

Abstract

Multidrug-resistant bacterial infections pose an ever-evolving threat to public health. Since the outset of the antibacterial age, bacteria have developed a multitude of diverse resistance mechanisms that suppress the effectiveness of current therapies. New drug entities, such as Novel Bacterial Topoisomerase Inhibitors (NBTIs), can circumvent this major issue. A computational docking model was employed to predict the binding to DNA gyrase of atypical NBTIs with novel pharmacophores. Synthesis of NBTIs based on computational docking and subsequent antibacterial evaluation against both Gram-positive and Gram-negative bacteria yielded congeners with outstanding anti-staphylococcal activity and varying activity against select Gram-negative pathogens.
Keywords: MRSA, gyrase, antimicrobial resistance, cleavage, efflux
Antibacterial resistance is an ever-growing threat to humanity. The essential bacterial topoisomerase enzymes, DNA gyrase1 and topoisomerase IV (TopoIV),2 are attractive targets, as exemplified by the fluoroquinolone class of antibacterials. However, a rapid increase in resistance in recent decades, driven primarily by mutations to the genes encoding the topoisomerase targets, has reduced their effectiveness.3,4 One strategy to attack such resistant bacteria is to target these essential enzymes at novel binding sites. Novel Bacterial Topoisomerase Inhibitors (NBTIs), such as gepotidacin (now known as a triazaacenaphthylene), elude fluoroquinolone resistance using this strategy and display a differentiated pharmacological profile.5
The pharmacophore of gepotidacin is typical for most NBTIs (see Figure 1).5,6 The left-hand side (LHS, blue) utilizes either a bi- or tricyclic ring system to bind bacterial DNA. The linker (black) connects the LHS and the right-hand side (RHS, red), a moiety binding to a hydrophobic pocket in the topoisomerase. The RHS secondary amine is prototypical and interacts with a highly conserved aspartate (D83, Staphylococcus aureus numbering) in the GyrA subunit of DNA gyrase, at the entrance to the binding pocket.7,8 A mono- or bicyclic ring system binds nonspecifically to this pocket. The binding mode identified by X-ray crystallographic studies has been further validated by selection for NBTI-resistant mutants, of which GyrA D83N is common.9 Extensive optimization efforts have varied each portion of the pharmacophore, with the aims of improving antibacterial activity, enhancing pharmacokinetic properties, and reducing key safety liabilities including hERG inhibition.5,6
Figure 1.

Examples of a fluoroquinolone (ciprofloxacin) and NBTI (gepotidacin) are shown, with the key elements of the NBTI pharmacophore highlighted.
In this study, we have combined experimental results from previously synthesized molecules with a computational model to predict binding affinities of novel structures, enabling evaluation and prioritization of unique RHS pharmacophores prior to synthesis. In doing so, we identified atypical NBTIs with potent Gram-positive antibacterial activity. Gram-negative activity has proven more challenging, with efflux posing a particular challenge, as has been observed previously.5,10
Our efforts began with a set of 96 compounds, for which we had experimentally determined IC50 values for inhibition of gyrase-mediated supercoiling, which were used as a rough surrogate for binding affinity. Using Schrödinger Glide,11 each compound was docked into seven receptors constructed from high-resolution DNA gyrase crystals with bound NBTIs: PDB IDs 2XCS, 4PLB, 5BS3, 5NPP, 6FM4, 6QTK, and 6QTP. An initial self-docking screening was conducted to evaluate the utility of each crystal as a docking receptor (Figure S1, Supporting Information [SI]). For each of these systems, we created four experimental binding affinity bins based on the ln IC50 (nM) values of the benchmark compounds with bounds at 4.6 (100 nM), 6.2 (500 nM), and 7.8 (2500 nM). We then calculated least-squares linear regressions from the Glide docking data and natural log IC50 values of each receptor. A second set of cutoffs to represent docking scores was then defined, where the regressions intersected the IC50 bounds. This gave us a total of 16 bins, into which the benchmark compounds fell (Figure 2). We could subsequently evaluate the performance of each system based on the number of benchmark compounds that had a matching docking score and IC50 bins. Those along the positive diagonal had consistent experimental and computational classifications, i.e., both values were ranked excellent or both poor. These compounds were categorized as “correctly predicted.” The inhibitors that fell outside of these diagonal bins, where the experimental and computational classifications did not agree, were “incorrectly predicted”. Depending on the number of bins by which the docking classification deviated from the ln IC50 classification, they were labeled as “within one”, “within two”, or “within three” of their correct bin.
Figure 2.

Assessing the predictive accuracy of docking results using binned classification: binning assessment of systems selected from benchmarking: (A) 6QTP, (B) 2XCS, and (C) 6FM4. For a series of known NBTIs, the predicted binding score was plotted versus the natural logarithm of the measured IC50 value from a S. aureus DNA gyrase supercoiling assay. Agreement between experimental values and average Glide docking score was measured to determine the predictive accuracy of each DNA gyrase system (green: correct; yellow: within one bin; orange: within two; red: within three).
Three systems were identified for downstream analysis based on a combination of their ability to predict compounds as “within one” or better and the R2 of their regression line: 6QTP (R2: 0.14, correct: 40.6%, within one bin: 72.9%), 2XCS (R2: 0.16, correct: 31.2%, within one bin: 78.1%), and 6FM4 (R2: 0.08, correct: 30%, within one bin: 74.0%). These were then used to screen all potential synthesis candidates as an initial evaluation. Docking score distributions of the 443 compounds tested are shown in Figure S2, SI. As docking scores cannot be directly compared across different structures, the results from all systems were normalized to Z-scores (see Equation S1, SI) as seen in previous publications.12−14 Compounds were selected for synthesis and experimental analysis based on the average of their Z-scores across the three systems mentioned above, with more negative values reflecting better predicted binding.
