

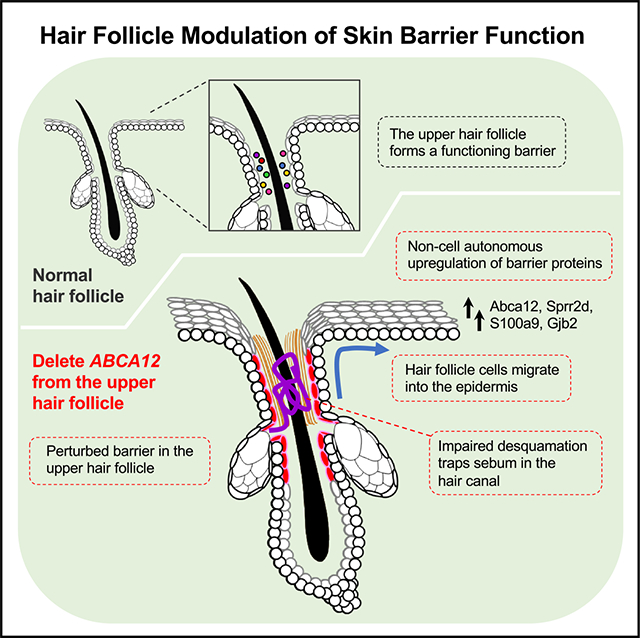
SUMMARY
Our skin provides a protective barrier that shields us from our environment. Barrier function is typically associated with the interfollicular epidermis; however, whether hair follicles influence this process remains unclear. Here, we utilize a potent genetic tool to probe barrier function by conditionally ablating a quintessential epidermal barrier gene, Abca12, which is mutated in the most severe skin barrier disease, harlequin ichthyosis. With this tool, we deduced 4 ways by which hair follicles modulate skin barrier function. First, the upper hair follicle (uHF) forms a functioning barrier. Second, barrier disruption in the uHF elicits non-cell-autonomous responses in the epidermis. Third, deleting Abca12 in the uHF impairs desquamation and blocks sebum release. Finally, barrier perturbation causes uHF cells to move into the epidermis. Neutralizing IL-17a, whose expression is enriched in the uHF, partially alleviated some disease phenotypes. Altogether, our findings implicate hair follicles as multi-faceted regulators of skin barrier function.
In brief
Skin usually comprises both follicular and interfollicular epithelia. Most studies on skin barrier function focus on the interfollicular epidermis without considering the role of the follicular epithelium. Using a targetable genetic tool to disrupt barrier function, Ford et al. identify multiple ways by which hair follicles modulate the skin’s barrier.
Graphical Abstract
INTRODUCTION
Our skin serves as a protective barrier that surrounds our internal organs while providing an interface for gas and liquid exchange. This barrier is formed when keratinocytes differentiate, stratify, and undergo cornification, a specialized form of cell death that gives rise to corneocytes.1,2 In the outermost layer of the skin, known as the stratum corneum, these corneocytes form a scaffold with other cross-linked structural proteins (“bricks”) and are surrounded by an amalgam of lipid-dominant lamellar sheets (“mortar”).3 Together, these bricks and mortar comprise the hydrophobic, insoluble barrier that is essential for life. With >100 inherited skin diseases linked to mutations in genes associated with barrier function, even subtle defects in this complicated process can have profound consequences.4
Ceramides are essential barrier lipids that constitute up to 50% of the total lipids by weight in the cornified envelope.5 These sphingolipids are synthesized by keratinocytes and transported by membrane-enclosed organelles known as lamellar granules (LGs).6 In granular layer keratinocytes, LGs travel from the Golgi apparatus to the apical plasma membrane, where they fuse and release their contents into the interstitial spaces of the overlying stratum corneum. In addition to lipids, LGs deliver a variety of cargo, including an assortment of lipid- and protein-processing enzymes, proteases for desquamation, structural proteins, and anti-microbial peptides.7 As such, LGs serve as the main secretory organelles in the skin.
Inherited ichthyoses encompass a large class of genetic disorders rooted in defective cornification.8 Common features include dysregulated barrier function, inflammation, and hyperkeratosis. Among all skin barrier diseases, harlequin ichthyosis (HI) is the most severe and is caused by mutations in ABCA12, which encodes an ATP-driven lipid transporter that localizes to LGs.9,10 In the absence of ABCA12, LGs fail to deliver lipids and desquamation enzymes to the stratum corneum.11,12 Consequently, HI patients possess scaly, plate-like skin that is paradoxically thickened but dysfunctional, lacking essential barrier lipids. Although neonatal intensive care and retinoid therapy can partially relieve symptoms, patients with HI suffer from debilitating, lifelong disfiguration and increased susceptibility to infection and dehydration.13,14
Most studies on skin barrier function and disease have focused on the interfollicular epidermis (IFE). However, the hair follicle epithelium is continuous with the IFE, and hair follicle openings constitute up to 10% of the skin’s total surface area at body sites such as the face.15 This upper hair follicle (uHF) domain, also known as the hair canal or infundibulum, is colonized by commensal microbes and secretes a variety of cytokines and chemokines to recruit immune cells.16–20 The uHF also provides a passageway for sebaceous gland (SG)-derived lipids to leave the follicle and enter the skin’s surface, where these oils can also modulate barrier function.21,22 Thus, given its unique anatomic location at the interface between the outer epidermis and the deeper hair follicle, the uHF likely serves as an active site of commerce in the skin.
Pharmacologically, the hair follicle is thought to be an important penetration route for topically applied drugs.15,23,24 Topical nanoparticles have also been reported to accumulate within the uHF,25 yet the influence of this domain on skin barrier function remains unclear.26 Here, we utilize a potent genetic strategy—targeted ablation of Abca12—to probe the consequences of disrupting a canonical epidermal barrier protein within the hair follicle.
RESULTS
The hair follicle forms a functional barrier
The epidermis and hair follicle are maintained by basal stem cell progenitors that give rise to differentiated suprabasal cells (Figure 1A). Previously, we reported that the inner, differentiated cells of the uHF express keratin (K) 79, a defining marker of this region27 (Figures 1B and 1C). To further compare the IFE and uHF compartments, we first assessed the localization of proteins associated with barrier function (Abca12, Rab11a),28 desquamation (kallikrein [Klk] 6 and 7),29 proteolysis (cathepsin D [Ctsd]),30 and differentiation (K10) in mouse dorsal skin. Although differentiated cells in both compartments express all markers, we noted that Klk6, Klk7, and Ctsd are especially enriched in K79+ uHF cells (Figures 1D and S1A). In contrast, the ceramide precursor glucosylceramide is abundant in the upper layers of the IFE but reduced in the uHF (Figure 1D). These findings indicate that barrier-associated proteins are found in both the IFE and uHF; however, these domains are also molecularly, and possibly functionally, distinct.
Figure 1. The hair follicle forms a functioning barrier.
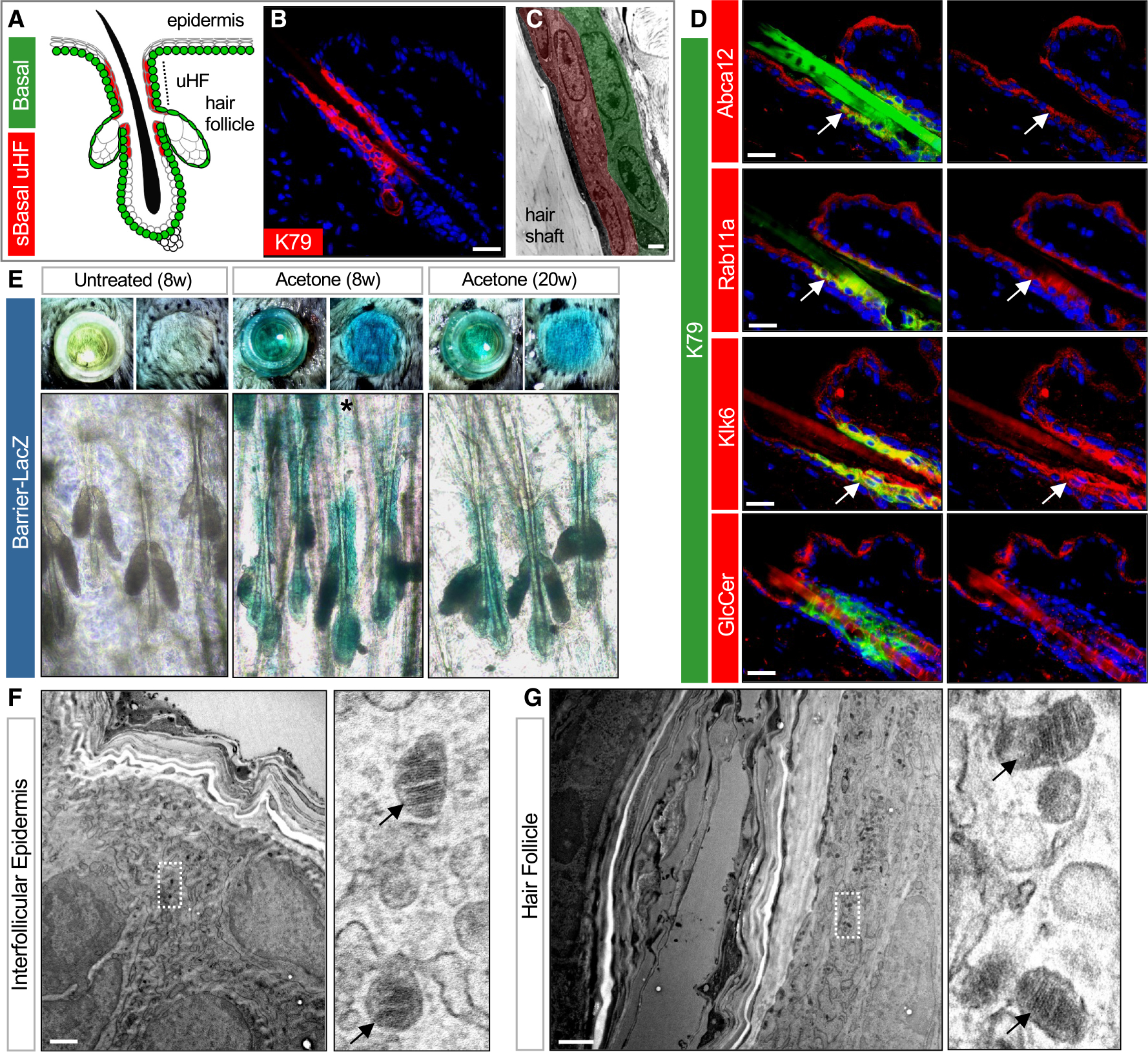
(A) Schematic of telogen hair follicle. Green, basal progenitors. Red, K79+ suprabasal (sBasal) cells in the upper hair follicle (uHF). Note that K79 is also expressed by sebocytes31 (not highlighted here).
(B) Immunohistochemical (IHC) staining for K79 (red).
(C) Transmission electron microscopy (TEM) of uHF showing basal (green) and suprabasal (red) hair follicle epithelium and hair shaft.
(D) IHC for barrier-associated proteins (red) and K79 (green). Right images show single-channel views of barrier-associated protein expression. Arrows, regions of overlap with K79+ uHF cells.
(E) Top images, overhead views of skin following incubation with X-gal in cloning cylinders (left) or with cylinders removed (right). Bottom images, whole mounts showing LacZ staining of untreated 8-week-old skin (left), acetone-treated 8-week-old skin (middle), and acetone-treated 20-week-old skin (right). Asterisk, occasional LacZ+ cells in the IFE.
(F) TEM of IFE. Right image is a magnified view showing LGs (arrows).
(G) TEM of uHF. Right image is a magnified view showing LGs (arrows).
Scale bars for (C), (F), and (G), 1 μm; all others, 50 μm.
See also Figure S1.
To visualize sites of barrier function, we next adapted an X-gal permeability assay originally described by Hardman et al.32 In this assay, barrier-competent skin excludes X-gal and remains unlabeled, whereas barrier-deficient skin turns blue from X-gal cleavage by endogenous β-galactosidase (LacZ). We exposed the surface of 8- or 20-week-old telogen mouse skin to X-gal loaded into cloning cylinders and observed no staining, indicating that the epidermis and hair follicle form a functional barrier (Figure 1E). However, when skin was pre-treated with acetone to perturb barrier lipids33 and then incubated with X-gal, extensive blue staining was seen in the hair follicle epithelium, with far less labeling in the epidermis, indicating selective barrier disruption in the follicle (Figure 1E).
Given these findings, we delved deeper into the ultrastructure of these domains and observed granular cells containing LGs, as well as secreted lipid lamellae, in both the IFE and uHF (Figures 1F, 1G, S1B, and S1C). Critically, whereas the IFE formed numerous cornified layers, these layers were less prominent toward the deeper follicle (Figure S1D). Altogether, these data suggest that while the hair follicle can enact a functioning barrier, the follicular barrier may be less robust than the epidermal barrier.
