

Abstract
Expression of the lama3 gene, encoding the laminin α3A chain, is restricted to specialized epithelia. We previously showed that lama3 gene expression is controlled by an epithelial enhancer through the cooperative effect of AP-1 binding sites. In fibroblasts, there is no lama3 expression because of the recruitment of a repressor complex absent or inactive in epithelial cells. In this paper, we show evidence that this repression of the lama3 gene is relieved by exogenous and UV-induced USF-1 through its interaction with a non-canonical E-box site. Using a chromatin immunoprecipitation assay, we find that UV stress induces USF to bind to the lama3 promoter in vivo. We further demonstrate that this loss of cell specificity is directly related to the accessibility of the E-box, resulting in a strong induction in fibroblasts, while expression remains constitutively high in keratinocytes. This accessibility appears to be dependent upon the recruitment of a fibroblastic repressor complex. Therefore, we speculate that anchorage of this repressor complex in fibroblasts modifies the enhancer geometry, allowing USF to interact under stress-inducing conditions with its heptameric binding site.
INTRODUCTION
Laminins are a family of glycoproteins that provide an integral part of the structural architecture of basement membranes (BM). Each laminin is a trimer assembled from α, β and γ chains, secreted and incorporated into cell-associated extracellular matrices. Through interactions with integrins, dystroglycan and other receptors, laminins contribute to cell proliferation, migration, differentiation and promotion of tissue survival (1–3). Laminins display a remarkable restricted expression profile, suggesting fine regulation of their genes (1,4–7).
Laminin-5, a heterotrimer of laminin α3A, β3 and γ2 chains, is found in many epithelial BM (8,9). It is the major anchoring filament ligand together with collagen XVII (BP180) permitting epidermal adhesion in skin (10,11). Mutations in the genes encoding the three chains of laminin-5 underlie the Herlitz type of junctional epidermolysis bullosa (H-JEB), a severe inherited bullous disease which leads to the early morbidity of the affected newborns (12–14). We provided the first experimental evidence of a phenotypic reversion of H-JEB keratinocytes by somatic gene therapy in vitro and demonstrated the feasibility of using a gene therapy approach for mild forms of skin blistering diseases or other inherited skin pathologies (13).
Growing lines of evidence suggest a role for laminin-5 in the re-epithelialization of wound skin repair. Laminin-5 supports migration of keratinocytes and forms the basis for migrating and proliferating basal keratinocytes in wound healing (15). Accordingly, laminin-5 is the first extracellular component to be expressed by migrating keratinocytes during wound healing in vivo (16) and upregulation of laminin α3 gene (LAMA3) expression is observed during cell migration and repair of the BM in vivo (1,10,17).
Accumulating data suggest that laminin-5 gene regulation might be associated with growth and migration of cancer cells (18,19). Laminin-5 was found to be expressed in invading tumor cells (20,21) and to be strongly active in promoting migration of some tumor cells (22,23). Regulation of laminin-5 gene expression by TGF-β, EGF and PDGF strengthens the potential role of this protein in wound healing processes and in tumor progression (23,24).
Because of their relevance in tissue remodeling, cancer metastasis and gene therapy, mechanisms of laminin-5 gene regulation need to be elucidated. We previously reported that cell-specific activity of lama3 promoter expression is controlled by a proximal cell-specific enhancer through the cooperative effect of AP-1 binding sites (25). We demonstrated further that a particular DNA conformation of the enhancer fragment bound by AP-1 complexes leads to the anchorage of non-DNA-binding fibroblastic cofactor(s) to the AP-1/DNA complex to form an inhibitory ternary complex (25). Since two E-box sequences are found to overlap the AP-1 binding sites on the lama3 promoter, we wished to evaluate the contribution of USF in the cell-specific regulation of lama3. In this work, we present evidence that (i) USF, but not other basic helix–loop–helix/leucine-zipper (bHLHzip) members of the Myc family, strongly stimulates the lama3 gene in fibroblast cells, while its effect on keratinocyte remains marginal; (ii) this effect, which is also observed in response to UV irradiation, occurs in vivo via the specific binding of USF to a non-canonical heptameric CAGCCTG site; and (iii) the accessibility of this unusual USF-binding site depends on the lama3 promoter conformation. This USF-opened conformation results from the interaction of a repressor complex with the AP-1 heterodimers in fibroblasts.
MATERIALS AND METHODS
Transient transfection
KHSV cell line (derived from normal human primary keratinocytes immortalized by SV40 infection) (26) are maintained in a mixture of Dulbecco’s modified Eagle’s/Ham’s F12 (2:1) medium supplemented with 10% fetal calf serum (Life Technologies, BRL, France), 4 mM glutamine, 0.4 µg/ml hydrocortisone, 10 ng/ml EGF and 2 mM choleratoxine. NIH-3T3 fibroblast and 804G bladder epithelial cell lines are cultured in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal calf serum. Cells (5 × 104) were plated in 24-well culture plates 24 h before transfection. Typically, 0.5 µg per well of DNA were used with either 2 µl of Fugene 6 (Roche Molecular Biochemicals) for KHSV and NIH-3T3 cell lines or 3 µl of Transfast reagent (Stratagene) for the 804G cell line, following the manufacturer’s instructions. Quantitations were performed 24 h post-transfection. Transfection efficiency was determined by cotransfection of plasmid CMVβgal. The values of luciferase expression data were normalized to the measured β-galactosidase activity. The enzyme activities were measured using a luminometer (Berthold Biolumat LB9500C). Before transfection, each DNA construct was purified using the silica columns from Quiagen 5 (Hylden, Germany). At least three separate experiments were performed. For gal4 experiments, we used plasmids supplied with the Two-hybrid Mammalian system (Clontech), including p53 fused to the Gal4 DNA-binding domain (pM-p53) and SV40 largeT fused to the activation domain of VP16 (pVP16-T), according to the manufactor’s instructions (Clontech).
