

Abstract
The first steps of animal speciation are thought to be the development of sexual isolating mechanisms. In contrast to recent progress in understanding the genetic basis of postzygotic isolating mechanisms, little is known about the genetic architecture of sexual isolation. Here, we have subjected Drosophila melanogaster to 29 generations of replicated divergent artificial selection for mating speed. The phenotypic response to selection was highly asymmetrical in the direction of reduced mating speed, with estimates of realized heritability averaging 7%. The selection response was largely attributable to a reduction in female receptivity. We assessed the whole genome transcriptional response to selection for mating speed using Affymetrix GeneChips and a rigorous statistical analysis. Remarkably, >3,700 probe sets (21% of the array elements) exhibited a divergence in message levels between the Fast and Slow replicate lines. Genes with altered transcriptional abundance in response to selection fell into many different biological process and molecular function Gene Ontology categories, indicating substantial pleiotropy for this complex behavior. Future functional studies are necessary to test the extent to which transcript profiling of divergent selection lines accurately predicts genes that directly affect the selected trait.
Species are groups of actually or potentially interbreeding natural populations, which are reproductively isolated from other such groups.
Recent studies by the students of animal behavior, as well as the revised interpretation of many earlier observations, indicate that behavior differences are among animals the most important factor in restricting random mating between closely related forms.
E. Mayr, 1942
One of the major challenges facing modern biology is to understand the genetic mechanisms causing speciation. Because sexual isolating mechanisms that act before fertilization [“ethological” isolating mechanisms (1)] are thought to precede the evolution of postzygotic isolating mechanisms (inviability and sterility), we need to understand the genetic basis of sexual isolation if we are to gain insight about the early stages of species formation. However, mating behaviors are complex traits, with variation attributable to multiple interacting loci with individually small effects, whose expression depends on the environment. Thus, understanding the genetic architecture of sexual isolation requires that we overcome the twin obstacles of mapping genes causing differences between organisms that, by definition, do not interbreed (2) and solving the problem of genetically dissecting complex behavioral traits (3).
Drosophila Mating Behavior
Drosophila species present an ideal model system in which to investigate the genetic basis of sexual isolation. Several species pairs are only partially reproductively isolated, producing fertile hybrids that can be backcrossed to one of the parental species to generate segregating backcross mapping populations. Furthermore, Drosophila melanogaster is a model organism with excellent genetic and genomic resources that are ideal for genetically dissecting complex traits, including the ability to clone chromosomes, replicate genotypes, and rear large numbers of individuals under uniform environmental conditions; publicly available mutations and deficiency stocks useful for mapping; abundant segregating variation in natural populations that can readily be selected in the laboratory to produce divergent phenotypes a complete well annotated genome sequence; and several platforms for whole-genome transcriptional profiling. Courtship behavior of Drosophila is composed of sequential actions that exchange auditory, visual, and chemosensory signals between males and females, allowing for individual components of the behavior to be quantified and separated (4, 5). Courtship is initiated when the male aligns himself with the female, using visual and olfactory signals for orientation. He then taps the female's abdomen with his foreleg, using pheromonal cues for gender and species recognition, followed by wing vibration to produce a species-specific courtship song. After courtship initiation, the male again uses pheromonal cues by licking the female's genitalia, after which he will attempt to copulate. The female can accept the male or reject him by moving away. Successful copulation is accompanied by the transfer of sperm and seminal fluids that stimulate the release of oocytes by the ovary (6) and reduce female receptivity to other males (7, 8). Components of the seminal fluids are associated with the reduced lifespan of mated females (9), setting up an intersexual conflict (10).
Given the complexity of Drosophila courtship behavior, it is not surprising that mutations in genes affecting multiple biological processes affect mating behavior (11, 12). These include mutations in genes required for normal morphology [white (13, 14), yellow (14), and curved (15)], as well as genes involved in learning and memory [Calcium calmodulin kinase II (16), dunce (17, 18), ruta-baga (19, 20), turnip (19, 21), and amnesiac (20, 22, 23)], circadian rhythm [period (18, 24-26)] and dopamine and serotonin synthesis [Dopa decarboxylase (27), pale (28, 29), tan (30, 31), and ebony (32-34)], sex determination [doublesex (35-37), transformer (38-43), fruitless (44-47), and sex lethal (48)], pheromone production [desaturase 2 (49)], and accessory gland-specific peptides (6-8, 50-52).
Sexual Isolation Among Species
Despite the wealth of knowledge regarding genetic mechanisms that affect Drosophila courtship behavior, we know virtually nothing of the genes that cause naturally occurring variation in mating behavior within and among species, their allelic effects, and their interactions. Are the loci that harbor naturally occurring variation a subset of loci identified by mutational analysis, or will the analysis of natural variants reveal novel loci? Is natural variation in mating behavior attributable to a few genes with large effects or many genes with small effects? Do the alleles at different loci interact additively or exhibit epistasis? Do the same genes that affect variation in courtship behavior within species account for sexual isolation between species? Answers to these questions require that we identify the quantitative trait loci (QTLs) affecting sexual isolation between species and variation in mating behavior within species.
Because QTLs often have small effects that are contingent on the environment, they can be mapped only by linkage to markers whose genotype can be scored unambiguously (53). Before the recent discovery of abundant polymorphic molecular markers, mapping the QTLs affecting sexual isolation between Drosophila species was confined to estimates of the effects of each chromosome arm (54-60).
Two recent studies addressed the genetic basis of variation in sexual isolation between Drosophila pseudoobscura and Drosophila persimilis (61) and between Drosophila simulans and Drosophila mauritiana (62) by linkage to molecular markers in large backcross populations. In the first species pair, sexual isolation is attributable to female discrimination against males of the sibling species; males readily court females of either species. QTLs affecting male traits against which D. pseudoobscura discriminate are located primarily on the left arm of the X chromosome, with minor contributions from the right arm of the X and second chromosomes. QTLs affecting male traits against which D. persimilis discriminate are located on the second chromosome (61).
D. mauritiana females rarely mate with D. simulans males. At least seven QTLs, mapping to all three chromosomes, affect the discrimination of D. mauritiana females against D. simulans males; and three QTLs, all on the third chromosome, affect the D. simulans male traits against which D. mauritiana females discriminate. QTLs for female choice are different from those for the male traits they are choosing against. Although D. simulans females mate with D. mauritiana males, copulations are abnormally short and often do not result in adequate sperm transfer (56). At least six autosomal QTLs affect the D. mauritiana male traits against which D. simulans females discriminate. No epistatic interactions were observed between QTLs affecting prezygotic isolation, in contrast to the genetic architecture of postzygotic isolation (2). Although a few QTLs with moderate effects affect prezygotic reproductive isolation in both of these species pairs, high-resolution recombination mapping will be necessary to identify individual genes.