To simplify the workflow, docking experiments were conducted with targeted variations in the RHS moiety while holding other components consistent (Figure 3 and Table 1). Docking of the “traditional” NBTI with a secondary amine (40) afforded an average Z-score of −0.949, and novel structures with superior (more negative) scores were prioritized for synthesis and experimental evaluation. In this research, our molecular design strategy focused on replacement of the secondary amine with other functional groups capable of engaging GyrA D83; four classes of analogues were of particular interest (alcohol 7a–c, syn- and anti-diols (15a–c and 16a–c; 22a–c and 23a–c), and primary amine (9a–c) (Figure 5). Primary amines have been associated with increased intracellular accumulation and reduced efflux in Gram-negative bacteria,10,15 and our chosen compound set enables a matched-pair comparison of primary amines with analogous alcohols and the secondary amine. Predicted docking poses (Figure 4) display interactions with the D83 residue at the entrance to the binding pocket of DNA gyrase. This prediction is in line with the known binding mode of traditional NBTIs.5−8 The diol (22/23a) and monohydroxyl (7a) form a hydrogen bonding interaction, while the amine (9a) forms an ionic interaction. Within each compound class, analogues with three diverse RHS moieties were synthesized, the hydrophobic para-toluyl group (a), the more polar benzodioxane possessing two potential hydrogen bond acceptors (b), and benzoxazinone (c) containing both hydrogen bond donor and acceptor moieties. The compounds shared similar synthetic routes. Moreover, antibacterial testing of various intermediates, namely, ketones (6a–c), (E)-olefins (13a–c), and (Z)-olefins (21a–c) as well as the readily accessible alkanes (14a–c) further expanded our understanding of structure–activity relationships.
Figure 3.

General structure of the targeted compounds.
Table 1. Predicted Binding Scores of Atypical NBTIs.

Figure 5.

Overview of synthesized compounds.
Figure 4.

Predicted docking poses and interactions with GyrA D83 in 2XCS of (A) monohydroxyl (7a) (1.6 Å to D83), (B) primary amine (9a) (1.8 Å to D83), and (C) anti-diol (22/23a) (1.6 and 1.9 Å to D83).
The tolyl and benzodioxane compounds were prepared by analogous routes (Schemes 1–3), while the oxazinones generally necessitated modified approaches (Schemes 4–7).
Scheme 1. Synthesis of Ketones and Monohydroxyls, and Primary Amines.
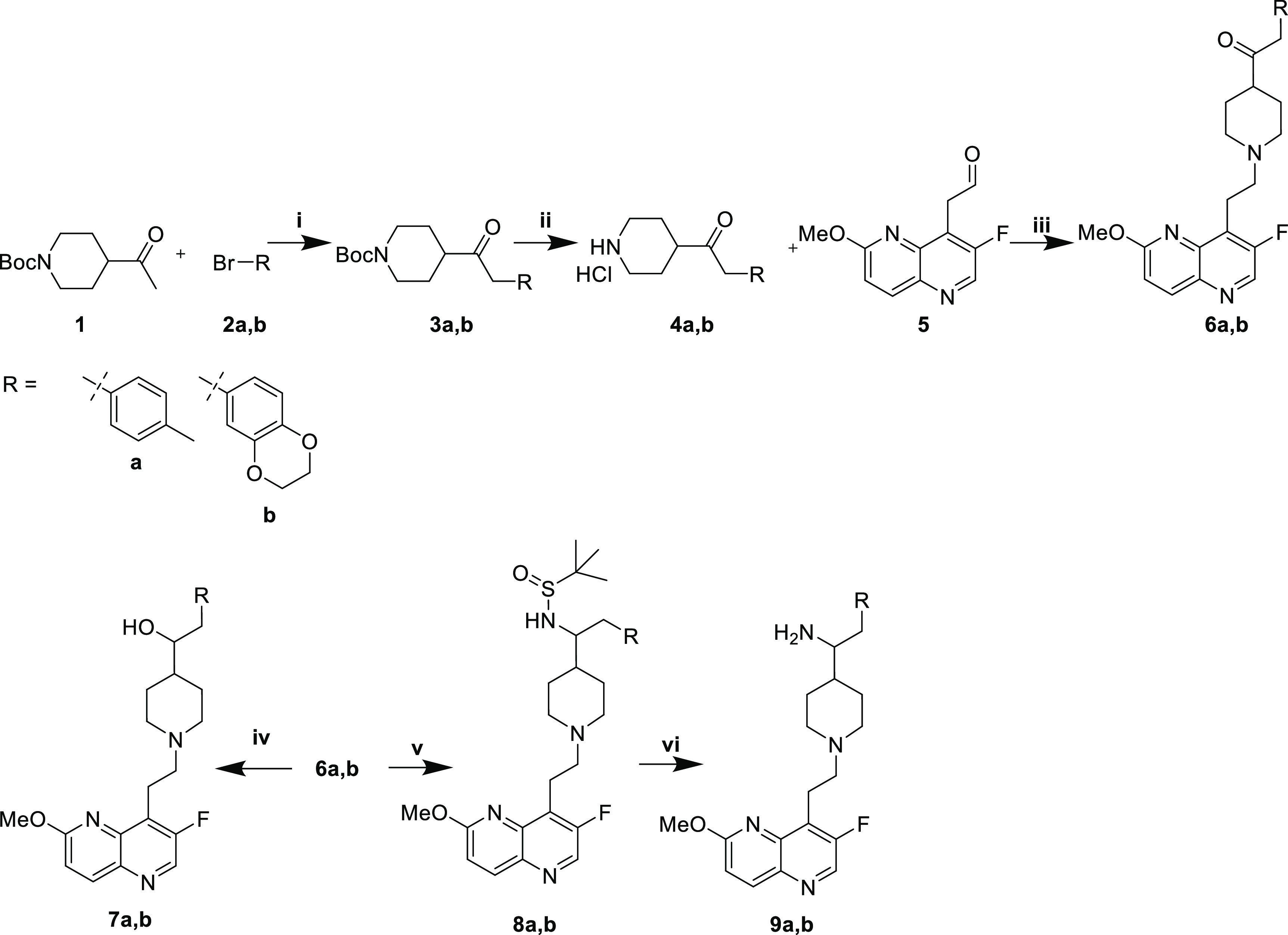
Reaction conditions: (i) NaOtBu, Pd2dba3, (±)-BINAP, THF, 70 °C; (ii) HCl, MeOH; (iii) 1. AcOH, THF/MeOH, 4 Å MS. 2. NaCNBH3; (iv) NaBH4, MeOH; (v) 1. H2NSOC(CH3)3, Ti(OEt)4, THF, 70 °C. 2. NaBH4, MeOH; (vi) HCl, MeOH.