Plasmid constructs
The different promoter constructs derived from the laminin α3A promoter fragment (PHA) and one of its deleted construct (6.2H) (25) using the Quick-Change™ site-directed mutagenesis kit protocol (Stratagene). The oligonucleotides used are listed in Table 1.
Table 1. Oligonucleotides used to generate the mutated and deleted promoter constructs.
| Name | Oligonucleotide sequences |
|---|---|
| mut Ebox1 |
5′-GCGCTCTGGCACAGGCTGACTCATGGATGAAGTTTAAAGGTGGGGCCCCTG-3′ |
| mut Ebox2 |
5′-CCGCAGACAGCCTTCCTTCCCTGAGTCAGGCAGGCCCGGGCACT-3′ |
| 6.2H +64/+218 |
5′-ATGTGTGAAGTTTAAAGGTGGGGATGGGATGGCTGAGGATCTTTGGG-3′ |
| 6.2H +64/+135 |
5′-CATGTGTGAAGTTTAAAGGTGGGGGAGGAAGAGGCAGAGGTTCCTGCG-3′ |
| 6.2H +175/+197 |
5′-TCCTGCGCACGCAGCGGACGTCCAGTGAGGCGGTCAGCCTCAGCCTGC-3′ |
| 6.2H +135/+175 |
5′-CACTGAGCAGGAAGGGCAGGTATAAAGGAACCGGGATGCCTCCAGCAGTG-3′ |
| Del +197/+218 |
5′-GTCCAGGAACCGGGATGCCTCCAGCATGGGATGGCTGAGGATCTTTGGG-3′ |
| mutEbox3 |
5′-GCCTCCAGCAGTGAGGCGGTGGATCCGCAGCATGGGATGGCTGAGGATCTT-3′ |
| Δ60 |
5′-TTCCCTGCCCGTGACTCAGCCTGTATTACAGAGCGGTGCCGTTACCTG-3′ |
| Δ109 |
5′-TTCCCTGCCCGTGACTCAGCCTGTCAGGCTGACTCATGTGTGAAGTTTA-3′ |
| 109 mix |
5′-CGCCCCACCTCACAGGAATTACAGCTTTGACCAACAAGAACGTGATTTTCGTTGCCGGTCTGGGCAGGCTGACTCATGTGTGAAGTTTAA-3′ |
| PHAgal4 |
5′-CAGGAACCGGGATGCCTCCAGAGCTTAGCCGGAAGACTCTCCTCCGACTAATGGGATGGCTGAGGATCTTTGGGG-3′ |
| mutABC |
5′-CAGGGCGTGTTCCCTGCCCGTGCCTTAGCCTGTGATTTAGGGCGTCTGCA-3′ |
| |
5′-GCGCTCTGGCACAGGCTGCCTTATGTGTGAAGTTTAAAGGTGGGGCCCC-3′ |
| 5′-TGCCGCAGACAGCCTTCTCACCTGCGTTAGGCAGGCCCGGGCACTGAGCA-3′ |
The letters in bold correspond to the mutated nucleotides.
Nuclear extract preparation and gel mobility shift assay
Nuclear proteins were extracted from KHSV and NIH-3T3 as reported elsewhere (25) after transient transfection of HA-tagged pRK7-USF1. In vitro binding reactions were performed in a total volume of 25 µl by mixing 200 000 c.p.m. of 32P-labeled Ebox3 probe (GTGAGGCGGTCAGCCTGCAGC) with 3 µg of nuclear extracts in the appropriate binding buffer (10 mM Tris–HCl pH 7.5, 4% glycerol, 0.5 mM EDTA, 0.5 mM DTT, 25 mM NaCl, 400 ng/µl salmon sperm). After 15 min of preincubation on ice, the probe was added and incubated for a further 20 min at 25°C. Then, DNA–protein complexes were resolved by electrophoresis on 4% polyacrylamide gels in TAE buffer (10 mM Tris, 9 mM Na-acetate/acetic acid, 275 mM EDTA) for 1 h at 400 V, dried and subjected to autoradiography. When indicated, a 20-fold excess of cold competitor oligonucleotides was added during preincubation. For antibodies supershift assays, nuclear extracts were preincubated with 0.5 µl of antisera against USF1 and HA (Santa-Cruz) for 15 min at 4°C before the binding reaction. Primers used: wild-type Ebox3, 5′-GTGAGGCGGTCAGCCTGCAGC-3′; mutated Ebox3, 5′-GTGAGGCGGTGGATCCGCAGC-3′ and canonical E-box, 5′-GTGAGGCGGTCACGTGCAGC-3′.
Chromatin immunoprecipitation
Live cells were treated with 1% formaldehyde for 1 h at 4°C to cross-link chromatin complexes. The cells were then harvested in the appropriate lysis buffer (20 mM HEPES pH 7.9, 350 mM NaCl, 20% glycerol, 1 mM MgCl2, 0.5 mM EDTA, 1% Nonidet P40, 0.5% sodium deoxycholate, 0.1% SDS). The chromatin was sheared by sonication and USF-1-containing complexes were recovered by immunoprecipitation using USF-1-specific serum (USF-1 antibody sc-229; Santa Cruz) or preimmune serum as a control. Identification of the captured lama3 regulatory sequences was performed by PCR analysis using primers 5′-CTGCCGCAGACAGCCTTC-3′ and 5′-GCTGCCCCAAAGATCCACA-3′. Thirty cycles of PCR were performed, using the captured fragments as templates and the Advantage-GC Genomic PCR (K1908-1; Clontech). The amplified products were analyzed on a 2% agarose gel.
RT–PCR
Total RNA were purified from NIH-3T3 cells using the TRIzol reagent (Life Technologies) according to the manufacturer’s protocol. When cells were UV-C irradiated (40 J/m2) in a Stratalinker (Stratagene), extractions were done 2 h after treatment. RT–PCR was performed with 200 ng of total RNA as template using the superscript RT–PCR kit reagent according to the manufacturer’s instructions (Life Technologies). Lama3A and control GAPDH mRNAs were reverse-transcribed and amplified using the specific primers 5′-TGTGGATCTTTGGGGCAG-3′ and 5′-TGGCAGCTACCTCCAAATT-3′; 5′-AACCATGAGAAGTATGACAAC-3′ and 5′-GTCATACCAGGAAATGAGCT-3′, respectively. The amplified products were resolved on a 2% agarose gel.