Variation in Mating Behavior Within D. melanogaster
Genetic variation for incipient sexual isolation has been implicated within populations of D. melanogaster by repeated observations that positive assortative mating can evolve as a correlated response to divergent artificial selection for sensory bristle numbers, geotaxis, phototaxis, and locomotor activity (63). Presumably, assortative mating evolves because genes affecting the selected traits are closely linked to genes affecting mating behavior or have pleiotropic effects on mating behavior. There is naturally occurring polymorphism for incipient sexual isolation within D. melanogaster. Females from populations in Zimbabwe (Z) exhibit strong preference for Z males when given a choice between Z and Cosmopolitan (C) males, but the reciprocal crosses exhibit weaker or no sexual isolation (64). Chromosome substitution analyses revealed that QTLs affecting the discrimination of Z females against C males, as well as QTLs affecting the attractiveness of Z males to Z females, reside on all major chromosomes, with the third chromosome having the greatest and the X chromosome the least effect (65). Recombination mapping of third-chromosome QTLs using visible morphological markers revealed at least four epistatic QTLs affecting Z male mating success and at least two QTLs affecting Z female mating preference (66).
Recently, QTLs affecting variation in male mating behavior between Oregon (Ore), a standard wild-type strain, and 2b, a strain selected for reduced male courtship and copulation latency, have been mapped with high resolution by linkage to molecular markers in a panel of 98 recombinant inbred lines derived from these strains (67). The initial genome scan revealed a minimum of one X chromosome and three autosomal QTLs affecting variation in male mating behavior between Ore and 2b. These QTLs mapped to relatively large genomic regions containing on average >600 genes. However, in D. melanogaster, one can readily map QTLs to subcM regions using deficiency complementation mapping (68) and identify candidate genes corresponding to the QTLs using quantitative complementation tests to mutations at the positional candidate genes (69, 70). The three autosomal QTLs fractionated into five QTLs containing 58 genes on average. Complementation tests to all 45 available mutations at the positional candidate genes delimited by deficiency mapping revealed seven novel candidate genes affecting male mating behavior: eagle, 18 wheeler, Enhancer-of-split, Polycomb, spermatocyte arrest, l (2)05510, and l (2)k02006. These genes are involved in spermatogenesis, chromatin and gene silencing, serotonin neuron fate determination, and nervous system development. None of these genes has been previously implicated in mating behavior, demonstrating that quantitative analysis of subtle variants can reveal novel pleiotropic effects of key developmental genes on behavior (67).
Our ability to map the genes affecting naturally occurring variation in mating behavior within D. melanogaster is compromised by two factors. First, the size of the mapping populations determines the minimum QTL effect that can be detected. Increasing the sample size will increase the numbers of mapped QTLs, because linked QTLs can be separated by recombination, and the minimum detectable effect decreases as the sample size increases. Second, any two strains used to map QTLs are limited samples of the existing variation (53). Recently, there has been great excitement about the utility of whole genome transcriptional profiling to identify candidate genes regulating complex traits by assessing changes in gene expression between lines selected for different phenotypic values of the trait (71). Here, we describe the results of 29 generations of replicated selection for increased and decreased mating speed from a large heterogeneous base population and the analysis of the whole genome transcriptional response to artificial selection.
Materials and Methods
Drosophila Selection Lines. The base population consisted of 60 isofemale lines collected in Raleigh, NC, in 2002 using fruit baits. The 60 lines were crossed in a round-robin design (♀1×♂2, ♀2×♂3,..., ♀60×♂1) in separate culture vials, with three females and three males per vial. After 2 days, one inseminated female from each cross was placed in each of two culture bottles to initiate replicate selection lines. The progeny from each replicate bottle were scored for copulation latency to initiate Generation 1 of selection. A total of 50 pairs of 4- to 7-day-old virgin males and females from each replicate bottle were placed in culture vials, and the time to copulation was scored for each pair, for a total of 3 h. The 20 fastest pairs from each replicate were placed in culture bottles to initiate the two Fast selection lines, and the 20 slowest pairs were placed in culture bottles to initiate the two Slow lines. Control lines were started from the 10 middle-scoring pairs from each line, plus 10 pairs of virgin males and females that were not scored. In the second and subsequent generations, 50 males and females from the six replicate lines were scored for copulation latency. The Fast lines were maintained by selecting the 20 fastest pairs each generation, and the Slow lines were maintained by selecting the 20 slowest pairs. The control lines were maintained by 20 pairs that were chosen at random with respect to copulation latency. Pairs that did not mate in the 3-h observation period were given a score of 180 min.
Flies were reared on standard cornmeal-molasses-agar medium and maintained in an incubator at 25°C and a 12:12 h light/dark cycle. Mating behavior was assessed for 3 h in the morning, 2 h after lights on.
Quantitative Genetic Analysis of Selection Response. Realized heritability of copulation latency was computed for each replicate from the regression of cumulated response (as a deviation from the control) on cumulated selection differential (72).
Male Mating Behavior. We assessed correlated responses in male mating behavior in response to selection for copulation latency from generations 21-23. Male mating behavior was assessed for 1 h, immediately after the flies were paired. Otherwise, the conditions were identical to those under which copulation latency was scored. Courtship latency is the time to initiate courtship behavior. We scored courtship intensity by observing individual males every minute after initiation of courtship until copulation occurred and recording whether they were engaged in courtship behavior. The measure of courtship intensity was the number of times they were observed courting divided by the total number of observations.
Transcriptional Profiling. At Generation 23, three replicate groups of 50 4- to 7-day-old virgin males and females were collected from the two Fast and two Slow replicate lines (i.e., the same age and mating status as the flies before selection). Total RNA was extracted independently for each of the 24 samples (four lines × two sexes × three replicates) by using the TRIzol reagent (GIBCO/BRL). The samples were treated with DNase and purified on Qiagen (Chatsworth, CA) RNeasy columns. Biotinylated cRNA probes were hybridized to high-density oligonucleotide Affymetrix Drosophila GeneChip 2.0 microarrays and visualized with a streptavidin-phycoerythrin conjugate, as described in the Affymetrix GeneChip Expression Analysis Technical Manual (2000), using internal references for quantification.
Micorarray Data Analysis. We normalized the expression data by scaling overall probe set intensity to 300 on each microarray using standard reference probe sets on each GeneChip for the normalization procedure. Every gene on the Affymetrix Drosophila GeneChip 2.0 is represented by a probe set consisting of 14 perfect match (PM) and 14 mismatch (MM) probe pairs. The quantitative estimate of expression of each probe set is the Signal (Sig) metric. Sig is computed by using the one-step Tukey's biweight estimate, which gives the weighted mean of the log(PM-MM) intensities for each probe set (AffymetrixMicroarray Suite, Ver. 5.0). A detection call (present, marginal, or absent) is also given for each probe set. We eliminated probe sets from consideration if over one-half were called absent. In practice, this retained probe sets with sex-specific expression and removed those with low and variable Sig values.