Scheme 3. Synthesis of (Z)-Olefins and Anti-Diols.

Reaction conditions: (i) KOtBu, THF, 0 °C; (ii) HCl, MeOH; (iii) 1. 5, AcOH, THF/MeOH, 4 Å MS. 2. NaCNBH3; (iv) AD-mix-α, tBuOH/water; (v) AD-mix-β, tBuOH/water.
Scheme 4. Synthesis of Alkane and Alkenes with Oxazinone RHS.

Reaction conditions: (i) 4-CZIPN, Co(dmgH)(dmgH2)Cl2, HK2PO4, Et3N, DMF, 440 nm light; (ii) HCl, MeOH; (iii) 1. 5, AcOH, THF/MeOH, 4 Å MS. 2. NaCNBH3; (iv) H2, 10% Pd/C, MeOH; (v) (Ir[dF(CF3)ppy]2(dtbpy))PF6, ACN, 456 nm light; (vi) HCl, MeOH.
Scheme 7. Synthesis of Secondary Amine.

Reaction conditions: (i) 1. 5, AcOH, THF/MeOH, 4 Å MS. 2. NaCNBH3; (ii) HCl, MeOH; (iii) AcOH, THF/MeOH, 4 Å MS. 2. NaCNBH3.
Ketone (6a,b), monohydroxyl (7a,b), and primary amine (9a,b) analogues were prepared from ketone 1 (Scheme 1). Preliminary studies (not shown) indicated little dependence of the antibacterial activity on the absolute stereochemistry of the monohydroxyl or primary amine compounds. Consequently, racemic mixtures were synthesized. Pd-catalyzed arylation with the requisite aryl bromides 2a,b afforded intermediates 3a,b. Boc deprotection with methanolic HCl yielded amines 4a,b as hydrochloride salts. Reductive amination of aldehyde 5 afforded ketones 6a,b. Reduction with NaBH4 provided racemic monohydroxyls 7a,b, whereas reductive amination with racemic tert-butylsulfinamide provided sulfinamide intermediates 8a,b. Acidic deprotection afforded racemic primary amines 9a,b.
The synthesis of (E)-alkene 13a,b, alkane 14a,b, and syn-diol compounds (15a,b and 16a,b) began with reductive amination of aldehyde 5 with 4-iodopiperidine 10 to afford intermediate 11 (Scheme 2). A Heck-type coupling under photocatalytic conditions with styrenes 12a,b produced the (E)-olefins 13a,b stereoselectively (dr >10:1).16 Hydrogenation afforded alkanes 14a,b. Alternatively, Sharpless asymmetric dihydroxylation of the (E)-olefins using commercially available AD-mix-α or -β provided syn-diols 15a,b and 16a,b, respectively.
Scheme 2. Synthesis of (E)-Olefins, Alkanes, and Syn-Diols.

Reaction conditions: (i) 1. 5, AcOH, THF/MeOH, 4 Å MS. 2. NaCNBH3; (ii) 4-CZIPN, Co(dmgH)(dmgH2)Cl2, HK2PO4, Et3N, DMF, 440 nm light; (iii) H2, 10% Pd/C, EtOH; (iv) AD-mix-α, tBuOH/water; (v) AD-mix-β, tBuOH/water.
The synthesis of (Z)-alkene 21a,b and anti-diol compounds (22a,b and 23a,b) began with Wittig reaction of 17 with commercially available aldehydes 18a,b to afford (Z)-alkene intermediates 19a,b in high stereoselectivity (dr ∼25:1, Scheme 3). Boc deprotection to afford 20a,b and subsequent reductive amination of aldehyde 5 yielded (Z)-alkenes 21a,b. Sharpless asymmetric dihydroxylation using AD-mix-α or -β delivered anti-diols 22a,b and 23a,b, respectively.
The synthesis of benzoxazinone-type analogues was generally more difficult owing to the presence of an abstractable proton, requiring modified routes. Heck-type coupling of iodide 24 and styrene 25 under photocatalytic conditions afforded (E)-alkene 26 (Scheme 4).16 Boc deprotection to afford 27, followed by reductive amination with 5, yielded (E)-olefin 13c, which was hydrogenated to provide 14c. Alternatively, photocatalytic isomerization of (E)-olefin 26 at 456 nm with iridium catalysis afforded (Z)-olefin 28. Boc deprotection to provide 29 and subsequent reductive amination of aldehyde 5 yielded (Z)-olefin 21c.
The syn- (15c, 16c) and anti-diols (22c, 23c) were prepared via Sharpless asymmetric dihydroxylation of the (E)- and (Z)-alkenes, respectively (Scheme 5).
Scheme 5. Synthesis of Syn- and Anti-Diols with Oxazinone RHS.

Reaction conditions: (i) AD-mix-α, tBuOH/water; (ii) AD-mix-β, tBuOH/water; (iii) AD-mix-α, tBuOH/water; (iv) AD-mix-β, tBuOH/water.