Validating a genetic tool to disrupt barrier function
We next used a genetic strategy to probe barrier function. Since HI is the most severe skin barrier disease, we reasoned that targeted deletion of Abca12, the causative gene mutated in HI,9,10 would provide us a tool to disrupt LGs and interrogate barrier function in different epithelial sub-compartments. We therefore first generated mice harboring a null allele of Abca12 that is constitutively disrupted by a LacZ insertion between exons 3 and 4, similar to mice previously described34 (Figures S2A and S2B). Newborn homozygous mutant pups (Abca12 knockout [Abca12-KO]) recapitulated HI features, including taut, shiny skin, thickened and compacted stratum corneum, and death from rapid dehydration, as has been reported for similar mouse models of HI35–37 (Figure 2A). We further confirmed the loss of Abca12 by immunohistochemistry (IHC) and validated by X-gal staining that the Abca12 promoter is active in both newborn epidermis and developing hair canals (Figure S2C).
Figure 2. Abca12 mutant mice provide a genetic tool to probe barrier function.
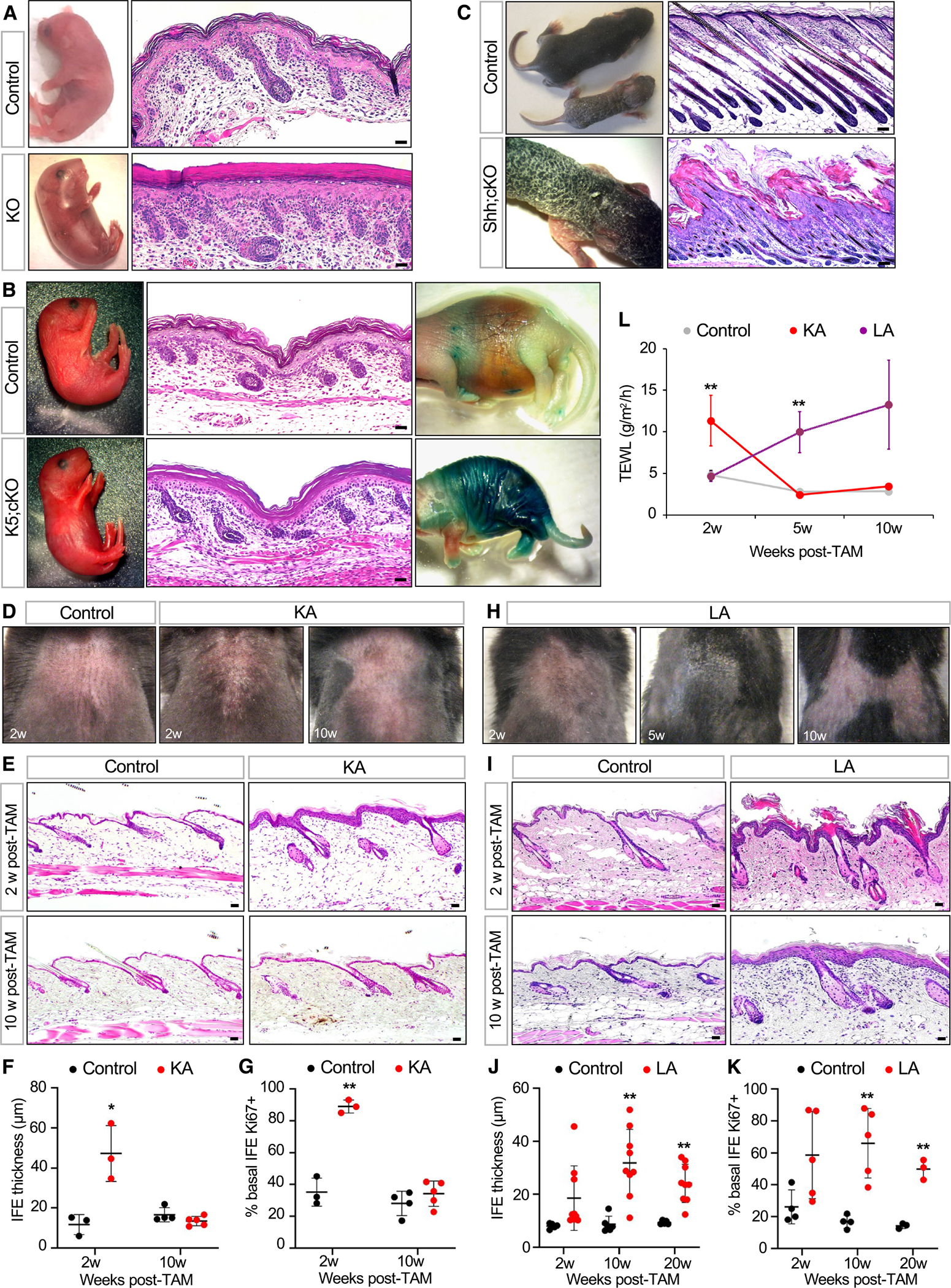
(A) Gross images of and skin histology from newborn Abca12 KO pup or littermate control.
(B) Similar to (A) but comparing newborn K5;cKO pup and littermate control. Rightmost images, LacZ barrier assay, with blue staining indicating dysfunctional skin barrier.32 Note that the cKO allele does not contain the LacZ transgene.
(C) Similar to (A) but comparing Shh;cKO pup and littermate control on post-natal days (P) 7–8.
(D) Gross images of shaved dorsal skin from control or KA mice at the indicated number of weeks post-TAM.
(E) Skin histology of control or KA mice at 2 weeks (top) or 10 weeks (bottom) post-TAM.
(F) Quantitation of IFE thickness in control (black) or KA (red) mice.
(G) Quantitation of basal IFE proliferation in control or KA mice.
(H) Gross images of shaved dorsal skin from LA mice at the indicated times post-TAM. See Figure S3B for an image of littermate control mouse.
(I) Skin histology of control or LA mice at 2 weeks (top) or 10 weeks (bottom) post-TAM.
(J) Quantitation of IFE thickness in control (black) or LA (red) mice.
(K) Quantitation of basal IFE proliferation in control or LA mice.
(L) TEWL measurements from control (gray), KA (red), and LA (purple) mice.
For (F), (G), (J), and (K): *p < 0.05 and **p < 0.01 by unpaired t test, comparing only samples from the same time point. n ≥ 3 mice per genotype per time point. Error bars indicate mean ± SD. For (L): **p < 0.01 by one-way ANOVA with post hoc Tukey test, comparing only samples from the same time point. For each time point, n = 4–5 KA mice, 6–9 LA mice, and ≥14 control mice. Data are represented as mean ± SE. Scale bar for (C), 100 μm; all others, 50 μm.
See also Figures S2 and S3.
Since early lethality precluded further studies with KO mice, we next generated a conditional allele of Abca12 (conditional KO [cKO]) (Figure S2A). When homozygous Abca12 cKO alleles were coupled with K5-promoter-driven Cre recombinase to delete Abca12 throughout the skin,38 these K5;cKO pups also exhibited severe barrier defects, validating this floxed deletion allele (Figures 2B and S2D).
Finally, we coupled homozygous Abca12 cKO alleles with Sonic hedgehog (Shh)-promoter-driven Cre recombinase, which targets genetic deletion to developing hair follicle progenitors but not to the epidermis.39 In striking contrast to Abca12-KO and K5;cKO mice, newborn Shh;cKO pups were indistinguishable from littermate controls up until post-natal day (P) 4. Subsequently, these mutants became runted and developed thick, scaly skin (Figure 2C). Histological analysis revealed that while follicular downgrowth was largely unaffected, hair follicle openings were plugged with keratotic material (Figure 2C). Since Shh;cKO mutants failed to thrive beyond ~P8, we grafted newborn mutant skin onto nude mice and found that hair protrusion was impaired even after several weeks (Figure S2E). These findings confirm that the Abca12 cKO allele can serve as a powerful genetic tool to disrupt at least 2 key features of normal skin—barrier function and desquamation.
Divergent outcomes after deleting Abca12 in different stem cell compartments
To probe the effects of deleting Abca12 in adult skin, we generated 2 additional tamoxifen (TAM)-inducible mouse strains. We and others have previously shown that K14-promoter-driven CreERT causes recombination primarily in IFE stem cells and less so in the follicle40,41 (Figure S3A). In contrast, Lrig1-promoter-driven CreERT2 induces recombination in Lrig1+ stem cells that maintain the uHF42,43 (Figure S3A). We therefore generated K14;Abca12-cKO (KA) and Lrig1;Abca12-cKO (LA) mice to compare the effects of deleting Abca12 in the IFE and uHF, respectively, in 8-week-old telogen skin.
In KA mice, we noticed the appearance of dry, flaky skin at 2 weeks post-TAM (Figure 2D). Unexpectedly, this phenotype disappeared by 10 weeks post-TAM (Figure 2D). These observations were confirmed histologically, which revealed that both epidermal thickness and proliferation increased sharply at 2 weeks post-TAM but then returned to normal by 10 weeks post-TAM (Figures 2E–2G).
In LA mice, we also observed dry, flaky skin starting at 2–5 weeks post-TAM (Figures 2H and S3B). Unlike KA mice, LA mutants displayed severe follicular hyperkeratosis, similar to Shh;cKO pups (Figure 2I). Although the overall appearance of some LA mice seemed to improve over time, all mutants retained chronically inflamed, reddish skin, and IHC revealed that the epidermis was hyperplastic and hyperproliferative even 20 weeks post-TAM (Figures 2J and 2K).
These trends—an immediate but reversible ichthyotic phenotype in KA mice versus longer-lasting effects in LA mutants—coincided with rates of transepidermal water loss (TEWL), a measure of inside-out skin barrier function. Whereas KA mice exhibited a temporary spike in TEWL, LA mutants displayed a more gradual sustained increase (Figure 2L). These findings demonstrate that deleting Abca12 in distinct IFE and hair follicle stem cell compartments causes divergent long-term outcomes in the skin. We explore the molecular and cellular basis for these differences below.
Disrupting the hair follicle barrier induces non-cell-autonomous responses
As mentioned above, the Abca12 KO allele contains a LacZ insertion that simultaneously disrupts gene function while reporting on endogenous Abca12 promoter activity (Figure S2A). In mice harboring both the KO and cKO alleles of Abca12—where one allele is constitutively inactivated by LacZ and the other is poised for deletion—targeted disruption of Abca12 in the adult IFE using K14-CreERT (KAL mice) caused a transient increase in Abca12 promoter activity in the hyperplastic epidermis, which later waned as the skin returned to normal (Figure 3A). Unexpectedly, when Abca12 was similarly deleted in the uHF using Lrig1-CreERT2 (LAL mice), this also caused increased Abca12 promoter activity in the IFE (Figure 3A). This effect was longer lasting, coincided with sustained epidermal hyperplasia, and occurred outside of the uHF, where Abca12 was initially deleted.
Figure 3. Abca12 deletion in the uHF induces non-cell-autonomous responses in the IFE.
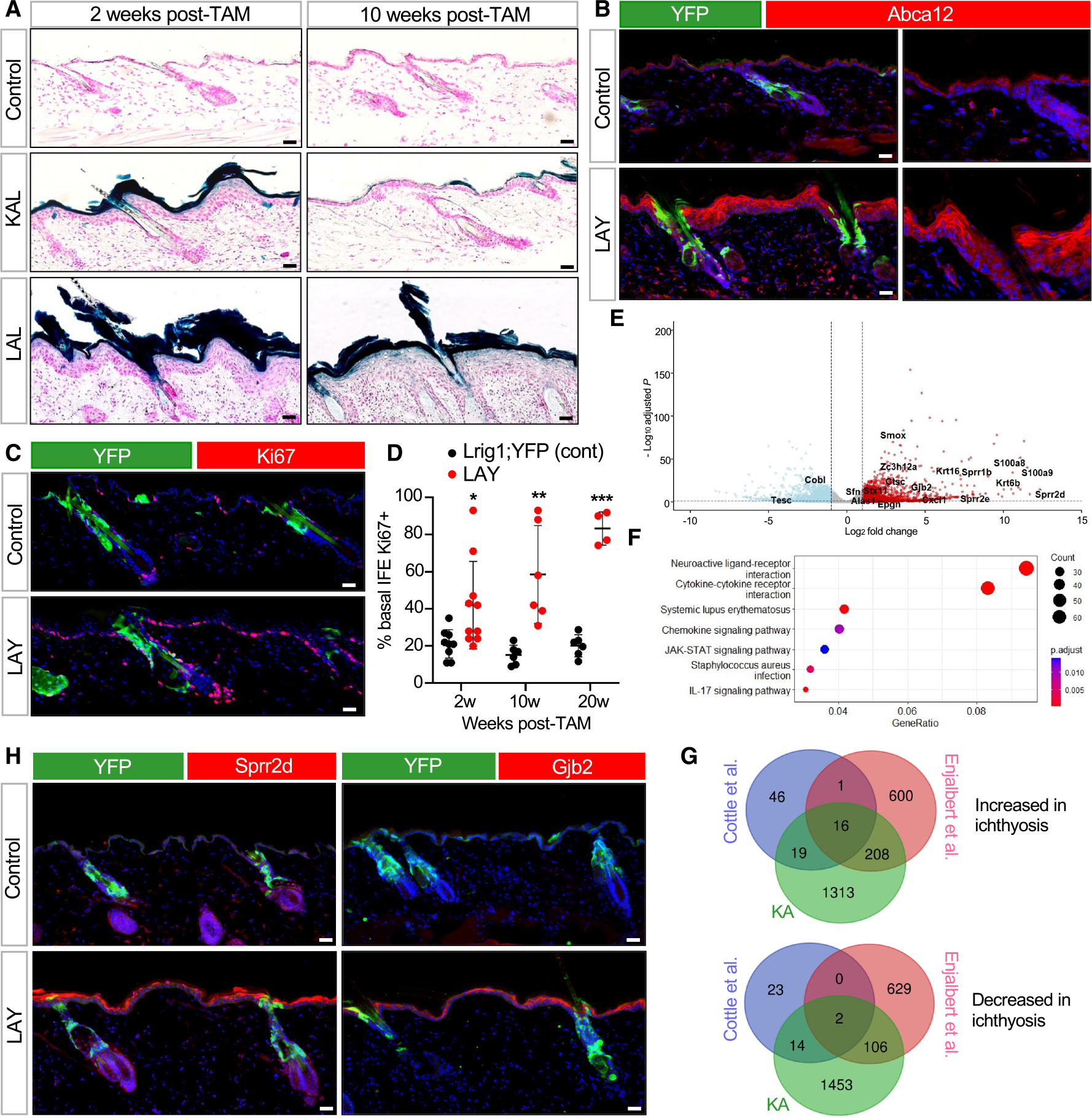
(A) LacZ staining, as a readout for Abca12 promoter activity, in K14-CreERT;Abca12-KO/cKO (KAL) or Lrig1-CreERT2;Abca12-KO/cKO (LAL) mice. Controls are Abca12-KO/cKO mice lacking Cre.
