

Keywords: Alzheimer’s disease, amyloid-β, APP/PS1 mice, cerebrovascular endothelial cells, cognitive deficits, gamma-glutamyl transferase 5, neurovascular unit, nuclear factor‐kappa B, synaptic plasticity, β-site APP cleaving enzyme 1
Abstract
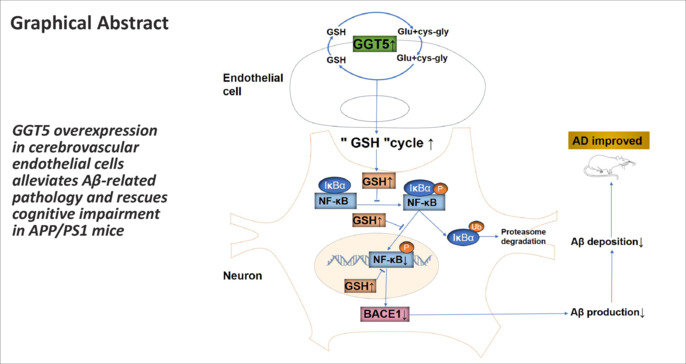
In patients with Alzheimer’s disease, gamma-glutamyl transferase 5 (GGT5) expression has been observed to be downregulated in cerebrovascular endothelial cells. However, the functional role of GGT5 in the development of Alzheimer’s disease remains unclear. This study aimed to explore the effect of GGT5 on cognitive function and brain pathology in an APP/PS1 mouse model of Alzheimer’s disease, as well as the underlying mechanism. We observed a significant reduction in GGT5 expression in two in vitro models of Alzheimer’s disease (Aβ1–42–treated hCMEC/D3 and bEnd.3 cells), as well as in the APP/PS1 mouse model. Additionally, injection of APP/PS1 mice with an adeno-associated virus encoding GGT5 enhanced hippocampal synaptic plasticity and mitigated cognitive deficits. Interestingly, increasing GGT5 expression in cerebrovascular endothelial cells reduced levels of both soluble and insoluble amyloid-β in the brains of APP/PS1 mice. This effect may be attributable to inhibition of the expression of β-site APP cleaving enzyme 1, which is mediated by nuclear factor-kappa B. Our findings demonstrate that GGT5 expression in cerebrovascular endothelial cells is inversely associated with Alzheimer’s disease pathogenesis, and that GGT5 upregulation mitigates cognitive deficits in APP/PS1 mice. These findings suggest that GGT5 expression in cerebrovascular endothelial cells is a potential therapeutic target and biomarker for Alzheimer’s disease.
Introduction
Alzheimer’s disease (AD) is the most common chronic neurodegenerative disease in elderly individuals, and its main clinical symptoms are progressive cognitive decline, memory deficits, and behavioral disorders. AD is characterized by the deposition of amyloid-β (Aβ) in the brain and hyperphosphorylation of tau protein, which leads to the formation of neurofibrillary tangles and neuronal loss (Serrano-Pozo et al., 2021). Aβ production and accumulation in the brain parenchyma are considered to be the main cause of AD, according to the widely accepted AD amyloid hypothesis (Stakos et al., 2020; Mary et al., 2023; Abyadeh et al., 2024). Abnormal Aβ aggregation leads to tau protein hyperphosphorylation, synaptic dysfunction, neuroinflammation, neuronal death, and ultimately dementia (De Strooper and Karran, 2016; Stakos et al., 2020). Hence, abnormal Aβ generation and accumulation in the brain are likely culprits in AD pathogenesis and inevitable consequences of AD progression, and targeting Aβ production is considered a potential strategy for treating AD (Stakos et al., 2020; Lao et al., 2021; Unnisa et al., 2023). Amyloid precursor protein (APP) undergoes sequential cleavage by β-secretase and γ-secretase to produce Aβ. β-Site APP cleaving enzyme 1 (BACE1) is the only β secretase and the rate-limiting step that initiates Aβ production (Lomoio et al., 2020; Lao et al., 2021). Patients with AD exhibit elevated BACE1 expression levels and enzymatic activity in the brain, and inhibiting BACE1 activity or deleting BACE1 in animal models of AD reduces pathological Aβ deposition, rescues memory deficits, and decreased defects in neurological function (Neumann et al., 2015; Das and Yan, 2017; Moussa-Pacha et al., 2020). These findings suggest that BACE1 plays a casual role in AD progression; and indeed, it is considered to be a promising therapeutic target for treating patients with AD (Lomoio et al., 2020; Moussa-Pacha et al., 2020). Pharmacological inhibition of BACE1 combined with therapy to counterbalance BACE1 inhibitor–mediated synaptic deficits has been proposed as a treatment strategy for AD (Das and Yan, 2019; Das et al., 2021). In addition, abnormal modulation of BACE1 expression at the transcriptional and translational levels may be involved in AD pathogenesis (Dobrowolska Zakaria and Vassar, 2018; Das and Yan, 2019). Increased levels of nuclear factor‐kappa B (NF-κB) and BACE1 expression have been reported in patients with sporadic AD (Kim et al., 2019). The transcription factor NF-κB regulates the expression of many genes, including BACE1 (Chen et al., 2012; Tamagno et al., 2012; Kim et al., 2019). Studies have shown that NF-κB activates BACE1 transcription (Snow and Albensi, 2016; Hou et al., 2019; Kim et al., 2019), resulting in Aβ production and aggregation. Aβ oligomers, in turn, activate NF-κB in neurons (Kim et al., 2019). Therefore, inhibiting NF‐κB–mediated BACE1 transcription attenuates Aβ production and accumulation in the brains of patients with AD.
Both patients with AD and animal models of AD exhibit disruptions to the neurovascular system, including alterations in blood vessel density, number, and diameter, which decreases brain perfusion and compromises the integrity of the blood‒brain barrier (Lau et al., 2020). Cerebrovascular endothelial cells mediate exchanges across the blood‒brain barrier to maintain the homeostasis of the neurovascular unit microenvironment, and are thus central to the neuropathological characteristics of AD (Shi et al., 2020; Zhang et al., 2022; Estudillo et al., 2023). Endothelial cell injury impairs neurovascular coupling and decreases cerebral oxygenation, which enhance BACE1 activity, resulting in increased amyloidogenic processing of APP and Aβ aggregation in the brain (Soto-Rojas et al., 2021; Fisher et al., 2022). Excessive Aβ production damages endothelial cell structure, impairs endothelial cell function, alters cerebral circulation, further aggravates neurovascular unit dysfunction, and drives AD progression (Liu et al., 2019; Su et al., 2021). Lau et al. (2020) showed that the expression of gamma-glutamyl transferase (GGT) 5 is downregulated in cerebrovascular endothelial cells in patients with AD. GGTs, glycosylated proteins that are partially embedded in the outer surface of the plasma membrane, are heavily expressed on the luminal side of capillary endothelial cells in the central nervous system (Varga et al., 1985; Heisterkamp et al., 2008). There are only two GGT genes in humans that encode functional proteins: GGT1 and GGT5 (Heisterkamp et al., 2008). GGTs break down extracellular glutathione (GSH) to release its constitutive amino acids, glutamate and cysteine-glycine, which are used for intracellular GSH resynthesis (Bachhawat and Yadav, 2018; Bjørklund et al., 2021). As such, GGT proteins play critical role in the GSH cycle, also known as the γ-glutamyl pathway (Bachhawat and Yadav, 2018; Ho et al., 2022), and help maintain intracellular and extracellular GSH homeostasis (Bachhawat and Kaur, 2017; Suzuki et al., 2020). A recent study found that GSH directly inhibits several steps within the NF-κB signaling pathway (Fraternale et al., 2021). Thus, GGT5 might play a critical role in AD etiology and pathogenesis by repressing NF-κB–mediated activation of BACE1 expression. However, the effect of GGT5 expression in cerebrovascular endothelial cells on AD onset and progression is unclear. In particular, it is unknown whether GGT5 upregulation can attenuate amyloid pathology and cognitive deficits in AD. Therefore, in this study we assessed the effects of GGT5 overexpression in cerebrovascular endothelial cells on cognitive function in APP/PS1 mice using multiple behavior tests, in vivo electrophysiology, and pathological examinations. Furthermore, mRNA sequencing was performed to determine the molecular mechanism by which GGT5 potentially attenuates Aβ accumulation and exerts neuroprotective effects.
Methods
Animals
The B6C3-Tg APPswe/PS1dE9 (APP/PS1) double transgenic mice used in this study express mutant human presenilin1 (DeltaE9) and human amyloid precursor protein with the Swedish mutation (APPswe). Male APP/PS1 mice (Stock# 004462, RRID: MMRRC_034829-JAX) (Jankowsky et al., 2001) and wild-type (WT) male littermates were purchased from Hangzhou Ziyuan Experimental Animal Technology Co., Ltd. (Zhejiang, China, license No. SCXK (Zhe) 2019-0004). We only used male APP/PS1 mice, similar to an earlier study (Xu et al., 2021a), to avoid the effect of female mouse hormones on performance in behavioral experiments and potential sex-based differences in AD pathology. The genotypes of all of the mice were confirmed by polymerase chain reaction (PCR) of toe tissue. All mice were housed in a specific pathogen–free facility under standard conditions (room temperature: 25 ± 2°C, air humidity: 50%–60%, 12/12-hour light-dark cycle) with ad libitum access to food and water. All animal experiments were approved by the Animal Ethics Committee of First Hospital of Shanxi Medical University (approval No. DWYJ-2023-004; March 2, 2023) and performed in strict accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
Eight-month-old APP/PS1 mice (n = 20) and their WT littermates (n = 20) were randomly divided into four groups: the WT vehicle group (WT + Vector) (n = 10), WT GGT5 overexpression group (WT + GGT5) (n = 10), APP/PS1 vehicle group (APP/PS1 + Vector) (n = 10), and APP/PS1 GGT5 overexpression group (APP/PS1 + GGT5) (n = 10). An adeno-associated virus (AAV) encoding GGT5 driven by the endothelial-specific promoter TIE (HBAAV2/9-TIE-m-Ggt5-3xflag-zsgreen, 100 μL/mouse, virus titer 1.5 × 1012 vector genomes per mL (vg/mL)) and a negative control AAV vector (HBAAV2/9-TIE-zsgreen, 100 μL/mouse, virus titer 1.4 × 1012 vg/mL) were administered to APP/PS1 mice by tail vein injection (Wu et al., 2021). Recombinant AAVs were constructed by Hanbio Biotechnology Co., Ltd. (Shanghai, China). One month after injection, behavioral tests were conducted to compare the performance of APP/PS1 mice and their WT littermates (Figure 1).
Figure 1.
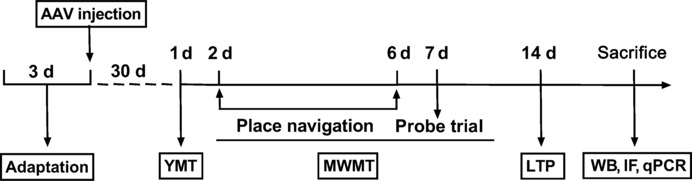
Experimental flowchart.
AAV: Adeno-associated virus; IF: immunofluorescence; LTP: long-term potentiation; MWMT: Morris water maze test; qPCR: quantitative polymerase chain reaction; WB: Western blot; YMT: Y-maze test.
Spontaneous Y-maze test
The spontaneous Y-maze test was used to evaluate the spatial working memory of mice (Kraeuter et al., 2019). The Y-maze (RWD Life Science Co., Ltd., Shenzhen, China) consisted of three gray plastic closed arms of equal length, at an angle of 120° from each other. Mice were placed in the central triangular area of the Y-maze and allowed to explore all three arms freely for 8 minutes. Entry into an arm was defined as the mouse placing all four of its paws completely inside one of the arms, and correct spontaneous alternation was defined as consecutive entry into each of the three different arms in sequence. Total arm entries and the percentage of correct spontaneous alternations (number of correct spontaneous alternations/(total number of arm entries – 2) × 100%) were recorded.
Morris water maze test
The Morris water maze test was used to assess long-term spatial learning and memory (Su et al., 2021). The water maze apparatus was a circular tank 120 cm in diameter and 50 cm in height filled with water (22 ± 2°C) that was opacified by the addition of nontoxic titanium dioxide to conceal anything under the water surface from the mice but allow video recording (Ethovision3.0, Noldus Information Technology, Wageningen, the Netherlands). The tank was divided into four virtual quadrants, with an escape platform (10 cm in diameter) hidden 1 cm beneath the water surface in the target quadrant. During the initial 5-day positioning navigation test, mice were placed in the water in different quadrants facing the wall of the tank and allowed to search for the hidden platform for 60 seconds. The escape latency was defined as the time that elapsed between entering the water and climbing onto the platform. When a mouse found and climbed onto the platform, it was allowed to stay on the platform for 5 seconds. If a mouse failed to find the platform within 60 seconds, it was guided to the platform and allowed to stay on the platform for 20 seconds (the escape latency was recorded as 60 seconds). In the probe trial performed on the 6th day, the platform was removed and the mice were allowed to swim freely for 60 seconds. Each mouse’s swimming path, escape latency, percentage of time spent swimming in the target quadrant, and number of platform crossings were recorded and analyzed using Ethovision XT10 software (Noldus Information Technology).