Ketone 6c, monohydroxyl 7c, and primary amine 9c were synthesized beginning with (E)-alkene 26 (Scheme 6). Reaction with NBS afforded bromohydrin 30, which was converted to epoxide 31 under basic conditions. Hydrogenolysis using 10% Pd/C provided alcohol 32. Deprotection with methanolic HCl afforded piperidine 33, and reductive amination of aldehyde 5 yielded monohydroxylated analogue 7c. Alternatively, the oxidation of 32 with Dess-Martin periodinane yielded ketone 34. Acidic Boc deprotection to provide piperidine 35 and reductive amination with aldehyde 5 afforded ketone analogue 6c. Reductive amination of the ketone with racemic tert-butylsulfinamide afforded 8c, and a final deprotection with methanolic HCl provided racemic primary amine 9c.
Scheme 6. Synthesis of Ketone, Monohydroxyl, and Monoamine with Oxazinone RHS.
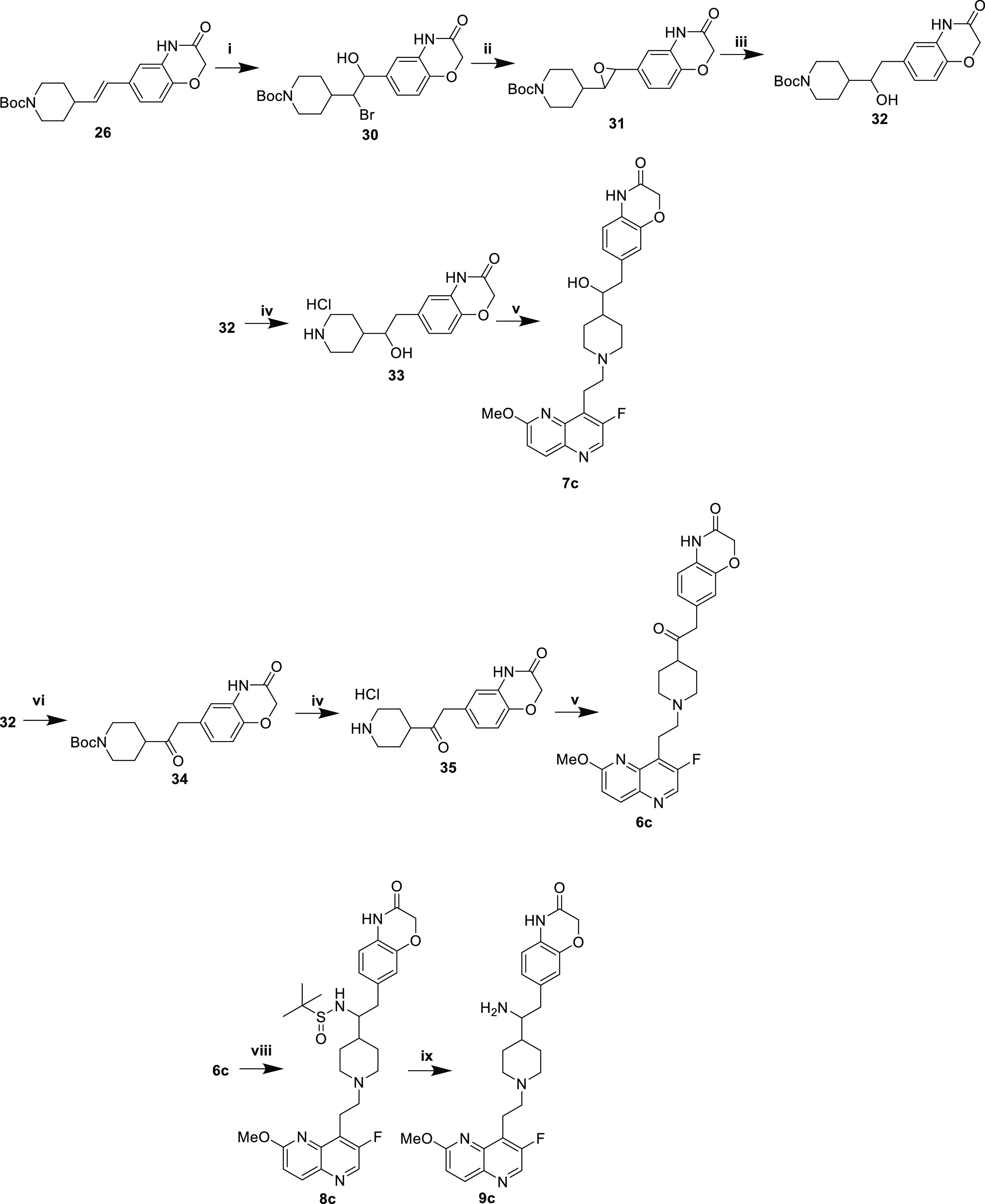
Reaction conditions: (i) NBS, ACN/water; (ii) K2CO3, MeOH; (iii) H2, 10% Pd/C, EtOH; (iv) HCl, MeOH; (v) 1. 5, AcOH, THF/MeOH, 4 Å MS. 2. NaCNBH3; (vi) DMP, DCM; (vii) HCl, MeOH; (viii) 1. H2NSOC(CH3)3, Ti(OEt)4, THF, 2. NaBH4, MeOH; (ix) HCl, MeOH..
A single secondary amine 40 was synthesized as a comparator representing the “traditional” NBTI scaffold (Scheme 7). Aldehyde 5 and piperidine 36 were reacted via reductive amination to afford Boc protected 37. Deprotection under acidic conditions and reductive amination with aldehyde 39 produced secondary amine 40.
Minimal inhibitory concentrations (MICs) were determined using three strains of S. aureus and two Gram-negative pathogens (Escherichia coli and Acinetobacter baumannii). The strains of S. aureus include one wildtype (ATCC 29213), one NBTI-resistant (GyrA D83N amino acid substitution17), and one fluoroquinolone-resistant MRSA (USA300).
