

Abstract
Measurement of overall survival (OS) remains the gold standard for interpreting the impact of new therapies for multiple myeloma in phase 3 trials. However, as outcomes have improved, it is increasingly challenging to use OS as the primary endpoint if timely approval of novel agents is to be ensured to enable maximum benefit for patients. Surrogate endpoints of OS, such as progression-free survival (PFS) and response to treatment, have contributed to approval decisions by the Food and Drug Administration (FDA) and European Medicines Agency as endpoints demonstrating clinical benefit, and the FDA has recently supported the use of minimal residual disease (MRD) as an accelerated approval endpoint in multiple myeloma. This review aims to address situations in which the use of PFS as a surrogate endpoint warrants careful interpretation especially for specific subgroups of patients and considers ways to ensure that studies can be designed to account for possible discordance between PFS and OS. The utility of subgroup analyses, including the potential for those not pre-specified, to identify target populations for new agents is also discussed.
Subject terms: Myeloma, Myeloma
Introduction
The overarching goal in improving treatment outcomes in cancer is to reduce symptom burden and prolong the length and quality of life (QoL) while limiting treatment-related toxicity. While measurement of overall survival (OS) remains the gold standard for interpreting the impact of new therapies for multiple myeloma in phase 3 clinical trials, as outcomes have improved, it has become increasingly challenging to wait for this endpoint to keep pace with the rapid progress being achieved in the field. Median OS, when short, can be compared directly between interventions. However, with improved myeloma therapies and increased survival duration, it is increasingly difficult to use OS as the primary endpoint to ensure timely approval of novel therapies for the benefit of patients. This difficulty is also reflective of the use of study designs that incorporate crossover after relapse and successful scientific advances over time, which for myeloma have consistently provided effective approved agents that have impacted outcomes after relapse both in and outside of clinical trials, ultimately resulting in greater reliance on surrogate endpoints of OS to provide reliable and validated indicators of clinical benefit of novel interventions.
Surrogate endpoints are defined by the National Institutes of Health as “biomarkers intended to substitute for a clinical endpoint” [1]. For surrogate endpoints to be used in the context of drug approval, the United States Food and Drug Administration (FDA) mandates that “clinical trials are needed to show that the surrogate endpoint can be relied upon to predict or correlate with clinical benefit in a context of use” [2]. Progression-free survival (PFS), defined as time from start of therapy to disease progression or death, has been used in this way by both the FDA and the European Medicines Agency (EMA) in many drug approvals to date [3, 4]. Second progression-free survival (PFS2), defined as time to second disease progression or death, has been used additionally to account for the impact of a drug/intervention on potential resistance after relapse [3]. Earlier surrogate endpoints predictive of long-term outcomes such as OS, including minimal residual disease (MRD) negativity, are increasingly used as research endpoints [5–7] but have previously been more challenging to regulatory agencies as primary endpoints of clinical trials [8]. The Oncologic Drugs Advisory Committee of the FDA has recently advised that current evidence supports the use of MRD as an accelerated approval endpoint in myeloma clinical trials [9]. This paves the way for accelerated approval based on MRD as an endpoint but with a requirement for ongoing follow-up and PFS/OS confirmation.
The aim of this review is to explore the current use of PFS as a surrogate endpoint for OS in myeloma, examine situations when this has not been reliable, and discuss ways to ensure that studies are planned to account for these situations, or at least mitigate their effect. This will improve the development of optimal study designs moving forward and increase acceptance of other surrogate endpoints such as MRD, while improving our ability to confidently establish the efficacy of novel agents/interventions in the treatment of patients with myeloma and rapidly translating this into approvals for patient access [10].
PFS is an accepted surrogate endpoint in myeloma
There are many examples of myeloma trials in which a demonstrable PFS benefit is associated with a similar benefit in OS [11–15]. Based on such data, PFS as a surrogate endpoint has contributed to many approval decisions by the EMA and FDA. Recent examples include the MAIA trial, which compared the combination of daratumumab, lenalidomide, and dexamethasone (DRd) to lenalidomide and dexamethasone (Rd) in patients with newly diagnosed myeloma (NDMM) considered ineligible for autologous stem cell transplant (ASCT). The trial showed a PFS benefit with DRd compared to Rd (hazard ratio [HR] 0.55 [95% CI, 0.45–0.67]; p < 0.0001) and an OS benefit (HR 0.66 [95% CI, 0.53–0.83]; p = 0.0003). The OS findings were consistent across subgroups, with the exception of patients with impaired baseline hepatic function, which favored Rd (HR 1.29 [95% CI, 0.64–2.60]), although patient numbers (DRd, n = 31; Rd, n = 29) were small [11]. Similar findings have been observed with combinations of agents from all anti-myeloma drug classes, in both transplant-eligible and -ineligible populations, as well as newly diagnosed and relapsed patient populations. These include the SWOG 0777 study that compared lenalidomide, bortezomib, and dexamethasone (RVd) to Rd in transplant-ineligible patients with NDMM and demonstrated that PFS benefit was accompanied by OS benefit [12], building on the efficacy shown for this combination in both transplant-eligible and -ineligible NDMM patients [13]. Similarly, in the setting of relapse, there are numerous examples, including the POLLUX (comparing DRd vs Rd) [14] and ASPIRE trials (comparing carfilzomib with Rd vs Rd) [15], where PFS and OS were consistently superior for the triplet vs the doublet. However, there are several recent studies where PFS does not act as a surrogate for OS, and the validity of using it as a surrogate endpoint has been questioned [16]. In some cases, adequately powering a study to identify an OS difference may be challenging due to sample size constraints, unless a large magnitude of effect is expected and may not be included as a co-primary endpoint. Exploring other reasons for lack of this translation and how this discrepancy might be addressed will help advance myeloma therapies at a greater pace by ensuring use of the earlier, more rapidly interpretable endpoints whilst preserving the validity of OS in reflecting improved outcomes overall.
Heterogeneity in populations can affect the ability to translate a PFS benefit to OS benefit
Recent examples of clinical trials in which a significant PFS benefit has not translated into an OS benefit for all patients has occurred in situations where specific subgroups have heterogeneous outcomes with respect to the endpoint. In some of these, subgroup heterogeneity has led to an apparent lack of OS benefit or even a detrimental effect on OS in the study population; however, subgroup analyses have led to the identification of groups that may benefit from the treatment being studied [17–19]. Important examples of this include differences based on heterogeneity by molecular subgroups, prior ASCT, and age.
Molecular heterogeneity
The BELLINI trial demonstrated a significant PFS benefit with the addition of the BCL-2 inhibitor venetoclax to bortezomib and dexamethasone (HR 0.63 [95% CI, 0.44–0.90]) but with a significantly worse OS (HR 2.03 [95% CI, 1.04–3.95]) in patients with relapsed/refractory myeloma (RRMM) [20]. This led to a partial clinical hold being imposed by the FDA on all venetoclax trials. However, on further analysis of the trial, there was evidence of significant heterogeneity in OS outcomes between patients with t(11;14) or high BCL2 expression (OS: HR 0.82 [95% CI, 0.40–1.70]) and those with no t(11;14) or low BCL2 expression (OS: HR 1.34 [95% CI, 0.81–2.20]), which likely accounted for the discordance between PFS and OS in the study population [18]. This heterogeneity was also evident in the PFS analysis (Fig. 1A). In those patients without t(11:14) and/or high BCL2 expression and lack of benefit from the use of venetoclax, additional toxicity may have led to the worsening in OS. Across the study, adverse events led to 12 deaths in the venetoclax arm (the majority due to infection) and only one in the placebo arm [18].
