

Abstract
The p16/RB/E2F regulatory pathway, which controls transit through the G1 restriction point of the cell cycle, is one of the most frequent targets of genetic alterations in human cancer. Any of these alterations results in the deregulated expression of the transcription factor E2F, one of the key mediators of cell cycle progression. Under these conditions, E2F1 also participates in the induction of apoptosis by a p53-dependent pathway, and independently of p53. Recently, we identified the p53-homolog p73 as a first direct target of p53-independent apoptosis. Here, we used a cDNA microarray to screen an inducible E2F1-expressing Saos-2 cell line for E2F1 target genes. Expression analysis by cDNA microarray and RT–PCR revealed novel E2F1 target genes involved in E2F1-regulated cellular functions such as cell cycle control, DNA replication and apoptosis. In addition, the identification of novel E2F1 target genes participating in the processes of angiogenesis, invasion and metastasis supports the view that E2F1 plays a central role in many aspects of cancer development. These results provide new insight into the role of E2F1 in tumorigenesis as a basis for the development of novel anti-cancer therapeutics.
INTRODUCTION
The balance between cell survival and cell death is critical for many aspects of the homeostasis of multicellular organisms. Compared with normal cells, tumor cells show a loss of these regulatory functions, which results in uncontrolled proliferation and genetic instability (1). In mammalian cells, the decision of whether to undergo DNA synthesis or to stop cell proliferation is made at the G1/S phase transition of the cell cycle (2). A number of cellular proteins such as the positively acting cyclins and cyclin-dependent kinases (CDKs), or the negatively acting cyclin-dependent kinase inhibitors (CDKIs) govern cell cycle progression by controlling the activity of the retinoblastoma (RB) protein through phosphorylation (3). Consistent with its role as a tumor suppressor, virtually all human cancers are associated with alterations in the RB pathway, either through inactivation of RB itself or the CDKI p16INK4a, or through overexpression of cyclin D1 and CDK4 oncoproteins (3–5). In this pathway, the E2F transcription factor is a key downstream target of RB. Hypophosphorylated RB binds E2F and thereby down-regulates E2F activity, suggesting a model in which RB restricts cell cycle progression by restraining E2F (6–8). In fact, the interaction of RB with E2F correlates with the capacity of RB to arrest cell growth in the G1 phase (9). On the other hand, loss of RB-mediated control of E2F activity leads to progression into DNA synthesis (10).
E2F DNA-binding sites have been identified in the promoter regions of many genes involved in DNA replication [e.g. dihydrofolate reductase (DHFR), DNA polymerase, thymidine kinase, thymidylate synthetase, ORC1 and CDC6] and cell cycle control [e.g. cyclin E (CCNE1), cyclin A, CDC2, CDC25A, p107, RB, c-Myc, N-Myc, B-Myb, E2F-1 and E2F-2] (8,11). So far, six members of the E2F family, E2F1–E2F6, have been cloned and molecularly characterized (12). All of them contain highly conserved regions encoding functional domains that are responsible for sequence-specific DNA-binding and heterodimerization with DP-family proteins. Association of E2Fs with one of the two DP proteins is necessary for high affinity, sequence-specific DNA binding, and in the case of E2F1–E2F5, binding to RB-family members (6,13). High-level expression of E2F or DP proteins can cause cell cycle progression and oncogenic transformation. Although the exact mechanism by which E2F activates transcription is still unknown, in vitro studies revealed that E2F1 can bind to TBP (14) and biochemical analysis showed an interaction between the transcriptional activation domain of E2F1 and CBP (15), potentially recruiting histone acetylase activity to the promoter. Overall, these studies argue that E2F plays a central role in orchestring cell cycle progression by integrating the processes that regulate G1/S phase transition with the transcription apparatus.
