suggest that binding of iMab to DNA oligonucleotides is governed by the presence of runs of at least two consecutive cytosines and is generally increased in acidic conditions, irrespectively of the capacity of the sequence to adopt, or not, an iM structure. Moreover, the results of the bulk-FRET assay indicate that interaction with iMab results in unfolding of iM structures even in acidic conditions, similarly to what has been observed with hnRNP K, well-studied single-stranded DNA binding protein. Taken together, our results strongly suggest that iMab actually binds to blocks of 2–3 cytosines in single-stranded DNA, and call for more careful interpretation of results obtained with this antibody.
Graphical Abstract
Graphical Abstract.

Introduction
i-Motifs (iMs, or i-DNA) are non-canonical, four-stranded secondary structures formed by cytosine-rich DNA sequences upon mutual intercalation of hemi-protonated cytosine–cytosine base pairs (CH+·C), predominantly at relatively low pH; temperature, ionic strength and the nature of metal cations may also influence the stability of these structures (1–7). iM structures are known for more than three decades and have been well-characterized in vitro (1,5,8–11); nevertheless, the debate about the presence, persistence, and possible functions of these structures in cells is still relevant. It has been suggested that iMs play functional roles as transcription regulators (3,12,13) and, therefore, could represent potential drug targets for a number of pathologies, including cancer and neurodegenerative diseases (13–16). The controversy regarding the presence of iMs in cells is mainly due to the strong pH dependence of their stability that, with a few exceptions (17,18), may preclude their persistence at physiological pH and temperature (19). To explain this controversy, the existence of iM-stabilizing factors present in cells, such as molecular crowding or specific protein partners, has been postulated (20–22). However, a very recent in-cell NMR study convincingly demonstrated that iM-forming sequences with in vitro transition pH (pHT) < 7 are predominantly unfolded in living HeLa cells, and that iM formation concerns only a tiny fraction (<1%) of putative iM-forming sequences (23).
The molecular toolset to detect the presence of iMs in cells or to interrogate their functions is sparse. Thus, despite a massive research effort, no bona fide (i.e., truly specific and strongly stabilizing) small-molecule iM ligand has been identified to date (24,25), which is in a stark contrast to the plethora of highly specific, biologically active G-quadruplex (G4) ligands. To address this gap, in 2018 the teams of D. Christ and M. Dinger raised an antibody (iMab) against the iM structure formed by the 22-mer C-rich DNA strand representing human telomeric sequence (CCC(TAACCC)3T, or hTeloC; hTeloC-22 in this work) (26). The initial characterization of iMab demonstrated high affinity for hTeloC-22 (KD = 59 nM at pH 6.0, as per biolayer interferometry (BLI) analysis) and excellent selectivity, as no binding to the mutated sequence unable to form an iM structure (hTeloC-mut), double-stranded and hairpin DNA, or G4 structures was observed by using ELISA and BLI assays. At the same time, reduced yet non-negligible binding of iMab to the hTeloC strand was observed at pH 7.0 and even 8.0, which is rather surprising considering the transition pH of this sequence (pHT = 6.11 for 24-mer hTeloC at 20 °C, in the presence of 110 mM Na+) (27) and the fact that iM ↔ single-strand transitions are highly cooperative, meaning that no significant fraction of this sequence could be present in the iM form at pH 7.0 or, even less likely, at pH 8.0 (5). Despite this inconsistency, immunostaining of human cells with iMab clearly evidenced the accumulation of iMab foci in the nuclei, which was interpreted as the presence of iM structures in cells (26).
Since this seminal work, iMab was commercialized and rapidly found numerous applications as a molecular biology tool to study the formation and dynamics of iM structures in cells by immunofluorescence (28) and to obtain genome-wide maps of iM-forming sequences. Along these lines, using DNA immunoprecipitation and sequencing, Christ et al. identified in the human genome over 650 000 sequences that were recognized by iMab and significantly enriched in cytosine; a biophysical validation of a subset of these sequences confirmed that most (but not all) were able to form iM structures in vitro, at pH 6.0 and 25 °C (29). At the same time, Richter et al., using CUT&Tag technique, found that most iMab peaks (85–94%) were located in promoter regions of genes and significantly co-localized with open-chromatin markers, which was consistent with the putative implication of iMs in transcription regulation (22). A biophysical validation confirmed that a number of sequences corresponding to iMab peaks were indeed able to form iM structures in vitro in acidic conditions (pH 5.4), but not in neutral (i.e., close to physiological) conditions, thus keeping the conundrum regarding the factors that could potentially stabilize these structures in cells and allow their immunodetection with iMab.
In parallel, Schneekloth et al. assessed the sequence preferences of iMab binding in vitro using a DNA microarray exposing over 11 000 various DNA sequences, and found that binding of iMab was correlated with the length of C-tracts in DNA sequences (typically linked to higher stability of the corresponding iMs) (30). Interestingly, the correlation analysis performed in that study indicated high degree of similarity (Pearson's r = 0.72) of the binding profile of iMab with that of hnRNP K, one of poly(C)-binding proteins (31,32). The structural details of poly(C) binding by hnRNP K are known since at least two decades: this protein binds to the runs of at least three cytosines in single-stranded DNA regions, by making highly specific hydrogens bonds with unpaired cytosine residues (33–35). Considering the fact that the cytosine residues that participate in the formation of the iM core are unavailable for additional hydrogen bonding, it is highly unlikely that hnRNP K could bind folded iMs. Accordingly, a recent single-molecule study by Wu et al. provided solid evidence that hnRNP k actually unfolds iMs in vitro, namely through the binding to the corresponding single-strands (36).
Considering all these observations, we questioned the actual conformation of DNA sequences recognized by iMab antibody. To shed light on this conundrum, we reassessed the interaction of hTeloC and its several variants, unable to adopt iM structures even in acidic conditions, with different forms of iMab (scFv-His6 and scFv-His6-FLAG) by using BLI and pull-down experiments. Our results provide firm evidence that binding of iMab to DNA oligonucleotides is governed by the presence of runs of at least two consecutive cytosines and is generally increased in acidic conditions, irrespectively of the capacity of the sequence to adopt, or not, an iM structure. Moreover, using bulk-FRET experiments, we demonstrate that, similarly to hnRNP K, iMab is able to induce unfolding of iM structures in acidic conditions. These results strongly suggest that iMab actually binds single-stranded cytosine-rich DNA motifs.