In vivo hippocampal long-term potentiation recording
After the behavioral tests were complete, the mice were anesthetized by intraperitoneal injection of 1% pentobarbital sodium solution (10 mL/kg; Lulong Biotechnology Co., Ltd. Shanghai, China) and fixed in a stereotaxic instrument (RWD Life Science, Shenzhen, China, 68016). As described previously (Li et al., 2022b), a 2-mm-diameter hole was made at 2 mm posterior to bregma and 1.5 mm lateral to the midline. A bound bipolar concentric stimulation/recording electrode (Alpha Omega Engineering Ltd., Shanghai, China) was implanted through the hole using stereotaxic coordinates and lowered by 20-µm increments to evoke field excitatory postsynaptic potentials (fEPSPs). fEPSPs recordings were taken from the stratum radiatum in the CA1 area of the hippocampus in response to stimulation of the ipsilateral Schaffer collateral pathway. Before each experiment, an input‒output curve was generated with different stimulus currents (0−0.2 mA) at a frequency of 0.033 Hz to determine the intensity of the current used during the long-term potentiation (LTP) experiment. The intensity of the test stimulus was set at a level that produced 30%−50% of the maximum fEPSP. Testing stimuli were applied at 30-second intervals over 30 minutes to measure basal synaptic transmission. Afterwards, paired pulse facilitation was induced by paired pulse stimulation (three pulse series with an interseries interval of 30 seconds and an interstimulus interval of 50 ms) to determine whether a presynaptic mechanism was involved. Finally, LTP was induced using a high-frequency stimulation (HFS) protocol consisting of square pulses (three series of 20 pulses at 200 Hz separated by 30 seconds) and recorded for 60 minutes.
Tissue processing
After the electrophysiological tests were complete, some of the mice in each group were used for western blot experiments, while the remaining mice were used for immunofluorescence staining. For western blotting, mice were anesthetized by intraperitoneal injection of 1% pentobarbital sodium solution (10 mL/kg), then transcardially perfused with normal saline (0.9%) and rapidly decapitated. The brains were removed quickly, and the cortical and hippocampal tissues were harvested on ice, frozen in liquid nitrogen, and stored at −80°C (Su et al., 2021). For immunofluorescence staining, mice were anesthetized by intraperitoneal injection of 1% pentobarbital sodium solution (10 mL/kg), transcardially perfused with normal saline (0.9%), and then perfused with 4% paraformaldehyde. Next, the brains were removed, fixed in 4% paraformaldehyde for 24 hours, and dehydrated in a 15% sucrose solution for 24 hours and a 30% sucrose solution for 48 hours (Su et al., 2021). Subsequently, continuous coronal sections (25-μm thick) were made with a cryostat (CM1950, Leica, Shanghai, China), and the sections were stored at −80°C.
Western blot assay
Hippocampal and cortical tissues and bEnd.3, hCMEC/D3, and HT22 cells were homogenized in precooled RIPA buffer containing protease inhibitor (Boster, Wuhan, China, AR1183) and phenylmethyl sulfonyl fluoride (AR1179, Boster) (10 mL radio immunoprecipitation assay buffer/1 g tissue) at a ratio of 100 RIPA:1 protease inhibitor: 1 PMSF. The homogenates were centrifuged at 4°C (13,800 × g) for 30 minutes, and the supernatants were collected. The protein concentration in the collected supernatants was detected using a bicinchoninic acid assay kit (AR0146, Boster), and the samples were diluted with sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) protein buffer (5×) (AR0131, Boster) to equivalent concentrations. The protein samples were then denatured at 100°C for 5 minutes, electrophoresed on 12% or 15% SDS‒PAGE gels (AR0138, Boster) and transferred to polyvinylidene fluoride membranes (0.45 μm or 0.22 μm, Millipore, Shanghai, China). The membranes were blocked with skim milk for 2 hours at 22 ± 2°C and then incubated with primary antibodies at 4°C overnight. The next day, after rinsing with Tris-buffered saline containing Tween, the membranes were incubated with the appropriate secondary antibodies for 90 minutes and then treated with enhanced chemiluminescence luminescent solution (AR1171, Boster). Bands were imaged on an imaging system (Bio-Rad, Hercules, CA, USA) and the optical density (expressed as a ratio to β-actin) was analyzed using ImageJ software V1.8.0. The primary antibodies used were as follows: anti-GGT5 (human, mouse, 1:500, ABclone, Wuhan, China, Cat# A14374, RRID: AB_2761241), anti-agrin (AGRN; human, mouse, 1:500, Santa Cruz Biotechnology, Santa Cruz, CA, USA, Cat# sc-374117, RRID: AB_10947251), anti-LRP1 (human, mouse, 1:5000, Abcam, Cambs, Cambridge, UK, Cat# ab92544, RRID: AB_2234877), anti-amyloid-β (D54D2) (mouse, 1:1000, Cell Signaling, Boston, MA, USA, Cat# 8243, RRID: AB_2797642), anti-amyloid-β (1–42) (D9A3A) (mouse, 1:1000, Cell Signaling, Cat# 14974, RRID: AB_2798671), anti-APP C-terminal fragment (CTF; mouse, 1:1000, Sigma, Munich, Bayern, Germany, Cat# A8717, RRID: AB_258409), anti-BACE1 (D10E5) (mouse, 1:1000, Cell Signaling, Cat# 5606, RRID: AB_1903900), anti-NF-κB p65 (F-6) (mouse, 1:200, Santa Cruz Biotechnology, Cat# sc8008, RRID: AB_628017), anti-phosphor-nuclear factor kappa B (p-NF-κB) p65 (Ser468) (mouse, 1:1000, Cell Signaling, Cat# 3039, RRID: AB_330579), anti-inhibitor-kappaBα (IκBα) (H-4) (mouse, 1:100, Santa Cruz Biotechnology, Cat# sc-1643, RRID: AB_627772), anti-neuronal pentraxin II (NPTX2) (mouse, 1:5000, Abcam, Cat# ab277523, RRID: AB_3075509), and anti-β-actin (human, mouse, 1:5000, Boster, Cat# BM0627, RRID: AB_2814866). The secondary antibodies used were goat anti-mouse IgG-horseradish peroxidase (HRP) conjugate (1:5000, Boster, Cat# BA1050, RRID: AB_10892412) and goat anti-rabbit IgG-HRP conjugate (1:5000, Boster, Cat# BA1054, RRID: AB_10892383).
Immunofluorescence staining
Brain sections were rinsed in phosphate-buffered saline (PBS) and blocked with 5% bovine serum albumin (AR1006, Boster) for 1 hour at room temperature. The sections were then incubated with primary antibodies at 4°C overnight, after which they were rinsed in PBS containing Tween and incubated with appropriate fluorescence-conjugated secondary antibodies at 37°C in the dark. Next, the sections were washed in PBS containing Tween, incubated with 4,6-diamino-2-phenyl indole (AR1176, Boster) for 6 minutes, and imaged using a fluorescence microscope (Olympus, Tokyo, Japan). The images were analyzed using ImageJ software. The primary antibodies used were anti-GGT5 (mouse, 1:250, Bioss, Beijing, China, Cat# bs-13349R, RRID: AB_3075527), anti-CD31/platelet endothelial cell adhesion molecule-1 (PECAM-1; mouse, 1:100, Santa Cruz Biotechnology, Cat# sc-376764, RRID: AB_2801330), and anti-β-amyloid (6E10; mouse, 1:1000, Biolegend, Santiago, CA, USA, Cat# 803015, RRID: AB_2565328). The secondary antibodies included goat anti-mouse IgG-Cy3 (1:100, Boster, Cat# BA1031, RRID: AB_10890402), goat anti-rabbit IgG-Cy3 (1:300, Boster, Cat# BA1032, RRID: AB_2716305), goat anti-rabbit IgG-Cy3 (1:500, Invitrogen, Shanghai, China, A10520, RRID: AB_10563288), and goat anti-mouse IgG-488 (1:300, Invitrogen, Cat# A32723, RRID: AB_2633275).
Cell culture and Aβ preparation
A murine cerebral microvascular endothelial cell line (bEnd.3; Cat# CL-0598; RRID: CVCL_0170) was purchased from Procell Life Science & Technology Co., Ltd. (Wuhan, China). An immortalized human cerebral microvascular endothelial cell line (hCMEC/D3; Cat# ZQ0961; RRID: CVCL_1985) was purchased from Shanghai Zhong Qiao Xin Zhou Biotechnology Co., Ltd. (Shanghai, China). A murine hippocampal neuronal cell line (HT22; Cat# CX0146; RRID: CVCL_0321) was purchased from Wuhan Boshide Bioengineering Co., Ltd. (Wuhan, China). All cells were cultured at 37°C with a humidified atmosphere containing 5% CO2 in complete growth medium (90% Dulbecco’s modified Eagle medium (DMEM) + 5%−10% fetal bovine serum + 1% 100 U/mL penicillin and 100 μg/mL streptomycin). Cells were seeded into six-well plates at an appropriate density and cultured for subsequent experiments, such as plasmid transfection and protein extraction.
One milligram of recombinant human Aβ1−42 peptide (ChinaPeptides, Shanghai, China) was dissolved in 60 μL of dimethyl sulfoxide (DMSO) (Li et al., 2022b) and then diluted to the final concentration (5 μM) with complete medium. After plasmid transfection, cells were cultured with Aβ1−42 for 48 hours.
Plasmid transfection and detection of GSH and oxidized glutathione contents
bEnd.3 cells were seeded into six-well plates at a density of 1 × 105 cells per well, and 4 μg of GGT5 plasmid (pCDNA3.1-CMV-GGT5-3flag-EF1-ZsGreen-T2A-Puro, NM_011820.5) or negative control vector plasmid DNA (pCDNA3.1-CMV-MCS-3flag-EF1-ZsGreen-T2A-Puro; both from Hanbio Biotechnology Co., Ltd.) mixed with 6 μL of LipoFiter3.0 (Hanbio Biotechnology Co., Ltd.) was added to each well.
After culturing for an appropriate amount of time, GGT5 expression was detected by western blotting, as described above. The intracellular and extracellular GSH and oxidized glutathione (GSSG) contents in each group were detected 12, 24, and 48 hours after transfection using GSH and GSSG detection kits (Beyotime Biotechnology, Shanghai, China), and the GSH/GSSG ratio was calculated.
Cell counting kit-8 assay
We used a cell counting kit-8 (CCK-8) (Boster) to measure HT22 cell viability. First, 1 × 105 HT22 cells were added to each well of a six-well plate. After culturing for 24 hours, the cells were treated with 3 μL DMSO + 2 mL conditioned medium from bEnd.3 cells transfected with the vector plasmid and 3 μL DMSO + 2 mL conditioned medium from bEnd.3 cells transfected with the GGT5 plasmid (vehicle group) or 3 μL Aβ1−42 + 2 mL conditioned medium from bEnd.3 cells transfected with the vector plasmid and 3 μL Aβ1−42 + 2 mL conditioned medium from bEnd.3 cells transfected with the GGT5 plasmid (Aβ1−42 intervention group). After incubating the cells for 24, 48, or 72 hours, 1 × 104 HT22 cells were added to each well of a 96-well plate. Three to five replicate wells were included for each group. 10 μL of CCK-8 reagent was added to 90 μL of DMEM, the mixture was added to each well, and the plates were reincubated for another 1−2 hours. The absorbance was then detected at 450 nm using an absorbance reader (BioTek, Burlington, VT, USA). The cell survival rate in each group was calculated as follows: (test well − blank well) optical density (OD) value/(vehicle well − blank well) OD value × 100%.
mRNA sequencing
hCMEC/D3 cells were cultured in a six-well plate with 3 μL of DMSO + 2 mL of complete growth medium (vehicle group) or 3 μL of Aβ1−42 + 2 mL of complete growth medium (Aβ1−42 intervention group). After 48 hours, 1 mL of TRIzol (Invitrogen) was added to each well to extract total RNA. Mouse tissue samples were harvested and processed as previously described (Li et al., 2022b), and total RNA was extracted from cortical tissues from the WT + Vector group, APP/PS1 + Vector group, and APP/PS1 + GGT5 group (n = 3/group) using TRIzol reagent according to the manufacturer’s instructions. RNA purity and concentration were assessed using a NanoPhotometer® spectrophotometer (Implen, Huntsville, AL, USA), and RNA integrity of was assessed using the RNA Nano 6000 analysis feature of the BioAnalyst 2100 system (Agilent Technologies, Santa Clara, CA, USA). Finally, mRNA sequencing (mRNA-seq), quantification, and differential gene expression analysis were performed by Novogene Biotechnology (Beijing, China).
Real-time polymerase chain reaction
Gene-specific primers for quantitative polymerase chain reaction (qPCR) were designed and synthesized by Sangon Biotech (Shanghai, China), and the sequences are shown in Tables 1 and 2. Total RNA was extracted from hCMEC/D3 cells and cortical tissue as described above. RNA purity and concentration were assessed, genomic DNA was removed using 4× gDNA wiper Mix according to the manufacturer’s instructions (Vazyme, Shanghai, China), and 1 μg total RNA was reverse transcribed to synthesize single-stranded complementary DNA (cDNA) using HiScriptIII RT SuperMix according to the manufacturer’s instructions (Vazyme). Each 20-µL qPCR reaction mixture contained 2 µL of cDNA, 10 µL of ChamQ Universal SYBR qPCR Master Mix (Vazyme), 0.4 µL (10 µM) each of forward and reverse primers, and 7.2 µL ddH2O. Real-time qPCR was performed on a Roche LightCycler 480IIreal-time PCR instrument (Roche, Basel, Switzerland). The reference genes used were glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (cells) and β-actin (tissues). The 2−ΔΔCt method was applied to analyze relative changes in gene expression (Flanigan et al., 2023).