Despite the clear importance in allowing cell cycle progression, several studies have suggested a role for E2F1 in apoptosis under conditions of deregulated expression, for example by deletion of RB (16–20). In mice, interference with the regulation of E2F1 provided by RB results in unregulated cell proliferation and apoptosis (21,22). In many cells the bulk of E2F1-induced apoptosis appears to be p53 dependent (16,20). Ectopic expression of E2F1 has been shown to lead to increased levels of p53 (23,24), as a result of E2F1-mediated induction of p19ARF that in turn blocks MDM2-associated degradation of p53 (19,25–27). However, E2F1-induced apoptosis occurs also independent of p53 in tissue culture and transgenic mice (8,28–30), and RB has been shown to protect p53-null cells from apoptosis in an E2F1-binding-dependent manner (31). Mapping studies revealed that the apoptotic functions of E2F1 in the absence of p53 requires the DNA-binding domain but not the transcriptional activation domain (28,29,32), suggesting that pro-apoptotic E2F1 target genes are activated by removal of E2F1/RB repression rather than direct transactivation (4,28,29,33). The role of E2F1 as a direct tumor suppressor was supported by the observation that E2F1-deficient mice show an impaired apoptotic function and increased incidence of cancer development (19,34,35). Overexpression of E2F in Drosophila revealed an induction of the ‘reaper’ gene, known as a regulator of cell death, which leads to apoptosis (36), implicating the existence of a number of E2F1-induced apoptosis genes also in mammalian cells. We have recently identified the p53-homolog p73 as a first target of p53-independent apoptosis, which is directly activated by E2F1 (37). Linking deregulated E2F1 activity to the activation of genes such as p73 might constitute a p53-independent, anti-tumorigenic safeguard mechanism that has direct implications for the development of novel anti-cancer therapeutics to treat cancer cells lacking functional p53.
To search for p53-independently activated E2F1 target genes, we used p53-negative Saos-2 cells to establish a 4-hydroxytamoxifen (4-OHT)-inducible cell line by fusion of E2F1 to the murine estrogen receptor (ER) ligand binding domain which permits conditional activation of E2F1 and allows us to distinguish between direct and indirect targets. cDNA-microarray screens combined with RT–PCR analysis revealed novel E2F1 target genes involved in multiple cellular functions such as cell cycle control and growth regulation, apoptosis, angiogenesis, invasion and metastasis. These results provide insight into the basis for a better understanding of the role of E2F1 in tumorigenesis as a basis for the development of novel anti-cancer therapeutics.
MATERIALS AND METHODS
Cell culture
Retrovirally infected Saos-2 ER–E2F1 cells which have been described previously (37) and VH6 human primary foreskin fibroblasts (obtained from M. Roggendorf, University of Essen) were maintained in Dulbecco’s modified Eagle medium (DMEM; Life Technologies, Karslruhe, Germany) supplemented with 10% fetal calf serum (FCS; Biochrom). Media were supplemented with 2 mM l-glutamine, 100 mg/ml penicillin and 100 U/ml streptomycin (LifeTechnologies, Berlin, Germany). For serum-starvation conditions cells were grown in media containig 0.1% FCS for 24 h. E2F1 activity was induced by the addition of 4-OHT at a final concentration of 1 µM or by fresh media containing 15% FCS. Cycloheximide (CHX; Sigma) was used at a final concentration of 10 µg/ml. VH6 fibroblasts were infected by AdER–E2F1 as described (38).
Microarray analysis
For microarray analysis total RNA was extracted from Saos-2 ER–E2F1 cells treated for 8 h with either 1 µM 4-OHT or ethanol as a control using RNeasy Mini Kit (Qiagen, Hilden, Germany). Poly(A+) RNA was purified with Oligotex™ (Qiagen). For hybridization the probes were labeled with [33P]dATP using the Strip-EZ™ RT kit according to the manufacturer’s protocol (Ambion, Austin, TX). Finally, two identical filters (Human LifeGrid™ 1.0) were hybridized according to the supplier’s protocol (Incyte Genomics, Palo Alto, CA). The labeling efficiency was determined using a Tri-Carb 2100 TR (Canberra-Packard GmbH; Dreieich, Gemany) scintillation counter and calculated as suggested by Incyte Genomics. Images were obtained on a Fujifilm BAS-1500 scanner and analysis was performed by Incyte Genomics using ArrayVision software.
Immunofluorescence
Cells were grown on coverslips to 60–80% confluence. Cell were serum-starved for 24 h, activation of the ER–E2F1 fusion protein was induced by 4-OHT for 8 h. For E2F1 staining, cells were subsequently fixed and permeabilized in –20°C cold methanol for 10 min. Coverslips were air dried and stained with the mouse monoclonal anti-HA antibody F-7 (Santa Cruz Biotechnology, Heidelberg, Germany), followed by a goat anti-mouse Cy3-conjugated antibody (Jackson ImmunoResearch Laboratories, Dianova, Hamburg, Germany).