Fig. 1. Heterogeneity in trial outcomes by molecular status and age.

A Heterogeneity by t(11:14) and BCL2 expression status in the BELLINI trial [18]. Heterogeneity in PFS and OS outcomes were seen between patients with t(11;14) or high BCL2 expression and those with no t(11;14) or low BCL2 expression. B Heterogeneity by age in the OCEAN trial [17]. Heterogeneity by age was observed for both PFS (p = 0.033het) and OS (p = 0.006het). Dex dexamethasone, mel melflufen, pom pomalidomide, pts patients, Vd bortezomib and dexamethasone, VenVd venetoclax, bortezomib, and dexamethasone.
This effect of heterogeneity between molecular subgroups was also identified in pre-clinical [21, 22] and early-phase studies [23]. However, the BELLINI trial was intentionally not restricted to the t(11;14) subtype of myeloma based on the premise that combination with dexamethasone [24] and bortezomib [25] could increase BCL-2 dependency even in patients without t(11;14) or high BCL2 expression, and therefore, all myeloma patients rather than a subset may benefit [26]. In phase 1b study investigating venetoclax, bortezomib, and dexamethasone in patients with RRMM, there were markedly improved responses in patients with high BCL2 expression compared to those with low BCL2 expression (94% vs 59%) [27], although it required the results of the phase 3 BELLINI study to demonstrate the negative interaction between PFS and OS outcomes in the non-t(11;14), low BCL2 subgroups [20, 28].
Designing the BELLINI study with recruitment of only patients with t(11;14) and/or high BCL2 expression or adequately powering this subgroup within the all-comers study may, in retrospect, have prevented the significant pause in development following the trial results and arguably led to the approval of venetoclax. However, the feasibility of recruiting the patient numbers required to achieve this was considered potentially prohibitive and reflects the reality of conducting clinical trials of selected subgroups within a meaningful time frame. Importantly, investigation of venetoclax, although delayed, was subsequently reinitiated. The phase 3 CANOVA study was designed to evaluate the safety and efficacy of venetoclax plus dexamethasone (Ven/dex) compared with pomalidomide plus dexamethasone (Pom/dex) in patients with RRMM and with t(11;14)-positive disease only, but did not meet its primary endpoint of PFS [29]. However, a post hoc sensitivity analysis of CANOVA that counted the start of a new line of anti-myeloma therapy as a PFS event (rather than being censored) demonstrated a significant PFS benefit with Ven/dex vs Pom/dex (HR 0.651 [95% CI, 0.487–0.870; p = 0.003). Of note, there was no significant difference in OS in this analysis although a trend in favor of Ven/dex was seen (Ven/dex: 32.4 months vs Pom/dex: 24.5 months; HR 0.697 [95% CI, 0.472–1.029]; p = 0.067) [29].
Importantly, this heterogeneity has not been observed in all studies, as it is usually determined by the degree of molecular targeting of the agent under study. During development of a new agent, it is therefore key to study molecular heterogeneity through allied translational research programmes to identify and validate potential populations that may benefit most. The results of such subgroup analyses would enable the definition of a “target population(s)” for specific therapies prior to the phase 3 trial design, which could benefit timely drug development. This suggested approach would be preferable to all-comers trials followed by subgroup analysis, which nonetheless may have validity, especially if these analyses are planned a priori, but may take longer, require a larger sample size, and may require further validation as part of the drug approval process.
Age-related heterogeneity
Heterogeneity in OS outcomes by age has recently been highlighted by results of the phase 3 OCEAN trial, where there was a significant PFS advantage with the novel peptide-drug conjugate melflufen and dexamethasone combination (melflufen/dex) compared to Pom/dex (HR 0.79 [95% CI, 0.64–0.98; p = 0.032]) in the overall population of patients with RRMM [17]. However, this improvement in PFS did not translate to an OS benefit (HR 1.10 [95% CI, 0.85–1.44; p = 0.47]) [17]. Examination of subgroups revealed evidence of heterogeneity by age for both PFS (phet = 0.033) and OS (phet = 0.006; Fig. 1B). There was also an apparent detriment to OS with melflufen/dex in patients <65 years (HR 1.71 [95% CI, 1.09–2.69]) compared to that with Pom/dex but a significant benefit in patients >75 years (HR 0.46 [95% CI, 0.23–0.92]) [17]. Whether this is related solely to age or to differences in prior treatment is of interest and may be a complex interaction of both, as there was also significant heterogeneity by previous ASCT with the use of high-dose alkylation as conditioning.
Heterogeneity by age was also seen in the Myeloma XI trial comparing lenalidomide maintenance to observation in patients of all ages [30]. The randomization of the lenalidomide and observation arms was conducted in transplant-eligible and -ineligible patients enrolled in the trial, with pre-planned analysis in both. Patients in the transplant-eligible pathway had a median age of 61 years vs 74 years for those in the transplant-ineligible pathway [31, 32]. In contrast to the OCEAN study, there was no heterogeneity by transplant eligibility for PFS. Overall, there was significant PFS benefit (HR 0.46 [95% CI, 0.41–0.53]; p < 0.0001), consistent in both pathways of the trial (transplant-eligible patients: HR 0.48 [95% CI, 0.40–0.58], p < 0.0001; transplant-ineligible patients: HR 0.44 [95% CI, 0.37–0.53], p < 0.0001). However, for OS, there was significant heterogeneity between pathways (transplant-eligible patients: HR 0.69 [95% CI, 0.52–0.93], p = 0.014; transplant-ineligible patients: HR 1.02 [95% CI, 0.80–1.29], phet = 0.0445) demonstrating a translation of PFS benefit to OS for younger and fitter patients, but not for older and/or less fit patients.
Whilst there are key differences between the OCEAN and Myeloma XI trials, including enrollment of RRMM patients in OCEAN compared to NDMM patients in Myeloma XI, taken together, these studies trigger the hypothesis that heterogeneity could be associated with reduced impact on OS for immunomodulatory drug (IMiD) use in older patients, compared to younger patients, perhaps from differences in immune effects and innate immune exhaustion associated with advancing age, and other potential confounders such as vascular health.
To better understand these differences, we examined heterogeneity by age groups seen within and between recently published phase 3 clinical trials in myeloma reporting PFS and OS (Supplementary Methods). To compare the PFS and OS impact from different classes of anti-myeloma therapies, only trials where the effect of specific agents belonging to one of the three major anti-myeloma drug classes (anti-CD38, proteasome inhibitors [PIs], or IMiDs) could be isolated were included. This included “add-on” trials of an agent or where it was compared to an inactive control (observation or placebo) but not a different class of agent. For example, where A is the agent in question, trials of AB vs B, ABC vs BC, and A vs observation/placebo were included while trials of A vs C or AC vs BC were not.