Materials and methods
Tris acetate (Tris-AcOH) and polysorbate 20 (P20) were purchased from Sigma-Aldrich. The oligonucleotides were purchased from IDT or Eurogentec (HPLC purity grade). Two variants of the iMab antibody (scFv-His6, #Ab01462-30.11 and scFv-His6-FLAG, #Ab01462-30.135) were purchased from Absolute Antibody. Recombinant hnRNPK protein (#NBP2-23120, lot 12220201) was purchased from Novus Biologicals. Anti-G4 antibody BG4 (#MABE917) was purchased from Sigma-Aldrich/Merck.
BLI measurements
All BLI experiments were performed at 20 °C using Octet Red 96 instrument (Sartorius) and BLI sensors coated with streptavidin (FortéBio SA sensors, Sartorius). All 3′-biotinylated DNA oligonucleotides were annealed at a concentration of 100 nM in the running buffer (50 mM Tris–AcOH, pH 6.0 or 7.5, with 0.05% v/v surfactant P20) for 5 min at 85 °C, then cooled to room temperature for at least 30 min so that DNA could adopt its equilibrium conformation. Prior to use, SA sensors were immerged for 10 min in the running buffer (pH 6.0 or 7.5) to remove the protective sucrose layer from the sensor surface, then dipped in solutions containing annealed, biotinylated DNA oligonucleotides (100 nM) during 900 s. The immobilization was stopped when the signal reached 0.2 nm, to improve the quality of the fitting. The functionalized sensors were then rinsed with the buffer for 10 min to remove unbound molecules. The functionalized sensors were next dipped in the iMab solutions at different concentrations for 30 min interspersed by a rinsing step in the buffer solution for 10 min. Reference sensors without DNA immobilization were used to subtract the non-specific adsorption on the SA layer. One sensor was used per concentration due to the weak dissociation and to avoid the use of regenerative solution. The dissociation and association equilibrium constants (KD and KA, respectively) were calculated from the binding rate constants as KD = koff/kon or KA = kon/koff (where koff and kon represent the dissociation kinetic constant and the association kinetic constant, respectively). The reported values are the means of representative independent experiments. Each experiment was repeated at least two times.
CD spectroscopy
Circular dichroism studies were performed on a Jasco J-1500 spectropolarimeter using a 0.5 cm path length quartz cuvette. CD spectra were recorded at 20 °C using wavelength range from 230 to 340 nm and were an average of four scans with a 0.5 s response time, 1 nm data pitch, 4 nm bandwidth and 200 nm min−1 scanning speed. Samples containing 2.5 μM of DNA oligonucleotides were annealed at 90 °C in 10 mM lithium cacodylate, 100 mM potassium chloride buffer with indicated pH and cooled slowly overnight prior to CD analysis. Potentiometric CD titrations were performed as described elsewhere (37).
Pull-down assay
5′-Biotinylated oligonucleotides were resuspended in 50 mM of 2-(N-morpholino)ethanesulfonic acid buffer (MES, pH 5.8 or pH 7.0) at a 1.5 μM final concentration, annealed at 95 °C for 5 min and cooled to room temperature overnight. Folded oligonucleotides were incubated with 100 ng of iMab (scFv-His6-FLAG) at 4 °C for 1 h in MES buffer with indicated pH (50 mM MES, 150 mM NaCl, 0.1% Triton and 1% BSA). After incubation, complexes were trapped with MES buffer-preequilibrated streptavidin-coated magnetic beads (Dynabeads™ M-280 Streptavidin, ThermoFisher Scientific, #11205D). Excess antibody was washed by incubating 3 × 15 min with MES buffer without BSA, then magnetic beads were incubated with Laemmli buffer, heat-denatured at 95 °C for 10 min, and the dissociated fraction was recovered. Western blot was performed with anti-FLAG antibody 1:1000 (Sigma Aldrich, #F3165) incubated overnight at 4 °C in PBS-T buffer (PBS, 0.1% Tween 20) containing 1% BSA. Images were acquired using ChemiDoc imaging system (Bio-Rad).
Bulk-FRET experiments
The bulk-FRET experiments were set up as described by Wu et al. (36). The sequences of oligonucleotides are listed in Figure 5A. A solution of Cy3- and Cy5-labelled strands (2 μM each) in a 50 mM potassium phosphate (KH2PO4/K2HPO4), 200 mM potassium chloride, pH 5.8 buffer supplemented with 20% (w/v) PEG2000, in a total volume of 100 μl, was prepared and annealed for 5 min at 95 °C before being slowly brought to room temperature. Samples containing 4 nM of iM substrates in a total volume of 400 μl, accounting for the volume of the added iMab solution, were diluted in the same buffer adjusted to the desired pH. Samples were placed in Sigmacote-treated quartz cells with optical path lengths of 10 × 2 mm and thermostated at 20 °C. Fluorescence spectra were recorded with a HORIBA Jobin–Yvon FluoroMax-3 spectrofluorimeter, using excitation wavelength of 532 nm, emissions wavelength range of 550–800 nm (1-nm increment), excitation and emission bandwidths of 5 nm, and integration time of 0.2 s. After an initial measurement with DNA substrate alone, iMab was added to the desired final concentration, and fluorescence emission spectra were recorded in 2-min increments. FRET efficiency (EFRET) was calculated according to Equation (1):
Figure 5.

(A) Oligonucleotide sequences and (B) principle of the bulk-FRET assay to assess the unfolding of iM structures. (C, D) Time-dependent change of EFRET upon addition of iMab scFv-His6 (0, 400, or 800 nM) to Py25 substrate at pH 5.8 (C) or pH 6.5 (D). (E) Time-dependent change of EFRET upon addition of iMab scFv-His6 (0, 400 or 800 nM) to hTeloC substrate at pH 5.8. (F) Comparison of effects of BG4 and iMab antibodies (800 nM each) on the unfolding of Py25 substrate at pH 5.8. In all experiments, the concentration of the iM substrate was 4 nM in 50 mM phosphate buffer containing 200 mM potassium chloride and 20% (w/v) PEG2000, temperature: 20 °C.
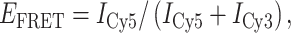 |
(1) |
where ICy5 and ICy3 are the intensities of the Cy5 (667 nm) and Cy3 (565 nm) emission peaks, respectively.
