

Abstract
Target protein degradation (TPD) has emerged as a revolutionary approach in drug discovery, leveraging the cell’s intrinsic machinery to selectively degrade disease-associated proteins. Nanoluciferase (nLuc) fusion proteins and the NanoBiT technology offer two robust and sensitive screening platforms to monitor the subtle changes in protein abundance induced by TPD molecules. Despite these advantages, concerns have arisen regarding potential degradation artifacts introduced by tagging systems due to the presence of lysine residues on them, prompting the development of alternative tools. In this study, we introduce HiBiT-RR and nLucK0, variants devoid of lysine residues, to mitigate such artifacts. Our findings demonstrate that HiBiT-RR maintains a similar sensitivity and binding affinity with the original HiBiT. Moreover, the comparison between nLucWT and nLucK0 constructs reveals variations in degradation patterns induced by certain TPD molecules, emphasizing the importance of choosing appropriate tagging systems to ensure the reliability of experimental outcomes in studying protein degradation processes.
Keywords: HiBiT, Nano luciferase, Tagging system, Targeted protein degradation (TPD), Proteolysis targeting chimeras (PROTACs), High-throughput screening
Targeted protein degradation (TPD) is a groundbreaking approach in drug discovery that utilizes the intrinsic machinery of the cell to selectively degrade disease-associated proteins. Unlike traditional therapeutics that inhibit protein function, TPD molecules (also known as degraders) modulate protein levels within the cell, providing a unique strategy to tackle diseases. A key player in the TPD field is proteolysis-targeting chimeras (PROTACs). PROTACs are heterobifunctional molecules that can provide body-wide phenocopy knockdown without modification on the genome. The warhead part can bind to the target protein, and the E3 ligand part can bind to E3 ubiquitin ligase to facilitate the formation of a ternary complex, leading to ubiquitination on surface-exposed lysine residues of the target protein, ultimately leading to targeted protein degradation.1,2 The ability of PROTACs to harness the natural protein degradation pathway has broadened the scope of “druggable” targets, offering new possibilities for therapeutic intervention. As of 2023, a total of 26 PROTAC projects worldwide have entered the clinical stage. The most rapid progression was observed in the ARV-471 project, a collaborative effort between Arvinas and Pfizer, which commenced phase III clinical trials in 2022.3−6 Simultaneously, several other degraders are either already in the initial stages of clinical trials or poised to enter them. The PROTAC field is flourishing with a multitude of degraders competing for a place in the clinical market.
To monitor the subtle target protein abundance change induced by the TPD molecules in the complex cellular environment, a highly sensitive and high-throughput compatible protein abundance assay with broad dynamic range is ideal for screening purposes. Nanoluciferase (nLuc), a small 19-kDa, highly stable, ATP-independent, bioluminescent protein engineered from the original deep-sea shrimp luciferase (Oplophorus-luciferin 2-monooxygenase), has been utilized to develop robust and ultrahigh sensitivity screening systems.7 As a commonly used chemical biology tool in the high throughput evaluation of degrader efficacy, a strong bioluminescence signal generated from the nLuc fusion protein could sensitively reflect the real-time fusion protein level in live cells or crude cell lysates, allowing for the kinetic monitoring of protein degradation events upon degrader treatment.8,9 To further minimize the tagging size, NanoBiT technology was utilized.10 This approach involves a complementing system in which an 11-amino acid peptide (HiBiT) is tagged onto the protein of interest, enabling interaction with an 18 kDa polypeptide (LgBiT). This interaction results in the formation of an active luciferase capable of producing light upon reaction with its luminogenic substrate, furimazine, or its stabilized version endurazine.8,10,11
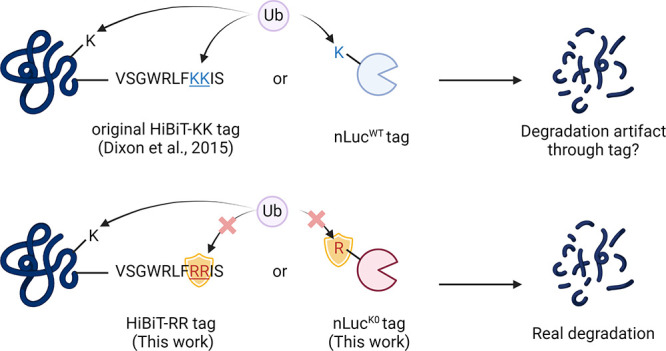
The efficient ubiquitination of a substrate lysine requires the appropriate spatial proximity between the substrate lysine and the narrow catalytic site on the E2 enzyme.12−14 This spatial arrangement is crucial for facilitating the ubiquitination reaction. Therefore, the accessibility and positioning of lysine residues play a critical role in determining their susceptibility to ubiquitination.15,16 It has been reported that lysine residues of the target protein located on the ubiquitin accessible band of the E3 ligase machinery will contribute more to the degradation.16−18 Therefore, recently, there have been increasing concerns that the introduced lysine residues on the tagging protein or peptide may contribute to degradation artifacts. A typical example was the successful degradation of GFP-KRASG12C but not the endogenous KRASG12C by XY-4-88.19 For those target proteins with only a few lysine residues on the surface, the artificially introduced lysine on the tag may even mislead the researchers for pursuing a nondegradable PROTAC molecule.