This search strategy identified 20 trials that were included in the analysis (Supplementary Fig. 1; Supplementary Table 1); of these, five isolated an anti-CD38 antibody effect, eight an IMiD effect, and seven a PI effect. Median age across all trials was 66 years (range, 59–73). In total, 19 of 20 trials reported significant PFS benefit in the overall population (five of five evaluating anti-CD38 antibodies, eight of eight evaluating IMiDs, and six of seven evaluating PIs), but only 10 of 20 trials reported significant OS benefit (four of five evaluating anti-CD38 antibodies, three of eight evaluating IMiDs, and three of seven evaluating PIs).
When adjusting for individual drug class effect, there was no evidence of a relationship between the HR and age for PFS (p = 0.607; Fig. 2A). For OS, however, there was a significant negative relationship between HR and median age of patients included in trials (p = 0.022). Notably, this relationship was observed for IMiDs and PIs, but not anti-CD38 antibodies (p = 0.017; Fig. 2B).
Fig. 2. Random effect model of progression-free survival and overall survival across trials isolating the effect of different anti-myeloma agents.
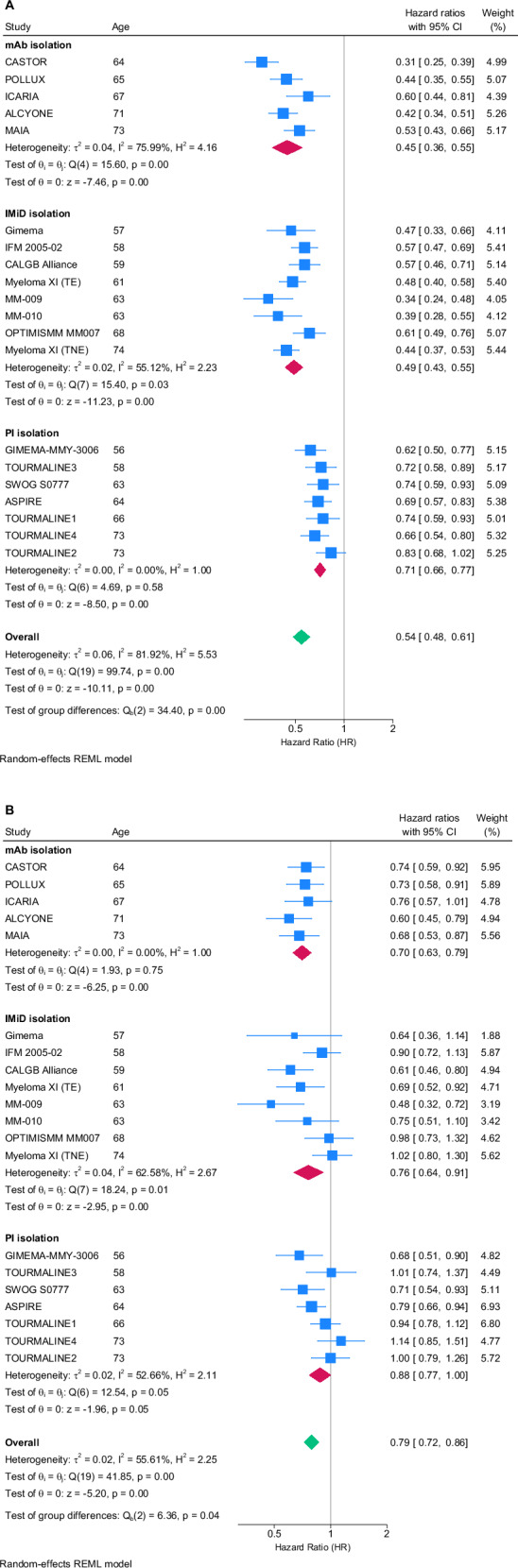
The PFS (A) and OS (B) data were extracted from the most recent publication or presentation related to each trial including hazard ratio (HR) and 95% CI as well as median age of patients enrolled. mAb monoclonal anti-CD38 antibody, IMiD immunomodulatory agent, PI proteasome inhibitor, REML restricted maximum likelihood.
For trials where subgroup analysis of HR for OS by age were reported, these intra-trial outcomes were plotted by age group (Supplementary Fig. 2). Lack of standardization in age groupings reported made the cross-trial analysis challenging. The clearest trend appeared in a prespecified analysis of the ICARIA-MM trial that analyzed OS with isatuximab plus pomalidomide and low-dose dexamethasone vs pomalidomide and low-dose dexamethasone in patients with RRMM, wherein a decrease in the HR was seen with increasing age [33].
On examining the effect of both IMiDs and PIs in this analysis, there appears to be a relationship between median age of participating patients and the HR for OS, but not PFS. It appears to be less likely for a PFS benefit to translate to an OS benefit in both IMiD and PI trials recruiting older patients, suggesting a differential effect of IMiDs and PIs on survival after progression in older patients that is not seen with anti-CD38 antibody therapies. Possible hypothesis for this effect could be an ongoing effect of IMiDs and PIs after relapse or lack of dosing optimization of these agents for older patients, leading to toxicity that continues into subsequent lines of therapy, thus impacting outcomes. Alternatively, both IMiDs and PIs may induce alterations in myeloma clonality and/or in the bone marrow stroma, which in turn may impair the efficacy and/or tolerance of subsequent treatments. Within all trials reported, dosing of the key agents was not adjusted based on age or frailty, which could perhaps have led to different outcomes. This approach of age- or frailty-based dosing is being prospectively studied in the ongoing FiTNEss (Myeloma XIV) trial, which is randomizing ASCT-ineligible patients to receive standard-of-care dosing and reactive dose modification in the event of toxicity or frailty score-adapted dosing (International Myeloma Working Group frailty score) with dose reductions determined by frailty groups [34].
Limitations and confounders of this analysis (Fig. 2) included an inability to examine agents separately within each class due to the limited number of trials. Specifically, within the PI group, there are many trials with ixazomib; however, there are only very few trials studying the addition of bortezomib or carfilzomib that also have OS data at the current time. In the IMiD groups, it is also difficult to interrogate differences between lenalidomide and pomalidomide, and these data do not include any trials of the next-generation cereblon E3 ligase modulatory drug (CELMoD) agents. It is, therefore, possible that the effects identified may not be a class effect but rather, driven by one or more of the individual agents within that class. The included trials also have different lengths of follow-up, which affects both the likelihood of OS data being available and the maturity of data. Importantly, differences in the way age subgroups were reported or lack of age subgroup outcome reporting limited the breadth of the intra-trial analyses.
These findings provide critical considerations for future trial designs, as well as interpretation of current study results. They highlight the critical importance of reporting subgroup analyses by age to ensure these interactions are understood and explored. For future trials, this analysis also raises the question of whether agents should be added in such a way that their individual outcomes can be studied (as in the studies included in our analysis) or used in more head-to-head studies. The latter clearly provides key information about an agent or combination in direct comparison with another, but when conducting head-to-head comparisons, it is important to understand the impact of subgroup heterogeneity based on prior studies such as those presented here that may otherwise be “hidden”. Recent examples of when this might be important include the DREAMM3 trial that compared belantamab mafodotin to Pom/Dex in patients with RRMM [35], which did not meet its primary endpoint of PFS. The data reported to date suggest that there was a subset of patients sensitive to belantamab mafodotin, with a median duration of response for belantamab mafodotin not yet reached vs 8.5 months with Pom/dex, consistent with the idea that heterogeneity in the population being studied may have prevented the overall PFS being significantly different from that of the control group. Similar considerations should be given to reporting other potential surrogate endpoints, with sub-analyses being especially valuable in identifying likely subgroups in which early endpoints are more or less likely to translate into OS benefit. Ongoing studies of belantamab in different populations may shed additional light on its true efficacy and are awaited with great interest.