RESULTS
We have previously shown that the laminin α3A regulatory region (PHA) is sufficient for cell-specific enhancer activity (25). Primer extension analysis indicates the presence of multiple start sites within a 217 bp window located at nucleotides 217, 177, 135 and 82 upstream of the translation initiator (Fig. 1B). Since PHA contains two canonical E-box motifs overlapping AP-1 binding sites at the position +40 (CATGTG) and +87 (CACCTG) relative to the most upstream transcription initiation site (Fig. 1A), the ability of USF to regulate lama3 gene expression was examined in this work. Keratinocytes and NIH-3T3 fibroblasts were transfected with PHA or 6.2H [a truncated version that is functionally equivalent (25)] reporter constructs together with a USF expression vector. Ectopically expressed USF slightly activates these promoter constructs in keratinocytes (Fig. 2). As expected from the cell-specific repression of the lama3 promoter activity in fibroblasts, the constructs are not active in NIH-3T3 cells. However, they are strongly stimulated by USF, raising the promoter activity in fibroblasts to a level similar to that observed in keratinocytes. We can conclude that USF releases the cell-specific repression of lama3 gene expression. Surprisingly, this activation is not mediated through the two E-box sites, since single and dual mutations of each site do not abolish the stimulation (Fig. 2). To identify the sequences responsive to USF, various portions of 6.2H were removed and the corresponding constructs were tested in transient transfections (Fig. 2). Removal of the sequences located between base pairs +64 and +218 abolishes responsiveness to USF. Similarly, deletion of the sequences located between base pairs +197 and +218 renders the promoter construct unresponsive. Examination of the nucleotide sequence of this 21 bp region reveals a non-consensus E-box (CAGCCTG), located at base pairs +207 to +213 in the lama3 promoter. Mutation of this site to a BamHI site (GGATCC) abolishes the USF-dependent stimulation of the 6.2H construct (mutEbox3). These data indicate that the lama3 heptameric E-box site is a target for USF regulation.
Figure 1.

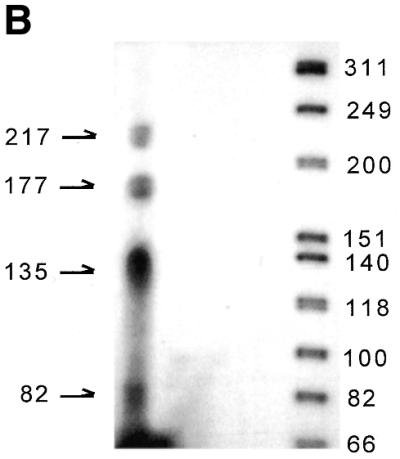
(A) Nucleotide sequence of the 3′ end of the lama3 regulatory region. Nucleotide numbering is relative to the first nucleotide of the transcription initiation site (+1). The three AP-1 binding sites are underlined, the two putative E-box motifs are open boxes and the USF responsive element (Ebox3) is the shaded box. The numbers refer to the position of the deleted sequences. Asterisks represent the multiple transcription start sites identified by primer extension analysis. The translation start site is identified by a dot. (B) Primer extension on lama3 mRNA isolated from human primary keratinocytes are shown. The primer used is indicated by an arrow.
Figure 2.

Mapping of the USF responsive element. Left, full-length PHA and its mutated or deleted constructs are represented. The numbers indicate the position of the deleted fragments relative to the start of translation. Right, the different constructs were transiently transfected into keratinocytes and NIH-3T3 cells together with or without a USF-1 expression vector. Luciferase activities were assayed as described in the Materials and Methods and are shown as absolute values. Error bars reflect the SD between at least three separate experiments performed in triplicate.
These observations are confirmed by gel-shift analysis (EMSA) (Fig. 3). A single retarded band is detected when an oligonucleotide representing the E-box motif of the lama3 promoter (Ebox3) is incubated with nuclear extracts made from NIH-3T3 fibroblasts (Fig. 3A) and keratinocytes (Fig. 3B) transfected by a HA-tagged USF-1-expressing vector. The retarded complex is specific for the non-canonical E-box since unlabeled wild-type Ebox3 (Fig. 3A, lane 3) and the canonical E-box (Fig. 3A, lane 10) probes both compete the retarded complex (Fig. 3A, lane 3, and B, lane 2), while a probe in which the Ebox3 site has been mutated (mt) does not (Fig. 3A, lane 9). No complex is formed with labeled mutated Ebox3 (mt; Fig. 3A, lane 1). To verify the identity of the protein binding to Ebox3, we performed a supershift assay using two different antibodies specific for HA-tagged USF-1 (anti-USF-1 and anti-HA) confirming that USF interacts with the lama3 heptameric E-box site (Fig. 3A and B).
Figure 3.

Identity of nuclear protein bound to the lama3 USF-responsive element. The 21 bp sequence containing the heptameric E-box (wt) is incubated with nuclear extracts from NIH-3T3 cells (A) and keratinocytes (B) transfected with HA-tagged USF-1. When indicated, nuclear extracts are incubated with a pre-immune (pi) serum or with antibodies raised against USF-1 or HA. Ebox3 probe in which the heptameric E-box was replaced by a BamHI site (mt) or the canonical E-box probe (Ebox) was incubated with USF-1-expressing NIH-3T3 nuclear extracts as indicated in lanes 9 and 10, respectively.