We performed two-way fixed-effect ANOVAs of the expression values for all remaining probe sets, according to the model Y = μ + S + L + S×L + E, where S and L are the crossclassified effects of sex and selection line (Fast replicate 1, Fast replicate 2, Slow replicate 1, and Slow replicate 2), respectively, and E is the variance between replicate arrays. P values were computed from F ratio tests of significance for each of the terms in the ANOVA. Because there are >18,000 probe sets on the array, this poses a huge multiple testing problem for determining the significance threshold using P values. Bonferroni corrections for multiple tests are too conservative, and a conventional 5% significance threshold will yield too many false positives. We used a Q = 0.001 false-discovery rate criterion (73) for the significance of any of the terms in the ANOVA model. Unlike the P value, which is the number of false positives expected when truly nothing is significant, the false discovery rate Q value controls the proportion of false positives among all terms declared significant (73).
Variation in transcript abundance between lines could be attributable to changes in gene frequency due to random drift or to changes in frequency of genes under selection. In the latter case, one would expect common alleles affecting variation in transcript abundance to have the same effect in both selection lines. Therefore, contrast statements were used to assess whether transcript abundance for probe sets with L and/or S×L terms at or below the Q = 0.001 threshold was significantly different between the two Fast lines and the two Slow lines, both pooled over sexes, and for each sex separately.
Statistical analyses were conducted by using SAS software (SAS Institute, Cary, NC). Cytological locations and biological process and molecular function gene ontologies were given by the NetAffyx (www.affymetrix.com/analysis/index.affx) database, supplemented by information from the FlyBase Consortium (74), current as of December 31, 2004.
Results
Phenotypic Response to Selection for Copulation Latency. The result of 29 generations of replicated selection for increased and decreased copulation latency is depicted in Fig. 1A. The selection response is highly asymmetrical in the direction of increased copulation latency. The Fast and Slow replicate lines were significantly diverged from Generation 25. We analyzed the mating speed data from generations 25-29 according to the mixed model ANOVA Y = μ + S + G + G×S + R(S) + G×R(S) + E, where μ is the overall mean; S and G are the crossclassified fixed effects of direction of selection (Fast vs. Slow) and generation, respectively; R is the random effect of replicate line; and E is the variance within lines. The effect of direction of selection was highly significant (F1, 2 = 617.71, P = 0.0016).
Fig. 1.

Phenotypic response to selection for copulation latency. (A) Mean mating speed of selection lines. ▴, Fast lines; ▿, Slow lines; ○, Control lines. (B) Regressions of cumulated response on cumulated selection differential for Fast and Slow selection lines. ▴, Replicate 1, Fast; ▵, Replicate 2, Fast; ▾, Replicate 1, Slow; fl, Replicate 2, Slow. (C) Regressions of cumulated response on cumulated selection differential for divergence between Fast and Slow selection lines. •, Replicate 1; ○, Replicate 2. (D) Mating speeds averaged over generations 18, 20, and 21 for Fast females paired with Fast males (FF), Fast females paired with Slow males (FS), Slow females paired with Slow males (SS), and Slow females paired with Fast males (SF). The subscripts denote Replicates 1 and 2, respectively. A, B, and C indicate the results of Tukey tests. Groups with the same letter are not significantly different. (E) Male courtship latency. Groups are the same as in D.(F) Male courtship intensity. Groups are the same as in D.
We computed realized heritabilities (h2) of mating speed from the regressions of cumulated response on cumulated selection differentials (ref. 72 and Fig. 1 B and C). Estimates of h2 (±SE of the regression coefficient) were h2 = 0.047 (0.025) and h2 = 0.011 (0.020) for Replicate 1 and 2 Fast lines, respectively; neither estimate is significantly different from zero. Estimates of h2 for the Replicate 1 and 2 Slow lines, respectively, were h2 = 0.059 (0.015, P = 0.0006) and h2 = 0.099 (0.016, P < 0.0001). Heritabilities estimated from the divergence were h2 = 0.056 (0.011, P < 0.0001) and h2 = 0.078 (0.012, P < 0.0001) for Replicates 1 and 2, respectively.
Reduced mating speed could be attributable to reduced male copulation latency, reduced female receptivity, or both. At generations 18, 20, and 21, we assessed copulation latency when Fast females of each replicate were paired with Slow males and when Slow females of each replicate were paired with Fast males. The results of these tests, as well as the responses of the selection lines in these generations, are shown in Fig. 1D. We analyzed the copulation latency data by the fixed-effects ANOVA model Y = μ + C + G + C×G + E, where C is cross, G is generation, and E is the variation within each cross and generation. The effect of cross was highly significant (F7, 1176 = 221.95, P < 0.0001). Post hoc Tukey tests revealed there was no significant difference in mating speed between Fast females of either replicate when paired with Fast or Slow males. However, Slow females were equally slow when paired with Slow or Fast males. Clearly, the rapid evolution of reduced copulation latency is attributable to reduced female receptivity: slow females are picky.
We assessed correlated responses in male behavior by measuring courtship latency and courtship intensity for each of the reciprocal pairs of selection lines (Fast females and Fast males, Fast females and Slow males, Slow females and Slow males, and Slow females and Fast males) for each replicate. The data were analyzed by ANOVA, as described above for copulation latency. There was no detectable difference in courtship latency of males in any of the crosses (F7, 143 = 1.54, P = 0.158; Fig. 1E). There were, however, highly significant differences in courtship intensity between the crosses (F7, 142 = 5.92, P < 0.0001; Fig. 1F). The courtship intensity of Fast males with Fast females was much greater than that with Slow males and Slow females. The courtship intensity of both replicates of Fast males with Slow females was not significantly different from that of these males with Fast females. However, the courtship intensity of Slow males from Replicate 1 with Fast females was as low as with Slow females, but the courtship intensity of Slow males from Replicate 2 with Fast females was as fast as the Fast males (Fig. 1F), indicating some divergence between the replicates in correlated male behaviors.
Transcriptional Response to Selection for Copulation Latency. We assessed transcript abundance at the time of selection for the Fast and Slow selection lines, using Affymetrix high-density oligonucleotide whole genome microarrays. Raw expression data are given in Table 4, which is published as supporting information on the PNAS web site. Statistically significant differences in transcript abundance were evaluated by factorial ANOVA (with line and sex the two crossclassified main effects) for each probe set. Using a false discovery rate of Q = 0.001 (i.e., one false positive in 1,000 among probe sets declared significant), 10,336 probe sets were significant for the main effect of sex, 4,420 were significant for the main effect of line, and 1,107 were significant for the line × sex interaction.
We used ANOVA contrast statements to detect probe sets that were up- or down-regulated in both Fast and Slow selection replicates, as would be expected if gene frequencies of the same common alleles changed in both selection lines. Remarkably, a total of 3,727 probe sets met this criterion (Table 5, which is published as supporting information on the PNAS web site). Of these, 836 were male-specific (505 of these probe sets were up-regulated in Fast males, and 331 were up-regulated in Slow males), 1,336 were female-specific (912 were up-regulated in Fast females, and 424 were up-regulated in Slow females), and 1,490 affected both sexes (575 were up-regulated in Fast lines, and 915 were up-regulated in Slow lines). In addition, transcript abundance for 65 probe sets had sexually antagonistic effects. Of these, 23 were up-regulated in Fast females and down-regulated in Fast males, and 42 were upregulated in Fast males and down-regulated in Fast females. Clearly, there has been a widespread transcriptional response to selection for mating speed. However, the magnitude of the changes of transcript abundance is not great, with the vast majority much less than 2-fold (Fig. 2).