Results
BLI response of oligonucleotides is pH-dependent
In their seminal paper, Christ et al. demonstrated, using BLI experiments, that iMab binds the 22-mer iM-forming sequence hTeloC-22 (5′-(CCCTTA)3CCCT-3′) with an equilibrium constant (KD) close to 60 nM at pH 6.0 (26). They suggested that iMab specifically recognizes the folded i-motif structure, because a gradual reduction of BLI signals was observed upon increasing pH above 6.0, interpreted as reduced binding of the antibody. However, using only the intensity of the BLI signal as a measure of binding can lead to a bias, since the intensity of BLI signals is known to strongly depend on the size and conformation of the interacting molecules, both the one attached on the surface on the sensor and the dissolved analyte. Since it is the induced change in optical thickness that gives rise to a BLI signal (38,39), its intensity depends not only on the molecular weight (MW) of the analyte, but also on the molecular shape and packing density of the bound partner. In other words, if two molecules of the same MW have very different structures, they may well give rise to different BLI signals by virtue of their shape and packing ability. As an example, this phenomena has been used to study the unfolding of proteins (40). In this regard, we were puzzled by the fact that in the sensograms obtained by Christ et al. at pH 6.0 (i.e., favorable for the formation of iMs) and at pH 8.0 (i.e., strongly unfavorable for iMs), the amplitude of the signals was different, but the shape was very similar in both the association and the dissociation steps. We anticipated that the difference of the signal amplitude could be not only due to the differences in binding of iMab, but also due to a different conformation of the DNA sequence in these conditions.
To verify this point, we immobilized the 3′-biotinylated 27-mer sequence hTeloC-27 (5′-TAA(CCCTAA)4-3′) on streptavidin-coated BLI sensors either at pH 6.0 (conditions in which this sequence is predominantly folded into an iM structure) or at 7.5 (conditions in which this sequence is unfolded at 20 °C, cf. Supplementary Figure S11), by dipping the sensors for 900 s into solutions containing 100 nM of hTeloC-27 (aiming to prevent the formation of intermolecular structures). Remarkably, the intensity of the BLI response during the immobilization step was strongly pH-dependent (Supplementary Figure S1), which reflects a change in optical thickness. Stronger signals were observed at pH 6.0, which is compatible with the formation of the iM structure by hTeloC (cf. Supplementary Figure S11). These results indicate that the amplitude of BLI signal is indeed strongly influenced by the secondary structure of the oligonucleotide attached to the biosensor surface. Consequently, the amplitude of BLI signals at a single concentration cannot be used to correctly estimate the affinity, and a precise KD measurement using several concentrations of the analyte is required. However, both the initial characterization of iMab (26) and the follow-up binding studies with various cytosine-rich sequences were performed with single analyte concentrations (29).
iMab exhibits moderate selectivity between iM-folded and unfolded hTeloC sequence
We first studied the binding of two variants of iMab (scFv-His6 and scFv-His6-FLAG) to hTeloC-27 sequence (41), which differs from the 22-mer sequence originally used for the development of iMab (26) only by slightly longer overhangs which may allow better separation from the sensor surface. A potentiometric titration of hTeloC-27 provided an average transition pH of this sequence pHT = 6.1 (Supplementary Figure S11A), in agreement with other studies (27). The titration isotherms suggest that at pH 6.0 and 20 °C, ∼80% of the oligonucleotide exists in the iM form; conversely, at pH 7.5, the iM structure is unfolded by >99%.
The raw sensograms obtained by dipping the sensor in solutions containing different concentrations of iMab are presented in Figure 1 (scFv-His6) and Supplementary Figure S2 (scFv-His6-FLAG). At pH 6.0, the amplitude of the BLI signal was similar to that observed in the work of Christ et al. (26), that is, close to 9 nm in the presence of 500 nM iMab. For the same concentrations of iMab, the response was slightly lower at pH 7.5 (Δλ = 7 nm), but the shape of the curves was very similar. To calculate the equilibrium dissociation constants KD, a fitting by using a 1:1 model was applied in order to determine the kinetics constants (kon and koff). A good agreement between the fitted and the experimental curves was observed.
Figure 1.

BLI analysis of binding of different concentrations of iMab scFv-His6 (62.5, 125, 250 and 500 nM) to hTeloC-27 at pH 6.0 (A) and pH 7.5 (B). Blue lines are experimental curves; red lines are the fits to the 1:1 model.
The kinetic and thermodynamic parameters obtained for the two variants of iMab (scFv-His6 and scFv-His6-FLAG) at pH 6.0 and 7.5 are summarized in Table 1. Interestingly, for both variants the kinetic constants and the KD values at pH 6.0 and 7.5 are essentially of the same order of magnitude (KD = 28–34 and 91–108 nM at pH 6.0 ant 7.5, respectively). The resulting, weak apparent selectivity (∼3.3-fold) is mainly due to lower kinetic dissociation constant values at pH 6.0. However, in both cases, the binding is non-negligible at pH 7.5, despite the fact that in the absence of iMab, the iM structure formed by hTeloC sequence is essentially unfolded in these conditions. Importantly, this binding cannot be attributed to the residual folded iM form (which represents <1% at pH 7.5), since it would be characterized by significantly less intense BLI signals with essentially the same kon and koff values; however, the results shown in Figure 1 clearly demonstrate that this is not the case.
Table 1.
Kinetics constants (kon and koff), calculated dissociation equilibrium constants (KD) and apparent selectivity for two variants of iMab antibody interacting with hTeloC-27 sequence at pH 6.0 and 7.5a
| iMab variant | pH 6.0 | pH 7.5 | Apparent selectivityb | ||||
|---|---|---|---|---|---|---|---|
|
k
on
(103 M−1 s−1) |
k
off
(10−4 s−1) |
K
D
(nM) |
k
on
(103 M−1 s−1) |
k
off
(10−4 s−1) |
K
D
(nM) |
||
| scFv-His6 | 3.7 (2.4; 5.0) | 1.2 (1.1; 1.2) | 34 (24; 44) | 2.3 (1.7; 3.0) | 2.4 (2.0; 2.8) | 108 (63; 153) | 3.2 |
| scFv-His6-FLAG | 11.8 (11.4; 12.4) | 3.3 (2.6; 3.7) | 28 (23; 31) | 13.0 (8.0; 18) | 8.2 (3.3; 13) | 91 (18; 164) | 3.3 |
aData are arithmetic means from several independent measurements; the individual values are given in parentheses.
bApparent selectivity = KD (at pH 7.5)/KD (at pH 6.0).
iMab shows no preferential binding to the constrained iM scaffold
Previously, we described a constrained analogue (I) of hTeloC iM, assembled on a rigid cyclopeptide scaffold (Figure 2A). The particularity of this constrained iM is its higher stability; thus, (I) is folded at pH 6.5, while the native sequence is unfolded by ∼90% in these conditions (Figure 2B and Supplementary Figure S11A) (24,42). The interaction of the constrained hTeloC (I) immobilized on SA sensor with iMab (scFv-His6-FLAG) was studied at pH 6.5 in comparison with the native sequence; despite a weaker signal, the recognition pattern for the iMab was similar to the previous experiments (Supplementary Figure S3). After the fitting of the sensograms, the kinetic constants were determined and the KD value was calculated (Table 2). Remarkably, iMab binds to the constrained hTeloC (I), which a fortiori adopts an iM structure at pH 6.5, with an affinity (KD = 155 nM) similar to the one observed with hTelo-27, which is chiefly unfolded in these conditions (KD = 54 nM). This suggests that binding of iMab is not preferentially driven by the more stable iM structure.