In this study, we present two alternative tools, HiBiT-RR and nLucK0, in which all of the lysine residues have been replaced by arginine residues from their original sequences to avoid potential risks from degradation artifacts. We show here that HiBiT-RR shows a comparable binding affinity toward LgBiT to the original HiBiT peptide, with no sacrifice on the luminescence intensity. The HiBiT-RR tag is as sensitive as HiBiT in detecting and quantifying drug-induced protein degradation. On the other hand, by comparing nLucWT and nLucK0 constructs, we report that some PROTAC molecules tested in this paper can trigger stronger degradation in the nLucWT fusion protein than the nLucK0 counterpart. This discovery suggests that degradation artifacts stemming from the tagging system might be more widespread than currently understood. It also highlights the importance of choosing the right tagging system to help minimize potential interference or artifacts in studying protein degradation processes, thereby enhancing the reliability and validity of the experimental results.
Lysine is the most common residue to accept ubiquitin transfer from E2 enzymes due to their reactivity. In order to deal with the potential artificial degradation of the target protein originating from the two lysine residues in the original version of HiBiT tag (VSGWRLFKKIS), referred as HiBiT-KK thereafter,10,11 we started to characterize the biophysical and enzymatic properties of the lysine-less version of the HiBiT (VSGWRLFRRIS), hereinafter referred to as HiBiT-RR, by substituting the two lysine with arginine residues (Figure 1A). We first characterized the binding between LgBiT protein and HiBiT-KK or HiBiT-RR. Fixing the amount of LgBiT, we titrated a series of HiBiT concentrations for both KK and RR variants. Both HiBiT-KK and HiBiT-RR showed low nM EC50 values and comparable luminescence signal intensities no matter whether our homemade His-LgBiT protein or the LgBiT protein from Promega Corp. was used (Figure 1B,C). A comparable level of luminescence signal is as expected since the KK to RR mutation site is distant from the luciferase catalytic pocket according to a recently deposited nLuc structure (PDB: 7SNT). However, the EC50 value we got from LgBiT-HiBiT interaction was ∼10 times weaker than previously reported 700 pM.11 Therefore, we performed a reverse titration of various concentrations of the LgBiT protein into a fixed amount of HiBiT variants. After subtracting the signal from LgBiT itself in the absence of any HiBiT peptide, both HiBiT-KK or RR still showed similar EC50 levels as are in the forward titration (Figure 1D,E). The EC50 levels remain unchanged for up to 6 h incubation, indicating the establishment of equilibrium (Figure S1).
Figure 1.

HiBiT-RR showed comparable LgBiT protein binding affinity and similar luminescence output with the original HiBiT-KK. A. Peptide sequences of the original HiBiT peptide (HiBiT-KK in this paper) from Promega and the lysine-less version (HiBiT-RR). B, C. The luminescence signal from 20 pM LgBiT protein (homemade His-LgBiT for B and Promega commercially available LgBiT protein for C) when titrating with various concentrations of HiBiT-KK or HiBiT-RR for 30 min. D. The luminescence signal from 20 pM HiBiT variants or PBS when titrating with various concentrations of His-LgBiT recombinant protein for 30 min. E. The background LgBiT luminescence from the PBS group were subtracted from panel D.
We also biophysically determined their affinity and binding kinetics in the absence of an enzyme substrate using grating-coupled interferometry (GCI). In parallel with enzymatic measurements, both HiBiT-KK and RR demonstrated an affinity at the single-digit nanomolar level toward His-LgBiT. Notably, HiBiT-RR exhibited a marginally superior performance compared to HiBiT-KK, displaying a slower dissociation rate (koff) under conditions of both high and low density of LgBiT (Figure 2).
Figure 2.

Grating coupled interferometry measurement of HiBiT-KK and RR showed comparable LgBiT protein binding kinetics. Various concentrations of HiBiT-KK (A, C) or HiBiT-RR (B, D) were flowed against immobilized His-LgBiT at high (A, B) or low (C, D) surface density. The association and dissociation events were monitored in real-time using grating-coupled interferometry.
All this evidence indicates that under such experimental conditions, the binding affinity between HiBiT and LgBiT in the presence of enzyme substrate may not be as tight as determined before in sub-nM range yet still tight enough for applications such as sensitively detecting HiBiT in cell lysates. Also, introducing the RR mutation into the established HiBiT sequence does not compromise the readout intensity, from which a robust luminescence assay with good sensitivity can be developed.
Protein degrader molecules like PROTACs or molecular glue are gathering increasing attention as both therapeutic modalities and chemical biology tools. The warhead component of these molecules binds to the target protein, while the E3 ligand portion interacts with the E3 ubiquitin ligase, facilitating the formation of a ternary complex. This complex, in turn, triggers ubiquitination, leading to eventual degradation of the target protein. A pivotal step in the protein degradation pathway is the transfer of ubiquitin onto lysine residues exposed on the protein surface, navigating the protein to the proteasome for degradation. There have been concerns since the release of the HiBiT-tagging system about the existing lysine in the HiBiT sequence, with apprehensions about whether these artificially introduced lysine residues could potentially contribute to the degradation of the target protein. This concern becomes particularly relevant in the case of protein degraders since the protein of interest has already been recruited and loaded onto the E3 ligase complex.