Fig. 2.

Relative log2 fold changes in transcript abundance in Fast vs. Slow selection lines. (A) Male-specific transcripts. (B) Female-specific transcripts. (C) Both sexes.
We assessed whether probe sets with significantly altered transcript abundance were randomly distributed among the five major chromosome arms. We counted the number of probe sets on each chromosome arm and used a χ2 goodness-of-fit test to check for departure from the expected number, computed based on the total fraction of the genome on each chromosome arm. We observed a nonrandom distribution of probe sets that were up-regulated in Fast relative to Slow males (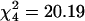 ; P = 0.0005) and for probe sets that were up-regulated in Slow relative to Fast males (
; P = 0.0005) and for probe sets that were up-regulated in Slow relative to Fast males (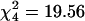 ; P = 0.0006) (Fig. 3). In both cases, a deficiency of up-regulated transcripts on the X chromosome contributed to the significant χ2 statistic. In addition, there was an excess of transcripts up-regulated in Slow relative to Fast males on chromosome 2L.
; P = 0.0006) (Fig. 3). In both cases, a deficiency of up-regulated transcripts on the X chromosome contributed to the significant χ2 statistic. In addition, there was an excess of transcripts up-regulated in Slow relative to Fast males on chromosome 2L.
Fig. 3.

Chromosomal distribution of transcripts on the major chromosome arms. *, χ24, P < 0.001.
We also assessed whether probe sets were nonrandomly distributed along each chromosome arm, as might be expected if selection caused linkage disequilibrium between selected loci and closely linked genes. We counted the number of probe sets in each major cytological division and used a χ2 goodness-of-fit test to check for departure from the expected number, based on the total fraction of genes on each chromosome arm per cytological division. Only 5 of the 30 χ2 statistics were significant at P < 0.05 and, of these, only one test statistic was significant based on a Bonferroni correction for multiple tests. This was for probe sets on chromosome 2L that were up-regulated in Fast relative to Slow females (χ219 = 50.638; P = 0.0001), where bands 25, 32, and 35 had fewer up-regulated probe sets than expected, and bands 29 and 31 had more upregulated probe sets than expected. Thus, there was little evidence for nonrandom distribution of probe sets with significantly altered transcript abundance within each chromosome arm.
The probe sets that were up-regulated in each comparison of Fast and Slow selection lines fell into all major biological process and molecular function Gene Ontology (GO) categories (Tables 1, 2, 3, 4). Comparison of the numbers of up-regulated probe sets in each GO category with the number expected based on representation on the microarray revealed that many categories were significantly over- or underrepresented. We hypothesize that GO categories that are overrepresented contain probe sets for which transcript abundance has been altered as a consequence of artificial selection, whereas natural selection opposes artificial selection for probe sets in GO categories that are underrepresented. For example, more probe sets than expected that are up-regulated in Fast relative to Slow females fall into the physiological biological process and binding molecular function categories. On the other hand, there are fewer probe sets than expected in the regulation biological process and transcription regulator categories that exhibit significant changes in transcript abundance in multiple comparisons of selection lines (Tables 1 and 2).
Table 1. Biological process GO categories.
| Male-specific, n
|
Female-specific, n
|
Both sexes, n
|
||||
|---|---|---|---|---|---|---|
| GO category | F > S | S > F | F > S | S > F | F > S | S > F |
| Behavior | 11 | 6 | 6 | 11 | 4 | 15 |
| (9.02 × 10−2) | (1.90 × 10−1) | (3.10 × 10−2) | (1.17 × 10−1) | (1.36 × 10−1) | (4.93 × 10−1) | |
| Cellular | 105 | 60 | 293 | 139 | 114 | 233 |
| (2.04 × 10−4) | (8.36 × 10−2) | (2.84 × 10−1) | (9.49 × 10−1) | (2.46 × 10−8) | (1.39 × 10−1) | |
| Development | 42 | 22 | 134 | 64 | 39 | 121 |
| (2.75 × 10−3) | (3.34 × 10−2) | (7.98 × 10−1) | (9.11 × 10−1) | (8.85 × 10−7) | (6.93 × 10−1) | |
| Physiological | 217 | 117 | 491 | 236 | 274 | 423 |
| (1.21 × 10−1) | (4.78 × 10−1) | (2.02 × 10−3) | (4.27 × 10−1) | (3.27 × 10−1) | (8.58 × 10−1) | |
| Regulation | 20 | 11 | 108 | 31 | 29 | 71 |
| (8.16 × 10−5) | (6.95 × 10−3) | (6.66 × 10−2) | (2.83 × 10−2) | (3.83 × 10−4) | (1.91 × 10−1) | |
Numbers are the numbers of up-regulated probe sets in each comparison. P values (in parentheses) are from χ2 tests of departure from expected numbers in each GO category, based on the frequency of probe sets in each category on the GeneChip. Italics denote fewer up-regulated probe sets than expected by chance; bold denotes more up-regulated probe sets than expected by chance.
Table 2. Molecular function GO categories.
| Male-specific, n
|
Female-specific, n
|
Both sexes, n
|
||||
|---|---|---|---|---|---|---|
| GO category | F > S | S > F | F > S | S > F | F > S | S > F |
| Transcription regulator activity | 13 | 6 | 73 | 16 | 25 | 41 |
| (1.24 × 10−4) | (7.31 × 10−3) | (1.44 × 10−1) | (6.79 × 10−3) | (1.68 × 10−2) | (5.31 × 10−3) | |
| Enzyme regulator activity | 13 | 4 | 32 | 8 | 9 | 22 |
| (8.49 × 10−1) | (2.86 × 10−1) | (1.67 × 10−1) | (2.07 × 10−1) | (1.14 × 10−1) | (5.36 × 10−1) | |
| Signal transducer activity | 33 | 10 | 41 | 32 | 20 | 68 |
| (1.72 × 10−1) | (2.04 × 10−1) | (6.51 × 10−6) | (4.03 × 10−1) | (8.66 × 10−6) | (3.96 × 10−1) | |
| Translation regulator activity | 2 | 1 | 10 | 1 | 5 | 1 |
| (4.31 × 10−1) | (6.00 × 10−1) | (1.32 × 10−1) | (2.28 × 10−1) | (6.08 × 10−1) | (4.25 × 10−1) | |
| Binding | 68 | 33 | 257 | 84 | 74 | 139 |
| (1.18 × 10−4) | (8.76 × 10−3) | (1.43 × 10−12) | (5.99 × 10−1) | (1.13 × 10−6) | (1.26 × 10−4) | |
| Antioxidant activity | 2 | 2 | 1 | 1 | 0 | 5 |
| (4.95 × 10−1) | (1.28 × 10−1) | (3.64 × 10−1) | (9.07 × 10−1) | (NA) | (1.05 × 10−1) | |
| Catalytic activity | 169 | 72 | 250 | 146 | 224 | 278 |
| (1.69 × 10−5) | (2.36 × 10−1) | (9.40 × 10−1) | (7.63 × 10−4) | (1.71 × 10−14) | (1.86 × 10−3) | |
| Structural molecule activity | 15 | 13 | 40 | 31 | 9 | 60 |
| (7.90 × 10−3) | (7.89 × 10−1) | (7.43 × 10−2) | (2.50 × 10−1) | (5.27 × 10−6) | (2.31 × 10−1) | |
| Motor activity | 2 | 1 | 5 | 3 | 2 | 3 |
| (5.54 × 10−1) | (6.98 × 10−1) | (7.96 × 10−1) | (8.44 × 10−1) | (4.06 × 10−1) | (2.72 × 10−1) | |
| Transporter activity | 49 | 28 | 34 | 45 | 51 | 81 |
| (6.28 × 10−2) | (1.87 × 10−2) | (1.75 × 10−6) | (4.65 × 10−2) | (2.44 × 10−1) | (9.00 × 10−2) | |
Numbers are the numbers of up-regulated probe sets in each comparison. P values (in parentheses) are from χ2 tests of departure from expected numbers in each GO category, based on the frequency of probe sets in each category on the GeneChip. Italics denote fewer up-regulated probe sets than expected by chance; bold denotes more up-regulated probe sets than expected by chance. NA, not applicable.