Several proteins belonging to the class of bHLHzip transcription factors can bind to DNA to sequences that contain the palindromic consensus CA–(–)TG (27). To test which bHLH protein transactivates the lama3 gene, NIH-3T3 cells were cotransfected with 6.2H construct and various bHLH-expressing vectors. As shown in Figure 4, Myc and Max are unable, either alone or in combination, to stimulate lama3 promoter like USF. This restricted behavior is specific to the lama3 promoter site and is not due to differences in protein stability since these constructs are equally able to transactivate a 3UBS-tk control promoter. This control promoter consists of three canonical E-box sites CACGTG with flanking sequences from the adenovirus major late promoter, inserted upstream of the thymidine kinase minimal promoter (Fig. 4). Identical results were obtained in transfected keratinocytes (data not shown).
Figure 4.

Among the bHLHzip members tested, only USF releases the cell-specific inhibition of lama3 gene expression. NIH 3T3 fibroblasts were transfected with the 6.2H or 3USB-tk constructs along with either USF-1, Max, Myc or Max/Myc and assayed as described in Materials and Methods. The values (fold stimulation) are derived from the ratio between the basal and the induced promoter activities. Error bars reflect the SD between three separate experiments performed in triplicate.
Recently, it has been demonstrated that the induction of endogenous USF-1 occurs as a stress response (28,29). We monitored the binding activity of endogenous USF-1 in NIH-3T3 fibroblasts at different times (from 30 min to 2 h) following UV irradiation (40 J/m2). As soon as 30 min after irradiation, the USF-1/DNA binding complex is clearly enhanced, and binding activity remains high for 2 h post-irradiation (Fig. 5A). This endogenous USF-1 induction perfectly correlates with the luciferase data obtained for the 6.2H promoter activity in a time course experiment after UV stimulation. Indeed, the promoter activity is significantly induced 30 min post-irradiation and reaches a 5-fold induction level 2 h post-irradiation (Fig. 5B). Mutation of the lama3 non-canonical E-box (mutEbox3) abolishes the UV stimulation of the promoter in NIH-3T3 fibroblasts but not in keratinocytes, suggesting that UV stimulation occurs in keratinocytes through a different pathway (Fig. 6A). These results demonstrate that in NIH-3T3 fibroblasts, UV irradiation stimulates the lama3 promoter through the binding of USF-1 to the lama3 heptameric non-canonical E-box.
Figure 5.

UV irradiation increases binding activity of USF-1 on the lama3 non-canonical E-box and induced 6.2H construct promoter activity. (A) USF binding analysis after UV irradiation. NIH-3T3 were UV irradiated (40 J/m2) and nuclear extracts were prepared at 30 min, 1, 2 and 3 h post-irradiation. Unstimulated and UV-irradiated nuclear extracts were then incubated with the radiolabeled Ebox3 probe. (B) 6.2H and empty vector control constructs were transiently transfected into NIH-3T3 fibroblasts. Twenty-four hours after transfection, the cells were UV irradiated (40 J/m2) and harvested at 30 min, 1, 2 and 3 h post-irradiation for luciferase activity measurements.
Figure 6.

UV and USF inducibility of the lama3 promoter in fibroblasts. (A) 6.2H and mutEbox3 constructs were transiently transfected into NIH-3T3 fibroblasts and keratinocytes stimulated by UV irradiation or transfected with a USF-1 expression vector. Luciferase activities were determined as described in Materials and Methods. Empty vector luciferase values correspond to the background expression of the pGL2 (no promoter) reporter gene. The luciferase activity values and error bars reflect the average and the SD between at least three separate experiments performed in triplicate. (B) USF-1 binds the lama3 promoter in vivo. Untreated NIH-3T3 cells (lanes 1 and 3) or UV-irradiated NIH-3T3 cells (lanes 2 and 4) were chemically cross-linked and immunoprecipitated with a specific USF-1 antibody or a preimmune control (pi) serum. Lane ct corresponds to the PCR control directly amplified from PHA. (C) Analysis of lama3 mRNA expression level by semi-quantitative RT–PCR assay using total RNA extracted from non-stimulated (lane 4), UV-stimulated (lane 3) and USF-1-transfected (lane 2) NIH-3T3 cells (3T3) as templates. The Lama3 mRNA expression level in keratinocytes (K) is shown as a positive control (lane 1). Bottom, amplification of GAPDH as an internal control.
In order to assess whether endogenous USF recognizes the non-canonical E-box of the lama3 promoter in vivo, chromatin immunoprecipitation experiments were performed. Non-irradiated and UV-irradiated NIH-3T3 cells were incubated with formaldehyde in order to directly cross-link proteins to DNA in living cells. Cross-linked DNA was immunoprecipitated with either a preimmune serum or USF antibodies. The immunoprecipitates were subjected to PCR using nested primers that amplify a fragment surrounding the lama3 E-box (Fig. 6B). We detected binding of USF to the lama3 E-box binding site in intact living cells only after UV irradiation. No amplification was obtained when a non-immune serum was used.
To test whether the UV-stimulated activity of the lama3 promoter in fibroblasts is relevant to endogenous gene regulation, total RNA was extracted from either control, USF-transfected or UV-irradiated NIH-3T3 cells. Semi-quantitative RT–PCR experiments were then performed with primers specific for the lama3 gene and GAPDH as an internal control (Fig. 6C). While mRNA expression of the lama3 gene remains almost undetectable in non-stimulated NIH-3T3 cells after 25 cycles (lane 4), it becomes clearly detectable after UV irradiation (lane 3) and is high in USF-1-transfected cells (lane 2). Lane 1 refers to the lama3 mRNA basal expression level in keratinocytes. Altogether, these results suggest that UV-induced USF-1 is able to release the cell-specific inhibition of the lama3 gene expression in fibroblasts through its interaction with a novel non-canonical E-box.
Our results in Figures 2 and 6 indicate that USF stimulates the lama3 promoter better in fibroblasts than in keratinocytes (10–15-fold versus 2-fold), whereas the synthetic 3UBS-tk promoter is similarly regulated in both cell types. The simplest explanation for this discrepancy would be that the lama3-USF responsive element is not as readily accessible in keratinocytes as it is in fibroblasts. Interestingly, we previously reported that the high keratinocyte-specific activity of the lama3 promoter stems from three AP1 sites found in a very precise sterical arrangement. We suggested that a non-DNA-binding repressor complex found in fibroblasts interacts with the AP1 complexes present on the lama3 promoter (25). This would result in the locking of the lama3 promoter in an inactive state. It is thus reasonable to hypothesize that the locked lama3 promoter configuration would render the heptameric E-box readily accessible, whereas that same E-box would be structurally hindered in the active promoter configuration found in keratinocytes.