Surprisingly, (E)-olefins 13a–c and alkanes 14a–c displayed anti-staphylococcal activity, suggesting that NBTIs do not absolutely require the prototypical interaction with D83 at the entrance of the DNA gyrase binding pocket if the appropriate binding vector is achieved. Meanwhile, (Z)-olefins 21a–c did not retain the same activity, perhaps due to the misorientation of the RHS by the bond angle produced by the (Z)-olefin. Monohydroxylated compounds 7a–c were more potent than the alkanes and alkenes and were comparable to other synthesized compounds when comparing Gram-positive MICs. This finding was also intriguing, as the monohydroxyls are incapable of forming an ionic interaction with D83 but can hydrogen bond, as predicted in Figure 4. Ketones (6a and 6c) were similarly active vs S. aureus, whereas compound 6b showed elevated MICs. Primary amines 9a–c were less potent than matched monohydroxyls 7a–c. The computational model (Table 1) predicted equivalent GyrA binding between diol enantiomers. Table 2 results confirmed this prediction: only minimal differences were observed for S. aureus [ATCC 29213, USA300 (MRSA)] MICs in head-to-head comparisons of 15a,b with 16a,b or 22a–c with 23a–c. Results for 15c and 16c were within the standard variability in the MIC assays. In contrast, docking results (Table 1) predicted a greater GyrA binding affinity for the anti- (22a–c and 23a–c) versus the syn-diols (15a–c and 16a–c). Results in Table 2 were consistent with this prediction, with S. aureus [ATCC 29213, USA300 (MRSA)] MICs values indicating improved activity for the anti- compared to the syn-diol analogues.
Table 2. Minimal Inhibitory Concentrations (μg/mL) of Atypical NBTIsa.
| Cmpd | S. aureus ATCC 29213 | S. aureus D83Nb | S. aureus USA300 (MRSA) | E. coli ATCC 25922 | A. baumannii ATCC 19606 |
|---|---|---|---|---|---|
| Ketones | |||||
| 6a | 0.13 | 4 | 0.13 | 16 | 32 |
| 6b | 4 | 32 | 4 | 64 | 64 |
| 6c | ≤0.13 | 16 | ≤0.13 | 2 | 4 |
| Monohydroxyls | |||||
| 7a | 0.06 | 8 | 0.25 | 16 | 32 |
| 7b | 0.25 | 4 | 0.25 | 8 | 8 |
| 7c | ≤0.13 | 16 | ≤0.13 | 4 | 4 |
| Primary amines | |||||
| 9a | 1 | 32 | 4 | 16 | 16 |
| 9b | 2 | ≥16 | 4 | 8 | 16 |
| 9c | 16 | 64 | 8 | 8 | 32 |
| (E)-Alkenes | |||||
| 13a | 0.25–0.5c | ≥32 | 0.5 | ≥64 | ≥64 |
| 13b | 0.25 | 2 | 0.5 | 8 | 8 |
| 13c | 0.5 | 16 | ≤0.25 | 4 | 8 |
| Alkanes | |||||
| 14a | 0.5 | 4 | 0.5 | 64 | 64 |
| 14b | 0.5 | 8 | 1 | 16 | 32 |
| 14c | 1 | 32 | 0.5 | 4 | 8 |
| (syn)-Diols | |||||
| 15a | 1–2c | ≥32 | 2–4c | 64 | 32–64c |
| 15b | 1 | ≥32 | 2 | 32 | 16 |
| 15c | 1 | ≥32 | 1 | 32 | 16 |
| 16a | 1 | ≥32 | 2 | 64 | 64 |
| 16b | 1–4c | 16 | 1–4c | 16 | 8–16c |
| 16c | 8 | ≥32 | 8 | 16 | 32 |
| (Z)-Alkenes | |||||
| 21a | 4–8c | 32 | 4–8c | ≥64 | ≥64 |
| 21b | 8 | ≥16 | 8 | ≥64 | 64 |
| 21c | ≥64 | ≥64 | 64 | 64 | 32 |
| (anti)-Diols | |||||
| 22a | 0.13 | 16 | 0.13 | 16 | 16 |
| 22b | ≤0.13 | 16 | 0.25 | 16 | 8 |
| 22c | 2 | 64 | 4 | 8 | 32 |
| 23a | 0.13 | ≥32 | 0.25 | 32 | 16 |
| 23b | 0.03 | 8 | 0.03 | 8 | 4 |
| 23c | 2 | 64 | 2 | 8 | 32 |
| Controls | |||||
| 40 | ≤0.25 | 16 | ≤0.25 | 4 | 4 |
| Gep.d | 0.125–1c | 8–32c | 0.125–0.5c | 1–4c | 8–32c |
| Cipro.d | 0.25–0.5c | 0.25–1c | 16–64c | 0.0075–0.25c | 1–2c |
All compounds lost activity in the NBTI-resistant strain (GyrA D83N) suggesting that the atypical NBTIs described here derive their antibacterial activity primarily from targeting DNA gyrase. The loss of activity for (E)-alkenes, alkanes, and (Z)-alkenes is particularly striking, given that they are incapable of direct interaction with D83. The precise origins of this effect are not fully understood. The analogous GyrA D82N mutation is in E. coli leads to diminished target inhibition by fluoroquinolones and imidazopyrazinones,19 neither of which directly contacts the aspartate. This observation has been attributed to reductions in the ability of gyrase to cleave DNA.
Compared to S. aureus, the MICs for these atypical NBTIs against Gram-negative pathogens were elevated. However, several structure–activity relationships were observed. Oxazinone-containing compounds often outperformed other RHS moieties (e.g., 6c, 7c, 13c, and 14c). Incorporating primary amines has previously been associated with a higher intracellular accumulation in E. coli.10,15 In contrast with their diminished anti-staphylococcal activity, primary amines (9a–c) displayed comparable E. coli MICs to the matched monohydroxyls (7a–c), potentially reflecting improved accumulation. Primary amine 9a was 4-fold less potent than the structure-matched secondary amine control (40) for both Gram-negative bacteria. As with S. aureus, MICs for (Z)-olefins (21a–c) were higher, whereas those of (E)-olefins (13b,c) were similar to other chemotypes. The syn- (15a–c and 16a–c) and anti-diols (22a–c and 23a–c) had comparable activity against Gram-negative pathogens. Overall, the compounds displayed more modest antibacterial activity against the studied Gram-negative pathogens, as is commonly seen with NBTIs,5,17 albeit with some exceptions.20 We suspect that the reduced activity may be due to lower intracellular accumulation as a result of efflux (see below).