Table 1.
Primer sequences used for qPCR in hCMEC/D3 cells
| Target genes | Forward primer sequences | Reverse primer sequences |
|---|---|---|
| GAPDH | 5'-CAG GAG GCA TTG CTG ATG AT-3' | 5'-GAA GGC TGG GGC TCA TTT-3' |
| AGRN | 5'-CCG CCA GGA GAA TGT CTT CAA-3' | 5'-TTT CGT AGG TGA CTC CGT CGT-3' |
| NAMPT | 5'-AGC AGA ACA CAG TAC CAT AAC A-3' | 5'-CCC ATA TTT TCT CAC ACG CAT T-3' |
| RPS27A | 5'-GGA CGT ACT TTG TCT GAC TAC A-3' | 5'-TAT TTC AGG ACA GCC AGC TTA A-3' |
| GGT5 | 5'-GTC AGC CTA GTC CTG CTG G-3' | 5'-GGA TGG CTC GTC CAA TAT CCG-3' |
| ARHGEF17 | 5'-TCC CTG TCA AAT CCA GAT ATC G-3' | 5'-GGA TGG GTA TGG GTT CAA GTT-3' |
| PLOD1 | 5'-AAG CCG GAG GAC AAC CTT TTA-3' | 5'-GCG AAG AGA ATG ACC AGA TCC-3' |
| MRC2 | 5'-CGT AGG GTT CTC TTA CCA CAA T-3' | 5'-GTG CTC AAA GAA CTT GTA CTC G-3' |
| TECR | 5'-AAG ACC CAT CCG CAG TGG TA-3' | 5'-CGT ACT CTG TTA GGA AGA CCG TC-3' |
| EFHD2 | 5'-GAT GTT CAA GCA GTA TGA TGC CGG-3' | 5'-AGA TCA GGA GGA ACT CCC GG-3' |
| MCAM | 5'-GTC AAT TTA ACC ACC CTC ACA C-3' | 5'-GCT TGC CCT TCT TAT AGA GGA A-3' |
| LRP1 | 5'-GTC TAC CAT CAC ACC TAC GAG-3' | 5'-CAG ACT GAG GGA GAT GTT GAT G-3' |
AGRN: Agrin; ARHGEF17: Rho guanine nucleotide exchange factor (GEF) 17; GAPDH: glyceraldehyde-3-phosphate dehydrogenase; GGT5: gamma-glutamyltransferase 5; LRP1: low density lipoprotein receptor related protein 1; MCAM: melanoma cell adhesion molecule; MRC2: mannose receptor C type 2; PLOD1: procollagen lysine-1,2-oxoglutarate-5-dioxygenase 1; qPCR: quantitative polymerase chain reaction; RPS27A: ribosomal protein S27A.
Table 2.
Primer sequences used for qPCR in cortex tissues of mice
| Target genes | Forward primer sequences | Reverse primer sequences |
|---|---|---|
| β-Actin | 5'-GTG CTA TGT TGC TCT AGA CTT CG-3' | 5'-ATG CCA CAG GAT TCC ATA CC-3' |
| Nptx2 | 5'-CAC GGT GGG AGG CAG ATT TGA TG-3' | 5'-TGG AGC AGT TGG CGA TGT TGA TG-3' |
| Nfkbia | 5'-CTG AAA GCT GGC TGT GAT CCT GAG-3' | 5'-CTG CGT CAA GAC TGC TAC ACT GG-3' |
| GGT5 | 5'-TGC TGG GTG TAG GTC TAG GT-3' | 5'-AAC ACC AGT ACA GAC CAG GG-3' |
GGT5: Gamma-glutamyltransferase 5; Nfkbia: nuclear factor of kappa light polypeptide gene enhancer in B-cells inhibitorα; Nptx2: neuronal pentraxin II; qPCR: quantitative polymerase chain reaction.
Statistical analysis
We used a resource equation approach and referred to previous studies (Arifin and Zahiruddin, 2017; Gong et al., 2023; Huang et al., 2023) to determine the appropriate sample size for the in vivo experiments, ultimately including 40 animals. The investigators were blinded to the group assignments while performing the experiments. The data were analyzed using SPSS Statistics for Windows, Version 26.0 (IBM Corp., Armonk, NY, USA) and GraphPad Prism version 8.0.0 for Windows (GraphPad Software, Boston, MA, USA, www.graphpad.com). All experimental data are expressed as mean ± standard error of the mean. The escape latency and swimming speed in the Morris water maze test were analyzed by two-way analysis of variance with repeated measures followed by post hoc Tukey’s multiple comparison tests, and other data were statistically analyzed by independent-samples t-test or one-way analysis of variance followed by Tukey’s post hoc test or least significant difference test. P < 0.05 indicated a statistically significant difference.
Results
GGT5 expression is decreased in AD mouse brains and AD model cells compared with controls
First, we performed mRNA-seq analysis of hCMEC/D3 cells treated with vehicle or Aβ1−42 and identified 178 differentially expressed genes (Figure 2A and Additional Table 1 (290.4KB, pdf) ). Comparing this list with an existing AD single-cell sequencing database yielded a list of 11 differentially expressed genes that are potentially related to AD (Lau et al., 2020; Figure 2B and Additional Table 1 (290.4KB, pdf) ). Nine of these genes were downregulated in Aβ1−42-treated cells compared with vehicle-treated cells, including GGT5, AGRN, LRP1, MCAM, EFHD2, TECR, PLOD1, MRC2, and ARHGEF17, while two were upregulated: NAMPT and RPS27A. qPCR analysis verified that, compared with the vehicle group, AGRN (P < 0.05), GGT5 (P < 0.05), ARHGEF17 (P < 0.05), and MCAM (P < 0.05) expression levels in hCMEC/D3 cells treated with Aβ1−42 were significantly lower, while LRP1 (P < 0.05) expression was significantly higher (Figure 2C). Western blot confirmed that GGT5 (P < 0.05), AGRN (P < 0.05), and LRP1 (P < 0.05) protein expression levels were lower in hCMEC/D3 cells treated with Aβ1−42 than in the vehicle group (Figure 2D and E). Similar results were obtained in bEnd.3 cells: that is, GGT5 expression was downregulated in bEnd.3 cells treated with Aβ1−42 (Figure 2F and G). Notably, GGT5 expression in the cortex and hippocampus of APP/PS1 mice was significantly lower (P < 0.05) than that seen in WT control mice (Figure 2H−K). These findings indicate that GGT5 downregulation may play a critical role in AD occurrence and development and suggest that increasing GGT5 expression in cerebrovascular endothelial cells could reduce pathology and improve cognitive ability in APP/PS1 mice.
Figure 2.
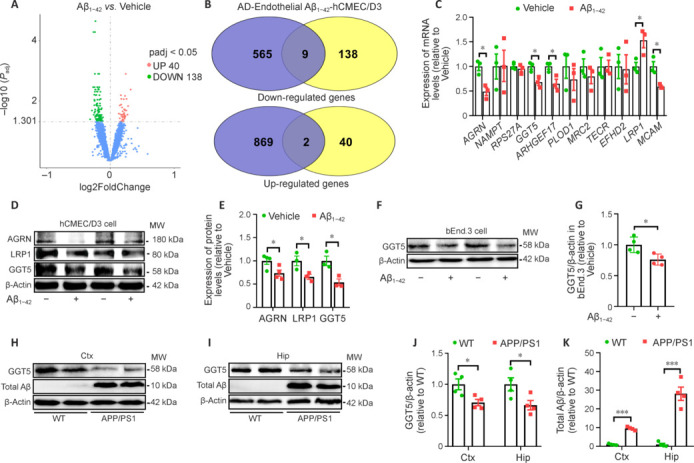
GGT5 expression is downregulated in Aβ 1–42 –treated hCMEC/D3 and bEnd.3 cells and in the brains of APP/PS1 mice.
(A) mRNA-seq analysis of hCMEC/D3 cells identified 178 genes that were differentially expressed between the vehicle and Aβ1–42 intervention groups. (B) Nine downregulated genes and two upregulated genes were selected by comparing the 178 differentially expressed genes with an existing AD single-cell sequencing database. (C) qPCR verification of the expression levels of the 11 selected genes (n = 3 per group). (D, E) Representative western blots (D) and quantitative analysis (E) of AGRN, LRP1, and GGT5 expression in hCMEC/D3 cells treated with vehicle or Aβ1–42 (each row is an image from a different blot) (n = 3–4 per group). (F, G) Representative western blots (F) and quantitative analysis (G) of GGT5 expression in bEnd.3 cells treated with vehicle or Aβ1–42 (n = 4 per group). (H–K) Representative western blots of GGT5 and Aβ in the Ctx (H) and Hip (I) and quantitative analysis of GGT5 (J) and Aβ (K) expression in the Ctx and Hip of mice (n = 4 per group). Data are expressed as mean ± SEM. *P < 0.05, ***P < 0.001 (one-way analysis of variance followed by Tukey’s post hoc test [J and K], or independent-samples t-test [C, E and G]). AD: Alzheimer’s disease; AGRN: Agrin; ARHGEF17: Rho guanine nucleotide exchange factor (GEF) 17; Aβ: amyloid-β; Ctx: cortex; GAPDH: glyceraldehyde-3-phosphate dehydrogenase; GGT5: gamma-glutamyltransferase 5; Hip: hippocampus; LRP1: low density lipoprotein receptor related protein 1; MCAM: melanoma cell adhesion molecule; MRC2: mannose receptor c type 2; PLOD1: procollagen lysine-1,2-oxoglutarate-5-dioxygenase 1; RPS27A: ribosomal protein S27A; WT: wide type.
GGT5 overexpression improves the learning and memory in APP/PS1 mice
First, we used immunofluorescence staining to verify stable transfection of the AAV9 construct and GGT5 overexpression in the cerebrovascular endothelial cells of APP/PS1 mice. The immunofluorescence results revealed that CD31-positive cerebrovascular endothelial cells were also positive for GGT5 staining in the brains of mice from all four experimental groups (Additional Figure 1 (3.4MB, tif) A and B). Quantification showed that the mean GGT5 fluorescence intensity in the WT + GGT5 group was stronger than that in the WT + Vector group (P < 0.05; Additional Figure 1 (3.4MB, tif) C). Similarly, the mean GGT5 fluorescence intensity was higher in the APP/PS1 + GGT5 group than that in the APP/PS1 + Vector group (P < 0.05; Additional Figure 1 (3.4MB, tif) C). These observations indicated that transfection was successful and GGT5 was stably overexpressed in the cerebrovascular endothelial cells of APP/PS1 mice.
To evaluate the effect of GGT5 overexpression on learning and memory in mice, we performed behavioral tests, including the spontaneous Y-maze and Morris water maze tests. In the spontaneous Y-maze test, there was no difference in total arm entry numbers among the groups (P > 0.05; Figure 3A). The percentage of correct spontaneous alternations in the APP/PS1 + Vector group was lower than that seen in the WT + Vector group (P < 0.05), whereas the APP/PS1 + GGT5 group displayed a significantly greater percentage of correct spontaneous alternations than the APP/PS1 + Vector group (P < 0.01; Figure 3B). These results suggest that GGT5 overexpression improved the short-term working memory of APP/PS1 mice. In the Morris water maze test, there were no differences in swimming speed among the four groups (P > 0.05; Figure 3C). In the positioning navigation test, the escape latency from the 3rd day to the 5th day was longer in the APP/PS1 + Vector group than in the WT + Vector group (3rd day: P < 0.05; 4th day: P < 0.05; 5th day: P < 0.01), while the escape latency in the APP/PS1 + GGT5 group (3rd day: P < 0.05; 4th day: P < 0.05; 5th day: P < 0.01) was shorter than that observed in the APP/PS1 + Vector group (Figure 3D). Representative swimming trajectories of mice from each group during the positioning navigation test performed on day 5 are shown in Figure 3E. In the probe trial performed on the 6th day, the percentage of time spent swimming in the target quadrant was lower in the APP/PS1 + Vector group than in the WT + Vector group (P < 0.001), and higher in the APP/PS1 + GGT5 group than in the APP/PS1 + Vector group (P < 0.05; Figure 3F). Similarly, the number of platform crossings in the APP/PS1 + Vector group was significantly decreased compared with that in the WT + Vector group (P < 0.05), and significantly increased in the APP/PS1 + GGT5 group (P < 0.05; Figure 3G). Representative swimming trajectories of mice from each group in the probe trial are shown in Figure 3H. These observations indicate that GGT5 overexpression improved spatial learning and memory in APP/PS1 mice.
Figure 3.