To test this, transient transfection assays were performed with USF-expressing vector and PHA constructs in which part of the sequences located between the AP-1 sites were deleted (Fig. 7). While deletion of 60 bp (Δ60) does not modify USF responsiveness in fibroblasts, removal of 109 bp between AP-1A and AP-1B sites (Δ109), known to prevent anchorage of the fibroblastic repressor complex, strongly inhibits the stimulation normally driven by USF. However, stimulation by USF is fully restored in fibroblasts when the 109 bp deleted sequence is replaced by irrelevant non-homologous sequences (109mix and pCDNA3 constructs). Interestingly, reinsertion of such a spacer sequence was shown to restore the interaction of the repressor complex (25). As expected, these deletions/insertions do not significantly modify USF responsiveness in keratinocytes (Fig. 7). But hindrance of AP-1 binding to PHA following mutagenesis (mutABC) renders the promoter strongly inducible by USF in both cell types (Fig. 7). To verify that this is not due to a new accessibility of the two E-box sites overlapping the AP-1 sites (E-box 1 and E-box 2, Fig. 1), E-box 1 and E-box 2 sites were mutated in the context of mutABC. The resulting construct is equally and strongly stimulated in both cell types (data not shown). Therefore, the strong USF responsiveness of mutABC is not the result of de novo accessibility of the canonical E-box sites, but due to the binding of USF to the non-canonical heptameric site.
Figure 7.

Functional analysis of the AP-1 binding sites and the intervening sequences within PHA. Full-length, truncated and AP-1-mutated PHA constructs are represented on the left. Keratinocytes and NIH-3T3 cells were transfected with pRK7 USF-1 along with different truncated/mutated constructs derived from PHA. Mut ABC construct corresponds to pHA in which the AP-1 binding sites have been mutated; Δ60 and Δ109 constructs correspond, respectively, to a 60 and 109 bp deletion between the AP1-A and AP1-B binding sites. 109 mix construct represents Δ60 in which 49 bp were inserted in a random order to respect the 109 bp spacing between AP-1 sites. The pCDNA3 construct corresponds to the substitution of 109 nt residing between AP1-A and AP1-B with a 109 bp heterologous sequence from the pCDNA3 polylinker plasmid.
Therefore, our results are in perfect agreement with the model depicted in Figure 8: in keratinocytes, the tridimensional structure of the active lama3 promoter following AP-1 binding blocks USF access to the heptameric box, whereas in cells where the lama3 gene is constitutively silenced due to the presence of the repressor complex within the AP-1 bound promoter, this same heptameric box remains accessible by USF.
Figure 8.

The differential cell-specific USF stimulation results from the accessibility of the heptameric E-box within the lama3 gene. The lama3 heptameric E-box has been mutated to a gal4 binding site within PHA to give the PHAgal4 construct. (A) Transfection of KHSV and 804G, two lama3-expressing cell lines, and NIH-3T3 cells expressing the transactivating factor pM-53/pVP16-T along with the gal4 luc positive control construct are shown. (B) Transfection of KHSV, 804G and NIH-3T3 cells with pM-53, pVP16-T and either PHA, PHAgal4 or PHAgal4 mut ABC constructs are shown. Induction folds are relative to activities in the absence of the transactivating constructs.
To further confirm that the cell-dependent USF driven induction of the lama3 gene is due to the accessibility of the heptameric E-box, and not to a cell-specific control of USF intrinsic activity, we substituted the heptameric E-box site with a consensus Gal4 binding site. The resulting PHA–Gal4 construct was co-transfected with the transcriptional activator system consisting of p53 fused to the Gal4 DNA-binding domain (pM-p53), which can interact with SV40 largeT fused to the activation domain of VP16 (pVP16-T). When both pM-p53 and pVP16-T are expressed, they strongly activate transcription through the Gal4 site. As shown in Figure 9, p53 and large T antigen activate transcription of PHA–Gal4 in fibroblasts but not in two laminin-5-expressing cell lines (804G and Ker). The mutation of the AP-1 binding sites within the PHA–Gal4 construct allows a high stimulation in keratinocytes and laminin-5-expressing epithelial cells (Fig. 9). These Gal4 constructs display the same behavior as the PHA constructs, demonstrating that access to the heptameric lama3 E-box, and not the nature of the transcription factor or its DNA binding site, is responsible for the cell-dependent stimulation.
Figure 9.

Model for the differential stimulation of lama3 gene by USF. The lama3 regulatory sequences are represented by their three AP-1 binding sites (A–C) bound by Jun/Fos proteins, the heptameric E-box site (dark box) and the transcription initiation site (diagonal arrow). (A) In keratinocytes, binding of the Jun/Fos heterodimers to the lama3 gene renders the heptameric E-box site inaccessible to USF which is therefore unable to stimulate the lama3 promoter activity. (B) The recruitment of the fibroblastic repressor complex in fibroblasts through binding to AP-1 (25) allows the heptameric E-box site to be accessible for USF. The resulting conformational modification breaks down the cell-specific repression of lama3 activity in fibroblasts. (C) One hypothesis is that the binding of USF to its site may modify the DNA geometry and thus release the repressor complex. However, it cannot be excluded that a repressor remains bound and that USF interacts with other transcription factors or even directly with the Pol II transcriptional machinery. Since the AP-1 mutant PHAmutABC construct activity remains high in fibroblasts after USF stimulation, AP-1 transactivation ability is not necessary for this stimulation.