Figure 2.

(A) Structure of the constrained iM (I). (B) CD spectra recorded at 20 °C for the constrained (I, black) and native hTeloC-27 (red) substrates at pH 6.5, as well as hTeloC-27 at pH 6.0 (magenta).
Table 2.
Kinetic constants (kon and koff) and calculated dissociation equilibrium constants (KD) for interaction of iMab (scFv-His6-FLAG) with the native and the constrained hTeloC substrates at pH 6.5 and 20 °Ca
| Substrate | k on (103 M−1 s−1) | k off (10−4 s−1) | K D (nM) |
|---|---|---|---|
| hTeloC-27 | 11.2 (11.0; 11.4) | 6.1 (5.4; 6.7) | 54 (49; 58) |
| Constrained hTeloC (I) | 4.6 (4.4; 4.9) | 7.2 (6.8; 7.6) | 155 (140; 171) |
aData are arithmetic means from two independent measurements; the individual values are given in parentheses.
iMab binds cytosine-rich sequences unable to adopt iM structures
We used BLI to assess the binding of iMab to a panel of 18 DNA oligonucleotides (Figure 3A). In this panel, 12 sequences have the same length as hTeloC-27 (i.e., 27 nt) but differ by the number and the arrangement of cytosine residues critical for the formation of iM structures; in particular, three variants (hTeloC-scr, hTeloC-3x4 and hTeloC-2x6T) have the same number of cytosine residues as the native sequence (i.e., 12). Additionally, we included the 22-mer iM-forming sequence originally used by Christ et al. (hTeloC-22) along with the corresponding negative controls (hTeloC-X3 and hTeloC-mut) (26), as well as one hairpin (hp-ATT) and a cytosine-poor single-stranded sequence (ss-DNA). A close inspection of the corresponding CD spectra (Supplementary Figure S12) revealed that among all these sequences, only three variants (hTeloC-4x5, hTeloC-3x4 and hTeloC-2x6T) were able to adopt iM structures similarly to hTeloC-27 and hTeloC-22, as indicated by the iM-characteristic CD bands at 283–285 nm observed at pH 6.0 and 5.5. While the formation of an iM structure by hTeloC-4x5 sequence was expected, hTeloC-3x4 and hTeloC-2x6T contain only three and two uninterrupted runs of cytosines, respectively, which is insufficient for the formation of intramolecular iM structures. Indeed, native gel electrophoresis analysis revealed the presence of bimolecular species in the case of hTeloC-2x6T and higher-order species in the case of hTeloC-3x4 (Supplementary Figure S13), suggesting the formation of intermolecular iM structures. In contrast, 13 other sequences were unable to form iM structures, as their CD spectra remained essentially identical upon decreasing pH from 7.5 to 6.0 and even to 5.5, except for hTeloC-3x3 and hTeloC-X3 (both having very similar sequence) whose CD spectra were minimally affected at pH 5.5 (but not at pH 6.0), allowing to suggest the presence of minor fractions of iM structures in these conditions.
Figure 3.

(A) Oligonucleotides used in BLI experiments and their capacity to adopt iM structures, as inferred from CD spectra (Supplementary Figure S12). (B, C) Comparison of KA (= 1/KD) values for binding of iMab scFv-His6 (B) and scFv-His6-FLAG (C) to the oligonucleotides listed in panel (A), at pH 6.0 (orange) and 7.5 (green bars). Data are mean values from two independent experiments (for individual values, cf. Supplementary Table S1).
The KD values obtained from BLI experiments with two variants of iMab (scFv-His6 and scFv-His6-FLAG) are summarized in Supplementary Table S1 (sensograms and 1:1 model fits, Supplementary Figures S4–S9), and the KA (= 1/KD) values are plotted on Figure 3B and C. Remarkably, at pH 6.0, the KD values obtained for most sequences were essentially in the same range (for scFv-His6, 27–65 nM; for scFv-His6-FLAG, 20–60 nM) with an exception for hTeloC-mut for which slightly higher KD values were observed (141 and 169 nM for scFv-His6 and scFv-His6-FLAG, respectively). Conversely, at pH 7.5, the affinity was strongly reduced for most sequences, with weak BLI response (indicative of weak or no binding) observed for hp-ATT, ss-DNA, hTeloC-mut, hTeloC-scr (i.e., sequences without consecutive cytosines) as well as hTeloC-3x2 and hTeloC-2x2 (i.e., sequences containing blocks of two consecutive cytosines).
We note that several sequences (hTeloC-3x4, hTeloC-2x6T and, to a much lesser extent, hTeloC-X3) could adopt intermolecular iM structures at pH 6.0, when folded at a concentration used for CD spectroscopy (i.e., 5 μM). However, the corresponding bands could not be detected when the oligonucleotides were annealed at a concentration used for BLI measurements, that is, 0.1 μM (Supplementary Figure S13). In addition, to verify that the formation of intermolecular structures directly on BLI sensors does not interfere with BLI measurements, we performed additional experiments with several representative sequences (Supplementary Table S2). Thus, after the immobilization of oligonucleotides performed as described above, BLI sensors were dipped into aqueous ammonia solution (pH 10) for 10 min at 50 °C, in order to denature putative intermolecular iM structures and leave only the immobilized strands. Then, the sensors were abundantly rinsed with the running buffer (Tris-AcOH, pH 6.0) and BLI measurements of interaction with iMab were performed. The results (Supplementary Figure S10 and Supplementary Table S2) demonstrate that the interaction with iMab was not significantly altered after ammonia treatment: thus, for all the sequences tested, the KD values were systematically inferior to 80 nM prior to, or after treatment with ammonia solution. In addition, no significant differences in kinetic constants were observed. These observations indicate that the putative formation of intermolecular iM structures on BLI sensors is not involved in antibody binding.
Taken together, the data presented in Figure 3 and Supplementary Table S1 indicate no significant difference, in terms of binding affinity, between the sequences capable of forming iM structures (hTeloC-27, hTeloC-22, hTeloC-4x5, hTeloC-3x4 and hTeloC-2x6T) and those unable of iM folding even in most favorable conditions. Instead, they suggest that (i) iMab preferentially binds DNA oligonucleotides containing two or, better, three consecutive cytosines, and (ii) for most oligonucleotides (whether they are able to adopt an iM structure or not), the binding is strongly increased in acidic conditions (pH 6.0).