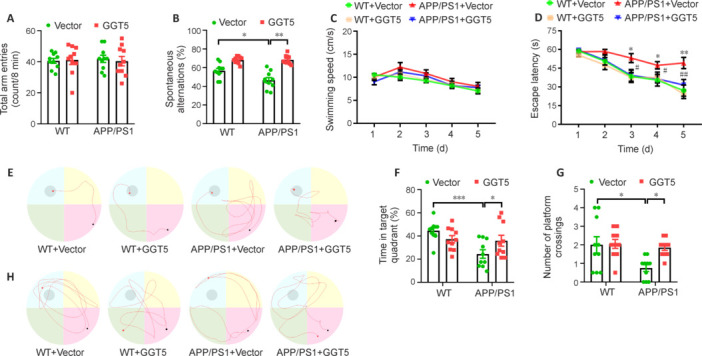
GGT5 overexpression in cerebrovascular endothelial cells improves learning and memory in APP/PS1 mice.
(A, B) Histograms of total arm entries (A) and the percentage of correct spontaneous alternations (B) within 8 minutes in the Y-maze. (C, D) The average swimming speed (C) and escape latency (D) during the positioning navigation test for the mice in each group. (E) Representative swimming trajectories of mice from each group in the positioning navigation test on the 5th training day. (F, G) Histograms of the percentage of time spent swimming in the target quadrant (F) and the number of platform crossings (in 60 seconds) (G) in the probe trial. (H) Representative swimming trajectories of mice from each group in the probe trial. Data are expressed as mean ± SEM (n = 10 per group). *P < 0.05, **P < 0.01, ***P < 0.001, APP/PS1 + Vector group vs. WT + Vector group; #P < 0.05, ##P < 0.01, APP/PS1 + GGT5 group vs. APP/PS1 + Vector group (in D) (one-way analysis of variance followed by Tukey’s post hoc test [A, B, F and G], two-way analysis of variance with repeated measures followed by Tukey’s post hoc test [C and D]). GGT5: Gamma-glutamyltransferase 5; WT: wide type.
GGT5 overexpression reverses hippocampal LTP impairment in APP/PS1 mice
Hippocampal LTP is crucial for learning and memory formation (Tan et al., 2020; Li et al., 2022a). There is extensive evidence that LTP impairment is strongly correlated with cognitive deficits in AD (Du et al., 2020; Li and Stern, 2022; Li et al., 2022b). To determine whether the improvement in cognitive performance induced by GGT5 overexpression was associated with improved LTP, we recorded changes in fEPSPs in the hippocampal CA1 region of mice (Figure 4A). The input‒output curves showed that fEPSP amplitude increased with stimulus intensity in each group, and there was no difference in fEPSP amplitude among groups at the same stimulation intensity (Figure 4B), suggesting that basal synaptic excitability was not affected by genotype or GGT5 overexpression in cerebrovascular endothelial cells. In addition, there was no significant difference in paired pulse facilitation ratios (fEPSP2/fEPSEP1) among the groups (P > 0.05; Figure 4C), indicating that presynaptic transmitter release was not affected by genotype or GGT5 overexpression in cerebrovascular endothelial cells. Furthermore, no significant difference was observed in fEPSP slopes during the 30 minutes before HFS, suggesting that basic hippocampal neuron synaptic transmission in the four groups was not impaired. Although the fEPSP slope increased immediately after HFS in all four groups, there was no significant difference among the groups, suggesting that LTP induction was not affected by genotype or GGT5 overexpression in cerebrovascular endothelial cells. However, 50–60 minutes after HFS, there were discernible variations in the fEPSP slope among the four groups. Compared with the WT + Vector group, the fEPSP slope in the APP/PS1 + Vector group was significantly lower (P < 0.01), while that in the APP/PS1 + GGT5 group was significantly higher (P < 0.05; Figure 4D−F). These results showed that GGT5 overexpression in cerebrovascular endothelial cells mitigates synaptic plasticity deficits in APP/PS1 mice.
Figure 4.
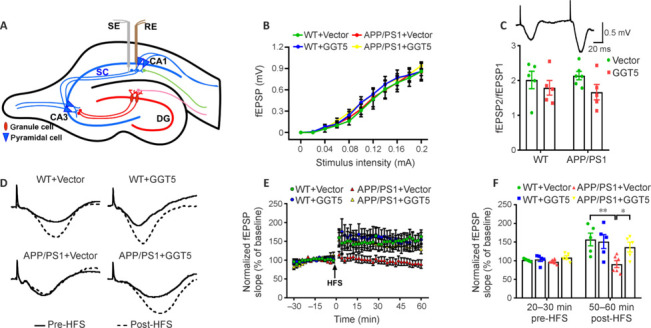
GGT5 overexpression in cerebrovascular endothelial cells enhances synaptic plasticity in the hippocampus of APP/PS1 mice.
(A) The location of the SE and RE in the hippocampus. Created with Adobe Illustrator CS5. (B) I–O curves recorded in the hippocampal CA1 region of mice in each group. (C) Histogram showing the PPF ratios in each group, and a representative fEPSP traces induced by PPF. (D) Sample fEPSP traces before HFS (solid line) and 60 minutes after HFS (dotted line) for each group. (E) Graph displaying the fEPSP slope in the hippocampal CA1 region before and after HFS for mice from each group. (F) Histogram showing the average fEPSP slope before HFS (20–30 minutes) and after HFS (50–60 minutes) in each group. Data are expressed as mean ± SEM (n = 5–6 per group). *P < 0.05, **P < 0.01 (one-way analysis of variance followed by the least significant difference test). CA: Cornu ammonis; DG: dentate gyrus; fEPSPs: field excitatory postsynaptic potentials; GGT5: gamma-glutamyltransferase 5; HFS: high frequency stimulation; I–O: input‒output; PPF: paired-pulse facilitation; RE: recording electrode; SE: stimulation electrode; WT: wide type.
Neuronal pentraxin2 (NPTX2) mediates excitatory synaptic transmission and is a powerful prognostic biomarker of AD progression (Pelkey et al., 2015). mRNA-seq analysis of mouse cortical tissue showed that Nptx2, which is associated with synaptic plasticity, exhibited the greatest change in expression out of a total of 277 differentially expressed genes. qPCR analysis confirmed a reduction in Nptx2 expression in the cortex of mice from the APP/PS1 + Vector group compared with the WT + Vector group (P < 0.05). Conversely, increased Nptx2 expression was observed in the cortex of mice from the APP/PS1 + GGT5 group (P < 0.05; Additional Figure 2 (1.3MB, tif) A). Similarly, western blot analysis revealed a decrease in Nptx2 expression in the hippocampus of mice from the APP/PS1 + Vector group compared with the WT + Vector group (P < 0.05), but an increased in NPTX2 expression in the APP/PS1 + GGT5 group (P < 0.05; Additional Figure 2 (1.3MB, tif) B and C). These data suggested that GGT5 overexpression enhanced hippocampal synaptic plasticity in APP/PS1 mice by upregulating NPTX2 expression.
GGT5 overexpression in cerebrovascular endothelial cells alleviates Aβ pathology in APP/PS1 mice
As APP/PS1 mice exhibit robust Aβ accumulation and plaque deposition (Fang et al., 2019; Su et al., 2021), we next asked whether the improvement in cognition and synaptic plasticity resulting from GGT5 overexpression correlated with alterations in Aβ-related pathology by detecting insoluble Aβ plaque deposition in the cortex and hippocampus of mice via immunofluorescence staining with the 6E10 antibody. No Aβ plaque staining was observed in the WT + Vector group or the WT + GGT5 group, while abundant Aβ deposits were seen in the APP/PS1 + Vector group and the APP/PS1 + GGT5 group (Figure 5A). Importantly, the APP/PS1 + GGT5 group exhibited a noticeable reduction in both the area and quantity of insoluble Aβ plaques in the cortex and hippocampus compared with the APP/PS1 + Vector group (P < 0.05; Figure 5B and C). These findings suggest that GGT5 overexpression in cerebrovascular endothelial cells effectively reduced amyloid deposition in the cortex and hippocampus of APP/PS1 mice.
Figure 5.
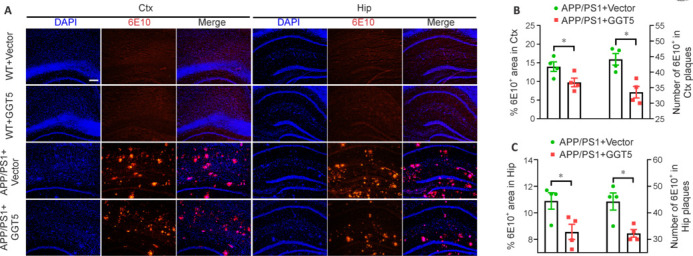
GGT5 overexpression in cerebrovascular endothelial cells alleviates Aβ-related pathology in APP/PS1 mice.
(A) Representative immunofluorescence images of Aβ plaques in the mouse Ctx and Hip. No positive staining for Aβ plaques was observed in the Ctx and Hip of mice in the WT + Vector group and the WT + GGT5 group. However, there were abundant Aβ deposits in the brains of mice in the APP/PS1 + Vector group and the APP/PS1 + GGT5 group. Importantly, the APP/PS1 + GGT5 group exhibited a noticeable reduction in the number of insoluble Aβ plaques compared with the APP/PS1 + Vector group. DAPI, blue; 6E10, red (Cy3). Scale bar: 100 µm. (B, C) Histograms showing the percent area and number of 6E10-positive Aβ plaques in the Ctx (B) and Hip (C). Data are expressed as mean ± SEM (n = 4 per group). *P < 0.05 (one-way analysis of variance followed by Tukey’s post hoc test). Aβ: Amyloid-β; Ctx: cortex; DAPI: 4,6-diamino-2-phenylindole; GGT5: gamma-glutamyltransferase 5; Hip: hippocampus; WT: wide type.
GGT5 overexpression in cerebrovascular endothelial cells decreases Aβ-related pathology by suppressing BACE1 in the brains of APP/PS1 mice
Aβ is produced from the cleavage of APP by β-secretase (BACE1) and γ-secretase, and BACE1 is the rate-limiting enzyme in Aβ production (Lao et al., 2021). Therefore, to investigate the potential impact of GGT5 overexpression on Aβ production through APP processing in APP/PS1 mice, western blotting was performed to quantify full-length APP (flAPP) and its proteolytic products CTF, BACE1, total Aβ oligomer, and Aβ1−42 in mouse cortical tissue lysates (Figure 6A). As expected, flAPP was expressed at higher levels in the cortex of APP/PS1 mice than WT mice, but there was no difference between the APP/PS1 + Vector group and the APP/PS1 + GGT5 group (P > 0.05; Figure 6B). The findings indicate that GGT5 overexpression in cerebrovascular endothelial cells had no impact on APP expression. However, CTF (P < 0.001; Figure 6C) and BACE1 (P < 0.05; Figure 6D) expression levels were increased in the APP/PS1 + Vector group compared with the WT + Vector group, while CTF (P < 0.05; Figure 6C) and BACE1 (P = 0.08; Figure 6D) expression levels were decreased in the APP/PS1 + GGT5 group compared with the APP/PS1+ Vector group. Similarly, compared with the WT + Vector group, the levels of soluble total Aβ oligomer (P < 0.001; Figure 6E) and Aβ1−42 (P < 0.01; Figure 6F) were noticeably increased in the cortex of the APP/PS1 + Vector group, while total Aβ oligomer (P < 0.01; Figure 6E) and Aβ1−42 (P < 0.05; Figure 6F) levels were decreased in the APP/PS1 + GGT5 group compared with the APP/PS1 + Vector group. Consistent with the results obtained from the cortex, BACE1 (P < 0.05; Figure 6G and H) and total Aβ (P < 0.001; Figure 6G and I) expression levels were markedly elevated in the hippocampus of mice from the APP/PS1 + Vector group compared with the WT + Vector group, but GGT5 overexpression decreased BACE1 (P < 0.05; Figure 6G and H) and total Aβ (P = 0.09; Figure 6G and I) levels in APP/PS1 mice. Taken together, these results suggest that GGT5 overexpression in cerebrovascular endothelial cells restricted the APP amyloidogenic pathway and reduced Aβ production by inhibiting BACE1 expression.
Figure 6.
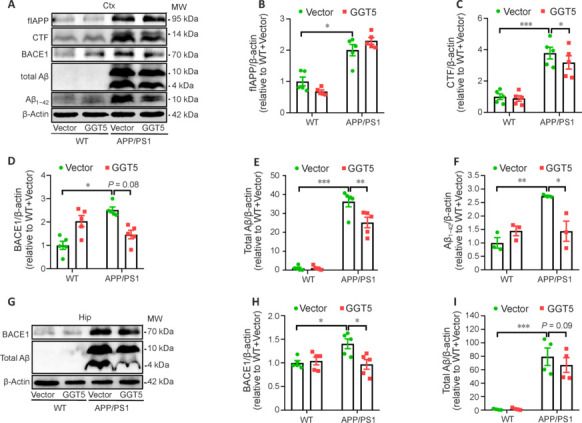
GGT5 overexpression in cerebrovascular endothelial cells reduces the Aβ load in the brains of APP/PS1 mice by inhibiting BACE1.
(A–F) Representative western blots (each row is an image from a different blot) (A) and quantitative analysis of flAPP (B), CTF (C), BACE1 (D), total Aβ (E), and Aβ1–42 (F) expression in the mouse cortex (n = 3–5 per group). (G–I) Representative western blots (each row is an image from a different blot) (G) and quantitative analysis of BACE1 (H) and total Aβ (I) expression in the mouse hippocampus (n = 4–5 per group). Data are expressed as mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001 (one-way analysis of variance followed by Tukey’s post hoc test). APP: Amyloid precursor protein; Aβ: amyloid-β; BACE1: β-site APP cleaving enzyme1; CTF: C-terminal fragment; Ctx: cortex; flAPP: full-length APP; GGT5: gamma-glutamyltransferase 5; Hip: hippocampus; WT: wide type.