(B) IHC for Abca12 in Lrig1-CreERT2;Abca12-c/c;ROSA-YFP (LAY) mice 2 weeks post-TAM. Control mice are similar to LAY mutants but possess one wild-type copy of Abca12 (Abca12-c/+). Right images are magnified, single-channel views showing non-cell-autonomous upregulation of Abca12 (red) in LAY mutant skin that does not overlap with YFP+ uHF cells (green).
(C) Same as (B) but with IHC for Ki67 (red) 10 weeks post-TAM.
(D) Quantitation of proliferation in YFP-negative, basal IFE cells.
(E) Volcano plot showing upregulated (red) and downregulated (blue) differentially expressed genes (DEGs) in KA mice compared to control Abca12-c/c mice lacking Cre.
(F) KEGG enrichment analysis of DEGs.
(G) Venn diagrams showing overlap of DEGs in KA mice, patients with HI,44 and Abca12-mutant embryonic mice.45 Overlapping genes from the 3 datasets are depicted in (E).
(H) Same as (B) but with IHC for Sprr2d (red, left) or Gjb2 (red, right) 2 weeks post-TAM.
For (D): *p < 0.05, **p < 0.01, and ***p < 0.001 by unpaired t test, comparing only samples from the same time point. n ≥ 4 mice per genotype per time point. Error bars indicate mean ± SD.
Scale bar, 50 μm.
We next confirmed these observations using Lrig1-CreERT2 mice harboring homozygous Abca12 cKO alleles and a YFP reporter to identify cells that had undergone Cre-mediated recombination (LAY mice). Here, IHC staining confirmed that Abca12 protein and cell proliferation were both increased in non-recombined, YFP-negative cells in the IFE, suggesting a non-cell-autonomous effect (Figures 3B–3D).
To broaden our scope, we performed bulk RNA sequencing (RNA-seq) to assess transcriptional changes in KA mice at 2 weeks post-TAM. This identified 1,556 upregulated genes, including numerous genes involved with epidermal differentiation, barrier function, and wound healing (Figure 3E; Data S1). Pathway analysis further revealed significant enrichment for signaling modules associated with lupus erythematosus, JAK-STAT, and interleukin (IL)-17 (Figure 3F). Finally, we overlapped our RNA-seq dataset with those previously generated by Cottle et al., from Abca12-mutant mouse embryos,45 and by Enjalbert et al., from patients with HI.44 This identified 16 common upregulated genes, including Sprr2d, S100a9, and Gjb2 (connexin 26), which is mutated in keratitis-ichthyosis-deafness syndrome46 (Figures 3E and 3G).
To determine whether these genes can also be induced non-cell autonomously, we performed IHC for Sprr2d, S100a9, and Gjb2 in LAY mice. Indeed, when Abca12 was deleted in the uHF, all 3 proteins were upregulated in non-recombined cells in the IFE (Figures 3H and S3C). These findings demonstrate that disrupting the follicular barrier causes the IFE to upregulate proteins involved with barrier function, possibly as a compensatory way to restore overall skin homeostasis.
Altered sebum secretion and hair loss after deleting Abca12 in the uHF
Our previous studies showed that disrupting epidermal differentiation indirectly causes the enlargement of SGs, which are hair follicle-associated appendages that release oily secretions known as sebum into the uHF.40 We therefore examined whether Abca12 mutants also possess expanded SGs and, indeed, found that SGs were enlarged in LA mice (Figures 4A and 4B). These effects are likely indirect, since deleting Abca12 in the IFE also expanded SGs in KA mice, consistent with our previously published findings (Figures 4C and 4D).
Figure 4. Impaired sebum secretion and hair loss following Abca12 deletion in the uHF.
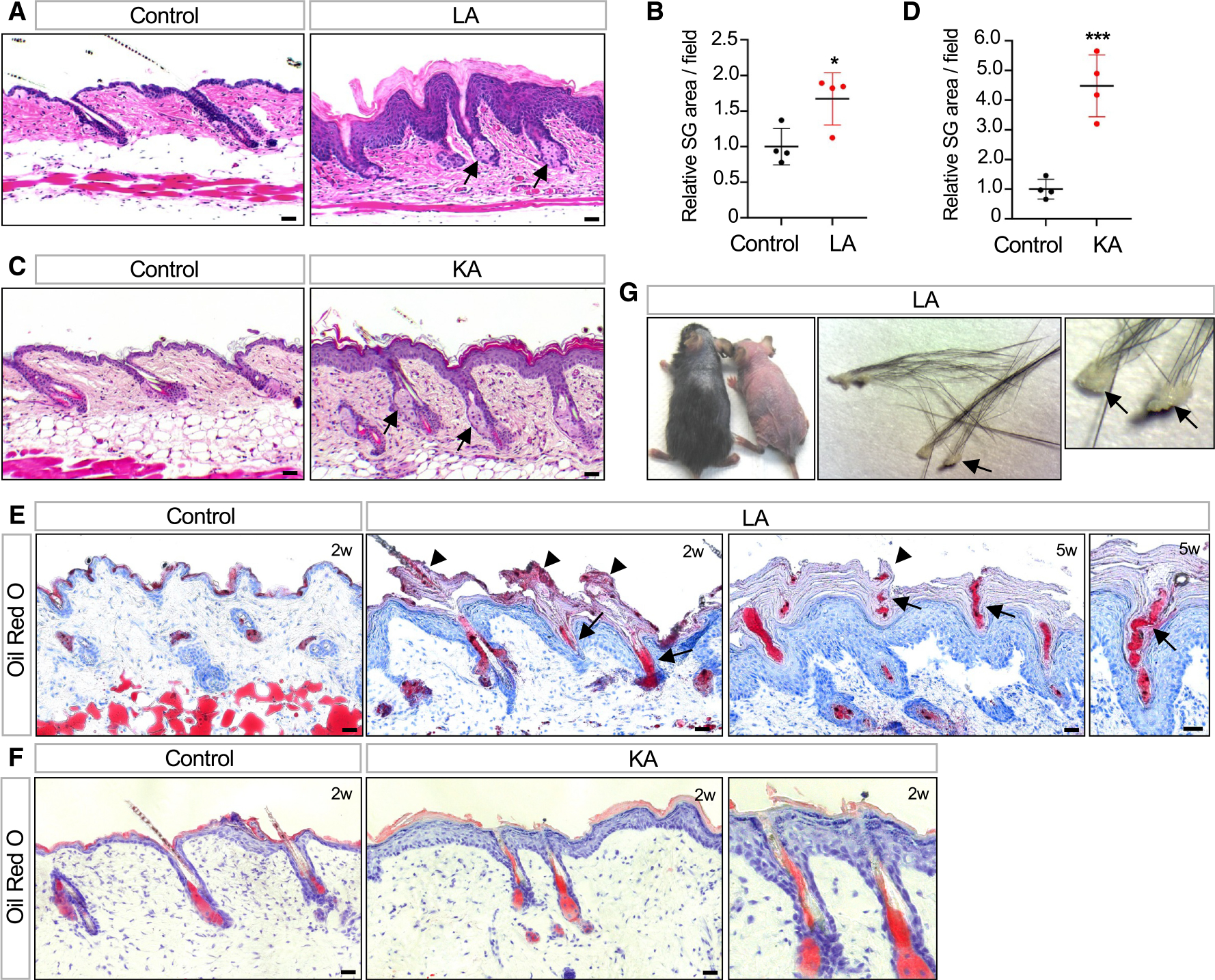
(A) Histology showing enlarged SGs (arrows) in LA skin.
(B) Quantitation of SG area in LA (red) or control (black) skin.
(C) Histology showing enlarged SGs (arrows) in KA skin.
(D) Quantitation of SG area in KA (red) or control (black) skin.
(E) Oil red O staining showing sebum lipids (arrows) trapped by hyperkeratotic material in the follicle (arrowheads) in LA mice 2–5 weeks post-TAM.
(F) Same as (E) but for KA mice 2 weeks post-TAM. Note that follicular hyperkeratosis and sebum occlusion are not observed even though the adjacent IFE is hyperplastic. Right image is magnified view of the center.
(G) Photo showing hair loss in two LA mutant mice, both 5 weeks post-TAM (left photo). Enlarged views of hair clumps with oily plugs (arrows) at the proximal end (middle and right photos).
For (B) and (D): *p < 0.05 and ***, p < 0.001 by unpaired t test. n = 4 mice per genotype. Error bars indicate mean ± SD.
Scale bar, 50 μm.
To monitor lipid output from these enlarged SGs, we next stained skin sections with the lipophilic dye oil red O. Notably, LA skin exhibited extensive follicular hyperkeratosis that blocked the outflow of sebum and caused entrapment of oils within the uHF (Figure 4E). In contrast, these phenotypes were rarely seen in KA skin, even though the IFE was hyperplastic (Figure 4F). These results indicate that the uHF specifically must undergo constant desquamation to allow unimpeded sebum release. When this process is blocked, a subset of LA and LAL mice also developed hair loss, with shed hairs retaining oily keratotic plugs, suggesting that fracturing had occurred near sites of occlusion in the uHF (Figure 4G).
Hair follicle-derived mutant cells enter the epidermis
Thus far, we have shown that hair follicles form a functioning barrier, modulate expression of barrier proteins in the IFE non-cell autonomously, and control the release of sebum, which also affects the skin’s barrier. However, a lingering question still remains: why does deleting Abca12 in the IFE cause transient ichthyotic phenotypes whereas deleting Abca12 in the uHF causes longer-lasting effects?
To answer this, we traced the fate of mutant (Abca12-c/c) or control (Abca12-c/+) cells over time, again using the Cre-inducible YFP reporter. Following Cre-mediated recombination in the IFE (KAY mice), we initially observed numerous YFP+ mutant cell clones in the epidermis after 2 weeks (Figures 5A and S3D). By 10 weeks post-TAM, however, the abundance of labeled mutant cells fell dramatically, coinciding with the phenotypic recovery seen in these animals (Figure 5B). In contrast, the abundance of YFP+ cells in control mice remained stable over time (Figures 5A and 5B).
Figure 5. Mutant uHF cells move into the IFE.