To confirm these—rather unexpected—conclusions, the interaction of iMab (scFv-His6-FLAG) with six variants of the telomeric sequence (hTeloC, hTeloC-3x3, hTeloC-3x4, hTeloC-3x2, hTeloC-scr and hTeloC-mut) was additionally assessed by a pull-down assay followed by the detection of the oligonucleotide-bound and subsequently released antibody by Western blotting. The results (Figure 4) demonstrated that in acidic condition (pH 5.8), iMab was binding to two sequence variants that were able to form iM structures (hTeloC and hTeloC-3x4), but also to two cytosine-rich sequences unable to form iMs: hTeloC-3x3 and, to a smaller extent, hTeloC-3x2. In contrast, we observed very weak binding to scrambled sequences (hTeloC-scr and hTeloC-mut). Remarkably, binding of iMab to all telomeric variants containing consecutive cytosine residues was also observed at pH 7.0, even though none of these sequences could adopt an iM structure. Of note, a comparison of band intensities at pH 5.8 and pH 7.0 (Figure 4A) suggests that binding of iMab to all sequences is actually enhanced at pH 7.0, which disagrees with BLI data (cf. Figure 3); however, this effect may also stem from imparted stability of iMab in acidic conditions during pull-down experiments. Altogether, these pull-down experiments confirm that binding of iMab to oligonucleotides is driven not by the formation of the iM structure, but merely by the presence of consecutive cytosine residues.
Figure 4.

(A) Representative results of a pull-down following by Western blot detection of iMab scFv-His6-FLAG, performed with 5′-biotinylated oligonucleotides at pH 5.8 and at pH 7.0. (B) Quantification of pull-down experiments (the band intensities were normalized with respect to hTeloC at a given pH); data are means ± s.d. from three independent experiments.
iMab unfolds iM structures in bulk-FRET experiments
Wu et al. recently demonstrated, using single-molecule and bulk-FRET experiments, that hnRNP K, a poly(C)-binding protein (33–35,43), rapidly unfolds an iM structure through a stepwise process driven by the binding of protein to the cytosine-rich single strand of DNA (36). We employed the bulk-FRET assay to assess the capacity of iMab to induce unfolding of iMs. In this assay, the iM-forming sequence is 5′-labeled with a donor fluorophore (Cy3) and hybridized, via a 17-nt 3′-overhang, to a complementary strand (Cy5-CS) labelled with Cy5 as an acceptor fluorophore in the vicinity of the iM–duplex junction (Figure 5A–B). In the folded state, the proximity between the fluorophores results in high efficiency of FRET (EFRET= 0.78–0.80); conversely, unfolding of the iM leads to a larger distance between the fluorophores and a decrease of EFRET value to 0.32–0.34, as demonstrated by CD and fluorescence spectra recorded at pH 5.5 and 7.5, respectively (Supplementary Figure S14). Using the same iM-forming sequence (Py25, derived from the MYC promoter, Figure 5A) and the same conditions as Wu et al., we first assessed the effect of recombinant hnRNP K (800 nM) at pH 5.8 and observed a rapid increase of Cy3 fluorescence with a concomitant decrease of Cy5 fluorescence, consistent with the reduction of EFRET from 0.74 to 0.36 after 3 min (Supplementary Figure S15), in agreement with the literature data (36). Importantly, the same effect was observed upon addition of iMab. Thus, addition of 400 nM iMab scFv-His6 to Py25 iM substrate resulted in a decrease of EFRET from 0.78 to 0.55 after 100 min. Addition of 800 nM iMab led to an even more rapid and stronger decrease of EFRET value to 0.35, i.e., close to that of the fully unfolded form, demonstrating that this effect is concentration-dependent (Figure 5C). Remarkably, the kinetics of the iMab-driven unfolding was dramatically accelerated at pH 6.5, even though the final EFRET values were not affected (Figure 5D). Similar unfolding, albeit with a slower rate, was observed with the hTeloC sequence at pH 5.8 (Figure 5E). Finally, the G4-specific antibody BG4 did not unfold Py25 iM (Figure 5F), giving evidence that the unfolding process is specific for the iMab antibody which behaves, in this regard, similarly to hnRNP K protein. Of note, addition of iMab to separate strands (Cy5-CS and Cy3-Py25) led to only marginal changes in fluorescence spectra (Supplementary Figure S16), suggesting that the antibody had no effects on the fluorophores and that the changes observed in Figure 5 result from disruption of the iM structure.
Discussion
In this work, using BLI analysis, we systematically investigated the binding of two commercially available variants of iMab antibody (scFv-His6 and scFv-His6-FLAG) to cytosine-rich DNA oligonucleotides. Initially, in the experiments with the hTeloC-27 sequence (differing from the 22-mer sequence, against which iMab was originally raised, only by longer overhangs), we observed limited selectivity for binding at pH 6.0 (where this sequence predominantly adopts an iM structure) comparing to pH 7.5 where this sequence is fully unfolded. This moderate selectivity (∼3-fold), already observed in the original study, is rather surprising considering the extremely weak binding of iMab to all other DNA structures (G4s, DNA hairpins and single-stranded RNA) (26). Two alternative scenarios could explain this moderate selectivity: (i) iMab binds and strongly stabilizes iM structures to the extent that they persist as such in the conditions of BLI experiments at pH 7.5 and 20 °C; (ii) iMab binds to the unfolded hTeloC sequence and thereby unfolds iM structures that are otherwise stable at pH 6.0. Further sets of experiments led us to favor the latter hypothesis.
First, we assessed the binding of iMab to the constrained iM structure (I) assembled on a cyclopeptide scaffold. Being significantly more stable than the native iM, this structure is nonetheless very similar to the iM formed by the native hTeloC sequence, featuring the same iM core consisting of six CH+·C base pairs and two AAT loops. To our surprise, the binding affinity of iMab to this structure was not significantly different with respect to the native hTeloC sequence (which is mainly unfolded in the conditions of this experiment, i.e., at pH 6.5), suggesting that a stable, folded iM structure is not the primary driver of iMab binding. Admittedly, one of the sides of (I) is hindered by the surface-linked cyclopeptide, which may prevent antibody binding. However, in BLI experiments the biotinylated, surface-bound 3′-end of the native hTelo iM is similarly unavailable for antibody binding, a fact which minimizes the differences between these two substrates.