Table 3. Genes with altered transcript abundance in lines selected for increased and decreased copulation latency.
| Trait | Gene* | Comparison | Fold |
|---|---|---|---|
| Olfactory-binding protein | Obp8a | F > S | 1.37 |
| Obp18a | S > F | 1.24 | |
| Obp19c | S ♀ > F ♀ | 1.51 | |
| Obp44a | S ♀ > F ♀ | 1.30 | |
| Obp50b | F ♂ > S ♂ | 1.39 | |
| Obp50c | F ♂ > S ♂ | 1.39 | |
| Obp51a | F ♂ > S ♂ | 1.28 | |
| Obp56a | S ♀ > F ♀ | 1.94 | |
| Obp56d | S ♀ > F ♀, F ♂ > S ♂ | 1.13, 1.25 | |
| Obp57a | S > F | 1.37 | |
| Obp57b | F > S | 1.39 | |
| Obp57c | S ♀ > F ♀ | 1.18 | |
| Obp83c | S ♀ > F ♀ | 1.75 | |
| Obp99b | S > F | 2.07 | |
| Obp99c | F > S | 1.09 | |
| Circadian rhythm | Pka-R2 | F ♀ > S ♀ | 1.10 |
| Cry | F ♂ > S ♂ | 1.20 | |
| Clk | F ♂ > S ♂ | 1.19 | |
| sgg | F ♂ > S ♂ | 1.19 | |
| tim | S > F | 1.48 | |
| S > F | 1.28 | ||
| Larval locomotion | sbb | S > F | 1.30 |
| for | S > F | 1.07 | |
| Learning and memory | Fas2 | S ♀ > F ♀ | 1.18 |
| Pka-R1 | S ♀ > F ♀ | 1.11 | |
| pum | F ♀ > S ♀ | 1.14 | |
| Olfaction | Van | F ♀ > S ♀ | 1.26 |
| Neurogenesis | pbl | F ♀ > S ♀ | 1.07 |
| stc | F ♀ > S ♀ | 1.04 | |
| Iola | S ♀ > F ♀ | 1.15 | |
| Ras85D | F ♀ > S ♀ | 1.11 | |
| robo | S ♀ > F ♀ | 1.35 | |
| Dl | F ♀ > S ♀ | 1.25 | |
| disco | S ♀ > F ♀ | 1.73 | |
| ab | S > F | 1.31 | |
| aay | S > F | 1.06 | |
| dlg1 | S > F | 1.17 | |
| sktl | S > F | 1.20 | |
| dally | S > F | 1.23 | |
| pnt | F > S | 1.51 | |
| elav | S > F | 1.35 | |
| numb | S > F | 1.43 | |
| cpo | S > F | 1.45 | |
| Catecholamine metabolism | Dat | S ♀ > F ♀ | 1.10 |
| Regulation of insulin receptor pathway | foxo | F ♂ > S ♂ | 1.28 |
| Hsp90 chaperone, stress response | Hsp83 | F ♂ > S ♂ | 1.14 |
| Protein folding, stress response | Hsp27 | F ♀ > S ♀ | 1.14 |
| Tryptophan synthesis serotonin metabolism | Hn | F ♂ > S ♂ | 1.12 |
| Tyrosine metabolism, defense response | Bc | S ♀ > F ♀ | 1.33 |
| Specification of segmental identity | tsh | S > F | 1.45 |
| Female gonad development | fz2 | S > F | 1.29 |
| Cell proliferation | I(2)gl | S > F | 1.29 |
See ref. 76 for full gene names and descriptions.
We can begin to build a picture of the transcriptional response to artificial selection by examining GO categories that are overrepresented in the various comparisons of selection lines (Tables 6 and 7, which are published as supporting information on the PNAS web site). Probe sets that are up-regulated in Fast relative to Slow females fall more often than expected in the biological processes categories of cell growth and maintenance (P = 1.55 × 10-7), oocyte maturation (P = 6.03 × 10-7), chromatin silencing (P = 7.50 × 10-9), sexual reproduction (P = 5.44 × 10-7), gene silencing (P = 2.63 × 10-9), RNA metabolism (P = 2.12 × 10-14), DNA metabolism (P = 1.66 × 10-26), and transcription (P = 1.73 × 10-4) and the molecular function categories of histone binding (P = 4.55 × 10-5), DNA replication origin binding (P = 1.44 × 10-23), chromatin binding (P = 9.45 × 10-14), RNA binding (P = 1.70 × 10-20), and helicase activity (P = 7.91 × 10-8). Probe sets involved in neurotransmitter catabolism (P = 3.53 × 10-13) and electron transport (P = 9.02 × 10-7) and that have NADH dehydrogenase activity (P = 1.87 × 10-7) are up-regulated more often than expected in Slow relative to Fast females. Probe sets involved in postmating behavior (P = 6.00 × 10-4), sperm storage (P = 4.67 × 10-7), lipid metabolism (P = 5.77 × 10-5), and defense response (P = 9.71 × 10-3), and that have hydrolase activity (P = 2.53 × 10-4) are up-regulated more often than expected in Fast relative to Slow males. Slow males are distinguished from Fast males by overrepresentation of up-regulated transcripts involved in postmating behavior (P = 1.19 × 10-12), insemination (P = 4.78 × 10-11), sperm displacement (P = 1.11 × 10-12), and steroid metabolism (P = 4.35 × 10-5).