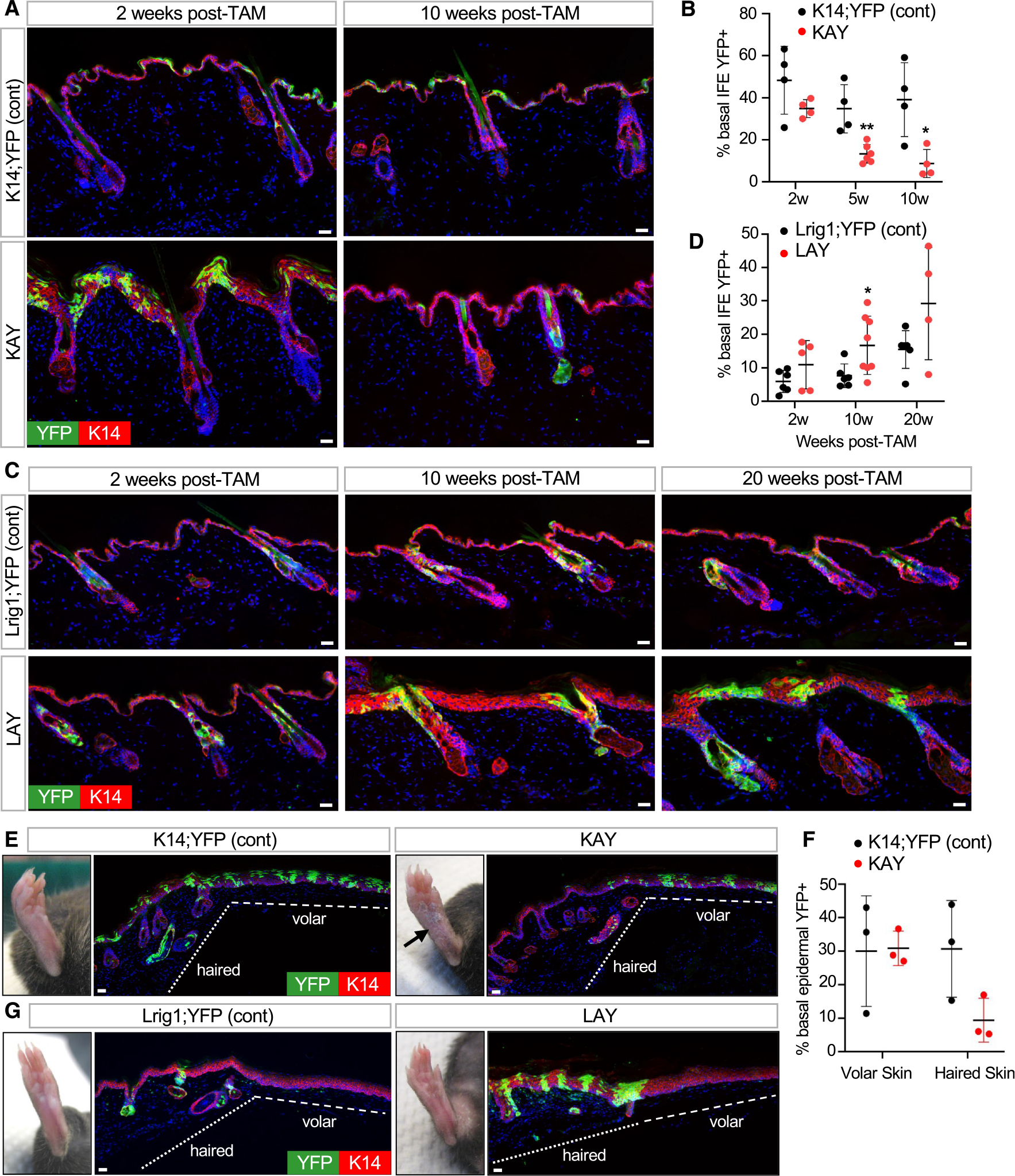
(A) IHC to trace recombined YFP+ cells (green) in K14-CreERT;Abca12-c/c;ROSA-YFP (KAY) mice. Control mice are similar to KAY mutants but possess one wild-type copy of Abca12 (Abca12-c/+).
(B) Quantitation of basal YFP+ cells in the IFE of KAY (red) or control (black) mice.
(C) IHC to identify recombined YFP+ cells (green) in LAY mice or controls. Genotypes are identical to those described in Figure 3B.
(D) Quantitation of basal YFP+ cells in the IFE of LAY (red) or control (black) mice.
(E) IHC for YFP+ cells (green) in KAY or control paw volar skin 15 weeks post-TAM. Arrow, persistent skin thickening in KAY volar skin.
(F) Quantitation of basal epidermal YFP+ cells in volar and adjacent haired skin from KAY (red) or control (black) mice. n = 3 mice per genotype.
(G) IHC for YFP+ cells (green) in LAY or control volar skin 10 weeks post-TAM. No recombination occurred in volar skin epidermis.
For (B) and (D): *p < 0.05 and **p < 0.01 by unpaired t test, comparing only samples from the same time point. n ≥ 4 mice per genotype per time point. Error bars indicate mean ± SD.
Scale bar, 50 μm.
See also Figure S3.
A dramatically different story emerged when we traced the fate of Abca12 mutant cells in the uHF (LAY mice). Here, mutant cells not only persisted long term in the uHF but also spread into the IFE (Figures 5C, 5D, and S3E). Labeled uHF cells in control mice also entered the IFE but did so more gradually (Figures 5C and 5D).
These divergent outcomes—loss of mutant cells from the IFE and expansion of mutant cells from the uHF—might be explained if Abca12 is required by epidermal cells to maintain competitive fitness. However, when we examined the fate of mutant cells in paw skin epidermis, a region devoid of hair follicles, these cells persisted up to 10 weeks post-TAM in KAY mice (Figures 5E–5G). This indicates that loss of Abca12-deficient cells occurs only in haired skin, arguing against a general requirement for Abca12 to maintain cell fitness. Instead, these observations suggest that Abca12 may possess critical skin site-specific functions. In haired skin IFE, mutant cells may become outcompeted by non-recombined neighboring cells or possibly diluted by cells originating from the uHF, as is seen after skin wounding.43,47 In LAY mice, mutant cells persist in the uHF and spread into the IFE to maintain the ichthyotic phenotype (Figures 5C and 5G). The movement of these mutant cells across compartments thereby suggests yet another mechanism by which hair follicles can modulate skin barrier function.
Disease features are partially driven by IL-17a
We next sought to identify factors that drive the pleiotropic ichthyotic phenotypes seen in our models. As mentioned above, our RNA-seq and functional enrichment analysis revealed that Il17 signaling is increased in KA mice at 2 weeks post-TAM, when peak disease features are observed (Figure 3F). This signature was partially driven by heightened expression of Il17a (log2 fold change of 8.24). By RNAscope in situ staining, we confirmed that Il17a is elevated throughout the IFE in both KA and LA mutants (Figure 6A). Unexpectedly, in both models, we noticed that Il17a RNA is especially abundant in the uHF and in SGs from LA mutants (Figure 6A). Il17a is also enriched in the uHF in non-ichthyotic control skin (Figure 6A).
Figure 6. IL-17a neutralization partially alleviates some disease phenotypes.
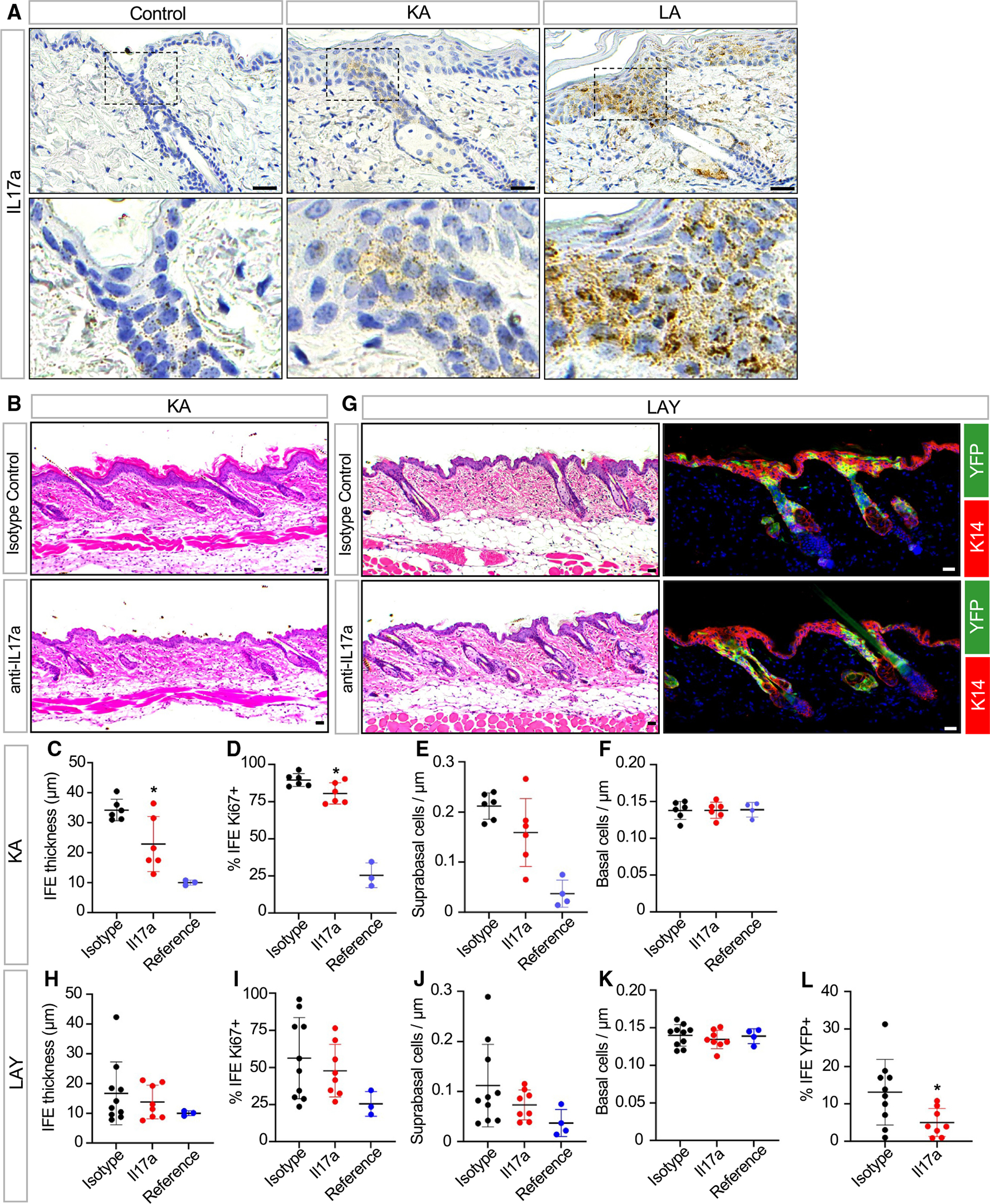
(A) RNAscope in situ staining for Il17a in control, KA, and LA skin 2 weeks post-TAM. Bottom images are magnified views of the uHF.
(B) Histology of KA skin treated with anti-IL-17a neutralizing antibody (bottom) or isotype control (top) 2 weeks post-TAM.
(C) Quantitation of IFE thickness in KA mice treated with anti-IL-17a neutralizing antibody (red) or isotype control (black) 2 weeks post-TAM. Measurements from untreated control mice (blue) are shown as a reference.
(D) Same as (C) but with quantitation for basal IFE proliferation.
(E) Same as (C) but with quantitation for suprabasal IFE cell abundance.
(F) Same as (C) but with quantitation for basal IFE cell abundance.
(G) Histology of LAY skin treated with anti-IL-17a neutralizing antibody (bottom) or isotype control (top) 2 weeks post-TAM (left). IHC for YFP+ mutant cells (green) in these samples (right).
(H) Quantitation of IFE thickness in LAY mice treated with anti-IL-17a neutralizing antibody (red) or isotype control (black) 2 weeks post-TAM. Measurements from untreated control mice (blue) are shown as a reference.
(I) Same as (H) but with quantitation for basal IFE proliferation.
(J) Same as (H) but with quantitation for suprabasal IFE cell abundance.
(K) Same as (H) but with quantitation for basal IFE cell abundance.
(L) Same as (H) but with quantitation for basal YFP+ mutant cells in the IFE.
*p < 0.05 by unpaired t test, comparing only samples from antibody-treated mice. For KA studies, n = 6 mice per treatment group. For LAY studies, n ≥ 8 mice per treatment group. Error bars indicate mean ± SD. Note that identical reference data are shown for (C)–(F) and (H)–(K), respectively, and that reference data were not used for statistical comparisons. n= 3–4 mice for reference data.
Scale bar, 50 μm.
To test the functional role of IL-17a in disease, we treated KA mutant mice with neutralizing antibodies against this cytokine. Relative to isotype control, IL-17a neutralization partially reduced IFE thickness and proliferation (Figures 6B–6D). Reduced epidermal thickness was associated with fewer differentiated suprabasal IFE cells, without changes in the abundance of underlying basal progenitors (Figures 6E and 6F).
We next attempted to extend these findings to LAY mutant mice. However, unlike KA mice, we observed no significant effect of IL-17a neutralization on IFE thickness, proliferation, or suprabasal cell counts, features that are increased non-cell autonomously in LAY mutants (Figures 6G–6K). Nonetheless, anti-IL-17a treatment significantly inhibited the expansion of YFP-labeled mutant cells from the uHF into the IFE (Figure 6L). Together, these findings further demonstrate that targeted barrier disruption in either the IFE or uHF causes complementary, but also distinct, skin phenotypes that are likely driven by non-overlapping disease mediators.
Selective barrier disruption in K79+ uHF cells causes ichthyosis-associated phenotypes
As a final question, we circled back to our original observation that the uHF is lined by K79+ suprabasal cells and asked whether even more precise ablation of Abca12 in only these differentiated cells—the exact cells that express Abca12 in the uHF (Figure 1D)—can cause ichthyotic phenotypes. Since Lrig1-CreERT2 induces genetic recombination in stem cells that maintain the entire uHF, we therefore turned instead to mice expressing constitutive Krt79-promoter-driven Cre (K79-Cre mice), which we previously generated and revalidated here (Figure 7A).48
Figure 7. Targeted deletion of Abca12 in K79+ uHF cells induces ichthyosis-associated phenotypes.