In line with these observations, additional BLI experiments did not reveal any significant difference in iMab binding between the sequences capable of forming an iM structure (hTeloC, hTeloC-4x5, hTeloC-3x4, and hTeloC-2x6T) and 10 other DNA sequences of similar length, but unable to form iMs even in most favorable conditions (i.e., at pH 5.5). The only sequence that demonstrated a significantly reduced iMab binding was hTeloC-mut, which was previously used as a negative control by Christ et al. However, the cytosine residues in this sequence are few (4 versus 12 in hTeloC) and spatially isolated. Remarkably, other sequence variants with similar number of cytosines, but assembled in blocks of two or three (hTeloC-1x3T, hTeloC-3x2, hTeloC-2x2) demonstrated KD values close to that of hTeloC at pH 6.0. In all cases, the binding of iMab was strongly reduced at pH 7.5, suggesting that it primarily depends on (i) pH (stronger binding in acidic conditions, independently of the capacity of the sequence to form iM) and (ii) the presence of at least two or, better, three consecutive cytosine residues. Furthermore, the results of the pull-down assay performed in two pH conditions also demonstrated the incapacity of iMab to discriminate between the sequences capable (hTeloC-27, hTeloC-3x4) or not (hTeloC-3x3, hTeloC-3x2) of folding into iM structures, further consolidating the above conclusions.
Finally, the results of bulk-FRET assay clearly indicate that iMab binds and unfolds iM structures even in acidic conditions, where iMs are otherwise thermodynamically stable. This implies that the free energy of iMab interaction with unfolded cytosine-rich sequences is superior to the thermodynamic stability of iMs. Altogether, all these observations can be rationalized by a model where iMab binds to blocks of cytosines in unfolded DNA sequences. This interaction may be similar to the binding mode of KH3 domains of hnRNP K to cytosine-rich DNA strands, but likely involves amino acid residues whose pKa is closer to the physiological conditions (pH 6–8), instead of two arginine residues in the KH3 domain of hnRNP K (34). In view of this model, the addition of iMab at pH 6.0 leads to unfolding of iMs both in bulk-FRET and BLI experiments; however, the binding is reduced at increased pH, presumably due to the deprotonation of the key amino acid residues in iMab, resulting in the ‘apparent’ selectivity as observed for hTeloC sequence. Interestingly, another protein, transcription and splicing regulator hnRNP LL, was shown to unfold iM structures via the formation of a stable complex with cytosine-rich single strands more than 10 years ago (44). In line, a recent study reported strong destabilization of iM structure by a tryptophan-rich cationic peptide (W5K5) (45).
Last but not least, our model in no way contradicts the experimental results observed in other studies that employed iMab, but rather calls for a different interpretation of such data. Thus, the microarray study of Schneekloth et al. included over 10 000 iM-forming sequences containing blocks of 2–10 cytosines, but only 500 ‘negative controls’ where C-tracts were replaced by adenines or thymines; it is thus unsurprising that the authors evidenced strong binding of iMab to cytosine-rich, and no binding to cytosine-poor sequences, whatever pH condition used (30). Likewise, the iMab peaks identified in the CUT&Tag analysis by Richter et al. were subject to a bioinformatics filter through a regular expression comprising four to five blocks of at least two cytosines. Expectedly, a number of recovered sequences were shown to fold iM structures in vitro (22). Interestingly, the high number of iMab peaks observed by Richter et al. in physiological conditions and the fact that iMab peaks are amplified in close proximity of BG4 peaks are both biologically plausible if one accepts that iMab binds single-stranded cytosine-rich regions that become available for the antibody binding, for example upon formation of G-quadruplexes on the opposite DNA strand in open chromatin. Likewise, the iMab foci observed in immunostaining experiments should be rather interpreted as an evidence of single-stranded, cytosine-rich DNA regions. In line with this hypothesis, the recent in-cell NMR study by Trantirek et al. convincingly demonstrated that iM-forming sequences with pHT < 7 are predominantly unfolded in cells, suggesting that only a minor fraction of iM-forming sequences could form iM structures in physiological conditions (23).
The quest for molecular tools capable of unbiased detection of iMs at the cellular level continues, and the question of their in vitro validation prior to the use in cellular systems is of utmost importance. In view of our findings, we strongly suggest that the efficiency and specificity of such tools be examined against a broad panel of bona fide cytosine-rich DNA sequences, both able and unable to form iM structures in a wide range of conditions. During the revision of this work, Dinger et al. described the production of anti-iM nanobody (iMbody); however, its specificity was assessed only with respect to prototypical iM-forming sequences (46). It is of interest for future investigations to determine whether DNA-binding properties of iMbody are similar to, or distinct than those of iMab. Finally, considering the highly specific binding of iMab to cytosine-rich, single-stranded DNA sequences described in our study, this antibody may find important applications for detection and visualization of such sequences at the cellular level.
Supplementary Material
Acknowledgements
The authors thank Dr. Jean-Louis Mergny (École Polytechnique, Palaiseau), Prof. Jean-François Riou and Congcong Li (MNHN, Paris) for stimulating discussions and critical reading of the manuscript. The NanoBio-ICMG platforms (UAR 2607) are acknowledged for their support. A part of graphical abstract was adapted from pictures provided by Servier Medical Art (Servier; https://smart.servier.com/), licensed under a Creative Commons Attribution (CC-BY) 4.0 International License.
Contributor Information
Joseph Boissieras, Chemistry and Modelling for Biology of Cancer (CMBC), CNRS UMR9187, INSERM U1196, Institut Curie, Université Paris Saclay, 91405 Orsay, France.
Hugues Bonnet, Département de Chimie Moléculaire (DCM), CNRS UMR5250, Université Grenoble-Alpes, 38000 Grenoble, France.
Maria Fidelia Susanto, Institut de Pharmacologie et Biologie Structurale (IPBS), CNRS UMR5089, Université Toulouse III – Paul Sabatier (UT3), Toulouse, France.
Dennis Gomez, Institut de Pharmacologie et Biologie Structurale (IPBS), CNRS UMR5089, Université Toulouse III – Paul Sabatier (UT3), Toulouse, France.
Eric Defrancq, Département de Chimie Moléculaire (DCM), CNRS UMR5250, Université Grenoble-Alpes, 38000 Grenoble, France.
Anton Granzhan, Chemistry and Modelling for Biology of Cancer (CMBC), CNRS UMR9187, INSERM U1196, Institut Curie, Université Paris Saclay, 91405 Orsay, France.
Jérôme Dejeu, Département de Chimie Moléculaire (DCM), CNRS UMR5250, Université Grenoble-Alpes, 38000 Grenoble, France; SUPMICROTECH, Université Franche-Comté, Institut FEMTO-ST, 25000 Besançon, France.