Because 21% of the probe sets on the array are implicated in the transcriptional response to selection, one expects the same fraction of loci in any pathway to be represented by chance. Nevertheless, it is gratifying that transcript levels of many genes that have previously been implicated in mating behavior have been altered by selection. These include several male-specific transcripts and accessory gland proteins (Acp26Aa, Acp26Ab, Acp29AB, Acp36CD, Mst35Bb, Mst57Da, Mst84Dd, and Mst89B) and genes involved in sex determination (doublesex, transformer, transformer 2, and fruitless), circadian rhythm/courtship song (nonA and period), and dopamine metabolism (ebony). In addition, transcript abundance of two of the genes identified by mapping QTLs that cause variation in mating behavior between Oregon and 2b and 18 wheeler and Enhancer of split (67) was also altered between these selection lines. Novel candidate genes affecting mating behavior implicated by changes in transcript abundance between selection lines include 15 of the 39 members of the predicted family of odorant binding proteins; genes involved in circadian rhythm, larval locomotion, learning and memory, and olfactory behavior; and genes involved in neurogenesis (Table 3).
Discussion
We have shown that Drosophila mating speed responds to artificial selection, and that response is largely attributable to an increase in female copulation latency (i.e., a reduction in female receptivity). Thus, there is naturally segregating variation for at least one component of mating behavior. The average divergence in mating speed from generations 25-29 is seemingly large at 113 min but is only 3.5 times the phenotypic standard deviation, which is a rather modest long-term selection response (72). To date, analysis of mating behavior in these lines has been confined to no-choice tests in which each female is paired with a single male. In the future, it will be of considerable interest to conduct choice mating tests to determine whether preference for Slow males has evolved as a correlated response to increased discrimination of Slow females (or vice versa) and to assess correlated responses of fertility, longevity, and other behavioral traits in the selection lines.
The response to selection for mating speed was highly asymmetrical, as is often observed for traits that are major components of fitness (72, 75), including previous studies selecting for divergent mating speed in Drosophila (76-79). Asymmetrical responses of fitness traits to selection are generally attributable to directional dominance and/or genetic asymmetry, such that alleles increasing fitness are at high frequency. Because we did not observe inbreeding depression for mating speed, as would be expected if deleterious alleles were recessive, we infer that the most likely cause of asymmetry was the segregation of low-frequency alleles affecting increased female copulation latency in the base population.
The transcriptional response to selection for mating speed was profound, with >3,700 probe sets (≈21% of the total number on the microarray) exhibiting a divergence in message levels between the Fast and Slow replicate lines, at a stringent false discovery rate of 0.001. In contrast, a previous study of transcriptional response to long-term selection for geotaxis behavior (71) found divergence in message levels for only 5% of the genes assessed. We speculate that this difference is attributable to a difference in criteria for declaring significance: Toma et al. (71) used a 2-fold change threshold, although we used a statistical test. We found that changes in transcript abundance of 10% or even less were often statistically significant.
The chromosomal locations of genes with male-specific changes in expression were nonrandom: the Drosophila X chromosome is depauperate for genes that are up-regulated in males. This is an apparently general phenomenon (80, 81). X chromosome demasculinization is perhaps attributable to selection against genes that are advantageous in males but deleterious to females (80).
The transcriptional response to selection is attributable to genes that have causally responded to selection and that are coregulated by these genes. Because the transcriptional response to single mutations with subtle phenotypic effects can involve >100 coregulated genes (82), the number of selected loci causing the changes in transcript abundance between the selection lines could well be rather modest. It will be necessary to map the QTLs causing divergence between the selection lines to disentangle causal vs. consequential transcriptional responses to selection.
Nevertheless, genes exhibiting parallel changes in transcript abundance between replicate Fast and Slow selected lines are candidate genes affecting mating behavior. Could 21% of the genome really be responsible for regulating mating speed? Recent studies assessing subtle quantitative effects of P element insertional mutations on numbers of sensory bristles (83) and resistance to starvation stress (84) have concluded that >20% of the genome affects each of these traits. These results imply massive pleiotropy: the same genes affect multiple complex traits. Thus, genes regulating mating behavior are as likely to be genes involved in neurogenesis, metabolism, development, and general cellular processes as genes with specific effects on behavior (85). In fact, the same loci may affect multiple behaviors. Pigment dispersing factor (Pdf) and cyrptochrome (cry) were defined based on the involvement in circadian rhythm but were up-regulated in lines selected for positive geotaxis and confirmed to affect geotaxis behavior in functional tests (71). We note that Pdf and cry are also differentially expressed between the Fast and Slow mating speed selection lines, implicating them in mating behavior.
Conclusion
In the future, functional studies will be required to test the extent to which transcript profiling of divergent selection lines accurately predicts genes that directly affect the selected trait. One such test is to assess whether mutations at candidate genes implicated by the analysis of differential transcript abundance affect the trait. A complication here is that mutational effects may be subtle, of the order of naturally occurring variation within and between strains. Many Drosophila mutant stocks have been generated in segregating genetic backgrounds, often containing multiple mutations. It is thus difficult to ascertain whether any difference in the phenotype of a complex trait and a wild-type control is attributable to the mutation in the candidate gene, ancillary mutations, or QTLs affecting the trait segregating between the mutant stock and the wild-type control. Multiple generations of backcrossing the mutation to a common control stock can abrogate this problem (71). A more convincing test is to obtain viable hypomorphic mutations that have been generated in a coisogenic background and compare their effect on the trait to the coisogenic control strain phenotype (83, 84). A subset of the P element insertion lines generated by the Berkeley Drosophila Gene Disruption Project (86) are in coisogenic backgrounds, as is the Exelixis collection of mutations (87). These resources will prove invaluable in testing the predictions of the expression analyses. Another functional test is to perform quantitative complementation tests of mutations at the candidate genes with the selection lines, to assess whether coregulation of transcription translates to epistasis at the level of trait phenotype. Mutations are not available for many of the genes with altered transcript abundance in response to selection (e.g., the odorant-binding proteins). In this case, one can use linkage disequilibrium mapping (88) to assess whether molecular polymorphisms in these candidate genes are associated with naturally occurring variations in mating behavior.
Supplementary Material
Acknowledgments
We are grateful to M. J. Zanis for his perl programming skills, which helped greatly in the organization of our statistical analyses. This work was supported by National Institutes of Health Grants F32 GM66603 (to T.J.M.), R01 GM 58260, R01 GM 45146, and P01 45344 (to T.F.C.M.). This is a publication of the W. M. Keck Center for Behavioral Biology.
This paper results from the Arthur M. Sackler Colloquium of the National Academy of Sciences, “Systematics and the Origin of Species: On Ernst Mayr's 100th Anniversary,” held December 16-18, 2004, at the Arnold and Mabel Beckman Center of the National Academies of Science and Engineering in Irvine, CA.
Abbreviations: QTLs, quantitative trait loci; Z, Zimbabwe; GO, Gene Ontology.
References
- 1.Mayr, E. (1942) Systematics and the Origin of Species (Columbia Univ. Press, New York).