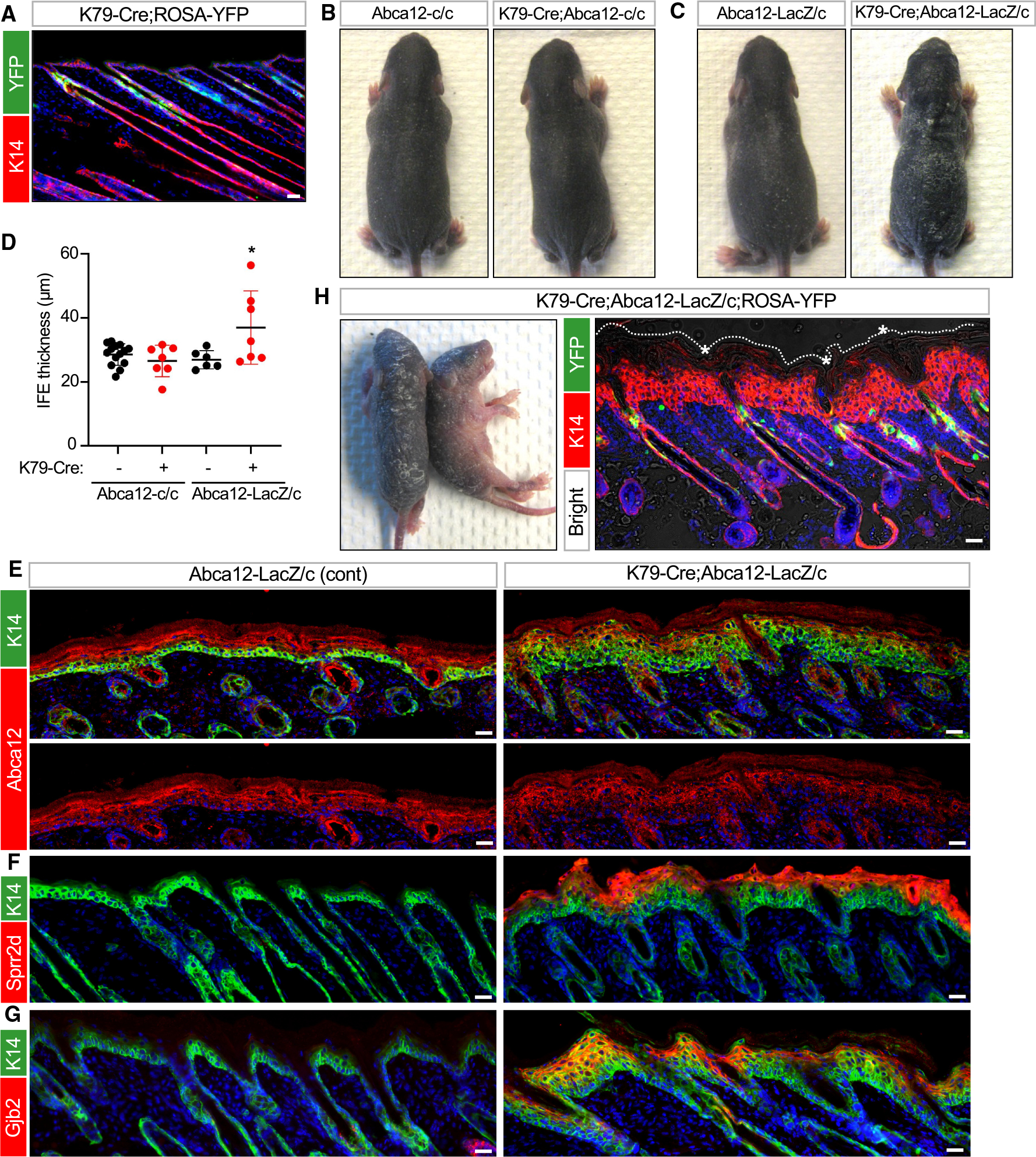
(A) IHC for YFP (green) confirming recombination in K79+ uHF cells in K79-Cre;ROSA-YFP mice at P12.
(B and C) Gross photos of P7 pups of the indicated genotypes. Only K79-Cre;Abca12-LacZ/c pups exhibited severe skin thickening and flakiness.
(D) Quantitation of epidermal thickness for P7 pups of the indicated genotypes. Each data point represents a single mouse.
(E) IHC for Abca12 (red) confirming that the protein was not ablated from the IFE of K79-Cre;Abca12-LacZ/c skin at P7. Bottom images are single-channel views of Abca12 staining.
(F) IHC for Sprr2d (red) in K79-Cre;Abca12-LacZ/c or control skin at P7.
(G) Same as (F) but with staining for Gjb2 (red).
(H) Left image, gross photos of two P6 mutant pups, both of genotype K79-Cre;Abca12-LacZ/c with ROSA-YFP. Right image, IHC showing YFP expression (green) in developing hair canals after Cre-mediated recombination and thickened epidermis (red). Asterisk, hyperkeratotic material obstructing hair canals, visible by bright-field overlay. Dotted line denotes the top of the epidermis. See Figure S4E for IHC of littermate control skin.
For (D): *p < 0.05 by one-way ANOVA with post hoc Tukey test, comparing K79-Cre;Abca12-LacZ/c pups against Abca12-LacZ/c or K79-Cre;Abca12-c/c pups. n ≥ 6 mice per genotype. Error bars indicate mean ± SD.
Scale bar, 50 μm.
See also Figure S4.
When K79-Cre was coupled with homozygous Abca12 cKO alleles, some mutant pups exhibited very subtle skin flaking at P4–P7, which disappeared within days (Figures 7B and S4A). Since this mild phenotype may be due to inefficient Cre-mediated recombination (Figure S4A), we generated K79-Cre mice harboring both the Abca12 KO (LacZ) and cKO (c) alleles, where deletion of only a single copy of Abca12 is needed to fully ablate its expression (K79-Cre;Abca12-LacZ/c mice). In a subset of these mutant pups, we now observed more severe skin thickening and flakiness between P4 and P7 (Figures 7C and 7D). We should note that some mutants did not exhibit overt phenotypes, while two mutants with thickened skin died just before P7 and were not included in our analysis. Finally, mutant animals that survived into adulthood appeared normal, suggesting that Cre inefficiency and variations in genetic background may have continued to cause incomplete penetrance (Figures S4B–S4D).
Focusing only on K79-Cre;Abca12-LacZ/c pups that exhibited ichthyotic features at P7, we confirmed that Abca12 was not deleted from the IFE, as expected (Figure 7E). Nonetheless, these pups possessed a hyperplastic epidermis that upregulated Sprr2d, S100a9, and Gjb2, similar to LAY mutants (Figures 7F–7G and S3F). As a final validation, we generated K79-Cre;Abca12-LacZ/c pups incorporating an additional YFP reporter allele. In these mutant pups, we again observed thickened epidermis and follicular hyperkeratosis. Critically, Cre-mediated recombination was highly restricted to developing hair canals, confirming K79-Cre targeting specificity (Figures 7H and S4E). In total, these findings firmly establish that K79+ cells in the uHF form a functioning barrier and that highly targeted barrier disruption in this domain is sufficient to induce widespread ichthyosis-associated phenotypes.
DISCUSSION
Most studies on skin barrier function have focused on IFE, with less consideration for whether hair follicles play a role. Nonetheless, studies in human HI fetuses have reported that hyperkeratosis initially appears prior to barrier formation and is most pronounced in developing hair canals.49 Concordantly, studies in ichq mice, which develop ichthyotic features due to a mutation in Cst6, have also found that disease onset coincides with the emergence of hair.50,51 These and other findings support the notion that the formation of the hair canal—an event characterized by extensive cellular remodeling, as well as shifts in immune and microbial composition—represents a seminal but also potentially vulnerable event in skin development.19,52,53
Our initial observation that K79 is only expressed in the uHF, but not in the IFE, indicated that these domains are molecularly distinct.27,48 Our results now argue that these domains are also functionally distinct. Previously, pharmacological studies have attempted to measure intrafollicular drug delivery, using varied approaches such as comparing haired versus non-haired skin, selectively blocking hair orifices, differential tape stripping, and imaging.15,24 These studies have suggested that hair follicles provide routes for drug penetration and may even act as drug reservoirs.23
Using a targetable genetic tool to disrupt LGs and perturb barrier function, here, we deduced at least 4 ways by which hair follicles modulate the skin’s barrier. First, we found that the uHF forms a functional, albeit “leakier,” barrier. Second, deleting Abca12 in the uHF causes non-cell-autonomous upregulation of barrier proteins in the IFE. Third, the uHF provides a passageway for sebum release, a process that requires constant desquamation in the hair canal. Fourth, uHF-derived mutant cells spread into the IFE. This last finding is contrasted by the observation that mutant cells in dorsal skin IFE are lost over time, leading to disease recovery. Differences in uHF versus IFE stem cell behavior, including proliferative potential, may account for these divergent outcomes.43,54,55
Additionally, we observed that the uHF is a site of enriched Il17a expression. IL-17a is a critical proinflammatory cytokine implicated in numerous skin disorders including psoriasis, atopic dermatitis, and ichthyosis.56,57 Previous studies have reported that in addition to IL-17a, uHF keratinocytes also secrete factors such as Il7, Il15, Ccl2, and Ccl20 to recruit leukocytes.18,19,58 Both T cells and Langerhans cells can be found in proximity to the uHF, further highlighting this domain as an important immune signaling center in the skin.18,58
Although HI is a skin-wide disease, our use of Abca12 cKO mice helps untangle the contributions of IFE and uHF keratinocytes to barrier function. Our findings also suggest that perturbing one epithelial compartment in the skin can elicit compensatory responses from the other. Indeed, previous studies have reported that barrier dysregulation can cause upregulation of barrier-associated genes,59 including Abca12,60,61 and stimulate processes associated with cornification, such as lipid synthesis and LG secretion.5,33,62 In keratinocytes, ceramide accumulation can act via PPAR nuclear receptors to upregulate Abca12 and promote differentiation.63–65 While these compensatory responses are typically regarded as cell-autonomous phenomena, a non-cell-autonomous effect acting broadly across different epithelial compartments, as seen here, has not been previously demonstrated.
Follicular hyperkeratosis and obstructed hair canals have been observed in several mouse mutants and human dermatoses. These include mice with mutations in genes encoding proteolytic enzymes such as matriptase, cathepsin L, and matrix metalloproteinase 9, suggesting that these proteases are critical for desquamation.66–68 Loss of phospholipase C and overexpression of Nrf2 have also been reported to induce ichthyotic phenotypes and occluded hair canals.69,70 In patients, follicular hyperkeratinization is seen in disorders such as acne, keratosis pilaris, ichthyosis follicularis with atrichia and photophobia, keratosis follicularis spinulosa decalvans, and pityriasis rubra pilaris.71–73 Follicular occlusion is also a unifying characteristic of a disease “tetrad” that includes hidradenitis suppurativa, acne conglobata, dissecting cellulitis, and pilonidal sinus. In our studies, we found that targeted deletion of Abca12 in the uHF, but not in the IFE, is sufficient to cause hair plugging. These effects are likely due, in part, to disrupted LG-mediated delivery of Klk6/7 and Ctsd, which are both normally enriched in K79+ cells of the uHF and serve to degrade corneodesmosomes.7,74 Subsequently, sebum becomes trapped within the hair canal, where these lipids are unable to fulfill their moisturizing and anti-microbial functions.75–77
Recent gene expression studies across multiple ichthyosis subtypes have indicated that these disorders share molecular similarities with psoriasis vulgaris, including upregulation of IL-17, IL-36, and tumor necrosis factor a signaling pathways.44,78 Moreover, disease severity correlated with IL-17A serum levels and IL-17 pathway gene expression.78 However, a recent clinical study reported that secukinumab, a monoclonal antibody against IL-17A, was ineffective at treating patients with ichthyosis.79 We also observed in mice that neutralizing IL-17a yielded only partial skin improvement, suggesting that targeting multiple inflammatory pathways may be critical.45,80 Finally, use of a NOS2 inhibitor or the JAK inhibitor tofacitinib has recently been shown to promote differentiation and barrier function in a 3D model of HI.44 These findings raise the possibility that drugs that restore barrier function may one day provide a novel treatment strategy for HI and other ichthyosis subsets.
Limitations of the study
Ichthyoses are inherited diseases that affect the entire skin. Because our studies seek to identify compartment-specific contributions to the skin’s barrier, the KA and LA mice used in our experiments are chimeric for Abca12 deletion and may not fully recapitulate all disease features. At the same time, the K14-CreERT and Lrig1-CreERT2 drivers that target Abca12 deletion to the adult IFE and uHF, respectively, can induce occasional recombination outside of their expected domains. Thus, we cannot formally rule out the possibility that these occasional recombination events may contribute to disease phenotypes. In KAY mice, our analyses of hairless volar skin suggest that losing Abca12 does not reduce the overall fitness of basal layer epidermal stem cells; nonetheless, we cannot rule out the possibility that Abca12 possesses vital skin site-specific functions, possibly explaining why mutant cells become lost from dorsal skin over time. Also currently unclear is why our use of the K79-Cre driver to delete Abca12 causes incomplete disease penetrance. A potential explanation is that Cre-mediated recombination occurs inefficiently in terminally differentiated cells. Finally, IL-17a neutralization yielded modest effects, especially in LAY mice. This may be partially due to phenotypic variability in this strain, although other disease mediators besides IL-17a are likely also involved and remain to be identified.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Sunny Wong (sunnyw@umich.edu).