GGT5 overexpression in cerebrovascular endothelial cells reduces Aβ production by inhibiting NF-κB–mediated BACE1 expression
To gain a deeper understanding of the mechanisms by which GGT5 overexpression in cerebrovascular endothelial cells suppresses BACE1 expression and subsequently reduces Aβ production, we first verified GGT5 expression levels in the mouse cortex by qPCR. As expected, GGT5 was expressed at lower levels in the APP/PS1 + Vector group than in the WT + Vector group, and at higher levels in the APP/PS1 + GGT5 group (P < 0.05; Figure 7A). mRNA-seq analysis of mouse cortical tissue showed that 133 genes were upregulated and 286 genes were downregulated in the APP/PS1 + Vector group compared with the WT + Vector group (Figure 7B). Additionally, 210 genes were upregulated and 107 genes were downregulated in the APP/PS1 + GGT5 group compared with the APP/PS1 + Vector group (Figure 7C). Furthermore, 277 differentially expressed genes were identified across the three groups (Figure 7D). Pathway analysis (Figure 7E) and a review of the literature showed that NF-κB binds to the BACE1 promoter to regulate its transcription (Snow and Albensi, 2016; Hou et al., 2019; Kim et al., 2019). Additionally, Nfkbia which encodes IκBα, a component of the NF-κB pathway, was one of the 277 differentially expressed genes identified as described above (Figure 7F). qPCR analysis verified that Nfkbia was expressed at lower levels in the cortex of mice from the APP/PS1 + Vector group compared with the WT + Vector group, and at higher levels in the cortex of mice from the APP/PS1 + GGT5 group (P < 0.05; Figure 7G). Similarly, western blot analysis showed that IκBα was expressed at lower levels in the cortex of mice from the APP/PS1 + Vector group compared with the WT + Vector group (P = 0.07), and at significantly higher levels in the APP/PS1 + GGT5 group (P < 0.05; Figure 7H and I). Furthermore, compared with the WT + Vector group, p-NF-κB/NF-κB expression was increased in the APP/PS1 + Vector group (P < 0.05), while p-NF-κB/NF-κB expression in the APP/PS1 + GGT5 group was decreased compared with the APP/PS1 + Vector group (P < 0.05; Figure 7H and J). Therefore, we concluded that GGT5 overexpression in cerebrovascular endothelial cells reduced cerebral Aβ production by suppressing NF-κB–mediated BACE1 expression.
Figure 7.
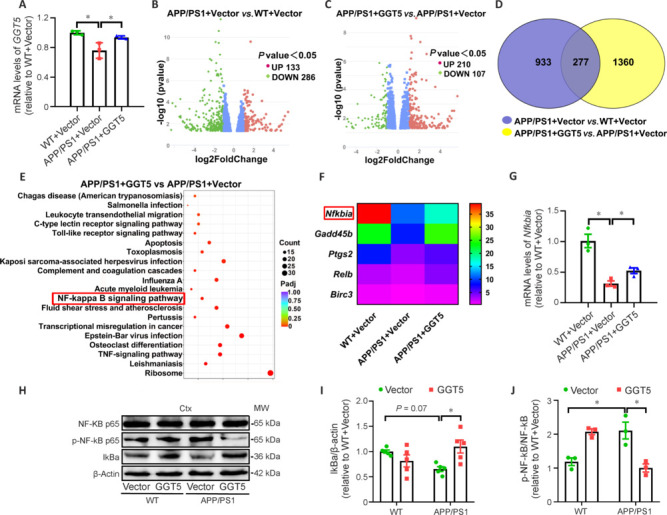
GGT5 overexpression in cerebrovascular endothelial cells suppresses the IκB/NF-κB signaling pathway.
(A) Histogram of GGT5 expression levels in the mouse cortex, as determined by qPCR (n = 3 per group). (B, C) Volcano plots showing differential gene expression based on mRNA-seq analysis in the mouse cortex in the APP/PS1 + Vector group vs. the WT + Vector group (B) and the APP/PS1 + GGT5 group vs. the APP/PS1 + Vector group (C). (D) Venn diagram showing that there were 277 differentially expressed genes in the mouse cortex in common between the WT + Vector group vs. the APP/PS1 + Vector group and the APP/PS1 + GGT5 group vs. the APP/PS1 + Vector group, as determined by mRNA-seq. (E) Pathway enrichment analysis of genes that were differentially expressed between the APP/PS1 + GGT5 group and the APP/PS1 + Vector group (the NF-κB pathway is highlighted). (F) Heatmap displaying the relative expression levels of differentially expressed genes involved in the NF-κB pathway. (G) Histogram of Nfkbia expression, as determined by qPCR, in the mouse cortex (n = 3 per group). (H–J) Representative western blots (each row is an image from a different blot) (H) and quantitative analysis of IκBα (I) and p-NF-κB/NF-κB (J) expression in the mouse cortex (n = 3–5 per group). Data are expressed as mean ± SEM. *P < 0.05 (one-way analysis of variance followed by Tukey’s post hoc test). Birc3: Baculoviral IAP repeat containing 3; Ctx: cortex; Gadd45b: growth arrest and DNA damage inducible protein beta; GGT5: gamma-glutamyltransferase 5; IκBα: inhibitor-kappaBα; NFKbia: nuclear factor of kappa light polypeptide gene enhancer in B-cells inhibitorα; NF-κB: nuclear factor‐kappa B; p-NF-κB: phosphor nuclear factor‐kappa B; Ptgs2: prostaglandin endoperoxide synthase 2; qPCR: real-time polymerase chain reaction; Relb: recombinant v-rel reticuloendotheliosis viral oncogene homolog B; TNF: tumor necrosis factor; WT: wide type.
GGT5 overexpression suppresses p-NF-κB expression by increasing GSH levels
GSH has been reported to hinder the classical NF-κB activation pathway (Fraternale et al., 2021), while GGT5 can increase intracellular GSH content by participating in the GSH cycle (Bachhawat and Yadav, 2018; Ho et al., 2022). Our in vitro experiments showed that GGT5 expression was significantly increased in bEnd.3 cells transfected with the GGT5 plasmid (P < 0.05; Additional Figure 3 (3.5MB, tif) A and B), and that intracellular (P < 0.05) and extracellular (P < 0.01) GSH/GSSG ratios were also increased 48 hours post-transfection (Additional Figure 3 (3.5MB, tif) C and D). Coculturing HT22 cells with conditioned medium from bEnd.3 cells transfected with the GGT5 plasmid, the viability of HT22 cells was observed to be enhanced at 72 hours in both the vehicle-treated and Aβ1−42-treated groups (P < 0.05; Additional Figure 3 (3.5MB, tif) E and F). In the Aβ1−42-treated group, NF-κB expression levels in HT22 cells cultured in conditioned medium from bEnd.3 cells transfected with vector and GGT5 plasmid did not exhibit any notable difference (P > 0.05; Additional Figure 3 (3.5MB, tif) G and H). However, p-NF-κB expression in HT22 cells treated with Aβ1−42 decreased when they were cultured in conditioned medium from bEnd.3 cells transfected with the GGT5 plasmid (P < 0.05; Additional Figure 3 (3.5MB, tif) G and I). These data suggest that GGT5 overexpression in cerebrovascular endothelial cells inhibits NF-κB pathway activation in neurons by enhancing GSH levels.
Discussion
Impaired cerebrovascular endothelial cell function is a prominent and possibly initial pathological manifestation of AD preceding the clinical presentation of dementia (Sweeney et al., 2019). Ameliorating cerebrovascular endothelial cell function may help alleviate the pathological and cognitive impairment associated with AD (Zenaro et al., 2017; Deng et al., 2022). However, the underlying molecular mechanism contributing to cerebral endothelial cell dysfunction in AD is still unclear. In recent years, progress in multiomics technologies has provided a new approach for exploring the pathogenesis of multiple complex diseases. A single-cell sequencing study performed by Lau et al. (2020) identified genes that were differentially expressed in cerebrovascular endothelial cells from patients with AD and healthy individuals. In the current study, we established an in vitro model of AD by treating hCMEC/D3 cells with Aβ1−42 and performed mRNA-seq to identify differentially expressed genes, then compared our in vitro findings with the in vivo findings from Lau et al. (2020). Both studies identified GGT5 as a potentially AD-related gene, and we found that the expression of GGT5 was downregulated in cerebrovascular endothelial cells from patients with AD and verified that the expression of GGT5 was downregulated in cerebrovascular endothelial cells (hCMEC/D3 and bEnd.3) treated with Aβ1−42. This finding implies a potential correlation between GGT5 downregulation in cerebrovascular endothelial cells and the progressive accumulation of pathological forms of Aβ in AD. Further verifying this hypothesis, we found that GGT5 expression levels were decreased in cerebrovascular endothelial cells from a classic mouse model of AD (APP/PS1 transgenic mice), as well as in the hippocampus and cortex of APP/PS1 mice. Hence, decreased GGT5 expression in cerebrovascular endothelial cells could potentially contribute to the initiation and progression of cognitive deficits in AD.
GGT is a membrane-bound glycoprotein consisting of two polypeptide chains that plays an important role in the GSH cycle, which is involved in GSH biosynthesis (Bachhawat and Kaur, 2017; Fraternale et al., 2021; Ho et al., 2022). Specifically, GGT recovers GSH precursors from extracellular pools and returns these constituents to cells (Bachhawat and Kaur, 2017). GGT deficiency in humans contributes to disrupted GSH homeostasis and tends to result in retarded mental development (Iida et al., 2005; Zhang and Forman, 2009). GGT-deficient mice manifest chronic mitochondrial GSH exhaustion, impaired oxidative phosphorylation, and disrupted adenosine triphosphate production, resulting in growth retardation and shortened lifespan (Will et al., 2000; Zhang and Forman, 2009). Abnormal GGT activity likely contributes to the pathology of diseases involving oxidative stress, such as AD (Zhang and Forman, 2009). The human GGT5 gene encodes a functional protein exhibiting GGT activity (Heisterkamp et al., 2008). However, the impact of GGT5 downregulation in cerebrovascular endothelial cells on AD pathogenesis remained unclear. Specifically, it was uncertain whether GGT5 upregulation could mitigate cognitive impairment and amyloid pathology in AD. Here, we overexpressed GGT5 from an endothelial cell–specific promoter in mouse cerebrovascular endothelial cells via injection with the adeno-associated virus AAV9. We found that CD31-positive vascular endothelial cells exhibited GGT5-specific staining, and that injection with the adenovirus increased GGT5 expression, indicating successful, stable GGT5 overexpression in the cerebrovascular endothelial cells of APP/PS1 mice. Then, we used the Y-maze and Morris water maze to confirm that GGT5 overexpression in cerebrovascular endothelial cells effectively improved short-term memory and long-term learning in APP/PS1 mice. Hippocampal synaptic plasticity is the crucial neural cytological foundation of learning, memory, and cognition (Palacios-Filardo and Mellor, 2019). LTP is an important form of synaptic plasticity and the main indicator of long-term learning and memory function (Li et al., 2022b). Impaired synaptic plasticity is closely linked with the decline in cognitive abilities seen in AD (He et al., 2019; Xu et al., 2021b). Therefore, hippocampal LTP recording was performed to explore the effects of GGT5 overexpression on synaptic plasticity in APP/PS1 mice. We demonstrated that GGT5 overexpression in cerebrovascular endothelial cells effectively rescued LTP suppression, thereby enhancing hippocampal synaptic plasticity in APP/PS1 mice. However, the mechanism by which GGT5 overexpression facilitates hippocampal synaptic plasticity in APP/PS1 mice remained unclear.
Interestingly, cortical tissue mRNA-seq analysis indicated that GGT5 overexpression in cerebrovascular endothelial cells enhances NPTX2 expression. NPTX2 serves as the hub molecule for synaptic remodeling. NPTX2 expression is downregulated in the brains of patients with AD, and its expression levels are strongly correlated with cognitive function and synaptic plasticity (Pelkey et al., 2015; Xiao et al., 2017). During the synaptic excitatory transmission process, pyramidal neurons express NPTX2 and secrete it from their axonal terminals. Subsequently, NPTX2 is released into the synaptic space via the presynaptic membrane and interacts with the AMPA-type glutamate receptor through its PTX domain (Lee et al., 2017). NPTX2 acts as a presynaptic factor and induces postsynaptic excitatory transmission (Pelkey et al., 2015; Libiger et al., 2021). Western blot analysis showed that GGT5 overexpression in cerebrovascular endothelial cells resulted in a significant increase in NPTX2 levels in the mouse hippocampus. Taken together with the LTP recording results, this observation suggests that GGT5 overexpression enhances synaptic plasticity in the hippocampal CA1 region by promoting NPTX2 expression. However, further research is needed to determine the mechanism by which endothelial cell GGT5 influences NPTX2 expression in neurons, which could involve intercellular interactions within the neurovascular unit.