Of note, in the presence of true heterogeneity of treatment effects, interpretation of the main treatment effect can be complex and may limit interpretation of the overall treatment effect. For example, given that IMiDs and PIs may have heterogeneous OS benefit across age groups (e.g., Myeloma XI trial [30]; Fig. 2), careful interpretation of observed HR heterogeneity in trials where these are used as a comparator is needed. If a treatment has an observed homogeneous HR benefit compared with a heterogeneous comparator, it paradoxically means that the experimental arm may in fact be heterogeneous as well. Similarly, observed heterogeneous HRs in comparison with a heterogenous comparator require caution in terms of interpretation of the treatment benefit of the experimental drug, as seen with ICARIA-MM or OCEAN [17, 33]. Taken together, these analyses further support the importance of evaluating subgroups in determination of clinical benefit.
Other situations where PFS benefit may not translate to OS: the issue of competing risks
The examples discussed above include trials exhibiting heterogeneity in outcomes between subgroups that could be contributing to the lack of translation of PFS benefit to OS benefit. Other trials have shown this lack of translation without distinct subgroup heterogeneity currently identified, key examples being the recent studies of early vs delayed ASCT examined in the EMN02 [36], IFM 2009 [37, 38], and DETERMINATION trials [39]. In the parallel IFM 2009 and DETERMINATION studies, there was a significant PFS benefit associated with early vs delayed ASCT (IFM 2009: HR 0.70 [95% CI, 0.59–0.83; p < 0.001] [38]; DETERMINATION: HR 1.53 [95% CI, 1.23–1.91; p < 0.001]). It should be noted that the HRs are calculated differently, such that they reflect a similar outcome in favor of ASCT in both trials. Despite these impressive PFS differences between early vs delayed ASCT groups in the IFM 2009 trial at 93 months follow-up [38, 40], there was no significant difference in OS (HR 1.03 [95% CI, 0.80–1.32; p = 0.81]). Similarly, in the DETERMINATION trial, at 76 months follow-up, there was no significant difference in OS between RVd plus ASCT vs RVd alone (HR 1.10 [95% CI, 0.73–1.65; p > 0.99]). This could be due to several reasons; of note, neither study was specifically powered to demonstrate an OS benefit, as the primary endpoint was PFS. While it is possible to hypothesize that an OS benefit would have been seen if the studies had been larger, notably, in other similarly sized studies not involving transplant, there has been a correlation between PFS and OS suggesting that transplant may be the confounding effect [11, 12, 41]. An alternative explanation could be that studies in transplant-eligible patients with NDMM now have long median PFS and OS, including the control arms of these studies and therefore, the influence of crossover and other therapeutic advances over time plays a larger role, with salvage therapy in particular proving increasingly effective. To this point, more patients in the IFM 2009 trial received a delayed transplant (77%) than in DETERMINATION (28%), suggesting that beyond crossover to ASCT, other factors such as the remarkable efficacy of next-generation novel agents have a favorable impact on long-term outcomes. In recent years the development of immunotherapies for myeloma including bispecific antibodies and chimeric antigen receptor T-cell therapies has moved at a rapid pace [42, 43]. These treatments challenge the old paradigm that patient remission will shorten with each line of treatment with many inducing deep and durable remissions at later lines of therapy. This confounds the follow-up of overall survival for clinical trials as longer-term outcomes may be driven by access to such therapies as opposed to the original trial randomization. In trials performed across multiple international centers, global heterogeneity in access to subsequent anti-myeloma drugs may also exacerbate this.
Importantly, the longer the median PFS and OS achieved, the greater the potential for competing risks affecting OS outcomes to become apparent. These competing risks may affect subgroups differently and reflect toxicity, side effects, and impact on the disease itself, such as mutational burden and signatures resulting from mutagenic treatments [40] as well as second primary malignancies, specifically, secondary myeloid leukemia and myelodysplastic syndrome [39]. Another example of the issue of competing risks is exemplified by a recent post-hoc analysis of DETERMINATION that explored outcomes with RVd alone vs RVd plus ASCT in African American and White patients which showed that while outcomes with RVd plus ASCT were similar in patients of both races, African American patients with high body mass index and female sex appeared to derive significantly more PFS benefit from RVd alone. This may be related to the Duffy-null genotype, a common variant in people of African and Middle Eastern genetic ancestry, that has a key role in cytokine homeostasis, inflammation and likely, myeloma pathobiology and was present in approximately 60% of African American patients tested to date in the study [44, 45].
Prior studies of transplant have shown consistent PFS with ASCT [46], but OS benefit has been less clear. Interestingly, in the EMN02 study that compared ASCT with bortezomib-melphalan-prednisone in patients with NDMM, ASCT was associated with PFS benefit (HR 0.73 [95% CI, 0.62–0.85]; p = 0.0001) compared with bortezomib-melphalan-prednisone, but there was no significant difference in OS (HR 0.9 [95% CI, 0.71–1.13]; p = 0.35). Conversely, tandem ASCT showed a benefit in both PFS (HR 0.74 [95% CI, 0.56–0.98]; p = 0.036) and OS (HR 0.62 [95% CI 0.41–0.93]; p = 0.022) compared with single ASCT [36]. This suggests that in the context of ASCT, the impact of high-dose melphalan and its effects on both the patient and their myeloma is key in understanding the complexity of these interactions, as well as their combined long-term effect on the outcome for either benefit or detriment in any particular patient through competing risk.
Does lack of translation of PFS benefit to OS benefit matter? Is OS the only important goal?
When examining the circumstances wherein an agent may have substantial PFS benefit without translation to OS, it is important to reflect on the impact of that situation on an individual patient. While prolonging life is of key importance, first remission is often when patients are feeling most well, with the fewest myeloma-related symptoms. Prolonging this time (before relapse) even in the context of no OS benefit may still, therefore, be of benefit to patients. An illustrative example is the transplant-ineligible pathway of the Myeloma XI trial in which the use of lenalidomide maintenance more than doubled median PFS from 11 months to 26 months (HR 0.44 [95% CI, 0.37–0.53]; p < 0.0001) in patients who had not undergone ASCT, but there was no significant OS difference [30]. Prolonging first remission, with only a single anti-myeloma agent rather than combination therapies required at relapse with more hospital attendances, may benefit patients’ QoL. It may also be more cost-effective for patients to continue single-agent maintenance therapy for longer, and thus need less expensive therapies for a shorter duration to achieve the same effect. Although this is an area of active study, recent data suggest that early ASCT may be more expensive than delayed therapy (or being kept in reserve), at least in the jurisdiction of US health care, challenging similar arguments used previously in favor of patients undergoing early vs deferred ASCT [47–50].