Here we characterized the degradation efficiency of four BTK PROTACs (RC-1,20 NX-2127,21,22 NRX-0492,23 and DD-03-17124) covering different target engagement mechanisms (RC-1 for reversible covalent PROTAC, others for reversible PROTAC) using the BTK kinase domain (residue 382–659, BTKKD) tagged with either HiBiT-KK or RR (Figure 3). Results show that the HiBiT-RR construct (Figure 3B, D) was able to reproduce those characteristic parameters of PROTACs observed in the traditional HiBiT-KK construct (Figure 3A, C) such as DC50, Dmax, and the strong hook effect observed in RC-1 at high doses. In our previous research, RC-1 did not show a very strong hook effect, and we believe it was due to the difference of the cooperativity between BTKKD and full length BTK. Indeed, in Ramos BTK-HiBiT-KK knock-in cell line, we observed a weaker hook effect and a deeper Dmax (Figure S2).
Figure 3.

Comparison of PROTAC degradation potency of HiBiT-KK or HiBiT-RR tagged BTK kinase domain in the HEK293T cell line. HEK293T cells transiently transfected with BTKKD-HiBiT-KK (A) or BTKKD-HiBiT-RR (B) were transferred into a 96-well plate for 20,000 cells per well. Cells were incubated with DMSO or indicated compounds at 0.064, 0.32, 1.6, 8, 40, 200, 1,000 nM in 1% DMSO for 24 h. A bioluminescence signal was generated by adding furimazine substrate and LgBiT purified protein in cell lysis buffer. C, D. Bioluminescence signals from A and B normalized to their corresponding DMSO groups.
With the robustness of the transiently expressed HiBiT-RR construct confirmed, we tested both systems in the context of HiBiT in situ knock-in cell lines. By CRISPR knocking in either HiBiT-KK or -RR at the N-terminus of BRD4 in HEK293 cells, MZ1 and dBET6, two classical BRD4 PROTACs,25,26 were further used to characterize the degradation potency difference in the HiBiT-KK or -RR knock-in cells. Similarly, no significant DC50 or Dmax difference was observed for MZ1 degrading HiBiT-BRD4 (Figure 4A–D). For dBET6, we observe a marginal difference in that HiBiT-RR BRD4 undergoes slower degradation kinetics than HiBiT-KK in high concentration dBET6 treatment, although a similar Dmax was observed, and the degradation potency when a plateau was achieved shows less than a 2-fold difference (Figure 4E–H). Altogether, our data indicate that HiBiT-RR also works as well as the original HiBiT-KK in the in situ genome knock-in systems.
Figure 4.

A HiBiT-KK CRISPR knock-in BRD4 in HEK293 cell line was compared to HiBiT-RR knock-in using dBET6 (A–D) and MZ1 (E-H). A–B, E–F. Degradation kinetics of HiBiT-KK or -RR BRD4 normalized to the DMSO group. C, G. HiBiT-KK or RR BRD4 degradation rate of dBET6 or MZ1 fitting from the initial phase (before 7 h). D, H. HiBiT-KK or RR BRD4 degradation potency comparison under dBET6 or MZ1 treatment when the plateau was achieved.
So far, we have validated HiBiT-RR as a feasible tool for protein abundance monitoring, which shows a sensitivity comparable to that of HiBiT-KK but ablated the potential pitfall of degradation artifacts. However, using the HiBiT-tag protein alone does not allow continuously monitoring the protein level changes in the time course study, unless an additional copy of LgBiT was also knocked into the genome.8 However, potential ubiquitination could still happen on the lysine residue of wild-type LgBiT. Ectopically expressing a nLuc fusion protein is a common way to evaluate degradation kinetics. Here we would also like to report our attempt of using a no-lysine version of nLuc (nLucK0), where all the lysine residues in the original nLuc sequence were mutated into arginine (sequence as shown in Figure 5A). By fusing nLuc WT or K0 to the N-terminus of the RIPK1 kinase domain (RIPK1KD) and expressing it in HEK293T cells, we head-to-head compared the degradation capacity of a series of RIPK1 degraders (Table S1) against these constructs (Figure 5B, C).100,101 For majority compounds, both nLuc WT and K0 constructs show similar maximal degradation (Dmax) and DC50 values, while the negative control compound 4172NC shows no degradation in both constructs. But for compound 5037, we observed a 3-fold worsening in DC50 when the nLucK0 construct was used (nLucWT 47 nM versus nLucK0 147 nM). A similar potency reduction was also observed in compound 5077, a CRBN-based RIPK1 PROTAC (nLucWT = 10 nM versus nLucK0 = 39 nM). This observed potency reduction could be due to the removal of the ubiquitin-able lysine residues on the nLuc surface, highlighting the potential degradation artifacts when ubiquitination happens on the nLuc protein. Transition to the K0 construct could avoid such degradation artifacts from the nLuc part. An immunoblot confirmed the degradation of both nLuc-RIPK1KD fusion proteins (Figure 5D).
Figure 5.