Semi-quantitative RT–PCR
RT–PCR was performed on total RNA prepared by RNeasy Mini Kit (Qiagen). Following DNase I treatment, 1 µg RNA was reverse transcribed using Omniscript RT (Qiagen) and Oligo-dT. PCR amplification was performed as described previously (37). A minimum amount of cycles was carried out to stay within the linear amplification process. Used primer sequences can be obtained on request.
Flow cytometry
For flow cytometry ER–E2F1-expressing Saos-2 cells were incubated in the absence or presence of 1 mM 4-OHT. Cells were harvested 48 h after induction, fixed in 70% ethanol and stained for DNA content with propidium iodide. Flow cytometric analysis was carried out (FACSVantage, Becton Dickinson) and analyzed as described (38) using CellQuest software (Becton Dickinson).
RESULTS
Characterization of the Saos-2 ER–E2F1-inducible cell line
We and others have previously shown that the post-translational regulation of E2F1 by fusion to the hormone-binding domain of the ER is a useful technique to analyze the functional consequences of deregulated E2F1 expression (37–40). The ER–E2F1 fusion protein is inactive in the absence of the synthetic ligand 4-OHT and becomes rapidly activated after addition of 4-OHT by allowing translocation from the cytosol to the nucleus (38,39). To identify E2F1 target genes, we generated a 4-OHT-inducible Saos-2 cell line by infection with a retrovirus encoding ER–E2F1 (37) (Fig. 1A). Ligand-dependent activation of E2F1 in the Saos-2 cell line was initially tested by immunofluorescence analyis of the subcellular localization of ER–E2F1. As shown in Figure 1B (top left), in the absence of ligand the fusion protein is located in the cytoplasm, while 8 h after addition of 4-OHT, ER–E2F1 was exclusively detected in the nucleus (Fig. 1B, top right), indicating that the ER–E2F1 fusion protein is correctly translocated from the cytosol to the nucleus upon activation. After 48 h of induction, morphological changes were observed only in the presence of 4-OHT, with cells rounding up at day 2 (Fig. 1B, middle right), characteristic for cells undergoing apoptosis. Flow cytometry analysis showed an increasing amount of cells with a sub-G1 DNA content (Fig. 1B, bottom right), indicative of apoptosis, whereas in the absence of 4-OHT, no significant increase in the sub-G1 population was observed (Fig. 1B, bottom left). Next, we determined whether E2F1 expression upon activation of the inducible Saos-2 cell line leads to the up-regulation of known E2F1 target genes. Semi-quantitative RT–PCR analysis revealed a strong mRNA increase of cyclin E (CCNE1) (6,8) and of the pro-apoptotic gene TP73, recently shown to be an E2F1 target (37,41). Expression was detected as early as 4 h after induction, reaching maximum levels by 8 h following activation (Fig. 1C). Based on these data, we have chosen the 8-h time point as the optimum duration of 4-OHT treatment before isolation of RNA for array hybridization.
Figure 1.

Regulation of E2F1 activity in the inducible Saos-2 ER–E2F1 cell line. (A) Schematic model of ER–E2F1 induction by 4-OHT. Upon ligand-dependent activation of ER–E2F1, constitutively expressed fusion proteins translocate from the cytoplasm into the nucleus. (B) Functional characterization of the inducible system. Cells were grown for 8 h (immunofluorescence) and 48 h (morphology and FACS), respectively, in the absence or presence of 4-OHT. Nuclear localization (top) was determined by using an anti-HA antibody (F-7). Induction of E2F1 is associated with morphological changes (middle) and by accumulation of cells with a sub-G1 DNA content in FACS analysis consistent with apoptosis. FACS profiles showing DNA content (x-axis) against cell number (y-axis). (C) Semi-quantitative RT–PCR analysis of cyclin E (CCNE1), TP73 and GAPDH mRNA levels in serum-starved Saos-2 ER–E2F1 grown in the presence of 4-OHT for the time indicated.
Identification of novel E2F1-regulated target genes
Given the central role of E2F in tumorigenesis, further elucidation of E2F1-regulated targets will help to better understand the molecular scenario controlled by E2F1 and possibly provide the basis for the identification of novel gene therapeutics for anti-cancer treatment. To assess changes in mRNA expression after E2F1 activation, we used the Human LifeGrid 1.0 cDNA microarray carrying 8400 cDNAs and ESTs. For microarray analysis, hybridization probes were prepared from total RNA isolated from Saos-2 ER–E2F1 cells treated for 8 h with 4-OHT or untreated cells as a control. We found that 470 genes were significantly up-regulated in Saos-2 cells following E2F1 activation. From these genes, we randomly selected four known E2F1 target genes (CCND1, CCNE1, CCNE2 and MAP3K5) as internal controls, and 30 additional potentially E2F1-regulated genes representing a spectrum from moderately activated (∼2-fold increase) to strongly activated (∼30-fold induction) for verification analysis by RT–PCR (Table 1).