Abnormal Aβ production and accumulation have been proposed to be the initial causative events in AD occurrence and development (Kim et al., 2019; Stakos et al., 2020; Mary et al., 2023). Aβ deposition is one of the primary features associated with progressive cognitive impairment in AD patients and mainly occurs in the cerebral cortex and hippocampus, regions that are intricately involved in cognition (Fisher et al., 2022; Liu, 2022; Tahami Monfared et al., 2022). The APP/PS1 transgenic mouse model simulates the pathological characteristics and cognitive behavior of patients with AD, and learning and memory in this model progressively deteriorate as cerebral Aβ accumulation occurs (Chen et al., 2021; Sasaguri et al., 2022). To test whether GGT5 overexpression in the cerebrovascular endothelial cells of APP/PS1 mice improved learning and memory by mitigating Aβ-related pathology, we fluorescently labeled Aβ plaques with an anti-6E10 antibody and found that GGT5 overexpression significantly reduced the number and size of insoluble Aβ plaques in the cortex and hippocampus. Furthermore, western blot analysis confirmed that GGT5 overexpression effectively decreased soluble Aβ1−42 levels in the cortex of APP/PS1 mice. APP is abnormally cleaved by BACE1, releasing the short extracellular domain Sappβ (N-terminal fragment) and retaining C99 (C-terminal fragment, CTF) on the membrane, where it is further cleaved by γ-secretase and hydrolyzed, releasing soluble Aβ peptides (Brothers et al., 2018; Tiwari et al., 2019). We found that flAPP expression in the mouse cortex was not affected by GGT5 overexpression in cerebrovascular endothelial cells, while BACE1, CTF, and total Aβ expression levels decreased, suggesting that GGT5 overexpression affected APP hydrolysis. Similarly, BACE1 and total Aβ expression levels were decreased in hippocampal tissues of APP/PS1 mice. These results suggest that GGT5 overexpression inhibits APP cleavage–mediated Aβ production by suppressing BACE1 expression. BACE1 is believed to be a rate-limiting enzyme for APP cleavage into Aβ, thereby exerting a significant influence on Aβ production. Elevated BACE1 expression levels and enhanced BACE1 activity have been reported in the brains of patients with sporadic AD (Kim et al., 2019; Hampel et al., 2021) and transgenic mouse models of AD (Du et al., 2016). Furthermore, the BACE1 upregulation in vascular endothelial cells causes endothelial dysfunction by promoting excessive lysis of tight junction proteins, and endothelial NO synthase dysfunction, which lead to phosphorylation of the tau protein in neurons (Chacón-Quintero et al., 2021). Disruption of cerebrovascular integrity and hyperphosphorylation of tau protein could lead to synaptic degeneration, neuron loss, and cognitive impairment (Zhou et al., 2022). Therefore, BACE1 inhibition is considered to be an attractive therapeutic strategy for alleviating Aβ-related pathology and improving cognitive function in patients with AD (Moussa-Pacha et al., 2020; Hampel et al., 2021; Patel et al., 2022).
Despite these results, the mechanism by which GGT5 overexpression inhibits BACE1 expression remained unclear. Our mRNA-seq analysis results suggested that GGT5 is involved in NF-κB signaling pathway regulation. The NF-κB signaling pathway plays an essential role in gene regulation and is strongly correlated with inflammation, oxidative stress, and apoptosis (Hou et al., 2019). Members of the NF-κB protein family include NF-κB1, NF-κB2, RelA, RelB, and c-Rel, and the N-terminus of each of these proteins contains a Rel homology domain. NF-κB1 encodes the precursor protein p105, which is processed by the ubiquitin proteasome pathway to produce the mature NF-κB subunit p50. The RelA gene encodes the p65 protein (Hayden and Ghosh, 2008; Yu et al., 2020). NF-κB forms both homologous and heterologous complexes, among which the P65/p50 heterodimer is the most common form (Sun et al., 2022). In the classical activation pathway, the NF-κB dimer P65/p50 binds to an IκB suppressor protein (IκBα is the most well studied) in the cytoplasm to form an inactive trimer. In response to external stimulation, IκB kinase rapidly activated IκB, IκB dissociates from the trimer complex, and undergoes proteasome-mediated degradation. This allows phosphorylation of the NF-κB dimer, yielding p-NF-κB, which unmasks its nuclear localization sequence, enabling it to quickly translocate to the nucleus from the cytoplasm and bind to specific nuclear DNA sequences to promote the transcription of related genes, such as BACE1 (Hayden and Ghosh, 2008; Morgan and Liu, 2011).
A previous study identified NF-κB binding elements within the BACE1 promoter (Buggia-Prevot et al., 2008), and abnormal Aβ aggregation has been shown to regulate BACE1 promoter activity through an NF-κB–dependent pathway (Chen et al., 2012; Kim et al., 2019). Indeed, increased NF‐κB activity has been observed in autopsied brain tissue from patients with AD (Chen et al., 2012; Kim et al., 2019). There are four hypothetical NF-κB binding elements in the human BACE1 gene promoter, two of which interact with NF-κB p65. Numerous studies have shown that the NF-κB signaling pathway promotes BACE1 promoter activity and BACE1 transcription, thereby enhancing APP processing and increasing Aβ production (Hayden and Ghosh, 2008; Kim et al., 2019; Yu et al., 2020; Sun et al., 2022).
On the basis of our sequencing results, we speculated that GGT5 overexpression in cerebrovascular endothelial cells mitigates Aβ generation by suppressing NF-κB–mediated BACE1 expression. To test this, we assessed the expression levels of IκBα, total NF-κB, and p-NF-κB, all components of the classical NF-κB activation pathway. GGT5 overexpression in APP/PS1 mouse cerebrovascular endothelial cells did not affect the level of total NF-κB but increased the level of IκBα and decreased the level of p-NF-κB. These results support our initial hypothesis, suggesting that GGT5 overexpression inhibits BACE1 expression by repressing activation of the NF-κB pathway.
GGT degrades extracellular GSH and supplies its precursor amino acids via the GSH cycle to promote intracellular GSH resynthesis (Bachhawat and Kaur, 2017; Corti et al., 2020; Suzuki et al., 2020). GGT overexpression increases the intracellular GSH content and promotes activation of the GSH cycle, which confers protection against oxidative stress (Ho et al., 2022). Fraternale et al. (Fraternale et al., 2021) demonstrated that GSH can inhibit the classical NF-κB activation pathway, not only by indirectly blocking NF-κB activation by scavenging oxidants but also by directly suppressing phosphorylation of the IκB/NF-κB complex. In addition, GSH can prevent NF-κB transport from the cytoplasm to the nucleus and impede its sequence-specific binding to promoter regions (Fraternale et al., 2021). Taken together, the findings from these studies suggest that GGT5 overexpression in cerebrovascular endothelial cells would increase GSH levels and suppress the NF-κB signaling pathway, ultimately decreasing BACE1 expression; indeed, we found that GGT5 overexpression in bEnd.3 cells increase the GSH content in the intracellular and extracellular microenvironment. Additionally, culturing HT22 cells treated with Aβ1−42 in conditioned medium from bEnd.3 cells transfected with the GGT5 plasmid enhanced their viability and inhibited p-NF-κB expression. Our results suggest that the mitigating effect of GGT5 overexpression on Aβ production is attributable to elevated GSH levels within the microenvironment, as well as inhibition of NF-κB–mediated BACE1 expression.
Despite these positive results, our study still had several limitations, and further research is needed to verify the effects and mechanisms of cerebrovascular endothelial cell GGT5 on cognitive function in APP/PS1 mice. First, the effects of GGT5 knockdown in cerebrovascular endothelial cells on AD-like pathology and cognition in APP/PS1 mice should be evaluated. Second, whether NF-κB signaling pathway activation can reverse the effects of GGT5 overexpression on APP/PS1 mice should be explored. Finally, whether inhibiting the NF-κB signaling pathway alleviates Aβ pathology and cognitive deficits in APP/PS1 mice should be investigated.
In summary, the present study revealed that GGT5 overexpression in cerebrovascular endothelial cells in APP/PS1 mice effectively rescued cognitive impairment and alleviated AD-like brain pathology, possibly by inhibiting NF-κB–mediated BACE1 expression, decreasing Aβ production, and alleviating Aβ-related pathology. These findings offer novel avenues for AD prevention and treatment.
Additional files:
Additional Figure 1 (3.4MB, tif) : Co-localization of GGT5 with cerebral vascular endothelial cells in the brains of mice.
Co-localization of GGT5 with cerebral vascular endothelial cells in the brains of mice.
(A, B) Representative immunofluorescence images of transverse sections (A) and longitudinal sections (B) of cerebral vessels in four groups of mice. The mean fluorescence intensity of vascular endothelial cells GGT5 in the mice of WT + GGT5 group was stronger than that in the WT + Vector group. And the mean fluorescence intensity of vascular endothelial cells GGT5 was higher in the APP/PS1 + GGT5 group than that in the APP/PS1 + Vector group. DAPI, blue; CD31 (green, marked by 488); GGT5 (red, marked by Cy3). Scale bars: 50 µm. (C) Histograms showing the mean fluorescence intensity of GGT5 in vascular endothelial cells in the cortex of mice (n = 4 per group). Data are expressed as mean ± SEM.*P < 0.05 (one-way analysis of variance followed by least significant difference test). Ctx: Cortex; DAPI: 4,6-diamino-2-phenyl indole; GGT5: gamma-glutamyltransferase 5; Hip: hippocampus; WT: wide type.
Additional Figure 2 (1.3MB, tif) : The expression of NPTX2 in the brains of mice.
The expression of NPTX2 in the brains of mice.
(A) Histogram showing Nptx2 gene expression in the cortex of mice by qPCR (n = 3 per group). (B, C) The representative Western blots (B) and quantitative analysis (C) for NPTX2 in the hippocampus of mice (n = 5 per group). Data are expressed as mean ± SEM. *P < 0.05 (one-way analysis of variance followed by Tukey's post hoc test). Ctx: Cortex; GGT5: gamma-glutamyltransferase 5; Hip: hippocampus; NPTX2: neuronal pentraxin II; qPCR: real-time polymerase chain reaction; WT: wide type.
Additional Figure 3 (3.5MB, tif) : Overexpression of GGT5 suppresses the expression of p-NF-κB by increasing GSH levels.
Overexpression of GGT5 suppresses the expression of p-NF-κB by increasing GSH levels.
(A, B) The representative Western blots (A) and quantitative analysis (B) for GGT5 in bEnd.3 cells transfected with vector and GGT5 plasmid (n = 3 per group). (C, D) Histogram showing the level of intracelluar (C) and extracelluar (D) GSH/GSSG in bEnd.3 cells transfected with vector and GGT5 plasmid (n = 3-4 per group). (E, F) The cell viability of HT22 cells cocultured with medium derived from bEnd.3 cells transfected with Vector and GGT5 plasmid in both the vehicle-treated (E) and Aβ1-42-treated (F) groups (n = 3 per group). (G-I) The representative Western blots (G) and quantitative analysis for NF-κB (H) and p-NF-κB (I) in HT22 cells cultured using the aforementioned methodology (n = 3-4 per group). Data are expressed as mean ± SEM. *P < 0.05, **P < 0.01 (one-way analysis of variance followed by Tukey’s post hoc test (H, I), or independent-samples t-test (B-F)). Aβ: Amyloid-β; Ctx: cortex; GGT5: gamma-glutamyltransferase 5; GSH: glutathione; GSSG: oxidized glutathione; Hip: hippocampus; NF-κB: nuclear factor-kappa B; p-NF-κB: phosphor nuclear factor-kappa B; WT: wide type.
Additional Table 1 (290.4KB, pdf) : The differential genes identified in our in vitro experiments and those identified in the in vivo experiments conducted by Lau et al.
The differential genes identified in our in vitro experiments and those identified in the in vivo experiments conducted by Lau et al.
Funding Statement
Funding: This study was supported by STI2030-Major Projects, No. 2021ZD 0201801 (to JG) and Shanxi Province Basic Research Program, No. 20210302123429 (to QS).
Footnotes
Conflicts of interest: The authors declare that no actual or potential conflict of interest concerning this manuscript.
Data availability statement: All data generated and/or analyzed in the current study are included in this manuscript and its Additional files and available from the corresponding author on reasonable request.