Materials availability
All reagents generated in this study are available from the lead contact.
Data and code availability
RNA-seq data generated for this study can be accessed through GEO: accession GSE254889.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this work is available from the lead contact upon request.
EXPERIMENTAL MODEL AND STUDY PARTICIPANT DETAILS
Animals
To generate Abca12 KO (tm1a) mice, embryonic stem cells from clone HEPD0708–3-F11 were purchased from EuComm and microinjected into blastocysts. Abca12tm1a mice were crossed with Flpo mice (B6J;B6N-Tg(CAG-Flpo)1Afst/Mmucd) to generate Abca12 cKO (tm1c) mice. To induce Abca12 deletion in adults, 8 week old mice were injected intraperitoneally with tamoxifen dissolved in corn oil, at a dose of 1 mg/40 g body weight for 3 consecutive days, and harvested between 2 and 20 weeks after the initial day of tamoxifen administration. For IL-17a neutralization studies, mice were injected intraperitoneally with 250 μg of anti-IL-17a antibody or IgG1 isotype control, diluted in 200 μL InVivoPure dilution buffer, once every 3 days starting 1 day after the final dose of tamoxifen, and then harvested 2 weeks after the initial day of tamoxifen administration. For grafting experiments, dorsal skin from Shh;Abca12-cKO or littermate control pups was removed and sutured onto the backs of female athymic NU/J mice. All Abca12 mutant mice were of a mixed genetic background. Littermate controls were used for comparisons whenever possible, and both genders were analyzed in roughly equal numbers for experiments. C57BL/6 mice of both genders were analyzed by immunohistochemistry and transmission electron microscopy at 8 weeks of age, and for barrier function at the ages indicated in the text. All mice were used in accordance with regulations established by the University of Michigan Unit for Laboratory Animal Medicine.
METHOD DETAILS
Immunohistochemistry and RNA in situ staining
Frozen sections were stained by IHC using the following antibodies: goat anti-K79 (1:300); rabbit anti-K79/Ag-719527 (1:500); goat anti-K14 (1:1,000); chicken anti-K14 (1:1,000); rabbit anti-K14 (1:1,000); rabbit anti-Abca12 (1:2,000)35; rabbit anti-Rab11a (1:100); goat anti-Klk6 (1:100); goat anti-Klk7 (1:100); goat anti-Ctsd (1:100); rabbit anti-GlcCer (1:1,000); rabbit anti-K10 (1:1,000); rabbit anti-Sprr2d (1:200); rabbit anti-Gjb2 (1:2,000); rabbit anti-S100a9 (1:500); chicken anti-GFP/YFP (1:1,000); and rabbit anti-Ki67 (1:500). Multiple IHC images from the same field were captured and merged using the Auto-Blend feature of Adobe Photoshop CS6 to maximize image sharpness automatically across multiple focal planes. For RNA in situ staining, 5 μm paraffin skin samples were stained with a probe against mouse Il17a (Mm-Il17a) using RNAscope 2.5 Brown kit, according to manufacture’s instructions.
Transmission electron microscopy
For TEM, skin was fixed in 2% paraformaldehyde, 2.5% glutaraldehyde, 0.1 M cacodylate and 2 mM calcium chloride for 2 h at room temperature. The fixative was replaced, and samples were incubated overnight at 4°C. The next day, samples were rinsed and stored in 0.1 M cacodylate buffer at 4°C. Samples were processed for TEM using standard protocols, sectioned at 70 nm thickness, and imaged using a JEOL JEM 1400 microscope at an HT voltage of 60 kV. Images were collected using AMT Capture software.
Barrier assays
Newborn pups were decapitated and submerged below the neck in 1 mg/mL X-gal diluted in 5 mM potassium ferrocyanide and 5 mM potassium ferricyanide, pH 4.5, overnight at 37°C. To assess barrier function in adults, mice were euthanized and immediately shaved. A ~2 × 2 cm region of telogen skin was swabbed with a Q-tip (paper cotton swab) repeatedly dipped in 100% acetone for 5 min. Both treated and untreated skin were then excised and flattened against a paper towel. A cloning cylinder was affixed to the skin surface with lubricant (white petrolatum), the chamber was filled with 200 μL X-gal solution, and samples were floated on PBS inside a 6-well plate and incubated overnight at 37°C. To visualize samples by whole-mount, cloning cylinders were removed the next day, and the samples were rinsed briefly in PBS. Skins were re-stretched on a paper towel, patted dry, covered with Elmer’s rubber cement, allowed to dry for 5 min, and covered with transparent tape. The samples were floated epidermis-side up for 6 h in 5 mM EDTA/PBS at 37°C, and the epidermis was peeled away from the dermis. Separated epidermis was rinsed in PBS, fixed in formalin for 30 min, rinsed again, and imaged. For TEWL, mice were anesthetized, dorsal skin was shaved, and loose hairs were gently removed with a Kimwipe. Mice were allowed to acclimate in an upright resting position for 5 min, and telogen skin was probed using a Tewameter TM300-MPA5 and CK Multi-Probe software. Littermate mice lacking Cre or possessing one wild-type copy of Abca12 were designated as controls.
RNA extraction and sequencing
RNA was harvested from 4 kA mice and 3 littermate controls, 2 weeks post-TAM. The epidermis was isolated from telogen skin, as previously described,40 and processed using RNeasy mini kit. RNA-seq was performed by Novogene Corporation, and data were processed using DESeq286 to identify differentially expressed genes (DEGs) with |log2 fold-change (FC)| > 1 and adjusted p-value <0.05. R package clusterProfiler85 was used to test statistical enrichment of DEGs in KEGG pathways. For overlap analysis, we used data from HI patients generated by Enjalbert et al.44 (GSE131903), |log2FC| > 1 and adjusted p-value <0.01; and data from E17.5 Abca12-mutant mouse embryos generated by Cottle et al.45 (GSE56125), FC > 1.6 and adjusted p-value <0.05. Analyses were performed using GEO2R. Venn diagrams were generated using http://bioinformatics.psb.ugent.be/webtools/Venn/
Abca12 genotyping
PCR was performed on DNA isolated from ear tissue. For tm1a and tm1c: WT-1F (5′-GCTCTCTCTCTCTCTTCCTCTTC-3′), CSD-F (5′-CACACCTCCCCCTGAACCTGAAAC-3′), and 3R (5′-GAACTTACTTGAATAAAGCATTGCAGGC-3′) primers were used together with an annealing temperature of 56°C and 40 amplification cycles. See also Figures S2A and S2B.
QUANTIFICATION AND STATISTICAL ANALYSIS
Quantitation
Frozen sections stained for Ki67 and YFP were quantitated for proliferation from 3 random fields at 20× magnification. Only YFP-negative, basal layer IFE cells were manually counted to assess non-cell autonomous effects. To quantitate basal and suprabasal cells, the number of DAPI+ nuclei located either at the lowest layer of the IFE, or above that layer, respectively, were counted and normalized by the length of the IFE. To quantitate labeled uHF cells that had entered the IFE in LAY mice, the number of YFP+ basal cells in the IFE was counted and normalized to total IFE basal cells from 3 random fields. To measure IFE thickness, the vertical length of the IFE was measured at 3 random locations per sample and averaged. For sebaceous gland quantitation, 10 representative H&E fields were imaged at 20× magnification, each gland was manually traced, and the total SG area was calculated in pixels using AxioVision software. For adult studies, only telogen skin samples were quantitated.
Statistics
Unpaired student’s t-test was performed at the following website: http://www.physics.csbsju.edu/stats/Index.html. One-way ANOVA was performed at the following website: https://www.socscistatistics.com/tests/anova/default2.aspx. All data-points represent independent mice, except in Figure 2L, where averages are depicted.
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| Rabbit anti-Abca12 | Zuo et al.35 | N/A |
| Goat anti-Ctsd | R&D Systems | Cat # AF1029 |
| Chicken anti-GFP | Abcam | Cat # ab13970 |
| Rabbit anti-Gjb2 | Fisher Scientific | Cat # 71–0500 |
| Rabbit anti-GlcCer | Glycobiotech | Cat # RAS_0011 |
| Rabbit anti-K10 | Covance | Cat # PRB-159P |
| Chicken anti-K14 | Biolegend | Cat # 906004 |
| Goat anti-K14 | Santa Cruz | Cat # sc-17104 |
| Rabbit anti-K14 | Covance | Cat # PRB-155P |
| Goat anti-K79 | Santa Cruz | Cat # sc-243156 |
| Rabbit anti-K79 | Abcam | Cat # ab7195 (Ag7195)27 |
| Rabbit anti-Ki67 | Cell Signaling | Cat # 12202S |
| Goat anti-Klk6 | R&D Systems | Cat # AF2008 |
| Goat anti-Klk7 | R&D Systems | Cat # AF2624 |
| Rabbit anti-Rab11a | Cell Signaling | Cat # 2413 |
| Rabbit anti-S100a9 | Cell Signaling | Cat # 73425 |
| Rabbit anti-Sprr2d | Proteintech | Cat # 23046–1-AP |
| InVivoMAb anti-mouse IL-17A | BioXCell | Cat # BE0173, clone 17F3 |
| InVivoMAb mouse IgG1 isotype control | BioXCell | Cat # BE0083, clone MOPC-21 |
|
| ||
| Biological samples | ||
|
| ||
| Mouse tissue samples, obtained in accordance with guidelines established by the University of Michigan Unit for Laboratory Animal Medicine | This manuscript | Study protocol # PRO00011782 |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| Acetone | Sigma | Cat # 650501 |
| Cacodylate solution | Electron Microscopy Sciences | Cat # 11652 |
| Calcium chloride solution | Electron Microscopy Sciences | Cat # 12340 |
| Corn oil | Sigma | Cat #C8267 |
| Glutaraldehyde solution | Electron Microscopy Sciences | Cat # 16300 |
| InVivoPure pH 7.0 Dilution Buffer | BioXCell | Cat # IP0070 |
| Paraformaldehyde solution | Electron Microscopy Sciences | Cat # 15700 |
| Potassium ferricyanide | Sigma | Cat # 702587 |
| Potassium ferrocyanide | Sigma | Cat #P9387 |
| Tamoxifen | Sigma | Cat #T5648 |
| X-Gal | Roche | Cat # 10651745001 |
|
| ||
| Critical commercial assays | ||
|
| ||
| RNAscope 2.5 HD Reagent Kit-BROWN | ACD | Cat #322310 |
| RNAscope 2.5 Pretreat Reagents-H202 and Protease Plus | ACD | Cat # 322330 |
| RNAscope Target Retrieval | ACD | Cat # 322000 |
| RNAscope Wash Buffer | ACD | Cat #310091 |
| RNeasy Mini Kit | Qiagen | Cat # 74104 |
|
| ||
| Deposited data | ||
|
| ||
| Data for RNA sequencing of KA skin | This study | GEO: GSE254889 |
| Data for E17.5 Abca12 mutant mouse embryos | Cottle et al.45 | GEO: GSE56125 |
| Data for human HI patients | Enjalbert et al.44 | GEO: GSE131903 |
|
| ||
| Experimental models: Organisms/strains | ||
|
| ||
| Mouse: Abca12tm1a (KO or “LacZ” allele) | EUCOMM | Clone # HEPD0708–3-F11 |
| Mouse: Abca12tm1c (cKO or “c” allele) | This study | N/A |
| Mouse: Lrig1tm1.1(cre/ERT2)Rjc (Lrig1-CreERT2) | The Jackson Laboratory42 | Cat # 018418 |
| Mouse: Tg(KRT5-cre)5132Jlj (K5-Cre) | Ramirez et al.38 | MGI:3050065 |
| Mouse: Tg(KRT14-cre/ERT)20Efu/J (K14-CreERT) | The Jackson Laboratory81 | Cat # 005107 |
| Mouse: Krt79-Cre (K79-Cre) | Mesler et al.48 | N/A |
| Mouse: Shhtm1(EGFP/cre)Cjt/J (Shh-Cre) | The Jackson Laboratory82 | Cat # 005622 |
| Mouse: Gt(ROSA)26Sortm1(EYFP)Cos (YFP reporter) | The Jackson Laboratory83 | Cat # 006148 |
| Mouse: B6J;B6N-Tg(CAG-Flpo)1Afst/Mmucd | Kranz et al.84 | MGI:4453967 |
| Mouse: C57BL/6J | The Jackson Laboratory | Cat # 000664 |
| Mouse: NU/J | The Jackson Laboratory | Cat # 002019 |
|
| ||
| Oligonucleotides | ||
|
| ||
| Genotyping primer WT-1F: 5'-GCTCTCTCTCTCTCTTCCTCTTC-3' | This manuscript | N/A |
| Genotyping primer CSD-F: 5' -CACACCTCCCCCTGAACCTGAAAC-3' | This manuscript | N/A |
| Genotyping primer 3R: 5'-GAACTTACTTGAATAAAGCATTGCAGGC-3' | This manuscript | N/A |
| In situ probe: Mouse IL-17a | ACD (RNAscope) | Cat # 319571 (Mm-Il17a) |
|
| ||
| Software and algorithms | ||
|
| ||
| Adobe Photoshop CS6 | Adobe Inc. | https://www.adobe.com |
| AxioVision, Version 4.8.2 | Carl Zeiss | https://www.zeiss.com/microscopy/us/products/microscope-software/axiovision.html |
| CK Multi-Probe software, Version 2.2.0.1/3/22/2016 | Courage + Khazaka GmbH | https://www.courage-khazaka.de/en/ |
| clusterProfiler | Yu et al.85 | https://bioconductor.org/packages/release/bioc/html/clusterProfiler.html |
| DESeq2 | Love et al.86 | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| Prism, Version 6 | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
|
| ||
| Other | ||
|
| ||
| Cloning cylinders | Bel-Art | Cat # 378470200 |
| Tewameter | Courage + Khazaka GmbH | Cat # TM300-MPA5 |
Highlights.