Comparison of nLucWT and nLucK0 tagged RIPK1 kinase domain degradation potency in a transfected HEK293T cell line. A. Sequence comparisons between nLucWT and nLucK0. Secondary structure information extracted from PDB 5IBO. B, C. HEK293T cells transiently transfected with nLucWT-RIPK1KD (B) or nLucK0-RIPK1KD (C) were transferred into a 96-well plate and incubated with DMSO or indicated compounds at 1.6, 8, 40, 200, 1,000 nM in 1% DMSO for 24 h. Bioluminescence signals were recorded in live cells using furimazine-containing OptiMEM. Unit for Bottom and Top: %, IC50: nM. D. Immunoblot of RIPK1 in HEK293T cells transfected with nLucWT/K0-RIPK1KD plasmid or empty vector after 24 h treatment of LD4172.
Since we observed a lower luminescence intensity when expressing the nLucK0 construct in the mammalian cell, we purified both His6-tagged nLuc WT and K0 protein from Escherichia coli BL21(DE3) (Figure S3A, B). Surprisingly, we found that nLucK0 protein had significantly decreased enzyme activity (up to 400-fold) compared to nLucWT under the same in vitro assay condition (Figure S3C). This result could explain the decrease in luminescence intensity from nLucK0 when overexpressed in mammalian cells. nLucK0 protein activity may heavily rely on the cellular environment to maintain the protein stability.
Target protein degradation (TPD) represents a revolutionary approach in drug discovery, harnessing the intrinsic machinery of the cell protein degradation pathway to selectively degrade disease-associated proteins. Unlike conventional therapeutics that inhibit protein function, TPD molecules, also known as degraders, modulate protein levels within cells, offering a unique strategy to combat diseases. However, amidst this progress, challenges exist in accurately monitoring subtle changes in target protein abundance induced by TPD molecules within complex cellular environments. The immunoblot has long been used as the gold standard to detect and quantify target protein degradation, but the long processing steps and the limited numbers of samples in each gel make the throughput very low, sometimes even leading to artifacts. Capillary electrophoresis immunoassay typically requires less sample consumption, involves simpler procedures, and shortens analysis time.27 It has been applied to monitor BTK level changes28 and BRD4 bromodomain ubiquitination levels29 after PROTAC treatment. Time-resolved fluorescence resonance energy transfer (TR-FRET) has recently been developed to quantify the protein level in the crude lysate in a high-throughput manner.30 However, all these methods depend on highly specific antibody–antigen interactions. In addition, the TR-FRET based assay also requires the development of tracer compounds. To address these problems, highly sensitive and high-throughput compatible protein abundance assays with broad dynamic ranges are indispensable for screening purposes.
Reporter assays represent a rapid and sensitive method to measure protein degradation based on fluorescence or bioluminescence. GFP has been used to quantitatively monitor the target protein degradation event for KRASG12C19 and BRD4 bromodomains.26 The development of a complementary luciferase based HiBiT tagging system offered another reporter assay to monitor protein degradation. The high affinity between HiBiT and LgBiT complexes allows immediate quantification of HiBiT tagged protein using bioluminescence readout. This system has been successfully applied in kinetically monitoring the potency of BRD2/3/4 PROTAC MZ18 and BRD7/9 PROTAC VZ185,31 IMiD molecular glues,32 kinases32,33 and fusion-tag PROTACs.34 However, there has been reported cases that fusion tags on the target protein may alter the degradability of the target protein by introducing artificial ubiquitination sites.19
Since there is currently no way to guarantee whether a fusion tag may contribute to the protein degradability by introducing novel ubiquitination sites, as an expansion of the current chemical biology toolbox, this study introduced two alternative tagging systems, HiBiT-RR and nLucK0, aimed at minimizing potential degradation artifacts during TPD molecule discovery processes. HiBiT-RR, a lysine-less version of the HiBiT tag, demonstrated comparable binding affinity to LgBiT protein with the original HiBiT-KK, ensuring reliable protein abundance monitoring without sacrificing assay sensitivity. Moreover, HiBiT-RR maintained similar degradation efficiency as HiBiT-KK when tested with various BTK and BRD4 PROTACs, indicating its suitability for evaluating degrader potency. Interestingly, we observed in the HiBiT-RR BRD4 knock-in cells only dBET6 showed a slightly slower degradation kinetics compared to HiBiT-KK, but not MZ1. This suggests that the fusion tag may or may not contribute to the degradation event depending on the compound used. It is plausible that the tag on the target protein adopts a conformation that is more favorable for accepting ubiquitin in the presence of certain compounds but not in others. This nuanced understanding underscores the complex interplay among the tagging system, compound-induced ternary complex conformation, and protein degradation mechanism.