The duration of first remission in the DETERMINATION trial was approximately 2 years longer with early vs delayed ASCT, with a median PFS of 67.5 months vs 46.2 months (HR 1.53 [95% CI, 1.23–1.91; p < 0.001]), but remarkably, no significant difference in OS [39]. However, for many patients, this would mean a prolonged time after ASCT when they may have regained a QoL not dissimilar to their pre-diagnosis state, perhaps returning to work, whilst taking oral maintenance therapy. This comes with the key caveat of significant loss of QoL during transplant, which may last for several months [51]. Conversely, there may clearly be patients for whom a delayed transplant is more appropriate, for example, if lifestyle factors favor early return to work after/during induction therapy, with ASCT delayed until first relapse or used later and only if needed. The risk-benefit balance in these scenarios are patient-specific and should therefore be carefully discussed, with personal and disease-related factors tailored to patient preference [52]. These issues highlight the critical importance of incorporating health-related QoL into clinical trial endpoints as well as parallel healthcare resource utilization and health economic analyses within studies to enhance optimal translation into clinical practice and for reimbursement discussions [53]. Efforts to encourage this approach are included in recommendations such as those in the European Society for Medical Oncology-Magnitude of Clinical Benefit Scale [54].
Similar to these arguments, the acceptance of the use of MRD as an endpoint for accelerated approval, does not mean that MRD negativity should be pursued at all costs. Particularly in the older patient population, the balance of efficacy and toxicity is critical and co-primary endpoints combining both should be considered to ensure the achievement of a deep response that does not come at too high a price for the patient.
Conclusions
Whilst PFS continues to be commonly used in FDA and EMA approvals as a surrogate endpoint for OS in myeloma, recent examples highlighted above demonstrate situations in which clinical benefit of PFS does not translate to OS. Understanding why this has occurred can be aided by understanding and utilizing subgroup analyses of clinical trials, potentially even if not pre-specified and new information has come to light, which may be of value to avoid overlooking a group that derives significant benefit from a particular therapy, or missing others for whom the benefit is less. Furthermore, this may help identify target populations for new agents that may better inform subsequent studies. The use of such analyses may require dialog with approval bodies such as the FDA and EMA to ensure such findings are accepted in drug approval reviews, as well as being patient-focused to optimize availability of novel agents to target populations in need. Whether or not subgroup analyses can contribute to approval processes may depend on whether there is a strong biological rationale for the subgroup effect, whether the analysis was prespecified and appropriately powered, and the magnitude of difference. Post-hoc analyses may provide a high degree of evidence but given the statistical risks associated with multiple testing and adequacy of powering of the study, further validation may be needed as part of the approval process. Importantly, for optimal patient access, this should not necessarily preclude initial approval, especially given the complexity of disease pathobiology and treatment effect as the therapeutic landscape expands, but critically, may require further validation as part of subsequent prospective studies. Additionally, ongoing follow-up of trials for OS (preferably also adequately powered) will ensure that a clear picture emerges over time. However, powering of endpoints for OS is likely to require additional patients, longer follow-up, and therefore, significant additional resources to implement. Translational analyses performed pre-clinically and during early-phase clinical trials are critical to understand how to better design later-phase studies. Novel trial designs such as adaptive studies in which subgroups are identified at interim analysis and expanded in the second part of the study may also help improve rapid integration of this approach.
Understanding the value of a long PFS, including the duration of progression-toxicity-free survival, and the translation (or otherwise) to OS for patients is best informed by concurrent patient-reported outcomes within key phase 3 trials. If feasible, this would need to be continued at least until the point estimate of PFS2 to capture patient experiences after first relapse, as well as continuing to assess OS, which remains the gold standard for outcome.
How can these findings be applied to implementation of other surrogate endpoints in myeloma, such as MRD? Recent acceptance of the use of MRD as an endpoint for accelerated approval is vital given the ongoing advances in myeloma therapies seen to date, to enable their rapid translation to real-world practice [53]. This means that even PFS is now too long to make this a meaningful endpoint and enable early adoption of novel therapies in the frontline setting. However, if MRD is used as an endpoint, it is equally important to ensure appropriate follow-up for PFS and OS. Subgroup analyses should be encouraged to avoid missing key groups that do, or do not, benefit unless a target population can be identified from prior knowledge of drug action, which requires both confirmation in randomized trials and careful attention to correlatives. In our view, implementing these measures will hopefully ensure timely drug approvals in myeloma and subsequently, further improvements in clinical benefit for all our patients.
Supplementary information
Acknowledgements
Editorial support was provided by Swati Ghatpande, PhD, Jayasri Srinivasan, MD, MBA (Team 9 Science) and Catie O’Connor, BA (Dana Farber Cancer Institute), under the direction of the authors and funded by Dana Farber Cancer Institute, in part supported by the RJ Corman Multiple Myeloma Research Fund and an unrestricted grant from Oncopeptides AB. They have nothing to disclose.
Author contributions
CP, FS, GJ, GM, and PGR were responsible for conceptualization and methodology. CP was responsible for data curation, visualization, and writing the full first draft of the paper with subsequent input from PGR, FS, DC, LW, VR, PS and GM. DC was responsible for statistical analysis in Fig. 2. All authors were responsible for reviewing and editing the drafted paper, verifying the underlying data, and the decision to submit the manuscript.
Data availability
All relevant data related to the study are already published, included in the article, or uploaded as supplementary information.
Competing interests
CP reports consultancy for AbbVie, Amgen, Celgene (Bristol Myers Squibb), Sanofi, iTEOS Therapeutics, and Pfizer; honoraria from Amgen, Janssen, Celgene (Bristol Myers Squibb), and Sanofi; and travel support from Amgen, Janssen, and Celgene (Bristol Myers Squibb). FHS has received grants or contracts from Celgene, GlaxoSmithKline, Janssen, Oncopeptides, Targovax, and Sanofi; payment or honoraria for lectures or speakers’ bureau participation from AbbVie, Amgen, Bristol Myers Squibb, Daiichi Sankyo, GlaxoSmithKline, Janssen, Novartis, Oncopeptides, Pfizer, Sanofi, SkylineDx, and Takeda; serving on a data safety monitoring or advisory board for AbbVie, Celgene, GlaxoSmithKline, Janssen, Oncopeptides, Sanofi, and Takeda. DAC reports grants and non-financial support from Celgene Corporation, Merck, Amgen, Takeda Oncology and Sanofi; travel support from Celgene Corporation; payment to institution for educational lecture from Janssen; participation on a Data Safety Monitoring Board for a multiple myeloma study; and personal payment for meeting attendance from European Myeloma Network, Rotterdam. LJW has no conflicts to disclose. FD reports consulting fees from Janssen and participation on an advisory board for Amgen, Bristol Myers Squibb, Celgene, GlaxoSmithKline, Janssen, Oncopeptides, Sanofi, and Takeda. ON reports honorarium from Pfizer; advisory board participation for BMS, Janssen, Takeda, Sanofi, GPCR Therapeutics; and research funding from Takeda, Janssen. HA reports advisory board participation for ADC Therapeutics and Genmab; grants and personal fees from Morphosys; grants from Genentech; and personal fees from Novartis, BMS, Amgen, and AbbVie. M-VM has received payment or honoraria for lectures or speakers’ bureau participation from Amgen, Bristol Myers Squibb/Celgene, GlaxoSmithKline, Janssen, Pfizer, Sanofi, and Takeda; and served on a data safety monitoring or advisory board for Amgen, BlueBird Bio, Bristol Myers Squibb/Celgene, GlaxoSmithKline, Janssen, Oncopeptides, Pfizer, Regeneron, Sanofi, and Takeda. JL has no conflicts to disclose. KW reports research grants from AbbVie, Amgen, Bristol Myers Squibb/Celgene, Janssen, GSK, Sanofi, and Takeda; honoraria from AbbVie, Amgen, Adaptive Biotech, AstraZeneca, Bristol Myers Squibb/Celgene, BeiGene, Janssen, GSK, Karyopharm, Novartis, Oncopeptides, Pfizer, Roche, Sanofi, Stemline, Takeda, and Menarini; and consulting fees from AbbVie, Amgen, Adaptive Biotech, Bristol Myers Squibb/Celgene, BeiGene, Janssen, GSK, Karyopharm, Oncopeptides, Pfizer, Roche, Sanofi, Takeda, and Minarini. HL reports grants or contracts from Takeda, Amgen, Sanofi; consulting fees from Celgene, Janssen-Cilag, Takeda, Amgen, BMS, Sanofi, GSK, Pfizer; honoraria from Celgene, Janssen-Cilag, Takeda, Amgen, BMS, Sanofi, GSK, Pfizer, AbbVie; and participation in data safety monitoring or advisory board of Celgene, Takeda, Amgen, BMS, Sanofi. SVR has no conflicts to disclose. PS has received grants or contracts from Amgen, Celgene, Janssen, Takeda, and SkylineDx. GJ has received honoraria and research funding from BMS/Celgene. GM has participated in advisory boards for Janssen, Bristol Myers Squibb, Celgene, Karyopharm and Oncopeptides. PGR has served on advisory committees for and received consulting fees from Astra Zeneca, Bristol Myers Squibb/Celgene, GlaxoSmithKline, Karyopharm, Oncopeptides, Regeneron, and Sanofi; and clinical research grants from Bristol Myers Squibb/Celgene, Karyopharm, and Oncopeptides.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors jointly supervised this work: Gareth Morgan, Paul G. Richardson.