DISCUSSION
Members of the bHLHzip family, which include USF, Myc, Max, USF and TFE3, interact with DNA as dimers and recognize E-box cis-acting elements characterized by a central hexamer CACGTG, CATGTG or CACATG sequence (30–33). In this report, we provide evidence that the lama3 promoter (PHA) is stimulated by USF. Although two canonical E-box sites are present on the lama3 promoter, mutation analyses ruled out their involvement in this USF-dependent stimulation. A systematic search for the USF-responsive element was therefore initiated within the PHA fragment and a heptameric non-canonical E-box sequence (CAGCCTG) was identified within the 5′-untranslated region of the lama3 gene. This sequence is specifically bound in vitro by USF and mutation of this site abolishes USF transactivation in living cells. In addition to binding to the canonical sites originally described for HLHzip proteins, a large number of different sequence combinations of non-canonical DNA sequences are bound with high affinity by these proteins (27). Moreover, heptameric sites have already been described for E2A proteins (34). Methylation interference experiments on the nucleotide at position +4 have shown that these sites behave as true hexamers (27). All these binding sites, including the non-canonical ones described up to now, share an internal CG or TG dinucleotide. This is not the case for the lama3 heptameric site. Therefore, and despite the presence of two canonical E-box sites, the promoter is stimulated by USF via a third unusual non-consensus heptameric site.
The lama3 gene encoding the laminin α3A polypeptide chain of laminin-5 is regulated in a developmental, inducible and tissue-specific manner (9,23). We have recently shown that the lama3 gene contains a strong epithelial enhancer which confers efficient expression in keratinocytes but not in fibroblasts, as expected from the endogenous lama3 gene regulation (25). Surprisingly, we show here that exogenous USF expression, as well as UV induction, drastically increases lama3 promoter activity in fibroblasts. By RT–PCR experiments, we further demonstrate that this occurs in vivo since the endogenous lama3 transcript is detected in fibroblasts only after USF or UV stimulation. In other words, UV-stimulated USF abolishes the cell-specific behavior of PHA by increasing lama3 promoter activity and mRNA levels in fibroblasts to a level similar to what is seen in keratinocytes. Activation of transcription has been observed following binding of USF to specific sites in many gene promoters (35,36), but an unlocking of cell specificity is, to our knowledge, unique.
It is intriguing that the stimulation of lama3 promoter by USF is more efficient in fibroblasts that do not produce laminin-5 than in laminin-5-producing keratinocytes. This feature is not due to differences in intrinsic DNA binding or transactivation abilities of USF since USF binds and transactivates a canonical E-box-containing reporter construct (3UBS-tk) with identical efficiency in both cell types. Furthermore, it can be ruled out that the weak stimulation of PHA in keratinocytes stems from a direct competition between USF and other transcription factors for the lama3 heptameric site, since the complex formed on the heptameric E-box-containing probe in keratinocyte nuclear extracts is quantitatively shifted in the presence of USF-1-specific antibodies (Fig. 3B).
In a recent study, we demonstrated that the full transcriptional activity of the lama3 promoter depends on the cooperative binding of three AP-1 binding sites (25). This synergistic activity is repressed in fibroblasts by the presence of a non-DNA binding repressor complex which interacts with AP-1 dimers leading to a particular DNA conformation. This interaction is abolished as soon as the appropriate distance between the AP-1 binding sites is modified (25). Mutation of the AP-1 binding sites on the PHA construct results in a dramatic increase of PHA activity in keratinocytes in response to USF while maintaining the strong USF stimulation in fibroblasts (Fig. 7). Furthermore, when the spacing between the AP-1 sites is reduced, the stimulation of PHA by USF in fibroblasts is basically abolished. This stimulation is recovered as soon as the AP-1 binding site spacing is restored, even by non-homologous DNA sequences. These results demonstrate that AP-1, together with the relative position of its DNA binding sites on the lama3 regulatory sequences are essential for USF stimulation. Therefore, we propose that USF activates the lama3 promoter through the heptameric box, which is only accessible when the promoter is in a locked configuration (Fig. 8). This locked configuration results from the interaction of a non-DNA binding repressor absent from keratinocytes with three AP-1 complexes present in a precise arrangement within the lama3 promoter. This model is further strengthened by the behavior of the PHA–Gal4 construct in which a Gal4 binding site has replaced the lama3 heptameric site (Fig. 9). This chimeric construct reproduces the restricted stimulation of PHA activity by USF: p53 and VP16-T stimulate the lama3 promoter much more efficiently in fibroblasts than in epithelial cells.
Altogether, our data demonstrate that the cell-dependent stimulation of PHA by USF is directly linked to the accessibility of this DNA region, but not to the nature of either the transcription factor or its DNA binding site.
We also show that USF binds to the heptameric E-box whereas other bHLHzip proteins, such as Myc and Max, do not. Consistent with this, only USF-1 transactivates the lama3 promoter in vivo via this non-canonical E-box site. The specificity of this new USF binding site presumably stands from both its non-canonical structure and its flanking sequences, as reported elsewhere (27,37,38). Alternatively, the position of the E-box with respect to the transcriptional start site has been suggested to be important in determining DNA binding specificity between bHLHzip proteins and regulation of gene expression in a post-DNA-binding mechanism (39). In this context, recent studies on the cad promoter demonstrated that even if Myc, USF and TFE3 are all able to bind an E-box site, directly or artificially via a gal4 DNA-binding domain, only Myc was able to transactivate the promoter (40).
In conclusion, the present studies suggest that a non-canonical E-box site contributes to a de novo expression of the lama3 gene through the action of stress-activated USF. Anchorage of the repressor complex to the enhancer fragment through AP-1 complexes in fibroblasts allows the accessibility of this unusual USF binding site. This results in the lama3 promoter to be efficiently stimulated by USF in normally lama3-silent cells. This provides a mechanism to fine tune the level of lama3 expression in different tissues, switching on de novo expression in non-permissive cells while preventing overexpression in laminin-5 producing cells.
Acknowledgments
ACKNOWLEDGEMENTS
This work was supported in parts by grants from Association pour la Recherche sur le Cancer (ARC), EEC Biomed2 (BMH4-CT972062), Fondation Coloplast pour la Qualité de la Vie and Institut National de la Santé et de la Recherche Médicale (INSERM). We thank Dr Kim Boulukos for her proofreading of the manuscript.