- 2.Orr, H. A. (2005) Proc. Natl. Acad. Sci. USA 102, 6522-6526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Anholt, R. R. H & Mackay, T. F. C. (2004) Nat. Rev. Genet. 5, 838-849. [DOI] [PubMed] [Google Scholar]
- 4.Hall, J. C. (1994) Science 264, 1702-1714. [DOI] [PubMed] [Google Scholar]
- 5.Greenspan, R. J. (1995) Sci. Am. 272 (4), 72-78. [DOI] [PubMed] [Google Scholar]
- 6.Heifetz, Y., Lung, O., Frongillo, E. A. & Wolfner, M. F. (2000) Curr. Biol. 10, 99-102. [DOI] [PubMed] [Google Scholar]
- 7.Clark, A. G., Aguade, M., Prout, T., Harshman, L. G. & Langley, C. H. (1995) Genetics 139, 189-201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wolfner, M. F., Harada, H. A., Bertram, M. J., Stelick, T. J., Kraus, K. W., Kalb, J. M., Lung, Y. O., Neubaum, D. M., Park, M. & Tram, U. (1997) Insect. Biochem. Mol. Biol. 27, 825-834. [DOI] [PubMed] [Google Scholar]
- 9.Chapman, T., Liddle, L. F., Kalb, J. M., Wolfner, M. F. & Partridge, L. (1995) Nature 373, 241-244. [DOI] [PubMed] [Google Scholar]
- 10.Rice, W. R., Linder, J. E., Friberg, U., Lew, T. A., Morrow, E. H. & Stewart, A. D. (2005) Proc. Natl. Acad. Sci. USA 102, 6527-6534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hall, J. C., Siegel, R. W., Tomkins, L. & Kyriacou, C. P. (1980) Stadler Genet. Symp. 12, 43-82. [Google Scholar]
- 12.Yamamoto D. & Nakano, Y. (1998) Biochem. Biophys. Res. Commun. 246, 1-6. [DOI] [PubMed] [Google Scholar]
- 13.Sturtevant, A. H. (1915) J. Anim. Behav. 5, 351-366. [Google Scholar]
- 14.Zhang, S. D. & Odenwald, W. F. (1995) Proc. Natl. Acad. Sci. USA 92, 5525-5529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Peixoto, A. & Hall, J. C. (1998) Genetics 148, 827-838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Joiner, M. A. & Griffith, L. C. (1997) J. Neurosci. 17, 9384-9391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kyriacou, C. P & Hall, J. C. (1985) Nature 314, 171-173. [DOI] [PubMed] [Google Scholar]
- 18.Kyriacou, C. P. (1990) Behav. Genet. 20, 191-211. [DOI] [PubMed] [Google Scholar]
- 19.Gailey, D. A., Jackson, F. R. & Siegel, R. W. (1984) Genetics 106, 613-623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kyriacou, C. P. & Hall, J. C. (1984) Nature 308, 62-65. [DOI] [PubMed] [Google Scholar]
- 21.Gailey, D. A., Jackson, F. R. & Siegel, R. W. (1982) Genetics 102, 771-782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Siegel, R. W. & Hall, J. C. (1979) Proc. Natl. Acad. Sci. USA 76, 3430-3434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ackerman, S. L. & Seigal, R. W. (1986) J. Neurogenet. 3, 111-123. [DOI] [PubMed] [Google Scholar]
- 24.Jackson, F. R., Gailey, D. A. & Siegel, R. W. (1983) J. Comp. Physiol. 151, 545-552. [Google Scholar]
- 25.Ewing, A. W. (1988) Anim. Behav. 36, 1091-1097. [Google Scholar]
- 26.Kyriacou, C. P. & Hall, J. C. (1994) Adv. Genet. 31, 139-186. [DOI] [PubMed] [Google Scholar]
- 27.Tempel, B. L., Livingstone, M. S. & Quinn, W. G. (1984) Proc. Natl. Acad. Sci. USA 81, 3577-3581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Buchner, E. (1991) J. Neurogenet. 7, 153-192. [DOI] [PubMed] [Google Scholar]
- 29.Neckameyer, W. S. (1998) J. Neurogenet. 12, 101-114. [DOI] [PubMed] [Google Scholar]
- 30.Cook, R. (1980) Biol. Cybern. 37, 41-51. [Google Scholar]
- 31.Tompkins, L., Gross, A. C., Hall, J. C., Gailey, D. A. & Siegel, R. W. (1982) Behav. Genet. 12, 295-307. [DOI] [PubMed] [Google Scholar]
- 32.Rendel, J. M. (1951) Evolution (Lawrence, Kans.) 5, 226-230. [Google Scholar]
- 33.Crossley, S. & Zuill, E. (1970) Nature 225, 1064-1065. [DOI] [PubMed] [Google Scholar]
- 34.Kyriacou, C. P., Burnet, B. & Connolly, K. (1978) Anim. Behav. 26, 1195-1206. [Google Scholar]
- 35.Jallon, J.-M. (1984) Behav. Genet. 14, 441-477. [DOI] [PubMed] [Google Scholar]
- 36.Villella, A. & Hall, J. C. (1996) Genetics 143, 331-344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Arthur, B. I., Jallon, J.-M., Caflisch, B., Choffat, Y. & Nothiger, R. (1998) Curr. Biol. 8, 1187-1190. [DOI] [PubMed] [Google Scholar]
- 38.Bernstein, A. S., Neumann, E. & Hall, J. C. (1992) J. Insect. Behav. 5, 15-36. [Google Scholar]
- 39.Taylor, B. J., Villella, A., Ryner, L. C., Baker, B. S. & Hall, J. C. (1994) Dev. Genet. 15, 275-296. [DOI] [PubMed] [Google Scholar]
- 40.O'Dell, K. M. & Kaiser, K. (1995) Neuron 15, 55-61. [DOI] [PubMed] [Google Scholar]
- 41.Ferveur, J.-F., Savarit, F., O'Kane, C. J., Sureau, G., Greenspan, R. J. & Jallon, J.-M. (1997) Science 276, 1555-1558. [DOI] [PubMed] [Google Scholar]
- 42.Finley, K. D., Edeen, P. T. Foss, M., Gross, E., Ghbeish, N., Palmer, R. H., Taylor, B. J. & McKeown, M. (1998) Neuron 21, 1363-1374. [DOI] [PubMed] [Google Scholar]
- 43.Waterbury, J. A., Jackson, L. L. & Schedl, P. (1999) Genetics 152, 1653-1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wheeler, D. A., Kulkarni, S. J., Gailey, D. A. & Hall, J. C. (1989) Behav. Genet. 19, 503-528. [DOI] [PubMed] [Google Scholar]
- 45.Gailey, D. A., Taylor, B. J. & Hall, J. C. (1991) Development (Cambridge, U.K.) 113, 879-890. [DOI] [PubMed] [Google Scholar]
- 46.Ryner, L. C., Goodwin, S. F., Castrillon, D. H., Anand, A., Villella, A., Baker, B. S., Hall, J. C., Taylor, B. J. & Wasserman, S. A. (1996) Cell 87, 1079-1089. [DOI] [PubMed] [Google Scholar]
- 47.Baker, B. S., Taylor, B. J. & Hall, J. C. (2001) Cell 105, 13-24. [DOI] [PubMed] [Google Scholar]