C-Editor: Zhao M; S-Editors: Yu J, Li CH; L-Editors: Crow E, Yu J, Song LP; T-Editor: Jia Y
References
- Abyadeh M, Gupta V, Paulo JA, Mahmoudabad AG, Shadfar S, Mirshahvaladi S, Gupta V, Nguyen CTO, Finkelstein DI, You Y, Haynes PA, Salekdeh GH, Graham SL, Mirzaei M. Amyloid-beta and tau protein beyond Alzheimer’s disease. Neural Regen Res. 2024;19:1262–1276. doi: 10.4103/1673-5374.386406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arifin WN, Zahiruddin WM. Sample size calculation in animal studies using resource equation approach. Malays J Med Sci. 2017;24:101–105. doi: 10.21315/mjms2017.24.5.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachhawat AK, Kaur A. Glutathione degradation. Antioxid Redox Signal. 2017;27:1200–1216. doi: 10.1089/ars.2017.7136. [DOI] [PubMed] [Google Scholar]
- Bachhawat AK, Yadav S. The glutathione cycle: glutathione metabolism beyond the γ-glutamyl cycle. IUBMB Life. 2018;70:585–592. doi: 10.1002/iub.1756. [DOI] [PubMed] [Google Scholar]
- Bjørklund G, Doşa MD, Maes M, Dadar M, Frye RE, Peana M, Chirumbolo S. The impact of glutathione metabolism in autism spectrum disorder. Pharmacol Res. 2021;166:105437. doi: 10.1016/j.phrs.2021.105437. [DOI] [PubMed] [Google Scholar]
- Brothers HM, Gosztyla ML, Robinson SR. The physiological roles of amyloid-β peptide hint at new ways to treat Alzheimer’s disease. Front Aging Neurosci. 2018;10:118. doi: 10.3389/fnagi.2018.00118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buggia-Prevot V, Sevalle J, Rossner S, Checler F. NFkappaB-dependent control of BACE1 promoter transactivation by Abeta42. J Biol Chem. 2008;283:10037–10047. doi: 10.1074/jbc.M706579200. [DOI] [PubMed] [Google Scholar]
- Chacón-Quintero MV, Pineda-López LG, Villegas-Lanau CA, Posada-Duque R, Cardona-Gómez GP. Beta-secretase 1 underlies reactive astrocytes and endothelial disruption in neurodegeneration. Front Cell Neurosci. 2021;15:656832. doi: 10.3389/fncel.2021.656832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CH, Zhou W, Liu S, Deng Y, Cai F, Tone M, Tone Y, Tong Y, Song W. Increased NF-κB signalling up-regulates BACE1 expression and its therapeutic potential in Alzheimer’s disease. Int J Neuropsychopharmacol. 2012;15:77–90. doi: 10.1017/S1461145711000149. [DOI] [PubMed] [Google Scholar]
- Chen Y, Zhao S, Fan Z, Li Z, Zhu Y, Shen T, Li K, Yan Y, Tian J, Liu Z, Zhang B. Metformin attenuates plaque-associated tau pathology and reduces amyloid-β burden in APP/PS1 mice. Alzheimers Res Ther. 2021;13:40. doi: 10.1186/s13195-020-00761-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corti A, Belcastro E, Dominici S, Maellaro E, Pompella A. The dark side of gamma-glutamyltransferase (GGT): Pathogenic effects of an ‘antioxidant’ enzyme. Free Radic Biol Med. 2020;160:807–819. doi: 10.1016/j.freeradbiomed.2020.09.005. [DOI] [PubMed] [Google Scholar]
- Das B, Yan R. Role of BACE1 in Alzheimer’s synaptic function. Transl Neurodegener. 2017;6:23. doi: 10.1186/s40035-017-0093-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das B, Yan R. A close look at BACE1 inhibitors for Alzheimer’s disease treatment. CNS Drugs. 2019;33:251–263. doi: 10.1007/s40263-019-00613-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das B, Singh N, Yao AY, Zhou J, He W, Hu X, Yan R. BACE1 controls synaptic function through modulating release of synaptic vesicles. Mol Psychiatry. 2021;26:6394–6410. doi: 10.1038/s41380-021-01166-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Strooper B, Karran E. The cellular phase of Alzheimer’s disease. Cell. 2016;164:603–615. doi: 10.1016/j.cell.2015.12.056. [DOI] [PubMed] [Google Scholar]
- Deng W, Guo S, van Veluw SJ, Yu Z, Chan SJ, Takase H, Arai K, Ning M, Greenberg SM, Lo EH, Bacskai BJ. Effects of cerebral amyloid angiopathy on the brain vasculome. Aging Cell. 2022;21:e13503. doi: 10.1111/acel.13503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobrowolska Zakaria JA, Vassar RJ. A promising, novel, and unique BACE1 inhibitor emerges in the quest to prevent Alzheimer’s disease. EMBO Mol Med. 2018;10:e9717. doi: 10.15252/emmm.201809717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du Y, Qu J, Zhang W, Bai M, Zhou Q, Zhang Z, Li Z, Miao J. Morin reverses neuropathological and cognitive impairments in APPswe/PS1dE9 mice by targeting multiple pathogenic mechanisms. Neuropharmacology. 2016;108:1–13. doi: 10.1016/j.neuropharm.2016.04.008. [DOI] [PubMed] [Google Scholar]
- Du Y, Fu M, Huang Z, Tian X, Li J, Pang Y, Song W, Tian Wang Y, Dong Z. TRPV1 activation alleviates cognitive and synaptic plasticity impairments through inhibiting AMPAR endocytosis in APP23/PS45 mouse model of Alzheimer’s disease. Aging Cell. 2020;19:e13113. doi: 10.1111/acel.13113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estudillo E, López-Ornelas A, Rodríguez-Oviedo A, Gutiérrez de la Cruz N, Vargas-Hernández MA, Jiménez A. Thinking outside the black box: are the brain endothelial cells the new main target in Alzheimer’s disease? Neural Regen Res. 2023;18:2592–2598. doi: 10.4103/1673-5374.373672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang EF, Hou Y, Palikaras K, Adriaanse BA, Kerr JS, Yang B, Lautrup S, Hasan-Olive MM, Caponio D, Dan X, Rocktäschel P, Croteau DL, Akbari M, Greig NH, Fladby T, Nilsen H, Cader MZ, Mattson MP, Tavernarakis N, Bohr VA. Mitophagy inhibits amyloid-β and tau pathology and reverses cognitive deficits in models of Alzheimer’s disease. Nat Neurosci. 2019;22:401–412. doi: 10.1038/s41593-018-0332-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher RA, Miners JS, Love S. Pathological changes within the cerebral vasculature in Alzheimer’s disease: new perspectives. Brain Pathol. 2022;32:e13061. doi: 10.1111/bpa.13061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flanigan ME, Hon OJ, D’Ambrosio S, Boyt KM, Hassanein L, Castle M, Haun HL, Pina MM, Kash TL. Subcortical serotonin 5HT(2c) receptor-containing neurons sex-specifically regulate binge-like alcohol consumption, social, and arousal behaviors in mice. Nat Commun. 2023;14:1800. doi: 10.1038/s41467-023-36808-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraternale A, Zara C, De Angelis M, Nencioni L, Palamara AT, Retini M, Di Mambro T, Magnani M, Crinelli R. Intracellular redox-modulated pathways as targets for effective approaches in the treatment of viral infection. Int J Mol Sci. 2021;22:3603. doi: 10.3390/ijms22073603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong Q, Li W, Ali T, Hu Y, Mou S, Liu Z, Zheng C, Gao R, Li A, Li T, Li N, Yu Z, Li S. eIF4E phosphorylation mediated LPS induced depressive-like behaviors via ameliorated neuroinflammation and dendritic loss. Transl Psychiatry. 2023;13:352. doi: 10.1038/s41398-023-02646-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hampel H, et al. The β-secretase BACE1 in Alzheimer’s disease. Biol Psychiatry. 2021;89:745–756. doi: 10.1016/j.biopsych.2020.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayden MS, Ghosh S. Shared principles in NF-kappaB signaling. Cell. 2008;132:344–362. doi: 10.1016/j.cell.2008.01.020. [DOI] [PubMed] [Google Scholar]
- He Y, Wei M, Wu Y, Qin H, Li W, Ma X, Cheng J, Ren J, Shen Y, Chen Z, Sun B, Huang FD, Shen Y, Zhou YD. Amyloid β oligomers suppress excitatory transmitter release via presynaptic depletion of phosphatidylinositol-4,5-bisphosphate. Nat Commun. 2019;10:1193. doi: 10.1038/s41467-019-09114-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heisterkamp N, Groffen J, Warburton D, Sneddon TP. The human gamma-glutamyltransferase gene family. Hum Genet. 2008;123:321–332. doi: 10.1007/s00439-008-0487-7. [DOI] [PubMed] [Google Scholar]
- Ho T, Ahmadi S, Kerman K. Do glutathione and copper interact to modify Alzheimer’s disease pathogenesis? Free Radic Biol Med. 2022;181:180–196. doi: 10.1016/j.freeradbiomed.2022.01.025. [DOI] [PubMed] [Google Scholar]
- Hou Y, Luo S, Zhang Y, Jia Y, Li H, Xiao C, Bao H, Du J. Contrasting effects of acute and long-term corticosterone treatment on amyloid-β, beta-secretase 1 expression, and nuclear factor kappa B nuclear translocation. J Integr Neurosci. 2019;18:393–400. doi: 10.31083/j.jin.2019.04.1172. [DOI] [PubMed] [Google Scholar]
- Huang L, Zhang Y, Fu H, Gu W, Mao J. A missense mutant of ocrl1 promotes apoptosis of tubular epithelial cells and disrupts endocytosis and the cell cycle of podocytes in Dent-2 disease. Cell Commun Signal. 2023;21:256. doi: 10.1186/s12964-023-01272-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iida M, Yasuhara T, Mochizuki H, Takakura H, Yanagisawa T, Kubo H. Two Japanese brothers with hereditary gamma-glutamyl transpeptidase deficiency. J Inherit Metab Dis. 2005;28:49–55. doi: 10.1007/s10545-005-4417-8. [DOI] [PubMed] [Google Scholar]
- Jankowsky JL, Slunt HH, Ratovitski T, Jenkins NA, Copeland NG, Borchelt DR. Co-expression of multiple transgenes in mouse CNS: a comparison of strategies. Biomol Eng. 2001;17:157–165. doi: 10.1016/s1389-0344(01)00067-3. [DOI] [PubMed] [Google Scholar]
- Kim HJ, Joe Y, Chen Y, Park GH, Kim UH, Chung HT. Carbon monoxide attenuates amyloidogenesis via down-regulation of NF-κB-mediated BACE1 gene expression. Aging Cell. 2019;18:e12864. doi: 10.1111/acel.12864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraeuter AK, Guest PC, Sarnyai Z. The Y-maze for assessment of spatial working and reference memory in mice. Methods Mol Biol. 20191916:105–111. doi: 10.1007/978-1-4939-8994-2_10. [DOI] [PubMed] [Google Scholar]
- Lao K, Zhang R, Luan J, Zhang Y, Gou X. Therapeutic strategies targeting amyloid-β receptors and transporters in Alzheimer’s disease. J Alzheimers Dis. 2021;79:1429–1442. doi: 10.3233/JAD-200851. [DOI] [PubMed] [Google Scholar]
- Lau SF, Cao H, Fu AKY, Ip NY. Single-nucleus transcriptome analysis reveals dysregulation of angiogenic endothelial cells and neuroprotective glia in Alzheimer’s disease. Proc Natl Acad Sci U S A. 2020;117:25800–25809. doi: 10.1073/pnas.2008762117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SJ, Wei M, Zhang C, Maxeiner S, Pak C, Calado Botelho S, Trotter J, Sterky FH, Südhof TC. Presynaptic neuronal pentraxin receptor organizes excitatory and inhibitory synapses. J Neurosci. 2017;37:1062–1080. doi: 10.1523/JNEUROSCI.2768-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li QQ, Chen J, Hu P, Jia M, Sun JH, Feng HY, Qiao FC, Zang YY, Shi YY, Chen G, Sheng N, Xu Y, Yang JJ, Xu Z, Shi YS. Enhancing GluN2A-type NMDA receptors impairs long-term synaptic plasticity and learning and memory. Mol Psychiatry. 2022a;27:3468–3478. doi: 10.1038/s41380-022-01579-7. [DOI] [PubMed] [Google Scholar]
- Li S, Stern AM. Bioactive human Alzheimer brain soluble Aβ: pathophysiology and therapeutic opportunities. Mol Psychiatry. 2022;27:3182–3191. doi: 10.1038/s41380-022-01589-5. [DOI] [PubMed] [Google Scholar]
- Li T, Su Q, Zhang Z, Zhang Y, Yang M, Wang Z, Guo J, Wang Z, Wu M, Cai H, Qi J. Ube2c-inhibition alleviated amyloid pathology and memory deficits in APP/PS1 mice model of AD. Prog Neurobiol. 2022b;215:102298. doi: 10.1016/j.pneurobio.2022.102298. [DOI] [PubMed] [Google Scholar]
- Libiger O, Shaw LM, Watson MH, Nairn AC, Umaña KL, Biarnes MC, Canet-Avilés RM, Jack CR, Jr., Breton YA, Cortes L, Chelsky D, Spellman DS, Baker SA, Raghavan N, Potter WZ. Longitudinal CSF proteomics identifies NPTX2 as a prognostic biomarker of Alzheimer’s disease. Alzheimers Dement. 2021;17:1976–1987. doi: 10.1002/alz.12353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu RM. Aging, cellular senescence, and Alzheimer’s disease. Int J Mol Sci. 2022;23:1989. doi: 10.3390/ijms23041989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Hou D, Lin F, Luo J, Xie J, Wang Y, Tian Y. The role of neurovascular unit damage in the occurrence and development of Alzheimer’s disease. Rev Neurosci. 2019;30:477–484. doi: 10.1515/revneuro-2018-0056. [DOI] [PubMed] [Google Scholar]