REFERENCES
- 1.Ryan M.C., Tizard,R., VanDevanter,D.R. and Carter,W.G. (1994) Cloning of the Lama3 gene encoding the alpha 3 chain of the adhesive ligand epiligrin. Expression in wound repair. J. Biol. Chem., 269, 22779–22787. [PubMed] [Google Scholar]
- 2.Streuli C.H., Schmidhauser,C., Bailey,N., Yurchenco,P., Skubitz,A.P., Roskelley,C. and Bissell,M.J. (1995) Laminin mediates tissue-specific gene expression in mammary epithelia. J. Cell Biol., 129, 591–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Colognato H. and Yurchenco,P.D. (2000) Form and function: the laminin family of heterotrimers. Dev. Dyn., 218, 213–234. [DOI] [PubMed] [Google Scholar]
- 4.Aberdam D., Virolle,T. and Simon-Assmann,P. (2000) Transcriptional regulation of laminin gene expression. Microsc. Res. Tech., 51, 228–237. [DOI] [PubMed] [Google Scholar]
- 5.Engel J. (1992) Laminins and other strange proteins. Biochemistry, 31, 10643–10651. [DOI] [PubMed] [Google Scholar]
- 6.Miner J.H., Patton,B.L., Lentz,S.I., Gilbert,D.J., Snider,W.D., Jenkins,N.A., Copeland,N.G. and Sanes,J.R. (1997) The laminin alpha chains: expression, developmental transitions and chromosomal locations of alpha1–5, identification of heterotrimeric laminins 8–11 and cloning of a novel alpha3 isoform. J. Cell Biol., 137, 685–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ekblom M., Falk,M., Salmivirta,K., Durbeej,M. and Ekblom,P. (1998) Laminin isoforms and epithelial development. Ann. N. Y. Acad. Sci., 857, 194–211. [DOI] [PubMed] [Google Scholar]
- 8.Carter W.G., Ryan,M.C. and Gahr,P.J. (1991) Epiligrin, a new cell adhesion ligand for integrin alpha 3 beta 1 in epithelial basement membranes. Cell, 65, 599–610. [DOI] [PubMed] [Google Scholar]
- 9.Aberdam D., Aguzzi,A., Baudoin,C., Galliano,M.F., Ortonne,J.P. and Meneguzzi,G. (1994) Developmental expression of nicein adhesion protein (laminin-5) subunits suggests multiple morphogenic roles. Cell. Adhes. Commun., 2, 115–129. [DOI] [PubMed] [Google Scholar]
- 10.Nguyen B.P., Ryan,M.C., Gil,S.G. and Carter,W.G. (2000) Deposition of laminin 5 in epidermal wounds regulates integrin signaling and adhesion. Curr. Opin. Cell Biol., 12, 554–562. [DOI] [PubMed] [Google Scholar]
- 11.Rousselle P., Lunstrum,G.P., Keene,D.R. and Burgeson,R.E. (1991) Kalinin: an epithelium-specific basement membrane adhesion molecule that is a component of anchoring filaments. J. Cell Biol., 114, 567–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Aberdam D., Galliano,M.F., Vailly,J., Pulkkinen,L., Bonifas,J., Christiano,A.M., Tryggvason,K., Uitto,J., Epstein,E.H.,Jr, Ortonne,J.P. et al. (1994) Herlitz’s junctional epidermolysis bullosa is linked to mutations in the gene (LAMC2) for the gamma 2 subunit of nicein/kalinin (LAMININ-5). Nature Genet., 6, 299–304. [DOI] [PubMed] [Google Scholar]
- 13.Vailly J., Gagnoux-Palacios,L., Dell’Ambra,E., Romero,C., Pinola,M., Zambruno,G., De Luca,M., Ortonne,J.P. and Meneguzzi,G. (1998) Corrective gene transfer of keratinocytes from patients with junctional epidermolysis bullosa restores assembly of hemidesmosomes in reconstructed epithelia. Gene Ther., 5, 1322–1332. [DOI] [PubMed] [Google Scholar]
- 14.Vidal F., Aberdam,D., Miquel,C., Christiano,A.M., Pulkkinen,L., Uitto,J., Ortonne,J.P. and Meneguzzi,G. (1995) Integrin beta 4 mutations associated with junctional epidermolysis bullosa with pyloric atresia. Nature Genet., 10, 229–234. [DOI] [PubMed] [Google Scholar]
- 15.Kainulainen T., Hakkinen,L., Hamidi,S., Larjava,K., Kallioinen,M., Peltonen,J., Salo,T., Larjava,H. and Oikarinen,A. (1998) Laminin-5 expression is independent of the injury and the microenvironment during reepithelialization of wounds. J. Histochem. Cytochem., 46, 353–360. [DOI] [PubMed] [Google Scholar]
- 16.Zhang K. and Kramer,R.H. (1996) Laminin 5 deposition promotes keratinocyte motility. Exp. Cell Res., 227, 309–322. [DOI] [PubMed] [Google Scholar]
- 17.Lampe P.D., Nguyen,B.P., Gil,S., Usui,M., Olerud,J., Takada,Y. and Carter,W.G. (1998) Cellular interaction of integrin alpha3beta1 with laminin 5 promotes gap junctional communication. J. Cell Biol., 143, 1735–1747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bonkhoff H. (1998) Analytical molecular pathology of epithelial–stromal interactions in the normal and neoplastic prostate. Anal. Quant. Cytol. Histol., 20, 437–442. [PubMed] [Google Scholar]
- 19.Borradori L. and Sonnenberg,A. (1999) Structure and function of hemidesmosomes: more than simple adhesion complexes. J. Invest. Dermatol., 112, 411–418. [DOI] [PubMed] [Google Scholar]
- 20.Pyke C., Salo,S., Ralfkiaer,E., Romer,J., Dano,K. and Tryggvason,K. (1995) Laminin-5 is a marker of invading cancer cells in some human carcinomas and is coexpressed with the receptor for urokinase plasminogen activator in budding cancer cells in colon adenocarcinomas. Cancer Res., 55, 4132–4139. [PubMed] [Google Scholar]