Abca12 is a canonical skin barrier gene mutated in harlequin ichthyosis
Conditional deletion of Abca12 provides a potent genetic tool to probe barrier function
Hair follicles exert multi-faceted control over the skin’s barrier
Deleting Abca12 in Krt79+ upper hair follicle cells causes ichthyosis-associated phenotypes
ACKNOWLEDGMENTS
We are grateful to the Dlugosz lab (University of Michigan) for insightful discussions and to Debra Crumrine, Dr. Jason Meyer (Vanderbilt), and Sasha Meshinchi (University of Michigan) for transmission electron microscopy advice. We also thank Drs. Michael Fitzgerald and Mason Freeman (Harvard Medical School) for the Abca12 antibody and Thomas Huyge (University of Michigan) for clerical assistance. S.Y.W. acknowledges support from the NIH (R01AR080654 and R01AR065409), the LEO Foundation (LF18017), and the UM Skin Biology and Disease Resource-based Center (P30AR075043).
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2024.114347.
REFERENCES
- 1.Candi E, Schmidt R, and Melino G (2005). The cornified envelope: a model of cell death in the skin. Nat. Rev. Mol. Cell Biol. 6, 328–340. [DOI] [PubMed] [Google Scholar]
- 2.Lippens S, Denecker G, Ovaere P, Vandenabeele P, and Declercq W (2005). Death penalty for keratinocytes: apoptosis versus cornification. Cell Death Differ. 12, 1497–1508. [DOI] [PubMed] [Google Scholar]
- 3.Elias PM, Gruber R, Crumrine D, Menon G, Williams ML, Wake-field JS, Holleran WM, and Uchida Y (2014). Formation and Functions of the Corneocyte Lipid Envelope (CLE). Biochim. Biophys. Acta 1841, 314–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lopez-Pajares V, Yan K, Zarnegar BJ, Jameson KL, and Khavari PA (2013). Genetic pathways in disorders of epidermal differentiation. Trends Genet. 29, 31–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Feingold KR, and Elias PM (2014). Role of lipids in the formation and maintenance of the cutaneous permeability barrier. Biochim. Biophys. Acta 1841, 280–294. [DOI] [PubMed] [Google Scholar]
- 6.Feingold KR, and Jiang YJ (2011). The mechanisms by which lipids coordinately regulate the formation of the protein and lipid domains of the stratum corneum: Role of fatty acids, oxysterols, cholesterol sulfate and ceramides as signaling molecules. Dermatoendocrinol. 3, 113–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Raymond AA, Gonzalez de Peredo A, Stella A, Ishida-Yamamoto A, Bouyssie D, Serre G, Monsarrat B, and Simon M (2008). Lamellar bodies of human epidermis: proteomics characterization by high throughput mass spectrometry and possible involvement of CLIP-170 in their trafficking/secretion. Mol. Cell. Proteomics. 7, 2151–2175. [DOI] [PubMed] [Google Scholar]
- 8.Schmuth M, Martinz V, Janecke AR, Fauth C, Schossig A, Zschocke J, and Gruber R (2013). Inherited ichthyoses/generalized Mendelian disorders of cornification. Eur. J. Hum. Genet. 21, 123–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Akiyama M, Sugiyama-Nakagiri Y, Sakai K, McMillan JR, Goto M, Arita K, Tsuji-Abe Y, Tabata N, Matsuoka K, Sasaki R, et al. (2005). Mutations in lipid transporter ABCA12 in harlequin ichthyosis and functional recovery by corrective gene transfer. J. Clin. Invest. 115, 1777–1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kelsell DP, Norgett EE, Unsworth H, Teh MT, Cullup T, Mein CA, Dopping-Hepenstal PJ, Dale BA, Tadini G, Fleckman P, et al. (2005). Mutations in ABCA12 underlie the severe congenital skin disease harlequin ichthyosis. Am. J. Hum. Genet. 76, 794–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Akiyama M (2014). The roles of ABCA12 in epidermal lipid barrier formation and keratinocyte differentiation. Biochim. Biophys. Acta 1841, 435–440. [DOI] [PubMed] [Google Scholar]
- 12.Hovnanian A (2005). Harlequin ichthyosis unmasked: a defect of lipid transport. J. Clin. Invest. 115, 1708–1710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Glick JB, Craiglow BG, Choate KA, Kato H, Fleming RE, Siegfried E, and Glick SA (2017). Improved Management of Harlequin Ichthyosis With Advances in Neonatal Intensive Care. Pediatrics 139, e20161003. [DOI] [PubMed] [Google Scholar]
- 14.Milstone LM, and Choate KA (2013). Improving outcomes for harlequin ichthyosis. J. Am. Acad. Dermatol. 69, 808–809. [DOI] [PubMed] [Google Scholar]
- 15.Meidan VM (2010). Methods for quantifying intrafollicular drug delivery: a critical appraisal. Expert Opin. Drug Deliv. 7, 1095–1108. [DOI] [PubMed] [Google Scholar]
- 16.Montes LF, and Wilborn WH (1970). Anatomical location of normal skin flora. Arch. Dermatol. 101, 145–159. [PubMed] [Google Scholar]
- 17.Conwill A, Kuan AC, Damerla R, Poret AJ, Baker JS, Tripp AD, Alm EJ, and Lieberman TD (2022). Anatomy promotes neutral coexistence of strains in the human skin microbiome. Cell Host Microbe 30, 171–182.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nagao K, Kobayashi T, Moro K, Ohyama M, Adachi T, Kitashima DY, Ueha S, Horiuchi K, Tanizaki H, Kabashima K, et al. (2012). Stress-induced production of chemokines by hair follicles regulates the trafficking of dendritic cells in skin. Nat. Immunol. 13, 744–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Scharschmidt TC, Vasquez KS, Pauli ML, Leitner EG, Chu K, Truong HA, Lowe MM, Sanchez Rodriguez R, Ali N, Laszik ZG, et al. (2017). Commensal microbes and hair follicle morphogenesis coordinately drive Treg migration into neonatal skin. Cell Host Microbe 21, 467–477.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Oulès B, Philippeos C, Segal J, Tihy M, Vietri Rudan M, Cujba AM, Grange PA, Quist S, Natsuga K, Deschamps L, et al. (2020). Contribution of GATA6 to homeostasis of the human upper pilosebaceous unit and acne pathogenesis. Nat. Commun. 11, 5067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schneider MR, and Paus R (2014). Deciphering the functions of the hair follicle infundibulum in skin physiology and disease. Cell Tissue Res. 358, 697–704. [DOI] [PubMed] [Google Scholar]
- 22.Zouboulis CC, Coenye T, He L, Kabashima K, Kobayashi T, Niemann C, Nomura T, Oláh A, Picardo M, Quist SR, et al. (2022). Sebaceous immunobiology - skin homeostasis, pathophysiology, coordination of innate immunity and inflammatory response and disease associations. Front. Immunol. 13, 1029818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lademann J, Richter H, Schanzer S, Knorr F, Meinke M, Sterry W, and Patzelt A (2011). Penetration and storage of particles in human skin: perspectives and safety aspects. Eur. J. Pharm. Biopharm. 77, 465–468. [DOI] [PubMed] [Google Scholar]
- 24.Patzelt A, and Lademann J (2020). Recent advances in follicular drug delivery of nanoparticles. Expert Opin. Drug Deliv. 17, 49–60. [DOI] [PubMed] [Google Scholar]
- 25.Patzelt A, Mak WC, Jung S, Knorr F, Meinke MC, Richter H, Rühl E, Cheung KY, Tran NBNN, and Lademann J (2017). Do nanoparticles have a future in dermal drug delivery? J. Control. Release 246, 174–182. [DOI] [PubMed] [Google Scholar]
- 26.Zorn-Kruppa M, Vidal-Y-Sy S, Houdek P, Wladykowski E, Grzybowski S, Gruber R, Gorzelanny C, Harcup J, Schneider SW, Majumdar A, and Brandner JM (2018). Tight Junction barriers in human hair follicles - role of claudin-1. Sci. Rep. 8, 12800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Veniaminova NA, Vagnozzi AN, Kopinke D, Do TT, Murtaugh LC, Maillard I, Dlugosz AA, Reiter JF, and Wong SY (2013). Keratin 79 identifies a novel population of migratory epithelial cells that initiates hair canal morphogenesis and regeneration. Development 140, 4870–4880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Reynier M, Allart S, Gaspard E, Moga A, Goudounèche D, Serre G, Simon M, and Leprince C (2016). Rab11a is essential for lamellar body biogenesis in the human epidermis. J. Invest. Dermatol. 136, 1199–1209. [DOI] [PubMed] [Google Scholar]
- 29.Borgoño CA, Michael IP, Komatsu N, Jayakumar A, Kapadia R, Clayman GL, Sotiropoulou G, and Diamandis EP (2007). A potential role for multiple tissue kallikrein serine proteases in epidermal desquamation. J. Biol. Chem. 282, 3640–3652. [DOI] [PubMed] [Google Scholar]
- 30.Turk V, Stoka V, Vasiljeva O, Renko M, Sun T, Turk B, and Turk D (2012). Cysteine cathepsins: from structure, function and regulation to new frontiers. Biochim. Biophys. Acta 1824, 68–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Veniaminova NA, Jia YY, Hartigan AM, Huyge TJ, Tsai SY, Grachtchouk M, Nakagawa S, Dlugosz AA, Atwood SX, and Wong SY (2023). Distinct mechanisms for sebaceous gland self-renewal and regeneration provide durability in response to injury. Cell Rep. 42, 113121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hardman MJ, Sisi P, Banbury DN, and Byrne C (1998). Patterned acquisition of skin barrier function during development. Development 125, 1541–1552. [DOI] [PubMed] [Google Scholar]
- 33.Grubauer G, Elias PM, and Feingold KR (1989). Transepidermal water loss: the signal for recovery of barrier structure and function. J. Lipid Res. 30, 323–333. [PubMed] [Google Scholar]
- 34.Cottle DL, Ursino GMA, Jones LK, Tham MS, Zylberberg AK, and Smyth IM (2020). Topical aminosalicylic acid improves keratinocyte differentiation in an inducible mouse model of harlequin ichthyosis. Cell Rep. Med. 1, 100129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zuo Y, Zhuang DZ, Han R, Isaac G, Tobin JJ, McKee M, Welti R, Brissette JL, Fitzgerald ML, and Freeman MW (2008). ABCA12 maintains the epidermal lipid permeability barrier by facilitating formation of ceramide linoleic esters. J. Biol. Chem. 283, 36624–36635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yanagi T, Akiyama M, Nishihara H, Sakai K, Nishie W, Tanaka S, and Shimizu H (2008). Harlequin ichthyosis model mouse reveals alveolar collapse and severe fetal skin barrier defects. Hum. Mol. Genet. 17, 3075–3083. [DOI] [PubMed] [Google Scholar]
- 37.Smyth I, Hacking DF, Hilton AA, Mukhamedova N, Meikle PJ, Ellis S, Satterley K, Collinge JE, de Graaf CA, Bahlo M, et al. (2008). A mouse model of harlequin ichthyosis delineates a key role for Abca12 in lipid homeostasis. PLoS Genet. 4, e1000192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ramirez A, Page A, Gandarillas A, Zanet J, Pibre S, Vidal M, Tusell L, Genesca A, Whitaker DA, Melton DW, and Jorcano JL (2004). A keratin K5Cre transgenic line appropriate for tissue-specific or generalized Cre-mediated recombination. Genesis 39, 52–57. [DOI] [PubMed] [Google Scholar]
- 39.Levy V, Lindon C, Harfe BD, and Morgan BA (2005). Distinct stem cell populations regenerate the follicle and interfollicular epidermis. Dev. Cell 9, 855–861. [DOI] [PubMed] [Google Scholar]
- 40.Veniaminova NA, Grachtchouk M, Doane OJ, Peterson JK, Quigley DA, Lull MV, Pyrozhenko DV, Nair RR, Patrick MT, Balmain A, et al. (2019). Niche-specific factors dynamically regulate sebaceous gland stem cells in the skin. Dev. Cell 51, 326–340.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang YV, Cheong J, Ciapurin N, McDermitt DJ, and Tumbar T (2009). Distinct self-renewal and differentiation phases in the niche of infrequently dividing hair follicle stem cells. Cell Stem Cell 5, 267–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Powell AE, Wang Y, Li Y, Poulin EJ, Means AL, Washington MK, Higginbotham JN, Juchheim A, Prasad N, Levy SE, et al. (2012). The pan-ErbB negative regulator Lrig1 is an intestinal stem cell marker that functions as a tumor suppressor. Cell 149, 146–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Page ME, Lombard P, Ng F, Göttgens B, and Jensen KB (2013). The epidermis comprises autonomous compartments maintained by distinct stem cell populations. Cell Stem Cell 13, 471–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Enjalbert F, Dewan P, Caley MP, Jones EM, Morse MA, Kelsell DP, Enright AJ, and O’Toole EA (2020). 3D model of harlequin ichthyosis reveals inflammatory therapeutic targets. J. Clin. Invest. 130, 4798–4810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cottle DL, Ursino GMA, Ip SCI, Jones LK, Ditommaso T, Hacking DF, Mangan NE, Mellett NA, Henley KJ, Sviridov D, et al. (2015). Fetal inhibition of inflammation improves disease phenotypes in harlequin ichthyosis. Hum. Mol. Genet. 24, 436–449. [DOI] [PubMed] [Google Scholar]