To address the question of efflux, compounds were tested against wildtype and efflux-deficient strains of Salmonella enterica serovar Typhimurium (Table 3), a clinically important Gram-negative pathogen causing salmonellosis.21 The tolC-knockout lacks a prominent efflux channel present in the wildtype.22 By comparing the MICs for both strains, we can estimate the impact of the efflux.
Table 3. Antibacterial Activity vs Salmonella Strainsa.
| Cmpd | S. enterica Typhimurium ATCC 14028 MIC (μg/mL) | S. enterica Typhimurium 14028 ΔtolC1::kanb MIC (μg/mL) | Fold Change |
|---|---|---|---|
| 6a | 64 | 0.5 | 128 |
| 6b | ≥64 | 1 to >1c | ≥64 |
| 6c | 16 | ≤0.125 | ≥128 |
| 7a | 64 | 1 | 64 |
| 7b | 32 | 0.25 | 128 |
| 7c | 16 | ≤0.125 | ≥128 |
| 9a | 64 | 4 | 16 |
| 9b | 32 | 2 | 16 |
| 9c | 32 | 4 | 8 |
| 13a | ≥64 | 0.5–1c | ≥64 |
| 13b | 64 | 0.125 | 512 |
| 13c | 16 | 0.25 | 64 |
| 14a | ≥64 | 1 | ≥64 |
| 14b | 64 | 0.25 | 256 |
| 14c | 16 | 0.5 | 32 |
| 15a | ≥64 | 1–2b | ≥64 |
| 15b | 64 | 1 | 64 |
| 15c | 32 | 0.25 | 128 |
| 16a | ≥64 | 0.5–1c | ≥128 |
| 16b | 64 | 0.5 | 128 |
| 16c | 64 | 1 | 64 |
| 21a | ≥64 | 8 | ≥8 |
| 21b | ≥64 | 4 | ≥16 |
| 21c | 64 | 16 | 4 |
| 22a | 64 | 1 | 64 |
| 22b | 32 | 0.25 | 128 |
| 22c | ≥64 | 8 | ≥8 |
| 23a | 64 | 1 | 64 |
| 23b | 32 | 0.125 | 256 |
| 23c | 32 | 0.5 | 64 |
| 40 | 32 | 0.5 | 64 |
| Gep.d | 2–8c | 0.125–0.5c | 4–16 |
| Cipro.d | ≤0.13 | ≤0.13 | N/A |
Minimal inhibitory concentrations (MICs) determined in triplicate (at a minimum) according to CLSI guidelines.18
Efflux-deficient strain EFB044. This strain was constructed by moving mutation ΔtolC1::kan from the McClelland mutant collection23 into wildtype strain 14028 using P22 transduction.24
Ranges of observed values given where appropriate.
Gepotidacin and ciprofloxacin employed as controls.
Whereas compounds displayed poor activity against the wildtype strain, MICs were significantly improved against the efflux-deficient strain. Primary amines 9a–c displayed reduced susceptibility to efflux, with 8- to 16-fold changes observed between strains, whereas changes for other compounds ranged as high as 512-fold. Thus, the primary amines are not effluxed as efficiently as other chemotypes,15 as has been previously observed in a structurally distinct series of NBTIs.10
Biochemical evaluations using isolated bacterial topoisomerases were conducted to validate the mechanism of action and further support the MIC results and the computational model. The inhibition of supercoiling by S. aureus DNA gyrase was measured as previously described.25 The secondary amine control (40), monohydroxyl (7a), monoamine (9a), and anti-diol (22a) were chosen as representative compounds, holding the LHS and RHS constant to assess the impact of the novel functionality. Secondary amine 40 afforded the most potent inhibition despite its predicted weaker binding (Table 4). For the novel compounds, the relative binding affinities predicted by the computational model (Table 1) were not always consistent with the inhibition of S. aureus DNA gyrase, with 9a being the least potent inhibitor, despite having the best predicted binding affinity. However, the relative whole cell anti-staphylococcal activity followed the same trend as gyrase inhibition (Table 2), supporting DNA gyrase as the principal target in S. aureus. Compounds were also evaluated by using E. coli DNA gyrase, where inhibition was considerably weaker. The correlation between binding affinities and biochemical inhibition was also less robust, perhaps attributable to the derivation of the docking model from S. aureus gyrase structures.
Table 4. Supercoiling IC50 against DNA Gyrase and Decatenation IC50 Against TopoIVa.
| Cmpd | S. aureus DNA gyrase IC50 (μM)a | S. aureus TopoIV IC50 (μM)b | S. aureus TopoIV/gyrase ratio | E. coli DNA gyrase IC50 (μM)a |
|---|---|---|---|---|
| 7a | 0.24 | 2.29 | 9.5 | 12.37 |
| 9a | 5.05 | 82.6 | 16.4 | >400 |
| 22a | 0.065 | 3.31 | 50.9 | 17.09 |
| 40 | 0.004 | 0.69 | 176.9 | NTc |
Inhibition of supercoiling activity (n = 3).
Inhibition of decatenation activity (n = 3).
Not tested.
Decatenation experiments with S. aureus TopoIV were conducted with the same select compounds to assess the potential for dual targeting of both bacterial topoisomerases. All compounds displayed elevated IC50 values for TopoIV over DNA gyrase, and compound 9a displayed the highest IC50 value (Table 4). Compounds 7a and 22a had comparable activity in the TopoIV assay, in contrast to their ∼4-fold differences in gyrase IC50 values, and 7a afforded the best TopoIV/gyrase ratio at 9.5. Despite the much higher TopoIV/gyrase ratio for 22a (51), its potent targeting of DNA gyrase nevertheless afforded excellent anti-staphylococcal activity (Table 2).