Furthermore, the study explored the potential of nLucK0, a no-lysine version of nLuc, in targeted protein degradation studies. While both nLucWT and nLucK0 constructs exhibited a similar degradation potency for most compounds tested, a notable reduction in degradation potency was observed for specific compounds with nLucK0, suggesting potential degradation artifacts stemming from ubiquitination on the lysine residues of nLucWT. Transitioning to the nLucK0 construct could mitigate such artifacts, enhancing the reliability of the degradation studies. However, it is worth noting that the relatively lower luminescence intensity observed with the nLucK0 construct when expressed in mammalian cells could be attributed to decreased enzyme activity compared to nLucWT. The enzyme activity difference was even more obvious using purified nLucWT and nLucK0 protein, which indicates the unstable nature of nLucK0 in a noncellular environment. But out of all the PROTACs we tested, we did not find any compounds that show more than a 2-fold stronger degradation potency in the nLucK0 than the nLucWT construct. This observation helps alleviate concerns regarding potential artificial degradation due to the instability of the nLucK0 protein. Although the luminescence signal decrease in the cellular assay is trivial and can be easily compensated by increasing instrument photomultiplier tube (PMT) gain setting, this finding underscores the importance of considering the influence on the cellular environment when overexpressing proteins with less stability, especially in the context of assay development and interpretation.
Overall, the study underscores the significance of choosing appropriate tagging systems, such as HiBiT-RR and nLucK0, to minimize potential interference and artifacts in studying protein degradation processes. By enhancing the reliability and validity of experimental results, these tools contribute to advancing drug discovery efforts utilizing TPD molecules, ultimately leading to more effective therapeutic interventions.
Acknowledgments
This research was supported in part by the National Institutes of Health (R01-CA250503 and R01-CA268518 to J.W.), the Cancer Prevention & Research Institute of Texas (CPRIT, RP220480 to J.W.), and Michael E. DeBakey, M.D., Professor in Pharmacology (to J.W.). We also appreciate all suggestions and advice from Dr. Chris Eggers at Promega and Dr. Ryan Potts at Amgen.
Glossary
Abbreviations
- BRD4
Bromodomain-containing protein 4
- BTK
Bruton’s tyrosine kinase
- DC50
Concentration of compound for inducing 50% protein degradation
- Dmax
Percentage of maximal protein degradation can be achieved
- GCI
Grating coupled interferometry
- HiBiT-KK
High affinity small subunit of nanoluciferase binary technology–with two Lys residues (peptide VSGWRLFKKIS)
- HiBiT-RR
High affinity small subunit of nanoluciferase binary technology–with two Arg residues (peptide VSGWRLFRRIS)
- K0
free of any lysine residues
- KD
Kinase domain
- LgBiT
Large subunit of nanoluciferase binary technology
- nLuc
Nano-luciferase
- PROTAC
Proteolysis targeting chimera
- RIPK1
Receptor-interacting serine/threonine-protein kinase 1
- TPD
Targeted protein degradation
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.4c00271.
Detailed experiment methods, biochemistry assay data, and RIPK1 degrader common structures (PDF)
The authors declare the following competing financial interest(s): J.W. is a co-founder of Chemical Biology Probes, LLC. and serves as a consultant for CoRegen Inc.
Supplementary Material
References
- Sakamoto K. M.; Kim K. B.; Kumagai A.; Mercurio F.; Crews C. M.; Deshaies R. J. Protacs: Chimeric Molecules That Target Proteins to the Skp1-Cullin-F Box Complex for Ubiquitination and Degradation. Proc. Natl. Acad. Sci. U. S. A. 2001, 98 (15), 8554–8559. 10.1073/pnas.141230798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scudellari M. Protein-Slaying Drugs Could Be the next Blockbuster Therapies. Nature 2019, 567 (7748), 298–300. 10.1038/d41586-019-00879-3. [DOI] [PubMed] [Google Scholar]