- 21.Sordat I., Bosman,F.T., Dorta,G., Rousselle,P., Aberdam,D., Blum,A.L. and Sordat,B. (1998) Differential expression of laminin-5 subunits and integrin receptors in human colorectal neoplasia. J. Pathol., 185, 44–52. [DOI] [PubMed] [Google Scholar]
- 22.Grassi M., Girault,J.M., Wang,W.P., Thiery,J.P. and Jouanneau,J. (1999) Metastatic rat carcinoma cells express a new retrotransposon. Gene, 233, 59–66. [DOI] [PubMed] [Google Scholar]
- 23.Mizushima H., Miyagi,Y., Kikkawa,Y., Yamanaka,N., Yasumitsu,H., Misugi,K. and Miyazaki,K. (1996) Differential expression of laminin-5/ladsin subunits in human tissues and cancer cell lines and their induction by tumor promoter and growth factors. J. Biochem. (Tokyo), 120, 1196–1202. [DOI] [PubMed] [Google Scholar]
- 24.Korang K., Christiano,A.M., Uitto,J. and Mauviel,A. (1995) Differential cytokine modulation of the genes LAMA3, LAMB3 and LAMC2, encoding the constitutive polypeptides, alpha 3, beta 3 and gamma 2, of human laminin 5 in epidermal keratinocytes. FEBS Lett., 368, 556–558. [DOI] [PubMed] [Google Scholar]
- 25.Virolle T., Djabari,Z., Ortonne,J.P. and Aberdam,D. (2000) DNA conformation driven by AP-1 triggers cell-specific expression via a strong epithelial enhancer. EMBO Rep., 4, 328–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Miquel C., Gagnoux-Palacios,L., Durand-Clement,M., Marinkovich,P., Ortonne,J.P. and Meneguzzi,G. (1996) Establishment and characterization of cell line LSV5 that retains the altered adhesive properties of human junctional epidermolysis bullosa keratinocytes. Exp. Cell Res., 224, 279–290. [DOI] [PubMed] [Google Scholar]
- 27.Blackwell T.K., Huang,J., Ma,A., Kretzner,L., Alt,F.W., Eisenman,R.N. and Weintraub,H. (1993) Binding of myc proteins to canonical and noncanonical DNA sequences. Mol. Cell. Biol., 13, 5216–5224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Malyankar U.M., Hanson,R., Schwartz,S.M., Ridall,A.L. and Giachelli,C.M. (1999) Upstream stimulatory factor 1 regulates osteopontin expression in smooth muscle cells. Exp. Cell Res., 250, 535–547. [DOI] [PubMed] [Google Scholar]
- 29.Galibert M.D., Carreira,S. and Goding,C.R. (2001) The Usf-1 transcription factor is a novel target for the stress-responsive p38 kinase and mediates UV-induced tyrosinase expression. EMBO J., 20, 5022–5031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ayer D.E., Kretzner,L. and Eisenman,R.N. (1993) Mad: a heterodimeric partner for Max that antagonizes Myc transcriptional activity. Cell, 72, 211–222. [DOI] [PubMed] [Google Scholar]
- 31.Beckmann H., Su,L.K. and Kadesch,T. (1990) TFE3: a helix–loop–helix protein that activates transcription through the immunoglobulin enhancer muE3 motif. Genes Dev., 4, 167–179. [DOI] [PubMed] [Google Scholar]
- 32.Blackwood E.M. and Eisenman,R.N. (1991) Max: a helix–loop–helix zipper protein that forms a sequence-specific DNA-binding complex with Myc. Science, 251, 1211–1217. [DOI] [PubMed] [Google Scholar]
- 33.Gregor P.D., Sawadogo,M. and Roeder,R.G. (1990) The adenovirus major late transcription factor USF is a member of the helix–loop–helix group of regulatory proteins and binds to DNA as a dimer. Genes Dev., 4, 1730–1740. [DOI] [PubMed] [Google Scholar]
- 34.Sun X.H. and Baltimore,D. (1991) An inhibitory domain of E12 transcription factor prevents DNA binding in E12 homodimers but not in E12 heterodimers. Cell, 64, 459–470. [DOI] [PubMed] [Google Scholar]
- 35.Luo X. and Sawadogo,M. (1996) Functional domains of the transcription factor USF2: atypical nuclear localization signals and context-dependent transcriptional activation domains. Mol. Cell. Biol., 16, 1367–1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Roy A.L., Du,H., Gregor,P.D., Novina,C.D., Martinez,E. and Roeder,R.G. (1997) Cloning of an inr- and E-box-binding protein, TFII-I, that interacts physically and functionally with USF1. EMBO J., 16, 7091–7104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bendall A.J. and Molloy,P.L. (1994) Base preferences for DNA binding by the bHLH-Zip protein USF: effects of MgCl2 on specificity and comparison with binding of Myc family members. Nucleic Acids Res., 22, 2801–2810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fisher D.E., Carr,C.S., Parent,L.A. and Sharp,P.A. (1991) TFEB has DNA-binding and oligomerization properties of a unique helix–loop–helix/leucine-zipper family. Genes Dev., 5, 2342–2352. [DOI] [PubMed] [Google Scholar]
- 39.Desbarats L., Gaubatz,S. and Eilers,M. (1996) Discrimination between different E-box-binding proteins at an endogenous target gene of c-myc. Genes Dev., 10, 447–460. [DOI] [PubMed] [Google Scholar]
- 40.Boyd K.E., Wells,J., Gutman,J., Bartley,S.M. and Farnham,P.J. (1998) c-Myc target gene specificity is determined by a post-DNA binding mechanism. Proc. Natl Acad. Sci. USA, 95, 13887–13892. [DOI] [PMC free article] [PubMed] [Google Scholar]