NBTIs typically lead to the accumulation of single-strand breaks (SSBs) with both DNA gyrase and TopoIV.5,7 Given the atypical structures of the NBTIs described herein, an assessment of cleavage activity was warranted. All compounds tested displayed typical NBTI behavior and resulted in an accumulation of SSBs, with minimal to no DSBs observed (<1%, data not shown). For S. aureus gyrase at 10 μM, lower levels of SSBs were observed for primary amine 9a and secondary amine 40 as compared to those of monohydroxyl 7a and anti-diol 22a (Figure 6A). Similar results were obtained for the TopoIV cleavage assay (Figure 6B).
Figure 6.

DNA cleavage assays with S. aureus DNA gyrase (left, A) and TopoIV (right, B).
In this study, we employed a computational docking model to prioritize compounds for the synthesis and testing. Diverse novel chemotypes delivered potent MICs with the Gram-positive pathogen S. aureus. Elevated MICs in the NBTI-resistant GyrA D83N mutant S. aureus and biochemical testing with isolated enzymes were consistent with gyrase as the primary antibacterial target. Using a test set of three novel chemotypes, we found predicted docking scores were consistent with relative inhibition of S. aureus DNA gyrase and with whole cell anti-staphylococcal activity. Antibacterial activity was significantly reduced in Gram-negative bacteria, including E. coli, A. baumannii, and S. enterica Typhimurium. Results from wildtype and efflux-deficient Salmonella strains demonstrated that efflux significantly compromised activity in the wildtype strain, indicating a need for continued optimization. In total, these results open the door for further chemical innovation of the NBTI class of antibacterials.
Acknowledgments
The authors thank Prof. Daniele Leonori of Aachen University and Dr. Fabio Juliá of Universidad Murcia for their correspondence on the photo-Heck coupling reaction, Drs. Erin Boulanger and Brian Ahmer of The Ohio State University for the tolC mutant strain of Salmonella, Dr. Yanran Lu for contributing to the biochemical assays, and Jose Orench-Benvenutti for support of MIC assays.
Glossary
Abbreviations
- NBTI
novel bacterial topoisomerase inhibitor
- TopoIV
topoiosomerase IV
- CLSI
Clinical and Laboratory Standards Institute
- SSB
single-strand break
- DSB
double-strand break
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.4c00162.
Assay methods, computational methods, synthesis and characterization of tested compounds. 1H NMR spectra of tested compounds, 13C NMR spectra, UPLC, and HRMS of selected compounds (PDF)
Funding was provided by the Dr. Ralph and Marian Falk Medical Research Trust (Transformational Award to M.M-F.) and the NIH (R21 AI148986 and R01 AI173072 to M.M-F. and R01 GM126363 and R01 AI170546 to N.O.).
The authors declare the following competing financial interest(s): MMF is a shareholder of Pfizer.
Supplementary Material
References
- Gellert M.; Mizuuchi K.; O’Dea M. H.; Nash H. A. DNA gyrase: An enzyme that introduces superhelical turns into DNA. Proc. Natl. Acad. Sci. U.S.A. 1976, 73 (11), 3872–3876. 10.1073/pnas.73.11.3872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato J.; Nishimura Y.; Imamura R.; Niki H.; Hiraga S.; Suzuki H. New topoisomerase essential for chromosome segregation in E. coli. Cell. 1990, 63 (2), 393–404. 10.1016/0092-8674(90)90172-B. [DOI] [PubMed] [Google Scholar]
- Munita J. M.; Arias C. A. Mechanisms of antibiotic resistance. Microbiol. Spectrum. 2016, 4 (2), VMBF-0016-2015. 10.1128/microbiolspec.VMBF-0016-2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aldred K. J.; Kerns R. J.; Osheroff N. Mechanism of Quinolone Action and Resistance. Biochemistry 2014, 53, 1565–1574. 10.1021/bi5000564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolarič A.; Anderluh M.; Minovski N. Two decades of successful SAR-grounded stories of the novel bacterial topoisomerase inhibitors (NBTIs). J. Med. Chem. 2020, 63 (11), 5664–5674. 10.1021/acs.jmedchem.9b01738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kokot M.; Anderluh M.; Hrast M.; Minovski N. The Structural Features of Novel Bacterial Topoisomerase Inhibitors that Define Their Activity on Topoisomerase IV. J. Med. Chem. 2022, 65 (9), 6431–6440. 10.1021/acs.jmedchem.2c00039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson E. G.; Bax B.; Chan P. F.; Osheroff N. Mechanistic and structural basis for the actions of the antibacterial gepotidacin against Staphylococcus aureus gyrase. ACS Infect. Dis. 2019, 5 (4), 570–581. 10.1021/acsinfecdis.8b00315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanden Broeck A.; Lotz C.; Ortiz J.; Lamour V. Cryo-EM structure of the complete E. coli DNA gyrase nucleoprotein complex. Nat. Commun. 2019, 10, 4935. 10.1038/s41467-019-12914-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Black M. T.; Stachyra T.; Platel D.; Girard A. M.; Claudon M.; Bruneau J. M.; Miossec C. Mechanism of action of the antibiotic NXL101, a novel nonfluoroquinolone inhibitor of bacterial type II topoisomerases. Antimicrob. Agents Chemother. 2008, 52 (9), 3339–3349. 10.1128/AAC.00496-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surivet J.-P.; Zumbrunn C.; Bruyère T.; Bur D.; Kohl C.; Locher H. H.; Seiler P.; Ertel E. A.; Hess P.; Enderlin-Paput M.; Enderlin-Paput S.; Gauvin J.-C.; Mirre A.; Hubschwerlen C.; Ritz D.; Rueedi G. Synthesis and Characterization of Tetrahydropyran-Based Bacterial Topoisomerase Inhibitors with Antibacterial Activity against Gram-Negative Bacteria. J. Med. Chem. 2017, 60 (9), 3776–3794. 10.1021/acs.jmedchem.6b01831. [DOI] [PubMed] [Google Scholar]