- 46.Richard G, Rouan F, Willoughby CE, Brown N, Chung P, Ryynänen M, Jabs EW, Bale SJ, DiGiovanna JJ, Uitto J, and Russell L (2002). Missense mutations in GJB2 encoding connexin-26 cause the ectodermal dysplasia keratitis-ichthyosis-deafness syndrome. Am. J. Hum. Genet. 70, 1341–1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Vagnozzi AN, Reiter JF, and Wong SY (2015). Hair follicle and interfollicular epidermal stem cells make varying contributions to wound regeneration. Cell Cycle 14, 3408–3417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mesler AL, Veniaminova NA, Lull MV, and Wong SY (2017). Hair follicle terminal differentiation is orchestrated by distinct early and late matrix progenitors. Cell Rep. 19, 809–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Akiyama M, Dale BA, Smith LT, Shimizu H, and Holbrook KA (1998). Regional difference in expression of characteristic abnormality of harlequin ichthyosis in affected fetuses. Prenat. Diagn. 18, 425–436. [PubMed] [Google Scholar]
- 50.Sundberg JP, Boggess D, Hogan ME, Sundberg BA, Rourk MH, Harris B, Johnson K, Dunstan RW, and Davisson MT (1997). Harlequin ichthyosis (ichq): a juvenile lethal mouse mutation with ichthyosiform dermatitis. Am. J. Pathol. 151, 293–310. [PMC free article] [PubMed] [Google Scholar]
- 51.Zeeuwen PLJM, van Vlijmen-Willems IMJJ, Hendriks W, Merkx GFM, and Schalkwijk J (2002). A null mutation in the cystatin M/E gene of ichq mice causes juvenile lethality and defects in epidermal cornification. Hum. Mol. Genet. 11, 2867–2875. [DOI] [PubMed] [Google Scholar]
- 52.Klufa J, Bauer T, Hanson B, Herbold C, Starkl P, Lichtenberger B, Srutkova D, Schulz D, Vujic I, Mohr T, et al. (2019). Hair eruption initiates and commensal skin microbiota aggravate adverse events of anti-EGFR therapy. Sci. Transl. Med. 11, eaax2693. [DOI] [PubMed] [Google Scholar]
- 53.Mesler AL, Benedeck RE, and Wong SY (2021). Preparing the hair follicle canal for hair shaft emergence. Exp. Dermatol. 30, 472–478. 10.1111/exd.14210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Roy E, Neufeld Z, Cerone L, Wong HY, Hodgson S, Livet J, and Khosrotehrani K (2016). Bimodal behaviour of interfollicular epidermal progenitors regulated by hair follicle position and cycling. EMBO J 35, 2658–2670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhang YV, White BS, Shalloway DI, and Tumbar T (2010). Stem cell dynamics in mouse hair follicles: a story from cell division counting and single cell lineage tracing. Cell Cycle 9, 1504–1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Liu T, Li S, Ying S, Tang S, Ding Y, Li Y, Qiao J, and Fang H (2020). The IL-23/IL-17 Pathway in Inflammatory Skin Diseases: From Bench to Bedside. Front. Immunol. 11, 594735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Paller AS, Renert-Yuval Y, Suprun M, Esaki H, Oliva M, Huynh TN, Ungar B, Kunjravia N, Friedland R, Peng X, et al. (2017). An IL-17-dominant immune profile is shared across the major orphan forms of ichthyosis. J. Allergy Clin. Immunol. 139, 152–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Adachi T, Kobayashi T, Sugihara E, Yamada T, Ikuta K, Pittaluga S, Saya H, Amagai M, and Nagao K (2015). Hair follicle-derived IL-7 and IL-15 mediate skin-resident memory T cell homeostasis and lymphoma. Nat. Med. 21, 1272–1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Koch PJ, de Viragh PA, Scharer E, Bundman D, Longley MA, Bickenbach J, Kawachi Y, Suga Y, Zhou Z, Huber M, et al. (2000). Lessons from loricrin-deficient mice: compensatory mechanisms maintaining skin barrier function in the absence of a major cornified envelope protein. J. Cell Biol. 151, 389–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhang L, Ferreyros M, Feng W, Hupe M, Crumrine DA, Chen J, Elias PM, Holleran WM, Niswander L, Hohl D, et al. (2016). Defects in Stratum Corneum Desquamation Are the Predominant Effect of Impaired ABCA12 Function in a Novel Mouse Model of Harlequin Ichthyosis. PLoS One 11, e0161465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhang H, Ericsson M, Weström S, Vahlquist A, Virtanen M, and Törmä H (2019). Patients with congenital ichthyosis and TGM1 mutations overexpress other ARCI genes in the skin: Part of a barrier repair response? Exp. Dermatol. 28, 1164–1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Demerjian M, Hachem JP, Tschachler E, Denecker G, Declercq W, Vandenabeele P, Mauro T, Hupe M, Crumrine D, Roelandt T, et al. (2008). Acute modulations in permeability barrier function regulate epidermal cornification: role of caspase-14 and the protease-activated receptor type 2. Am. J. Pathol. 172, 86–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Jiang YJ, Uchida Y, Lu B, Kim P, Mao C, Akiyama M, Elias PM, Holleran WM, Grunfeld C, and Feingold KR (2009). Ceramide stimulates ABCA12 expression via peroxisome proliferator-activated receptor {delta} in human keratinocytes. J. Biol. Chem. 284, 18942–18952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Jiang YJ, Lu B, Kim P, Paragh G, Schmitz G, Elias PM, and Feingold KR (2008). PPAR and LXR activators regulate ABCA12 expression in human keratinocytes. J. Invest. Dermatol. 128, 104–109. [DOI] [PubMed] [Google Scholar]
- 65.Amen N, Mathow D, Rabionet M, Sandhoff R, Langbein L, Gretz N, Jäckel C, Gröne HJ, and Jennemann R (2013). Differentiation of epidermal keratinocytes is dependent on glucosylceramide:ceramide processing. Hum. Mol. Genet. 22, 4164–4179. [DOI] [PubMed] [Google Scholar]
- 66.List K, Haudenschild CC, Szabo R, Chen W, Wahl SM, Swaim W, Engelholm LH, Behrendt N, and Bugge TH (2002). Matriptase/MT-SP1 is required for postnatal survival, epidermal barrier function, hair follicle development, and thymic homeostasis. Oncogene 21, 3765–3779. [DOI] [PubMed] [Google Scholar]
- 67.Tobin DJ, Foitzik K, Reinheckel T, Mecklenburg L, Botchkarev VA, Peters C, and Paus R (2002). The lysosomal protease cathepsin L is an important regulator of keratinocyte and melanocyte differentiation during hair follicle morphogenesis and cycling. Am. J. Pathol. 160, 1807–1821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sharov AA, Schroeder M, Sharova TY, Mardaryev AN, Peters EMJ, Tobin DJ, and Botchkarev VA (2011). Matrix metalloproteinase-9 is involved in the regulation of hair canal formation. J. Invest. Dermatol. 131, 257–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Nakamura Y, Fukami K, Yu H, Takenaka K, Kataoka Y, Shirakata Y, Nishikawa SI, Hashimoto K, Yoshida N, and Takenawa T (2003). Phospholipase Cdelta1 is required for skin stem cell lineage commitment. EMBO J 22, 2981–2991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Schäfer M, Willrodt AH, Kurinna S, Link AS, Farwanah H, Geusau A, Gruber F, Sorg O, Huebner AJ, Roop DR, et al. (2014). Activation of Nrf2 in keratinocytes causes chloracne (MADISH)-like skin disease in mice. EMBO Mol. Med. 6, 442–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Oeffner F, Fischer G, Happle R, König A, Betz RC, Bornholdt D, Neidel U, Boente M.d.C., Redler S, Romero-Gomez J, et al. (2009). IFAP syndrome is caused by deficiency in MBTPS2, an intramembrane zinc metalloprotease essential for cholesterol homeostasis and ER stress response. Am. J. Hum. Genet. 84, 459–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Gruber R, Sugarman JL, Crumrine D, Hupe M, Mauro TM, Mauldin EA, Thyssen JP, Brandner JM, Hennies HC, Schmuth M, and Elias PM (2015). Sebaceous gland, hair shaft, and epidermal barrier abnormalities in keratosis pilaris with and without filaggrin deficiency. Am. J. Pathol. 185, 1012–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zeeuwen PLJM, Cheng T, and Schalkwijk J (2009). The Biology of Cystatin M/E and its Cognate Target Proteases. J. Invest. Dermatol. 129, 1327–1338. [DOI] [PubMed] [Google Scholar]
- 74.Ishida-Yamamoto A, Deraison C, Bonnart C, Bitoun E, Robinson R, O’Brien TJ, Wakamatsu K, Ohtsubo S, Takahashi H, Hashimoto Y, et al. (2005). LEKTI is localized in lamellar granules, separated from KLK5 and KLK7, and is secreted in the extracellular spaces of the superficial stratum granulosum. J. Invest. Dermatol. 124, 360–366. [DOI] [PubMed] [Google Scholar]
- 75.Lovászi M, Szegedi A, Zouboulis CC, and Törőcsik D (2017). Sebaceous-immunobiology is orchestrated by sebum lipids. Dermatoendocrinol. 9, e1375636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Shi VY, Leo M, Hassoun L, Chahal DS, Maibach HI, and Sivamani RK (2015). Role of sebaceous glands in inflammatory dermatoses. J. Am. Acad. Dermatol. 73, 856–863. [DOI] [PubMed] [Google Scholar]
- 77.Dahlhoff M, Camera E, Schäfer M, Emrich D, Riethmacher D, Foster A, Paus R, and Schneider MR (2016). Sebaceous lipids are essential for water repulsion, protection against UVB-induced apoptosis, and ocular integrity in mice. Development 143, 1823–1831. [DOI] [PubMed] [Google Scholar]
- 78.Malik K, He H, Huynh TN, Tran G, Mueller K, Doytcheva K, Renert-Yuval Y, Czarnowicki T, Magidi S, Chou M, et al. (2019). Ichthyosis molecular fingerprinting shows profound TH17 skewing and a unique barrier genomic signature. J. Allergy Clin. Immunol. 143, 604–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lefferdink R, Rangel SM, Chima M, Ibler E, Pavel AB, Kim H, Wu B, Abu-Zayed H, Wu J, Jackson K, et al. (2023). Secukinumab responses vary across the spectrum of congenital ichthyosis in adults. Arch. Dermatol. Res. 315, 305–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Paller AS (2019). Profiling Immune Expression to Consider Repurposing Therapeutics for the Ichthyoses. J. Invest. Dermatol. 139, 535–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Vasioukhin V, Degenstein L, Wise B, and Fuchs E (1999). The magical touch: genome targeting in epidermal stem cells induced by tamoxifen application to mouse skin. Proc Natl Acad Sci USA 96, 8551–8556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Harfe BD, Scherz PJ, Nissim S, Tian H, McMahon AP, and Tabin CJ (2004). Evidence for an expansion-based temporal Shh gradient in specifying vertebrate digit identities. Cell 118, 517–528. [DOI] [PubMed] [Google Scholar]
- 83.Srinivas S, Watanabe T, Lin CS, William CM, Tanabe Y, Jessell TM, and Costantini F (2001). Cre reporter strains produced by targeted insertion of EYFP and ECFP into the ROSA26 locus. BMC Dev. Biol. 1, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kranz A, Fu J, Duerschke K, Weidlich S, Naumann R, Stewart AF, and Anastassiadis K (2010). An improved Flp deleter mouse in C57Bl/6 based on Flpo recombinase. Genesis 48, 512–520. [DOI] [PubMed] [Google Scholar]
- 85.Yu G, Wang LG, Han Y, and He QY (2012). clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS 16, 284–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNA-seq data generated for this study can be accessed through GEO: accession GSE254889.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this work is available from the lead contact upon request.