Supplementary information
The online version contains supplementary material available at 10.1038/s41408-024-01109-4.
References
- 1.Biomarkers Definitions Working Group. Biomarkers and surrogate endpoints: preferred definitions and conceptual framework. Clin Pharm Ther. 2001;69:89–95. 10.1067/mcp.2001.113989 [DOI] [PubMed] [Google Scholar]
- 2.FDA-NIH Biomarker Working Group BEST (Biomarkers, EndpointS, and other Tools) Resource [Internet] (National Institutes of Health (US) 2017).
- 3.EMA. Guideline on the clinical evaluation of anticancer medicinal products. 2019. Available from: https://www.ema.europa.eu/en/documents/scientific-guideline/draft-guideline-evaluation-anticancer-medicinal-products-man-revision-6_en.pdf. Accessed 9 January 2024.
- 4.US FDA. Clinical Trial Endpoints for the Approval of Cancer Drugs and Biologics: Guidance for Industry. 2018. Available from: https://www.fda.gov/media/71195/download. Accessed 9 January 2019.
- 5.Anderson KC, Auclair D, Adam SJ, Agarwal A, Anderson M, Avet-Loiseau H, et al. Minimal residual disease in myeloma: application for clinical care and new drug registration. Clin Cancer Res. 2021;27:5195–212. 10.1158/1078-0432.CCR-21-1059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kumar S, Paiva B, Anderson KC, Durie B, Landgren O, Moreau P, et al. International Myeloma Working Group consensus criteria for response and minimal residual disease assessment in multiple myeloma. Lancet Oncol. 2016;17:e328–e46. 10.1016/S1470-2045(16)30206-6 [DOI] [PubMed] [Google Scholar]
- 7.Anderson KC, Auclair D, Kelloff GJ, Sigman CC, Avet-Loiseau H, Farrell AT, et al. The role of minimal residual disease testing in myeloma treatment selection and drug development: current value and future applications. Clin Cancer Res. 2017;23:3980–93. 10.1158/1078-0432.CCR-16-2895 [DOI] [PubMed] [Google Scholar]
- 8.US FDA. Hematologic malignancies: regulatory considerations for use of minimal residual disease in development of drug and biological products for treatment—guidance for industry. 2020. Available from: https://www.fda.gov/media/134605/download. Accessed 9 January 2024.
- 9.Serani S. FDA’s ODAC votes 12 to 0 that MRD is a viable end point in myeloma trials. 2024. Available from: https://www.targetedonc.com/view/fda-odac-meeting-april-12-mrd-myeloma-trials. Accessed - May 2024.
- 10.Munshi NC, Avet-Loiseau H, Anderson KC, Neri P, Paiva B, Samur M, et al. A large meta-analysis establishes the role of MRD negativity in long-term survival outcomes in patients with multiple myeloma. Blood Adv. 2020;4:5988–99. 10.1182/bloodadvances.2020002827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Facon T, Kumar SK, Plesner T, Orlowski RZ, Moreau P, Bahlis N, et al. Daratumumab, lenalidomide, and dexamethasone versus lenalidomide and dexamethasone alone in newly diagnosed multiple myeloma (MAIA): overall survival results from a randomised, open-label, phase 3 trial. Lancet Oncol. 2021;22:1582–96. 10.1016/S1470-2045(21)00466-6 [DOI] [PubMed] [Google Scholar]
- 12.Durie BGM, Hoering A, Abidi MH, Rajkumar SV, Epstein J, Kahanic SP, et al. Bortezomib with lenalidomide and dexamethasone versus lenalidomide and dexamethasone alone in patients with newly diagnosed myeloma without intent for immediate autologous stem-cell transplant (SWOG S0777): a randomised, open-label, phase 3 trial. Lancet. 2017;389:519–27. 10.1016/S0140-6736(16)31594-X [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Richardson PG, Weller E, Lonial S, Jakubowiak AJ, Jagannath S, Raje NS, et al. Lenalidomide, bortezomib, and dexamethasone combination therapy in patients with newly diagnosed multiple myeloma. Blood. 2010;116:679–86. 10.1182/blood-2010-02-268862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dimopoulos MA, Oriol A, Nahi H, San-Miguel J, Bahlis NJ, Usmani SZ, et al. Daratumumab, lenalidomide, and dexamethasone for multiple myeloma. N Engl J Med. 2016;375:1319–31. 10.1056/NEJMoa1607751 [DOI] [PubMed] [Google Scholar]
- 15.Stewart AK, Rajkumar SV, Dimopoulos MA, Masszi T, Spicka I, Oriol A, et al. Carfilzomib, lenalidomide, and dexamethasone for relapsed multiple myeloma. N Engl J Med. 2015;372:142–52. 10.1056/NEJMoa1411321 [DOI] [PubMed] [Google Scholar]
- 16.Etekal T, Koehn K, Sborov DW, McClune B, Prasad V, Haslam A, et al. Time-to-event surrogate end-points in multiple myeloma randomised trials from 2005 to 2019: a surrogacy analysis. Br J Haematol. 2023;200:587–94. 10.1111/bjh.18568 [DOI] [PubMed] [Google Scholar]
- 17.Schjesvold FH, Dimopoulos MA, Delimpasi S, Robak P, Coriu D, Legiec W, et al. Melflufen or pomalidomide plus dexamethasone for patients with multiple myeloma refractory to lenalidomide (OCEAN): a randomised, head-to-head, open-label, phase 3 study. Lancet Haematol. 2022;9:e98–e110. 10.1016/S2352-3026(21)00381-1 [DOI] [PubMed] [Google Scholar]
- 18.Kumar S, Harrison SJ, Cavo M, de la Rubia J, Popat R, Gasparetto CJ, et al. Final overall survival results from BELLINI, a phase 3 study of venetoclax or placebo in combination with bortezomib and dexamethasone in relapsed/refractory multiple myeloma. Blood. 2021;138:84. 10.1182/blood-2021-145757 [DOI] [Google Scholar]
- 19.Sonneveld P, Richardson PG, Ludwig H, Dimopoulos M-A, Schjesvold FH, Hájek R, et al. Benefit versus risk assessment of melflufen and dexamethasone in relapsed/refractory multiple myeloma: analyses from longer follow-up of the OCEAN and HORIZON studies. Clin Lymphoma Myeloma Leuk. 2023;23:P687–96. 10.1016/j.clml.2023.05.004 [DOI] [PubMed] [Google Scholar]