Table 1. Classification of E2F1 target genes verified by RT–PCR.

GenBank numbers are as given by the array supplier. Unigene entries and gene symbols are given as found at http://www.ncbi.nlm.nih.gov/UniGene
+/– 4-OHT ratio refers to the microarray measurements; d, direct target; i, indirect target; n.d., not determined; n.a., not applicable.
*TP73 was not present on the microarray; it has been used a positive control for verification experiments.
Verification of microarray analysis by RT–PCR
This group of known and putative E2F1-regulated genes was confirmed by RT–PCR using gene-specific primers. For RT–PCR total RNA was prepared from the ER–E2F1 expressing Saos-2 cell line at 0, 4, 8 and 24 h after 4-OHT treatment. All of the known target genes and 24 of the 30 randomly selected putative genes (80%) that showed increased expression levels in the microarray screen were confirmed as significantly up-regulated by E2F1 (mRNA levels of 13 representative targets are shown in Fig. 2; see also Table 1). Six genes (CLU, CRADD, FAT, HLA-DMA, MAPK12 and MAPK14) could not be verified by RT–PCR. As shown in Figure 2, the fold induction of gene expression as calculated by the ArrayVision software parallels the levels of induction observed by RT–PCR as detected for KIA0767, KIA0455, RAD52, STK15, MAP3K14, MMP16, RFC3, FGF-2, IFNA2, BAK1, PAWR, BAD and BID at 8 h after ER–E2F1 activation. None of the genes with an induction of <2-fold in the microarray screen could be verified as an E2F1 target gene by RT–PCR, suggesting that a cut-off of 2-fold is a reliable limit to ensure a maximum number of positive genes.
Figure 2.

Verification of microarray analysis by RT–PCR. Serum-starved Saos-2 cells stably transfected with ER–E2F1 were grown in the presence of 4-OHT for the time indicated. Semi-quantitative RT–PCR analysis of potential E2F1 targets including TP73 as a control and GAPDH expression was carried out under linear amplification conditions.
Selection of direct and indirect E2F1 targets
As an advantage of the post-translational regulatory system, it enables us to measure the effect of E2F1 activation in the absence of de novo protein synthesis, thus allowing the identification of directly activated transcripts (8). To test whether the genes identified as E2F1 targets are directly or indirectly regulated by E2F1, we performed RT–PCR on ER–E2F1-expressing Saos-2 cells grown in the presence of the protein synthesis inhibitor CHX alone, CHX plus 4-OHT or 4-OHT alone. An analysis of a subset of genes is shown in Figure 3. By addition of 4-OHT, no increase in expression intensity was observed for KIAA0455, STK15, MMP16 and RFC3 in cells grown in the presence of CHX, suggesting that in these cases E2F1-induced synthesis of other proteins is required for the stimulation of gene expression (compare lanes 2 and 4). As shown for the primary E2F1 targets, cyclin E (CCNE1) and TP73 (6,8,37), a significant induction of RNA expression was observed for KIAA0767, RAD52 and MAP3K14 by addition of 4-OHT even in the presence of CHX. Based on these data, these genes appear to be direct targets of E2F1 (Fig. 3). However, the addition of CHX alone leads to an increase in mRNA levels, which is consistent with previous data and is most likely due to a stabilization of RNA.
Figure 3.