- Campone M.; Ma C. X.; De Laurentiis M.; Iwata H.; Hurvitz S. A.; Wander S. A.; Danso M. A.; Lu D. R.; Perkins Smith J.; Liu Y.; Tran L.; Anderson S.; Hamilton E. P. VERITAC-2: A Global, Randomized Phase 3 Study of ARV-471, a Proteolysis Targeting Chimera (PROTAC) Estrogen Receptor (ER) Degrader, vs Fulvestrant in ER+/Human Epidermal Growth Factor Receptor 2 (HER2)- Advanced Breast Cancer. JCO 2023, 41 (16_suppl), TPS1122–TPS1122. 10.1200/JCO.2023.41.16_suppl.TPS1122. [DOI] [Google Scholar]
- Wang X.; Qin Z.-L.; Li N.; Jia M.-Q.; Liu Q.-G.; Bai Y.-R.; Song J.; Yuan S.; Zhang S.-Y. Annual Review of PROTAC Degraders as Anticancer Agents in 2022. Eur. J. Med. Chem. 2024, 267, 116166. 10.1016/j.ejmech.2024.116166. [DOI] [PubMed] [Google Scholar]
- Garber K. The PROTAC Gold Rush. Nat. Biotechnol. 2022, 40, 12. 10.1038/s41587-021-01173-2. [DOI] [PubMed] [Google Scholar]
- Mullard A. Targeted Protein Degraders Crowd into the Clinic. Nat. Rev. Drug Discov 2021, 20 (4), 247–250. 10.1038/d41573-021-00052-4. [DOI] [PubMed] [Google Scholar]
- Hall M. P.; Unch J.; Binkowski B. F.; Valley M. P.; Butler B. L.; Wood M. G.; Otto P.; Zimmerman K.; Vidugiris G.; Machleidt T.; Robers M. B.; Benink H. A.; Eggers C. T.; Slater M. R.; Meisenheimer P. L.; Klaubert D. H.; Fan F.; Encell L. P.; Wood K. V. Engineered Luciferase Reporter from a Deep Sea Shrimp Utilizing a Novel Imidazopyrazinone Substrate. ACS Chem. Biol. 2012, 7 (11), 1848–1857. 10.1021/cb3002478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riching K. M.; Mahan S.; Corona C. R.; McDougall M.; Vasta J. D.; Robers M. B.; Urh M.; Daniels D. L. Quantitative Live-Cell Kinetic Degradation and Mechanistic Profiling of PROTAC Mode of Action. ACS Chem. Biol. 2018, 13 (9), 2758–2770. 10.1021/acschembio.8b00692. [DOI] [PubMed] [Google Scholar]
- Riching K. M.; Caine E. A.; Urh M.; Daniels D. L. The Importance of Cellular Degradation Kinetics for Understanding Mechanisms in Targeted Protein Degradation. Chem. Soc. Rev. 2022, 51 (14), 6210–6221. 10.1039/D2CS00339B. [DOI] [PubMed] [Google Scholar]
- Dixon A. S.; Schwinn M. K.; Hall M. P.; Zimmerman K.; Otto P.; Lubben T. H.; Butler B. L.; Binkowski B. F.; Machleidt T.; Kirkland T. A.; Wood M. G.; Eggers C. T.; Encell L. P.; Wood K. V. NanoLuc Complementation Reporter Optimized for Accurate Measurement of Protein Interactions in Cells. ACS Chem. Biol. 2016, 11 (2), 400–408. 10.1021/acschembio.5b00753. [DOI] [PubMed] [Google Scholar]
- Schwinn M. K.; Machleidt T.; Zimmerman K.; Eggers C. T.; Dixon A. S.; Hurst R.; Hall M. P.; Encell L. P.; Binkowski B. F.; Wood K. V. CRISPR-Mediated Tagging of Endogenous Proteins with a Luminescent Peptide. ACS Chem. Biol. 2018, 13 (2), 467–474. 10.1021/acschembio.7b00549. [DOI] [PubMed] [Google Scholar]
- Cardote T. A. F.; Gadd M. S.; Ciulli A. Crystal Structure of the Cul2-Rbx1-EloBC-VHL Ubiquitin Ligase Complex. Structure 2017, 25 (6), 901–911. 10.1016/j.str.2017.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J.; Purser N.; Liwocha J.; Scott D. C.; Byers H. A.; Steigenberger B.; Hill S.; Tripathi-Giesgen I.; Hinkle T.; Hansen F. M.; Prabu J. R.; Radhakrishnan S. K.; Kirkpatrick D. S.; Reichermeier K. M.; Schulman B. A.; Kleiger G. Cullin-RING Ligases Employ Geometrically Optimized Catalytic Partners for Substrate Targeting. Mol. Cell 2024, 84 (7), 1304–1320. 10.1016/j.molcel.2024.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crowe C.; Nakasone M. A.; Chandler S.; Tatham M. H.; Makukhin N.; Hay R. T.; Ciulli A. Mechanism of Degrader-Targeted Protein Ubiquitinability. bioRxiv 2024, 2024.02.05.578957. 10.1101/2024.02.05.578957. [DOI] [Google Scholar]
- Dixon T.; MacPherson D.; Mostofian B.; Dauzhenka T.; Lotz S.; McGee D.; Shechter S.; Shrestha U. R.; Wiewiora R.; McDargh Z. A.; Pei F.; Pal R.; Ribeiro J. V.; Wilkerson T.; Sachdeva V.; Gao N.; Jain S.; Sparks S.; Li Y.; Vinitsky A.; Zhang X.; Razavi A. M.; Kolossváry I.; Imbriglio J.; Evdokimov A.; Bergeron L.; Zhou W.; Adhikari J.; Ruprecht B.; Dickson A.; Xu H.; Sherman W.; Izaguirre J. A. Predicting the Structural Basis of Targeted Protein Degradation by Integrating Molecular Dynamics Simulations with Structural Mass Spectrometry. Nat. Commun. 2022, 13 (1), 5884. 