- Lomoio S, Willen R, Kim W, Ho KZ, Robinson EK, Prokopenko D, Kennedy ME, Tanzi RE, Tesco G. Gga3 deletion and a GGA3 rare variant associated with late onset Alzheimer’s disease trigger BACE1 accumulation in axonal swellings. Sci Transl Med. 2020;12:eaba1871. doi: 10.1126/scitranslmed.aba1871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mary A, Eysert F, Checler F, Chami M. Mitophagy in Alzheimer’s disease: molecular defects and therapeutic approaches. Mol Psychiatry. 2023;28:202–216. doi: 10.1038/s41380-022-01631-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan MJ, Liu ZG. Crosstalk of reactive oxygen species and NF-κB signaling. Cell Res. 2011;21:103–115. doi: 10.1038/cr.2010.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moussa-Pacha NM, Abdin SM, Omar HA, Alniss H, Al-Tel TH. BACE1 inhibitors: current status and future directions in treating Alzheimer’s disease. Med Res Rev. 2020;40:339–384. doi: 10.1002/med.21622. [DOI] [PubMed] [Google Scholar]
- Neumann U, Rueeger H, Machauer R, Veenstra SJ, Lueoend RM, Tintelnot-Blomley M, Laue G, Beltz K, Vogg B, Schmid P, Frieauff W, Shimshek DR, Staufenbiel M, Jacobson LH. A novel BACE inhibitor NB-360 shows a superior pharmacological profile and robust reduction of amyloid-β and neuroinflammation in APP transgenic mice. Mol Neurodegener. 2015;10:44. doi: 10.1186/s13024-015-0033-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palacios-Filardo J, Mellor JR. Neuromodulation of hippocampal long-term synaptic plasticity. Curr Opin Neurobiol. 2019;54:37–43. doi: 10.1016/j.conb.2018.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel S, Bansoad AV, Singh R, Khatik GL. BACE1: a key regulator in Alzheimer’s disease progression and current development of its inhibitors. Curr Neuropharmacol. 2022;20:1174–1193. doi: 10.2174/1570159X19666211201094031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelkey KA, Barksdale E, Craig MT, Yuan X, Sukumaran M, Vargish GA, Mitchell RM, Wyeth MS, Petralia RS, Chittajallu R, Karlsson RM, Cameron HA, Murata Y, Colonnese MT, Worley PF, McBain CJ. Pentraxins coordinate excitatory synapse maturation and circuit integration of parvalbumin interneurons. Neuron. 2015;85:1257–1272. doi: 10.1016/j.neuron.2015.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaguri H, Hashimoto S, Watamura N, Sato K, Takamura R, Nagata K, Tsubuki S, Ohshima T, Yoshiki A, Sato K, Kumita W, Sasaki E, Kitazume S, Nilsson P, Winblad B, Saito T, Iwata N, Saido TC. Recent advances in the modeling of Alzheimer’s disease. Front Neurosci. 2022;16:807473. doi: 10.3389/fnins.2022.807473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serrano-Pozo A, Das S, Hyman BT. APOE and Alzheimer’s disease: advances in genetics, pathophysiology, and therapeutic approaches. Lancet Neurol. 2021;20:68–80. doi: 10.1016/S1474-4422(20)30412-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi H, Koronyo Y, Rentsendorj A, Regis GC, Sheyn J, Fuchs DT, Kramerov AA, Ljubimov AV, Dumitrascu OM, Rodriguez AR, Barron E, Hinton DR, Black KL, Miller CA, Mirzaei N, Koronyo-Hamaoui M. Identification of early pericyte loss and vascular amyloidosis in Alzheimer’s disease retina. Acta Neuropathol. 2020;139:813–836. doi: 10.1007/s00401-020-02134-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snow WM, Albensi BC. Neuronal gene targets of NF-κB and their dysregulation in Alzheimer’s disease. Front Mol Neurosci. 2016;9:118. doi: 10.3389/fnmol.2016.00118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soto-Rojas LO, Campa-Córdoba BB, Harrington CR, Salas-Casas A, Hernandes-Alejandro M, Villanueva-Fierro I, Bravo-Muñoz M, Garcés-Ramírez L, De La Cruz-López F, Ontiveros-Torres M, Gevorkian G, Pacheco-Herrero M, Luna-Muñoz J. Insoluble vascular amyloid deposits trigger disruption of the neurovascular unit in Alzheimer’s disease brains. Int J Mol Sci. 2021;22:3654. doi: 10.3390/ijms22073654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stakos DA, Stamatelopoulos K, Bampatsias D, Sachse M, Zormpas E, Vlachogiannis NI, Tual-Chalot S, Stellos K. The Alzheimer’s disease amyloid-beta hypothesis in cardiovascular aging and-disease: JACC focus seminar. J Am Coll Cardiol. 2020;75:952–967. doi: 10.1016/j.jacc.2019.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su Q, Li T, He PF, Lu XC, Yu Q, Gao QC, Wang ZJ, Wu MN, Yang D, Qi JS. Trichostatin A ameliorates Alzheimer’s disease-related pathology and cognitive deficits by increasing albumin expression and Aβ clearance in APP/PS1 mice. Alzheimers Res Ther. 2021;13:7. doi: 10.1186/s13195-020-00746-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun E, Motolani A, Campos L, Lu T. The pivotal role of NF-kB in the pathogenesis and therapeutics of Alzheimer’s disease. Int J Mol Sci. 2022;23:8972. doi: 10.3390/ijms23168972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki H, Fukuyama K, Kumagai H. Bacterial γ-glutamyltranspeptidases, physiological function, structure, catalytic mechanism and application. Proc Jpn Acad Ser B Phys Biol Sci. 2020;96:440–469. doi: 10.2183/pjab.96.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweeney MD, et al. Vascular dysfunction-the disregarded partner of Alzheimer’s disease. Alzheimers Dement. 2019;15:158–167. doi: 10.1016/j.jalz.2018.07.222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tahami Monfared AA, Byrnes MJ, White LA, Zhang Q. Alzheimer’s disease: epidemiology and clinical progression. Neurol Ther. 2022;11:553–569. doi: 10.1007/s40120-022-00338-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamagno E, Guglielmotto M, Monteleone D, Vercelli A, Tabaton M. Transcriptional and post-transcriptional regulation of β-secretase. IUBMB Life. 2012;64:943–950. doi: 10.1002/iub.1099. [DOI] [PubMed] [Google Scholar]
- Tan HL, Chiu SL, Zhu Q, Huganir RL. GRIP1 regulates synaptic plasticity and learning and memory. Proc Natl Acad Sci U S A. 2020;117:25085–25091. doi: 10.1073/pnas.2014827117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiwari S, Atluri V, Kaushik A, Yndart A, Nair M. Alzheimer’s disease: pathogenesis, diagnostics, and therapeutics. Int J Nanomedicine. 2019;14:5541–5554. doi: 10.2147/IJN.S200490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unnisa A, Greig NH, Kamal MA. Nanotechnology-based gene therapy as a credible tool in the treatment of Alzheimer’s disease. Neural Regen Res. 2023;18:2127–2133. doi: 10.4103/1673-5374.369096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varga V, Török K, Feuer L, Gulyás J, Somogyi J. gamma-Glutamyltransferase in the brain and its role in formation of gamma-L-glutamyl-taurine. Prog Clin Biol Res. 1985;179:115–125. [PubMed] [Google Scholar]
- Will Y, Fischer KA, Horton RA, Kaetzel RS, Brown MK, Hedstrom O, Lieberman MW, Reed DJ. gamma-glutamyltranspeptidase-deficient knockout mice as a model to study the relationship between glutathione status, mitochondrial function, and cellular function. Hepatology. 2000;32:740–749. doi: 10.1053/jhep.2000.17913. [DOI] [PubMed] [Google Scholar]
- Wu WP, Zhou MY, Liu DL, Min X, Shao T, Xu ZY, Jing X, Cai MY, Xu S, Liang X, Mo M, Liu X, Xiong XD. circGNAQ, a circular RNA enriched in vascular endothelium, inhibits endothelial cell senescence and atherosclerosis progression. Mol Ther Nucleic Acids. 2021;26:374–387. doi: 10.1016/j.omtn.2021.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao MF, Xu D, Craig MT, Pelkey KA, Chien CC, Shi Y, Zhang J, Resnick S, Pletnikova O, Salmon D, Brewer J, Edland S, Wegiel J, Tycko B, Savonenko A, Reeves RH, Troncoso JC, McBain CJ, Galasko D, Worley PF. NPTX2 and cognitive dysfunction in Alzheimer’s disease. Elife. 2017;6:e23798. doi: 10.7554/eLife.23798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu L, Zhou Y, Hu L, Jiang H, Dong Y, Shen H, Lou Z, Yang S, Ji Y, Ruan L, Zhang X. Deficits in N-methyl-D-aspartate receptor function and synaptic plasticity in hippocampal CA1 in APP/PS1 mouse model of Alzheimer’s disease. Front Aging Neurosci. 2021a;13:772980. doi: 10.3389/fnagi.2021.772980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X, Sun Y, Cen X, Shan B, Zhao Q, Xie T, Wang Z, Hou T, Xue Y, Zhang M, Peng D, Sun Q, Yi C, Najafov A, Xia H. Metformin activates chaperone-mediated autophagy and improves disease pathologies in an Alzheimer disease mouse model. Protein Cell. 2021b;12:769–787. doi: 10.1007/s13238-021-00858-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu H, Lin L, Zhang Z, Zhang H, Hu H. Targeting NF-κB pathway for the therapy of diseases: mechanism and clinical study. Signal Transduct Target Ther. 2020;5:209. doi: 10.1038/s41392-020-00312-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zenaro E, Piacentino G, Constantin G. The blood-brain barrier in Alzheimer’s disease. Neurobiol Dis. 2017;107:41–56. doi: 10.1016/j.nbd.2016.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Forman HJ. Redox regulation of gamma-glutamyl transpeptidase. Am J Respir Cell Mol Biol. 2009;41:509–515. doi: 10.1165/rcmb.2009-0169TR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang YL, Wang J, Zhang ZN, Su Q, Guo JH. The relationship between amyloid-beta and brain capillary endothelial cells in Alzheimer’s disease. Neural Regen Res. 2022;17:2355–2363. doi: 10.4103/1673-5374.335829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou H, Gao F, Yang X, Lin T, Li Z, Wang Q, Yao Y, Li L, Ding X, Shi K, Liu Q, Bao H, Long Z, Wu Z, Vassar R, Cheng X, Li R, Shen Y. Endothelial BACE1 impairs cerebral small vessels via tight junctions and eNOS. Circ Res. 2022;130:1321–1341. doi: 10.1161/CIRCRESAHA.121.320183. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Co-localization of GGT5 with cerebral vascular endothelial cells in the brains of mice.
(A, B) Representative immunofluorescence images of transverse sections (A) and longitudinal sections (B) of cerebral vessels in four groups of mice. The mean fluorescence intensity of vascular endothelial cells GGT5 in the mice of WT + GGT5 group was stronger than that in the WT + Vector group. And the mean fluorescence intensity of vascular endothelial cells GGT5 was higher in the APP/PS1 + GGT5 group than that in the APP/PS1 + Vector group. DAPI, blue; CD31 (green, marked by 488); GGT5 (red, marked by Cy3). Scale bars: 50 µm. (C) Histograms showing the mean fluorescence intensity of GGT5 in vascular endothelial cells in the cortex of mice (n = 4 per group). Data are expressed as mean ± SEM.*P < 0.05 (one-way analysis of variance followed by least significant difference test). Ctx: Cortex; DAPI: 4,6-diamino-2-phenyl indole; GGT5: gamma-glutamyltransferase 5; Hip: hippocampus; WT: wide type.
The expression of NPTX2 in the brains of mice.
(A) Histogram showing Nptx2 gene expression in the cortex of mice by qPCR (n = 3 per group). (B, C) The representative Western blots (B) and quantitative analysis (C) for NPTX2 in the hippocampus of mice (n = 5 per group). Data are expressed as mean ± SEM. *P < 0.05 (one-way analysis of variance followed by Tukey's post hoc test). Ctx: Cortex; GGT5: gamma-glutamyltransferase 5; Hip: hippocampus; NPTX2: neuronal pentraxin II; qPCR: real-time polymerase chain reaction; WT: wide type.
Overexpression of GGT5 suppresses the expression of p-NF-κB by increasing GSH levels.
(A, B) The representative Western blots (A) and quantitative analysis (B) for GGT5 in bEnd.3 cells transfected with vector and GGT5 plasmid (n = 3 per group). (C, D) Histogram showing the level of intracelluar (C) and extracelluar (D) GSH/GSSG in bEnd.3 cells transfected with vector and GGT5 plasmid (n = 3-4 per group). (E, F) The cell viability of HT22 cells cocultured with medium derived from bEnd.3 cells transfected with Vector and GGT5 plasmid in both the vehicle-treated (E) and Aβ1-42-treated (F) groups (n = 3 per group). (G-I) The representative Western blots (G) and quantitative analysis for NF-κB (H) and p-NF-κB (I) in HT22 cells cultured using the aforementioned methodology (n = 3-4 per group). Data are expressed as mean ± SEM. *P < 0.05, **P < 0.01 (one-way analysis of variance followed by Tukey’s post hoc test (H, I), or independent-samples t-test (B-F)). Aβ: Amyloid-β; Ctx: cortex; GGT5: gamma-glutamyltransferase 5; GSH: glutathione; GSSG: oxidized glutathione; Hip: hippocampus; NF-κB: nuclear factor-kappa B; p-NF-κB: phosphor nuclear factor-kappa B; WT: wide type.
The differential genes identified in our in vitro experiments and those identified in the in vivo experiments conducted by Lau et al.