- Schrodinger, LLC . Glide, Version 86013; Schrodinger, LLC: New York, NY, 2020. [Google Scholar]
- Kim S. S.; Aprahamian M. L.; Lindert S. Improving inverse docking target identification with Z-score selection. Chem. Biol. Drug Des. 2019, 93 (6), 1105–1116. 10.1111/cbdd.13453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casey F. P.; Pihan E.; Shields D. C. Discovery of small molecule inhibitors of protein-protein interactions using combined ligand and target score normalization. J. Chem. Inf. Model. 2009, 49 (12), 2708–2717. 10.1021/ci900294x. [DOI] [PubMed] [Google Scholar]
- Liu S.; Fu R.; Zhou L.-H.; Chen S.-P. Application of consensus scoring and principal component analysis for virtual screening against beta-secretase (BACE-1). PLoS One 2012, 7 (6), e38086 10.1371/journal.pone.0038086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter M. F.; Drown B. S.; Riley A. P.; Garcia A.; Shirai T.; Svec R. L.; Hergenrother P. J. Predictive compound accumulation rules yield a broad-spectrum antibiotic. Nature 2017, 545 (7654), 299–304. 10.1038/nature22308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Constantin T.; Zanini M.; Regni A.; Sheikh N. S.; Juliá F.; Leonori D. Aminoalkyl radicals as halogen-atom transfer agents for activation of alkyl and aryl halides. Science 2020, 367 (6481), 1021–1026. 10.1126/science.aba2419. [DOI] [PubMed] [Google Scholar]
- Lu Y.; Vibhute S.; Li L.; Okumu A.; Ratigan S. C.; Nolan S.; Papa J. L.; Mann C. A.; English A.; Chen A.; Seffernick J. T.; Koci B.; Duncan L. R.; Roth B.; Cummings J. E.; Slayden R. A.; Lindert S.; McElroy C. A.; Wozniak D. J.; Yalowich J.; Mitton-Fry M. J. Optimization of TopoIV Potency, ADMET Properties, and hERG Inhibition of 5-Amino-1,3-dioxane-Linked Novel Bacterial Topoisomerase Inhibitors: Identification of a Lead with In Vivo Efficacy against MRSA. J. Med. Chem. 2021, 64 (20), 15214–15249. 10.1021/acs.jmedchem.1c01250. [DOI] [PubMed] [Google Scholar]
- Clinical and Laboratory Standards Institute (CLSI) . Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria That Grow Aerobically; Approved Standard, 11the ed.; CLSI standard M07; CLSI: Wayne, PA, 2018. [Google Scholar]
- Germe T.; Vörös J.; Jeannot F.; Taillier T.; Stavenger R. A.; Bacqué E.; Maxwell A.; Bax B. D. A New Class of Antibacterials, the Imidazopyrazinones, Reveal Structural Transitions Involved in DNA Gyrase Poisoning and Mechanisms of Resistance. Nucleic Acids Res. 2018, 46 (8), 4114–4128. 10.1093/nar/gky181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cumming J. G.; Kreis L.; Kühne H.; Wermuth R.; Vercruysse M.; Kramer C.; Rudolph M. G.; Xu Z. Discovery of a series of Indane-containing NBTIs with activity against multidrug-resistant gram-negative pathogens. ACS Med. Chem. Lett. 2023, 14 (7), 993–998. 10.1021/acsmedchemlett.3c00187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fàbrega A.; Vila J. Salmonella enterica Serovar Typhimurium Skills to Succeed in the Host: Virulence and Regulation. Clin. Microbiol. Rev. 2013, 26 (2), 308–341. 10.1128/CMR.00066-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akshay S. D.; Deekshit V. K.; Raj J. M.; Maiti B. Outer membrane proteins and efflux pumps mediated multi-drug resistance in Salmonella: Rising threat to antimicrobial therapy. ACS Infect. Dis. 2023, 9 (11), 2072–2092. 10.1021/acsinfecdis.3c00408. [DOI] [PubMed] [Google Scholar]
- Porwollik S.; Santiviago C. A.; Cheng P.; Long F.; Desai P.; Fredlund J.; Srikumar S.; Silva C. A.; Chu W.; Chen X.; Canals R.; Reynolds M. M.; Bogomolnaya L.; Shields C.; Cui P.; Guo J.; Zheng Y.; Endicott-Yazdani T.; Yang H.-J.; Maple A.; Ragoza Y.; Blondel C. J.; Valenzuela C.; Andrews-Polymenis H.; McClelland M. Defined Single-Gene and Double-Gene Deletion Mutant Collections in Salmonella enterica sv Typhimurium. PLoS One 2014, 9 (7), e99820 10.1371/journal.pone.0099820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maloy S. R.; Stewart V. J.; Taylor R. K.. Genetic analysis of pathogenic bacteria: a laboratory manual; Cold Spring Harbor Laboratory Press: Plainville, NY, 1996. [Google Scholar]
- Li L.; Okumu A.; Nolan S.; English A.; Vibhute S.; Lu Y.; Hervert-Thomas K.; Seffernick J. T.; Azap L.; Cole S. L.; Shinabarger D.; Koeth L. M.; Lindert S.; Yalowich J. C.; Wozniak D. J.; Mitton-Fry M. J. 1,3-dioxane-linked bacterial topoisomerase inhibitors with enhanced antibacterial activity and reduced hERG inhibition. ACS Infect. Dis. 2019, 5 (7), 1115–1128. 10.1021/acsinfecdis.8b00375. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.