10.1038/s41467-022-33575-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W.; Roy Burman S. S.; Chen J.; Donovan K. A.; Cao Y.; Shu C.; Zhang B.; Zeng Z.; Gu S.; Zhang Y.; Li D.; Fischer E. S.; Tokheim C.; Shirley Liu X. Machine Learning Modeling of Protein-Intrinsic Features Predicts Tractability of Targeted Protein Degradation. Genomics, Proteomics & Bioinformatics 2022, 20 (5), 882–898. 10.1016/j.gpb.2022.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lv D.; Pal P.; Liu X.; Jia Y.; Thummuri D.; Zhang P.; Hu W.; Pei J.; Zhang Q.; Zhou S.; Khan S.; Zhang X.; Hua N.; Yang Q.; Arango S.; Zhang W.; Nayak D.; Olsen S. K.; Weintraub S. T.; Hromas R.; Konopleva M.; Yuan Y.; Zheng G.; Zhou D. Development of a BCL-xL and BCL-2 Dual Degrader with Improved Anti-Leukemic Activity. Nat. Commun. 2021, 12 (1), 6896. 10.1038/s41467-021-27210-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai N.; Riching K. M.; Makaju A.; Wu H.; Acker T. M.; Ou S.-C.; Zhang Y.; Shen X.; Bulloch D. N.; Rui H.; Gibson B. W.; Daniels D. L.; Urh M.; Rock B. M.; Humphreys S. C. Modeling the CRL4A Ligase Complex to Predict Target Protein Ubiquitination Induced by Cereblon-Recruiting PROTACs. J. Biol. Chem. 2022, 298 (4), 101653. 10.1016/j.jbc.2022.101653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng M.; Xiong Y.; Safaee N.; Nowak R. P.; Donovan K. A.; Yuan C. J.; Nabet B.; Gero T. W.; Feru F.; Li L.; Gondi S.; Ombelets L. J.; Quan C.; Jänne P. A.; Kostic M.; Scott D. A.; Westover K. D.; Fischer E. S.; Gray N. S. Exploring Targeted Degradation Strategy for Oncogenic KRASG12C. Cell Chem. Biol. 2020, 27 (1), 19–31. 10.1016/j.chembiol.2019.12.006. [DOI] [PubMed] [Google Scholar]
- Guo W.-H.; Qi X.; Yu X.; Liu Y.; Chung C.-I.; Bai F.; Lin X.; Lu D.; Wang L.; Chen J.; Su L. H.; Nomie K. J.; Li F.; Wang M. C.; Shu X.; Onuchic J. N.; Woyach J. A.; Wang M. L.; Wang J. Enhancing Intracellular Accumulation and Target Engagement of PROTACs with Reversible Covalent Chemistry. Nat. Commun. 2020, 11 (1), 4268. 10.1038/s41467-020-17997-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montoya S.; Bourcier J.; Noviski M.; Lu H.; Thompson M. C.; Chirino A.; Jahn J.; Sondhi A. K.; Gajewski S.; Tan Y. S. May; Yung S.; Urban A.; Wang E.; Han C.; Mi X.; Kim W. J.; Sievers Q.; Auger P.; Bousquet H.; Brathaban N.; Bravo B.; Gessner M.; Guiducci C.; Iuliano J. N.; Kane T.; Mukerji R.; Reddy P. J.; Powers J.; Sanchez Garcia de los Rios M.; Ye J.; Barrientos Risso C.; Tsai D.; Pardo G.; Notti R. Q.; Pardo A.; Affer M.; Nawaratne V.; Totiger T. M.; Pena-Velasquez C.; Rhodes J. M.; Zelenetz A. D.; Alencar A.; Roeker L. E.; Mehta S.; Garippa R.; Linley A.; Soni R. K.; Skånland S. S.; Brown R. J.; Mato A. R.; Hansen G. M.; Abdel-Wahab O.; Taylor J. Kinase-Impaired BTK Mutations Are Susceptible to Clinical-Stage BTK and IKZF1/3 Degrader NX-2127. Science 2024, 383 (6682), eadi5798 10.1126/science.adi5798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbins D. W.; Noviski M. A.; Tan Y. S.; Konst Z. A.; Kelly A.; Auger P.; Brathaban N.; Cass R.; Chan M. L.; Cherala G.; Clifton M. C.; Gajewski S.; Ingallinera T. G.; Karr D.; Kato D.; Ma J.; McKinnell J.; McIntosh J.; Mihalic J.; Murphy B.; Panga J. R.; Peng G.; Powers J.; Perez L.; Rountree R.; Tenn-McClellan A.; Sands A. T.; Weiss D. R.; Wu J.; Ye J.; Guiducci C.; Hansen G.; Cohen F. Discovery and Preclinical Pharmacology of NX-2127, an Orally Bioavailable Degrader of Bruton’s Tyrosine Kinase with Immunomodulatory Activity for the Treatment of Patients with B Cell Malignancies. J. Med. Chem. 2024, 67 (4), 2321–2336. 10.1021/acs.jmedchem.3c01007. [DOI] [PubMed] [Google Scholar]
- Zhang D.; Harris H. M.; Chen J.; Judy J.; James G.; Kelly A.; McIntosh J.; Tenn-McClellan A.; Ambing E.; Tan Y. S.; Lu H.; Gajewski S.; Clifton M. C.; Yung S.; Robbins D. W.; Pirooznia M.; Skånland S. S.; Gaglione E.; Mhibik M.; Underbayev C.; Ahn I. E.; Sun C.; Herman S. E. M.; Noviski M.; Wiestner A. NRX-0492 Degrades Wild-Type and C481 Mutant BTK and Demonstrates in Vivo Activity in CLL Patient-Derived Xenografts. Blood 2023, 141 (13), 1584–1596. 10.1182/blood.2022016934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobrovolsky D.; Wang E. S.; Morrow S.; Leahy C.; Faust T.; Nowak R. P.; Donovan K. A.; Yang G.; Li Z.; Fischer E. S.; Treon S. P.; Weinstock D. M.; Gray N. S. Bruton Tyrosine Kinase Degradation as a Therapeutic Strategy for Cancer. Blood 2019, 133 (9), 952–961. 