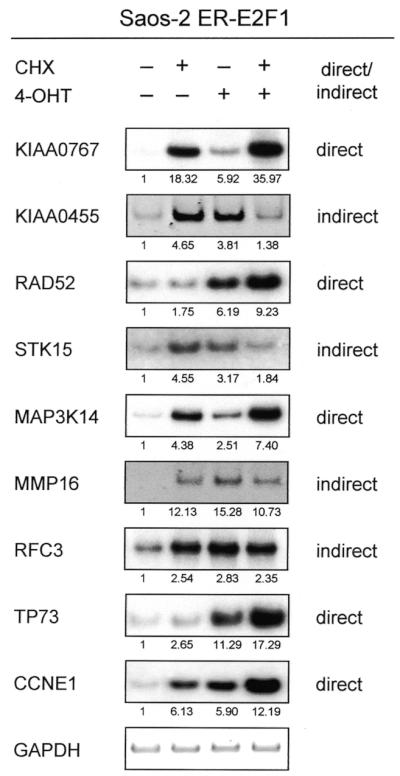
Classification of E2F1 targets as direct and indirect. Semi-quantitative RT–PCR analysis of different E2F1 regulated genes in serum-starved Saos-2 cells expressing ER–E2F1 grown in the presence of 4-OHT and/or CHX as indicated. All targets are shown 4 h after induction. KIAA0455, STK15, MMP16 and RFC3 are indirect targets, whereas KIAA0767, RAD52 and MAP3K14, as well as the controls TP73 and CCNE1 are direct targets. PCR products were quantified in relative software units by the Bio-Imaging-Analyzer (Fuji) using the TINA program version 2.09 (shown as fold induction). The data were normalized to GAPDH values and the untreated control was set as 1.
Effect of E2F1 in primary cells and by endogenous E2F1
To exclude that the effects seen with overexpressed E2F1 in Saos-2 cells are limited to this system or tumor cells, we infected primary VH6 fibroblasts with an adenoviral vector encoding the ER–E2F1 fusion protein as described previously (38). Subsequently, RT–PCR was performed on infected fibroblasts that had been serum starved and then induced with 4-OHT using primers for a subset of genes that were found to be up-regulated by ER–E2F1 in Saos-2 cells (Fig. 4). In summary, 9 of 11 selected genes induced by Saos-2 ER–E2F1 cells were also significantly up-regulated in primary cells, implying that E2F1 has a general impact on gene expression independent of whether or not cells are transformed, resulting in the modulation of a common pattern of targets. Moreover, to ensure that the identified targets are not an effect of E2F1 overexpression but can also be induced by endogenous E2F1 levels, transcripts were analyzed in Saos-2 ER–E2F1 cells grown in the absence of serum and following serum induction with 15% FCS (Fig. 5). As shown for all target genes analyzed, expression can be substantially induced by serum to levels comparable with the fold changes calculated from the array screen following ER–E2F1 activation by 4-OHT. However, it should be mentioned that the serum induction experiment provides a hint, but certainly no firm evidence, that the identified genes are regulated by endogenous E2F1 proteins.
Figure 4.

E2F1-induced changes in transcript levels in normal cells. Semi-quantitative RT–PCR analysis of KIAA0767, KIAA0455, RAD52, STK15, MAP3K14, MMP16, RFC3, ARGHGAP4 and VEGF-B expression in VH6 fibroblasts. Cells were serum-starved, infected with 100 MOI of AdERE2F-1, and grown in the presence of 4-OHT for indicated time points. TP73 was used as a positive control.
Figure 5.
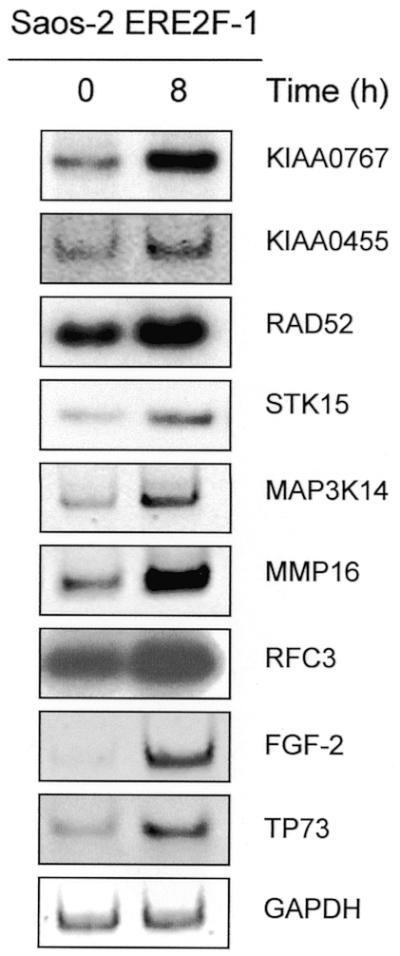
Activation of endogenous E2F1 is sufficient to induce target genes. Semi-quantitative RT–PCR analysis on total RNA of candidate E2F1 target genes (and TP73 as a control) following induction of endogenous E2F1 with serum. Cells were serum-starved for 24 h and subsequently grown in the presence of 15% FCS as indicated.