- 20.Kumar SK, Harrison SJ, Cavo M, de la Rubia J, Popat R, Gasparetto C, et al. Venetoclax or placebo in combination with bortezomib and dexamethasone in patients with relapsed or refractory multiple myeloma (BELLINI): a randomised, double-blind, multicentre, phase 3 trial. Lancet Oncol. 2020;21:1630–42. 10.1016/S1470-2045(20)30525-8 [DOI] [PubMed] [Google Scholar]
- 21.Touzeau C, Dousset C, Le Gouill S, Sampath D, Leverson JD, Souers AJ, et al. The Bcl-2 specific BH3 mimetic ABT-199: a promising targeted therapy for t(11;14) multiple myeloma. Leukemia. 2014;28:210–2. 10.1038/leu.2013.216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bodet L, Gomez-Bougie P, Touzeau C, Dousset C, Descamps G, Maïga S, et al. ABT-737 is highly effective against molecular subgroups of multiple myeloma. Blood. 2011;118:3901–10. 10.1182/blood-2010-11-317438 [DOI] [PubMed] [Google Scholar]
- 23.Kumar S, Kaufman JL, Gasparetto C, Mikhael J, Vij R, Pegourie B, et al. Efficacy of venetoclax as targeted therapy for relapsed/refractory t(11;14) multiple myeloma. Blood. 2017;130:2401–9. 10.1182/blood-2017-06-788786 [DOI] [PubMed] [Google Scholar]
- 24.Matulis SM, Gupta VA, Nooka AK, Hollen HV, Kaufman JL, Lonial S, et al. Dexamethasone treatment promotes Bcl-2 dependence in multiple myeloma resulting in sensitivity to venetoclax. Leukemia. 2016;30:1086–93. 10.1038/leu.2015.350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Qin JZ, Ziffra J, Stennett L, Bodner B, Bonish BK, Chaturvedi V, et al. Proteasome inhibitors trigger NOXA-mediated apoptosis in melanoma and myeloma cells. Cancer Res. 2005;65:6282–93. 10.1158/0008-5472.CAN-05-0676 [DOI] [PubMed] [Google Scholar]
- 26.Punnoose EA, Leverson JD, Peale F, Boghaert ER, Belmont LD, Tan N, et al. Expression profile of BCL-2, BCL-XL, and MCL-1 predicts pharmacological response to the bcl-2 selective antagonist venetoclax in multiple myeloma models. Mol Cancer Ther. 2016;15:1132–44. 10.1158/1535-7163.MCT-15-0730 [DOI] [PubMed] [Google Scholar]
- 27.Moreau P, Chanan-Khan A, Roberts AW, Agarwal AB, Facon T, Kumar S, et al. Promising efficacy and acceptable safety of venetoclax plus bortezomib and dexamethasone in relapsed/refractory MM. Blood. 2017;130:2392–400. 10.1182/blood-2017-06-788323 [DOI] [PubMed] [Google Scholar]
- 28.Moreau P, Harrison S, Cavo M, De La Rubia J, Popat R, Gasparetto C, et al. Updated analysis of Bellini, a phase 3 study of venetoclax or placebo in combination with bortezomib and dexamethasone in patients with relapsed/refractory multiple myeloma. Blood. 2019;134:1888. 10.1182/blood-2019-126015 [DOI] [PubMed] [Google Scholar]
- 29.Mateos M-V, Beksac M, Dimopoulos MA, Gatt, ME, Gay F, Jo J-C, et al. Results from the randomized, open-label phase 3 CANOVA study of venetoclax-dexamethasone versus pomalidomide-dexamethasone in patients with t(11;14)-positive relapsed/refractory multiple myeloma. Presented at IMS 2023. September 27–30, 2023. Abstract OA-52.
- 30.Jackson GH, Davies FE, Pawlyn C, Cairns DA, Striha A, Collett C, et al. Lenalidomide maintenance versus observation for patients with newly diagnosed multiple myeloma (Myeloma XI): a multicentre, open-label, randomised, phase 3 trial. Lancet Oncol. 2019;20:57–73. 10.1016/S1470-2045(18)30687-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Graham HJ, Faith ED, Charlotte P, David AC, Alina S, Corinne C, et al. Lenalidomide before and after autologous stem cell transplantation for transplant-eligible patients of all ages in the randomized, phase III, Myeloma XI trial. Haematologica. 2020;106:1957–67. 10.3324/haematol.2020.247130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jackson GH, Pawlyn C, Cairns DA, Striha A, Collett C, Waterhouse A, et al. Optimising the value of immunomodulatory drugs during induction and maintenance in transplant ineligible patients with newly diagnosed multiple myeloma: results from Myeloma XI, a multicentre, open-label, randomised, Phase III trial. Br J Haematol. 2021;192:853–68. 10.1111/bjh.16945 [DOI] [PubMed] [Google Scholar]
- 33.Richardson PG, Perrot A, San-Miguel J, Beksac M, Spicka I, Leleu X, et al. Isatuximab plus pomalidomide and low-dose dexamethasone versus pomalidomide and low-dose dexamethasone in patients with relapsed and refractory multiple myeloma (ICARIA-MM): follow-up analysis of a randomised, phase 3 study. Lancet Oncol. 2022;23:416–27. 10.1016/S1470-2045(22)00019-5 [DOI] [PubMed] [Google Scholar]
- 34.Coulson AB, Royle KL, Pawlyn C, Cairns DA, Hockaday A, Bird J, et al. Frailty-adjusted therapy in Transplant Non-Eligible patients with newly diagnosed Multiple Myeloma (FiTNEss (UK-MRA Myeloma XIV Trial)): a study protocol for a randomised phase III trial. BMJ Open. 2022;12:e056147. 10.1136/bmjopen-2021-056147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hungria VT, Radinoff A, Delimpasi S, Mikala G, Masszi T, Li J, et al. A phase 3, open-label, randomized study to evaluate the efficacy and safety of single-agent belantamab mafodotin (belamaf) compared to pomalidomide plus low-dose dexamethasone (Pd) in patients (pts) with relapsed/refractory multiple myeloma (RRMM): DREAMM-3. J Clin Oncol. 2023;41:8007. 10.1200/JCO.2023.41.16_suppl.8007 [DOI] [PubMed] [Google Scholar]
- 36.Cavo M, Gay F, Beksac M, Pantani L, Petrucci MT, Dimopoulos MA, et al. Autologous haematopoietic stem-cell transplantation versus bortezomib-melphalan-prednisone, with or without bortezomib-lenalidomide-dexamethasone consolidation therapy, and lenalidomide maintenance for newly diagnosed multiple myeloma (EMN02/HO95): a multicentre, randomised, open-label, phase 3 study. Lancet Haematol. 2020;7:e456–e68. 10.1016/S2352-3026(20)30099-5 [DOI] [PubMed] [Google Scholar]