10.1182/blood-2018-07-862953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zengerle M.; Chan K.-H.; Ciulli A. Selective Small Molecule Induced Degradation of the BET Bromodomain Protein BRD4. ACS Chem. Biol. 2015, 10 (8), 1770–1777. 10.1021/acschembio.5b00216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowak R. P.; DeAngelo S. L.; Buckley D.; He Z.; Donovan K. A.; An J.; Safaee N.; Jedrychowski M. P.; Ponthier C. M.; Ishoey M.; Zhang T.; Mancias J. D.; Gray N. S.; Bradner J. E.; Fischer E. S. Plasticity in Binding Confers Selectivity in Ligand-Induced Protein Degradation. Nat. Chem. Biol. 2018, 14 (7), 706–714. 10.1038/s41589-018-0055-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu X.; Lin H.; Li F.; Wang J.; Lu D. Development of Biochemical and Cellular Probes to Study RIPK1 Target Engagement. ACS Med. Chem. Lett. 2024, 15 (6), 906–916. 10.1021/acsmedchemlett.4c00104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu X.; Lu D.; Qi X.; Lin H.; Holloman B. L.; Jin F.; Xu L.; Ding L.; Peng W.; Wang M. C.; Chen X.; Wang J.. Development of a First-in-Class RIPK1 Degrader to Enhance Antitumor Immunity.bioRxiv 2024 10.1101/2024.03.25.586133 [DOI] [Google Scholar]
- Yeung W. S. B.; Luo G. A.; Wang Q. G.; Ou J. P. Capillary Electrophoresis-Based Immunoassay. J. Chromatogr B Analyt Technol. Biomed Life Sci. 2003, 797 (1–2), 217–228. 10.1016/S1570-0232(03)00489-6. [DOI] [PubMed] [Google Scholar]
- Zorba A.; Nguyen C.; Xu Y.; Starr J.; Borzilleri K.; Smith J.; Zhu H.; Farley K. A.; Ding W.; Schiemer J.; Feng X.; Chang J. S.; Uccello D. P.; Young J. A.; Garcia-Irrizary C. N.; Czabaniuk L.; Schuff B.; Oliver R.; Montgomery J.; Hayward M. M.; Coe J.; Chen J.; Niosi M.; Luthra S.; Shah J. C.; El-Kattan A.; Qiu X.; West G. M.; Noe M. C.; Shanmugasundaram V.; Gilbert A. M.; Brown M. F.; Calabrese M. F. Delineating the Role of Cooperativity in the Design of Potent PROTACs for BTK. Proc. Natl. Acad. Sci. U. S. A. 2018, 115 (31), E7285-E7292 10.1073/pnas.1803662115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vieux E. F.; Agafonov R. V.; Emerson L.; Isasa M.; Deibler R. W.; Simard J. R.; Cocozziello D.; Ladd B.; Lee L.; Li H.; Archer S.; Fitzgerald M.; Michael R.; Nasveschuk C. G.; Park E. S.; Kern G.; Proia D. A.; Phillips A. J.; Fisher S. L. A Method for Determining the Kinetics of Small-Molecule-Induced Ubiquitination. SLAS Discov 2021, 26 (4), 547–559. 10.1177/24725552211000673. [DOI] [PubMed] [Google Scholar]
- Payne N. C.; Maksoud S.; Tannous B. A.; Mazitschek R. A Direct High-Throughput Protein Quantification Strategy Facilitates Discovery and Characterization of a Celastrol-Derived BRD4 Degrader. Cell Chem. Biol. 2022, 29 (8), 1333–1340. 10.1016/j.chembiol.2022.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoppi V.; Hughes S. J.; Maniaci C.; Testa A.; Gmaschitz T.; Wieshofer C.; Koegl M.; Riching K. M.; Daniels D. L.; Spallarossa A.; Ciulli A. Iterative Design and Optimization of Initially Inactive Proteolysis Targeting Chimeras (PROTACs) Identify VZ185 as a Potent, Fast, and Selective von Hippel-Lindau (VHL) Based Dual Degrader Probe of BRD9 and BRD7. J. Med. Chem. 2019, 62 (2), 699–726. 10.1021/acs.jmedchem.8b01413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riching K. M.; Mahan S. D.; Urh M.; Daniels D. L. High-Throughput Cellular Profiling of Targeted Protein Degradation Compounds Using HiBiT CRISPR Cell Lines. JoVE (Journal of Visualized Experiments) 2020, (165), e61787 10.3791/61787. [DOI] [PubMed] [Google Scholar]
- Riching K. M.; Schwinn M. K.; Vasta J. D.; Robers M. B.; Machleidt T.; Urh M.; Daniels D. L. CDK Family PROTAC Profiling Reveals Distinct Kinetic Responses and Cell Cycle-Dependent Degradation of CDK2. SLAS Discovery 2021, 26 (4), 560–569. 10.1177/2472555220973602. [DOI] [PubMed] [Google Scholar]
- Caine E. A.; Mahan S. D.; Johnson R. L.; Nieman A. N.; Lam N.; Warren C. R.; Riching K. M.; Urh M.; Daniels D. L. Targeted Protein Degradation Phenotypic Studies Using HaloTag CRISPR/Cas9 Endogenous Tagging Coupled with HaloPROTAC3. Current Protocols in Pharmacology 2020, 91 (1), e81 10.1002/cpph.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.