Data availability
The data underlying this article are available in the article and in its online Supplementary material.
Supplementary data
Supplementary Data are available at NAR Online.
Funding
Agence Nationale de la Recherche [ANR-21-CE44-0005-02]; CNRS through the MITI interdisciplinary program (PRIME′80, project LiDNA); University Grenoble Alpes Graduate School [ANR-17-EURE-0003]; EIPHI Graduate School [ANR-17-EURE-0002]. Funding for open access charge: Agence Nationale de la Recherche [ANR-21-CE44-0005-02].
Conflict of interest statement. None declared.
References
- 1. Mergny J., Lacroix L., Han X., Leroy J.-L., Hélène C.. Intramolecular folding of pyrimidine oligodeoxynucleotides into an i-DNA Motif. J. Am. Chem. Soc. 1995; 117:8887–8898. [Google Scholar]
- 2. Benabou S., Aviñó A., Eritja R., González C., Gargallo R.. Fundamental aspects of the nucleic acid i-motif structures. RSC Adv. 2014; 4:26956–26980. [Google Scholar]
- 3. Assi H.A., Garavís M., González C., Damha M.J.. I-motif DNA: structural features and significance to cell biology. Nucleic Acids Res. 2018; 46:8038–8056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Day H.A., Pavlou P., Waller Z.A.E.. I-Motif DNA: structure, stability and targeting with ligands. Bioorg. Med. Chem. 2014; 22:4407–4418. [DOI] [PubMed] [Google Scholar]
- 5. Cheng M., Chen J., Ju H., Zhou J., Mergny J.L.. Drivers of i-DNA formation in a variety of environments revealed by four-dimensional UV melting and annealing. J. Am. Chem. Soc. 2021; 143:7792–7807. [DOI] [PubMed] [Google Scholar]
- 6. Kim S.E., Hong S.C.. Two opposing effects of monovalent cations on the stability of i-Motif structure. J. Phys. Chem. B. 2023; 127:1932–1939. [DOI] [PubMed] [Google Scholar]
- 7. Nguyen T., Fraire C., Sheardy R.D.. Linking pH, temperature, and K+ concentration for DNA i-Motif formation. J. Phys. Chem. B. 2017; 121:7872–7877. [DOI] [PubMed] [Google Scholar]
- 8. Gehring K., Leroy J.-L., Guéron M.. A tetrameric DNA structure with protonated cytosine-cytosine base pairs. Nature. 1993; 363:561–565. [DOI] [PubMed] [Google Scholar]
- 9. Reilly S.M., Morgan R.K., Brooks T.A., Wadkins R.M.. Effect of interior loop length on the thermal stability and pKa of i-motif DNA. Biochemistry. 2015; 54:1364–1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Školáková P., Renčiuk D., Palacký J., Krafčík D., Dvořáková Z., Kejnovská I., Bednářová K., Vorlíčková M.. Systematic investigation of sequence requirements for DNA i-motif formation. Nucleic Acids Res. 2019; 9:2177–2189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Iaccarino N., Cheng M., Qiu D., Pagano B., Amato J., Di Porzio A., Zhou J., Randazzo A., Mergny J.L.. Effects of sequence and base composition on the CD and TDS profiles of i-DNA. Angew. Chem. Int. Ed. 2021; 60:10295–10303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Brooks T.A., Kendrick S., Hurley L.. Making sense of G-quadruplex and i-motif functions in oncogene promoters. FEBS J. 2010; 277:3459–3469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Luo X., Zhang J., Gao Y., Pan W., Yang Y., Li X., Chen L., Wang C., Wang Y.. Emerging roles of i-motif in gene expression and disease treatment. Front. Pharmacol. 2023; 14:1136251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Amato J., Iaccarino N., Randazzo A., Novellino E., Pagano B.. Noncanonical DNA secondary structures as drug targets: the prospect of the i-motif. ChemMedChem. 2014; 9:2026–2030. [DOI] [PubMed] [Google Scholar]
- 15. Brown S.L., Kendrick S.. The i-Motif as a molecular target: more than a complementary DNA secondary structure. Pharmaceuticals. 2021; 14:96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Debnath M., Fatma K., Dash J.. Chemical regulation of DNA i-Motifs for nanobiotechnology and therapeutics. Angew. Chem. Int. Ed. 2019; 58:2942–2957. [DOI] [PubMed] [Google Scholar]
- 17. Wright E.P., Huppert J.L., Waller Z.A.E.. Identification of multiple genomic DNA sequences which form i-motif structures at neutral pH. Nucleic Acids Res. 2017; 45:2951–2959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Fleming A.M., Ding Y., Rogers R.A., Zhu J., Zhu J., Burton A.D., Carlisle C.B., Burrows C.J.. 4n-1 Is a “Sweet Spot” in DNA i-Motif folding of 2′-deoxycytidine homopolymers. J. Am. Chem. Soc. 2017; 139:4682–4689. [DOI] [PubMed] [Google Scholar]
- 19. Cheng M., Qiu D., Tamon L., Ištvánková E., Víšková P., Amrane S., Guédin A., Chen J., Lacroix L., Ju H.et al.. Thermal and pH stabilities of i-DNA: confronting in vitro experiments with models and in-cell NMR data. Angew. Chem. Int. Ed. 2021; 60:10286–10294. [DOI] [PubMed] [Google Scholar]
- 20. Takahashi S., Sugimoto N.. Stability prediction of canonical and non-canonical structures of nucleic acids in various molecular environments and cells. Chem. Soc. Rev. 2020; 49:8439–8468. [DOI] [PubMed] [Google Scholar]
- 21. Dzatko S., Krafcikova M., Hänsel-Hertsch R., Fessl T., Fiala R., Loja T., Krafcik D., Mergny J.L., Foldynova-Trantirkova S., Trantirek L.. Evaluation of the stability of DNA i-Motifs in the nuclei of living mammalian cells. Angew. Chem. Int. Ed. 2018; 57:2165–2169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zanin I., Ruggiero E., Nicoletto G., Lago S., Maurizio I., Gallina I., Richter S.N.. Genome-wide mapping of i-motifs reveals their association with transcription regulation in live human cells. Nucleic Acids Res. 2023; 51:8309–8321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Víšková P., Ištvánková E., Ryneš J., Džatko Š., Loja T., Živković M.L., Rigo R., El-Khoury R., Serrano-Chacón I., Damha M.J.et al.. In-cell NMR suggests that DNA i-motif levels are strongly depleted in living human cells. Nat. Commun. 2024; 15:1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bonnet H., Morel M., Devaux A., Boissieras J., Granzhan A., Elias B., Lavergne T., Dejeu J., Defrancq E.. Assessment of presumed small-molecule ligands of telomeric i-DNA by biolayer interferometry (BLI). Chem. Commun. 2022; 58:5116–5119. [DOI] [PubMed] [Google Scholar]