- 37.Attal M, Lauwers-Cances V, Hulin C, Leleu X, Caillot D, Escoffre M, et al. Lenalidomide, bortezomib, and dexamethasone with transplantation for myeloma. N Engl J Med. 2017;376:1311–20. 10.1056/NEJMoa1611750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Perrot A, Lauwers-Cances V, Cazaubiel T, Facon T, Caillot D, Clement-Filliatre L, et al. Early versus late autologous stem cell transplant in newly diagnosed multiple myeloma: long-term follow-up analysis of the IFM 2009 trial. Blood. 2020;136:39. 10.1182/blood-2020-134538 [DOI] [Google Scholar]
- 39.Richardson PG, Jacobus SJ, Weller EA, Hassoun H, Lonial S, Raje NS, et al. Triplet therapy, transplantation, and maintenance until progression in myeloma. N Engl J Med. 2022;387:132–47. 10.1056/NEJMoa2204925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Maura F, Weinhold N, Diamond B, Kazandjian D, Rasche L, Morgan G, et al. The mutagenic impact of melphalan in multiple myeloma. Leukemia. 2021;35:2145–50. 10.1038/s41375-021-01293-3 [DOI] [PubMed] [Google Scholar]
- 41.Mateos MV, Cavo M, Blade J, Dimopoulos MA, Suzuki K, Jakubowiak A, et al. Overall survival with daratumumab, bortezomib, melphalan, and prednisone in newly diagnosed multiple myeloma (ALCYONE): a randomised, open-label, phase 3 trial. Lancet. 2020;395:132–41. 10.1016/S0140-6736(19)32956-3 [DOI] [PubMed] [Google Scholar]
- 42.Lee H, Neri P, Bahlis NJ. BCMA- or GPRC5D-targeting bispecific antibodies in multiple myeloma: efficacy, safety, and resistance mechanisms. Blood. 2024;143:1211–7. 10.1182/blood.2023022499 [DOI] [PubMed] [Google Scholar]
- 43.Rasche L, Hudecek M, Einsele H. CAR T-cell therapy in multiple myeloma: mission accomplished? Blood. 2024;143:305–10. 10.1182/blood.2023021221 [DOI] [PubMed] [Google Scholar]
- 44.Zonder JA, Khan A, Jacobus ST, Hassoun H, Anderson Jr LD, Merz LE, et al. Treatment outcomes and prognostic factors with lenalidomide, bortezomib, and dexamethasone (RVd) alone versus Rvd plus autologous stem cell transplantation (ASCT) in African American (AA) patients (pts) with newly diagnosed multiple myeloma (NDMM) in the Determination phase 3 trial. Presented at American Society of Hematology Annual Meeting; December 9–12, 2023; San Diego, CA. Abstract 4762.
- 45.Richardson PG, Zonder JA, Khan AM, Jacobus SJ, Hassoun H, Anderson Jr LD, et al. Outcomes in African-American and White patients with newly diagnosed multiple myeloma receiving RVd-alone vs RVd+ASCT in DETERMINATION: efficacy, safety, and second primary malignancies (SPMs). Presented at EBMT 2024; April 14-17, 2024. Abstract OS10-05.
- 46.Koreth J, Cutler CS, Djulbegovic B, Behl R, Schlossman RL, Munshi NC, et al. High-dose therapy with single autologous transplantation versus chemotherapy for newly diagnosed multiple myeloma: a systematic review and meta-analysis of randomized controlled trials. Biol Blood Marrow Transpl. 2007;13:183–96. 10.1016/j.bbmt.2006.09.010 [DOI] [PubMed] [Google Scholar]
- 47.Pandya C, Hashmi S, Khera N, Gertz MA, Dispenzieri A, Hogan W, et al. Cost-effectiveness analysis of early vs. late autologous stem cell transplantation in multiple myeloma. Clin Transplant. 2014;28:1084–91. 10.1111/ctr.12421 [DOI] [PubMed] [Google Scholar]
- 48.Gandolfi S, Vekstein C, Laubach JP, O’Brien A, Masone K, Munshi NC, et al. The evolving role of transplantation in multiple myeloma: the need for a heterogeneous approach to a heterogeneous disease. Clin Adv Hematol Oncol. 2018;16:564–74. [PubMed] [Google Scholar]
- 49.Kelkar A, Midha S, Richardson P, Anderson K, Munshi N, Laubach J, et al. Cost-effectiveness of early versus delayed autologous stem cell transplantation (ASCT) for multiple myeloma (MM). Presented at the 49th Annual Meeting of the European Society for Blood and Marrow Transplantation; April 23–26, 2023; Paris Hybrid. Abstract OS02-07.
- 50.Hashmi S, Pandya C, Khera N, Gertz M, Dispenzieri A, Hogan W, et al. Cost effectiveness decision tree analysis of early versus late autologous stem cell transplantation (ASCT) in multiple myeloma (MM) in the United States (US). Biol Blood Marrow Transplant. 2013;19:S130–S1. 10.1016/j.bbmt.2012.11.065 [DOI] [Google Scholar]
- 51.Roussel M, Hebraud B, Hulin C, Perrot A, Caillot D, Stoppa AM, et al. Health-related quality of life results from the IFM 2009 trial: treatment with lenalidomide, bortezomib, and dexamethasone in transplant-eligible patients with newly diagnosed multiple myeloma. Leuk Lymphoma. 2020;61:1323–33. 10.1080/10428194.2020.1719091 [DOI] [PubMed] [Google Scholar]
- 52.Hassoun H, Jacobus SJ, Voorhees PM, Zonder JA, Lonial S, Raje N, et al. Response rates and outcomes in newly-diagnosed multiple myeloma (NDMM) patient subgroups receiving RVD+ASCT Plus lenalidomide maintenance until progression in the phase 3 DETERMINATION trial. Bone Marrow Transplant. 2023;58:20–152. abstract O12136167906 [Google Scholar]
- 53.Richardson PG, San Miguel JF, Moreau P, Hajek R, Dimopoulos MA, Laubach JP, et al. Interpreting clinical trial data in multiple myeloma: translating findings to the real-world setting. Blood Cancer J. 2018;8:109. 10.1038/s41408-018-0141-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Oosting SF, Barriuso J, Bottomley A, Galotti M, Gyawali B, Kiesewetter B, et al. Methodological and reporting standards for quality-of-life data eligible for European Society for Medical Oncology-Magnitude of Clinical Benefit Scale (ESMO-MCBS) credit. Ann Oncol. 2023;34:431–9. 10.1016/j.annonc.2022.12.004 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All relevant data related to the study are already published, included in the article, or uploaded as supplementary information.