- 48.Tompkins, L. & McRobert, S. P. (1995) J. Neurogenet. 9, 219-226. [DOI] [PubMed] [Google Scholar]
- 49.Takahashi, A., Tsaur, S.-C., Coyne, J. A. & Wu, C.-I. (2001) Proc. Natl. Acad. Sci. USA 98, 3920-3925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fleishmann, I., Dauwalder, B., Chapman, T., Cotton, B. & Kubli, E. (1995) J. Neurogenet. 10, 26-27. [Google Scholar]
- 51.Nakayama, S., Kaiser, K. & Aigaki, T. (1997) Mol. Gen. Genet. 254, 449-455. [DOI] [PubMed] [Google Scholar]
- 52.Lung, Y. O. & Wolfner, M. F. (2001) Insect. Biochem. Mol. Biol. 31, 543-551. [DOI] [PubMed] [Google Scholar]
- 53.Mackay, T. F. C. (2001) Annu. Rev. Genet. 35, 303-339. [DOI] [PubMed] [Google Scholar]
- 54.Zouros, E. (1981) Genetics 97, 703-718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Coyne, J. A. (1989) Proc. Natl. Acad. Sci. USA 86, 5464-5468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Coyne, J. A. (1993) Evolution (Lawrence, Kans.) 47, 778-788. [DOI] [PubMed] [Google Scholar]
- 57.Coyne, J. A. (1996) Genetics 143, 353-364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Coyne, J. A. (1996) Genet. Res. 68, 211-220. [DOI] [PubMed] [Google Scholar]
- 59.Noor, M. A. F. (1997) Evolution (Lawrence, Kans.) 51, 809-815. [DOI] [PubMed] [Google Scholar]
- 60.Coyne, J. A., Kim S. Y., Chang, A. S., Lachaise, D. & Elwyn, S. (2002) Evol. Int. J. Org. Evol. 56, 2424-2434. [DOI] [PubMed] [Google Scholar]
- 61.Noor, M. A. F., Grams, K. L., Bertucci, L. A., Almendarez, Y., Reiland, J. & Smith, K. R. (2001) Evolution (Lawrence, Kans.) 55, 512-521. [DOI] [PubMed] [Google Scholar]
- 62.Moehring, A. J., Li, J., Schug, M. D., Smith, S. G., DeAngelis M., Mackay, T. F. C. & Coyne, J. A. (2004) Genetics 167, 1265-1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Speith, H. T. & Ringo, J. M. (1983). in The Genetics and Biology of Drosophila, eds. Ashburner, M., Carson, H. L. & Thompson, J. N. (Academic, London), Vol. 3c, pp. 223-284. [Google Scholar]
- 64.Wu, C.-I., Hollocher, H., Begun, D. J., Aquadro, C. F., Xu, Y. & Wu, M.-L. (1995) Proc. Natl. Acad. Sci. USA 92, 2519-2523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hollocher, H., Ting, C.-T., Wu, M.-L. & Wu, C.-I. (1997) Genetics 147, 1191-1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ting, C.-T., Takahashi, A. & Wu, C.-I. (2001) Proc. Natl. Acad. Sci. USA 98, 6709-6713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Moehring, A. J. & Mackay, T. F. C. (2004) Genetics 167, 1249-1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Pasyukova, E. G., Vieira, C. & Mackay, T. F. C. (2000) Genetics 156, 1129-1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Long, A. D., Mullaney, S. L., Mackay, T. F. C. & Langley, C. H. (1996) Genetics 144, 1497-1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Mackay, T. F. C. & Fry, J. D. (1996) Genetics 144, 671-688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Toma, D. P., White, K. P., Hirsch, J. & Greenspan, R. J. (2002) Nat. Genet. 31, 349-353. [DOI] [PubMed] [Google Scholar]
- 72.Falconer, D. S. & Mackay, T. F. C. (1996) Introduction to Quantitative Genetics (Addison-Wesley-Longman, Harlow, Essex, U.K.), 4th Ed.
- 73.Storey, J. D. & Tibshirani, R. (2003) Proc. Natl. Acad. Sci. USA 100, 9440-9445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.The FlyBase Consortium (2003) Nucleic Acids Res. 31, 172-175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Frankham, R. (1990) Genet. Res. 56, 35-42. [Google Scholar]
- 76.Manning, A. (1961) Anim. Behav. 9, 82-92. [Google Scholar]
- 77.Manning, A. (1963) Anim. Behav. 11, 116-120. [Google Scholar]
- 78.Sherwin, R. N. (1975) Evolution (Lawrence, Kans.) 29, 519-530. [DOI] [PubMed] [Google Scholar]
- 79.Spuhler, K. P., Crumpacker, D. W., Williams J. S. & Bradley, B. P. (1978) Genetics 89, 729-749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Parisi, M., Nuttall, R., Naiman, D., Bouffard, G., Malley, J., Andrews, J., Eastman, S. & Oliver, B. (2003) Science 299, 697-700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Rantz, J. M., Castillo-Davis, C. I., Meiklejohn, C. D. & Hartl, D. L. (2003) Science 300, 1742-1745. [DOI] [PubMed] [Google Scholar]
- 82.Anholt, R. R. H., Dilda, C. L., Chang, S., Fanara, J. J., Kulkarni, N. H., Ganguly, I., Rollmann, S. M., Kamdar, K. P. & Mackay, T. F. C. (2003) Nat. Genet. 35, 180-184. [DOI] [PubMed] [Google Scholar]
- 83.Norga, K. K., Gurganus, M. C., Dilda, C. L., Yamamoto, A., Lyman, R. F., Patel, P. H., Rubin, G. M., Hoskins, R. A., Mackay, T. F. C. & Bellen, H. J. (2003) Curr. Biol. 13, 1388-1397. [DOI] [PubMed] [Google Scholar]
- 84.Harbison, S. T., Yamamoto, A. H., Fanara, J. J., Norga, K. K. & Mackay, T. F. C. (2004) Genetics 166, 1807-1823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Sokolowski, M. B. (2001) Nat. Rev. Genet. 2, 879-890. [DOI] [PubMed] [Google Scholar]
- 86.Bellen, H. J., Levis, R. W., Liao, G., He, Y., Carlson, J. W., Tsang, G., Evans-Holm, M., Hiesinger, R, Schultze, K, Rubin, G. M., et al. (2004) Genetics 167, 761-781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Parks, A. L., Cook, K. R., Belvin, M., Dompe, N. A., Fawcett, R., Huppert, K., Tan, L. R., Winter, C. G., Bogart, K. P., Deal, J. E., et al. (2004) Nat. Genet. 36, 288-292. [DOI] [PubMed] [Google Scholar]
- 88.Risch, N. & Merikangas, K. (1996) Science 273, 1516-1517. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.