- 25. Berthiol F., Boissieras J., Bonnet H., Pierrot M., Philouze C., Poisson J.-F., Granzhan A., Dejeu J., Defrancq E.. Novel synthesis of IMC-48 and affinity evaluation with different i-Motif DNA sequences. Molecules. 2023; 28:682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zeraati M., Langley D.B., Schofield P., Moye A.L., Rouet R., Hughes W.E., Bryan T.M., Dinger M.E., Christ D.. I-motif DNA structures are formed in the nuclei of human cells. Nat. Chem. 2018; 10:631–637. [DOI] [PubMed] [Google Scholar]
- 27. Wright E.P., Lamparska K., Smith S.S., Waller Z.A.E.. Substitution of cytosine with guanylurea decreases the stability of i-Motif DNA. Biochemistry. 2017; 56:4879–4883. [DOI] [PubMed] [Google Scholar]
- 28. King J.J., Irving K.L., Evans C.W., Chikhale R.V., Becker R., Morris C.J., Peña Martinez C.D., Schofield P., Christ D., Hurley L.H.et al.. DNA G-quadruplex and i-Motif structure formation is interdependent in human cells. J. Am. Chem. Soc. 2020; 142:20600–20604. [DOI] [PubMed] [Google Scholar]
- 29. David C., Martinez P., Zeraati M., Rouet R., Mazigi O., Gloss B., Chan C.-L., Bryan T.M., Smith N.M., Dinger M.E.et al.. Human genomic DNA is widely interspersed with i-motif structures. 2022; bioRxiv doi:14 April 2022, preprint: not peer reviewed 10.1101/2022.04.14.488274. [DOI]
- 30. Yazdani K., Seshadri S., Tillo D., Yang M., Sibley C.D., Vinson C., Schneekloth J.S.. Decoding complexity in biomolecular recognition of DNA i-motifs with microarrays. Nucleic Acids Res. 2023; 51:12020–12030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lacroix L., Liénard H., Labourier E., Djavaheri-Mergny M., Lacoste J., Leffers H., Tazi J., Hélène C., Mergny J.L.. Identification of two human nuclear proteins that recognise the cytosine-rich strand of human telomeres in vitro. Nucleic Acids Res. 2000; 28:1564–1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wang Z., Qiu H., He J., Liu L., Xue W., Fox A., Tickner J., Xu J.. The emerging roles of hnRNPK. J. Cell. Physiol. 2020; 235:1995–2008. [DOI] [PubMed] [Google Scholar]
- 33. Braddock D.T., Baber J.L., Levens D., Clore G.M.. Molecular basis of sequence-specific single-stranded DNA recognition by KH domains: solution structure of a complex between hnRNP K KH3 and single-stranded DNA. EMBO J. 2002; 21:3476–3485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Backe P.H., Messias A.C., Ravelli R.B.G., Sattler M., Cusack S.. X-ray crystallographic and NMR studies of the third KH domain of hnRNP K in complex with single-stranded nucleic acids. Structure. 2005; 13:1055–1067. [DOI] [PubMed] [Google Scholar]
- 35. Du Z., Lee J.K., Tjhen R., Li S., Pan H., Stroud R.M., James T.L.. Crystal structure of the first KH domain of human poly(C)-binding protein-2 in complex with a C-rich strand of human telomeric DNA at 1.7 Å. J. Biol. Chem. 2005; 280:38823–38830. [DOI] [PubMed] [Google Scholar]
- 36. Wu W.Q., Zhang X., Bai D., Shan S.W., Guo L.J.. Mechanistic insights into poly(C)-binding protein hnRNP K resolving i-motif DNA secondary structures. J. Biol. Chem. 2022; 298:102670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Boissieras J., Granzhan A.. Potentiometric titrations to study ligand interactions with DNA i-motifs. Methods Enzymol. 2024; 695:233–254. [DOI] [PubMed] [Google Scholar]
- 38. Concepcion J., Witte K., Wartchow C., Choo S., Yao D., Persson H., Wei J., Li P., Heidecker B., Ma W.et al.. Label-free detection of biomolecular interactions using biolayer interferometry for kinetic characterization. Comb. Chem. High Throughput Screen. 2009; 12:791–800. [DOI] [PubMed] [Google Scholar]
- 39. Apiyo D.O. Biolayer interferometry (Octet) for label-free biomolecular interaction sensing. Handbook of Surface Plasmon Resonance. 2017; The Royal Society of Chemistry; 356–397. [Google Scholar]
- 40. Naik S., Kumru O.S., Cullom M., Telikepalli S.N., Lindboe E., Roop T.L., Joshi S.B., Amin D., Gao P., Middaugh C.R.et al.. Probing structurally altered and aggregated states of therapeutically relevant proteins using GroEL coupled to bio-layer interferometry. Protein Sci. 2014; 23:1461–1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Rogers R.A., Fleming A.M., Burrows C.J.. Unusual Isothermal Hysteresis in DNA i-Motif pH Transitions: a Study of the RAD17 Promoter Sequence. Biophys. J. 2018; 114:1804–1815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Devaux A., Bonnat L., Lavergne T., Defrancq E.. Access to a stabilized i-motif DNA structure through four successive ligation reactions on a cyclopeptide scaffold. Org. Biomol. Chem. 2020; 18:6394–6406. [DOI] [PubMed] [Google Scholar]
- 43. Du Z., Lee J.K., Fenn S., Tjhen R., Stroud R.M., James T.L.. X-ray crystallographic and NMR studies of protein–protein and protein–nucleic acid interactions involving the KH domains from human poly(C)-binding protein-2. RNA. 2007; 13:1043–1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kang H., Kendrick S., Hecht S.M., Hurley L.H.. The transcriptional complex between the BCL2 i-Motif and hnRNP LL is a molecular switch for control of gene expression that can be modulated by small molecules. J. Am. Chem. Soc. 2014; 136:4172–4185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ghosh D., Pratihar S., Govindaraju T.. Designer tryptophan-rich peptide modulates structural dynamics of HIF-1α DNA i -motif DNA. J. Pept. Sci. 2024; 30:e3601. [DOI] [PubMed] [Google Scholar]
- 46. Zeraati M., Ross S.E., Aghaei B., Rajal A.G., King C., Dinger M.E.. Protocol for the production and purification of an i-Motif-specific nanobody. STAR Protoc. 2023; 4:102729. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data underlying this article are available in the article and in its online Supplementary material.