E2F1 regulates genes with multiple cellular functions
E2F plays an essential role in monitoring conditions of cell cycle progression, but also has apoptotic activity in response to perturbation of normal cell proliferation, consistent with its tumor suppressor function (6,28,29). Among the genes known to be involved in cell cycle control and DNA replication, we identified ARHGAP4, RFC3, RAD52, STK15 and TNFSF9 as E2F1 target genes. Regarding the role of E2F1 in apoptosis, we found that cellular factors such as the pro-apoptotic Bcl-2 family members BAD, BID and BAK1, or the PRKC apoptosis WT1 regulator PAWR, as well as the apoptosis inhibitors MAP3K14 (Fig. 2) and CFLAR (FLIP) might be involved in E2F1-regulated death. Besides these regulatory properties, our study supports the idea that E2F1 fulfils additional cellular functions important for tumorigenesis. For example, several genes associated with the process of angiogenesis showed an increased expression following E2F1 activation (e.g. FGF-2, VEGF-B) (Figs 2 and 4). Another interesting E2F1 target gene is the matrixmetalloproteinase MMP16, as shown in Figures 2, 3 and 5. Twenty-nine verified E2F1 target genes classified according to their cellular functions are shown in Table 1. To be undoubtedly verified as an E2F target, however, our findings by microarray analysis and RT–PCR have to be confirmed by other methods such as chromatin immunoprecipitation (ChIP) in preferentially primary untransfected cells.
DISCUSSION
Over the past decade, a large number of studies revealed the central role of the RB pathway in the regulation of G1/S transition and the control of cell proliferation by modulating the activity of the transcription factor E2F. From these studies it has become clear that E2F drives cell cycle progression through the induction of S-phase genes that encode cell cycle regulatory functions and DNA replication activities (6,7). In addition, it has been shown that the RB pathway is linked to events involved in an anti-tumor safeguard mechanism through the induction of apoptosis which is largely mediated by E2F1. E2F1 promotes apoptosis in several systems, either in association with p53 (16,18) or independent of p53 (28,29). Our group and others have recently shown that part of the apoptotic activity of E2F1 reflects the ability to induce p73 transcription (37,41,42). However, given the important role of individual activities within the pathway affecting cell growth control and cancer development, it is reasonable to assume that E2F1 likely participates in additional cellular functions, implying that the majority of E2F1 target genes is still unknown.
Supporting this view, our cDNA microarray screen using RNA from a 4-OHT-inducible Saos-2 cell line as a probe revealed that increased E2F1 activity leads to the induction of the expression of a large number of genes. These include genes that encode cell cycle regulatory activities and those which are involved in DNA replication and DNA repair. Moreover, from our analysis it becomes clear that E2F1 is specifically involved in additional cellular activities such as apoptosis and angiogenesis, as well as in the process of invasion and metastasis. We found that increased expression of E2F1 in Saos-2 cells resulted in a reproducible up-regulation of 24 genes, so far not known as E2F1-regulated targets.
A variety of results pointed to E2F1 as a critical determinant of the G1/S-phase transition during the cell cycle. E2F1 directly transactivates genes that are necessary for DNA synthesis, and promotes cell cycle progression via the induction of cyclin D1 (CCND1) and cyclin E (CCNE1) (43–45). The expression of both genes was also found to be induced in our screen following E2F1 activation. Besides these known E2F1 targets involved in cell cycle regulation, we detected an increase in the expression of cyclin E2 (CCNE2), which has recently been shown to be up-regulated by all three E2Fs (E2F1–E2F3) in human U2OS cells (40). In addition, we identified several novel target genes coding for components of the DNA replication and/or repair machinery [e.g. replication factor C 3 (RFC3) and RAD52]. The human replication factor C (RFC) is a five-subunit DNA polymerase accessory protein that functions as a structure-specific, DNA-dependent ATPase. Through the action of RFC, the proliferating cell nuclear antigen (PCNA) is loaded onto DNA in an ATP-dependent reaction (46,47). A previous analysis of the interaction between the five subunits suggested a cooperative mechanism in the assembly of the RFC complex between a three-subunit core complex, consisting of p36, p37 and p40, and the large p140 subunit (46). From this study it appears that the RFC3 p38 subunit, which we identified as a target of E2F1, is essential for the core/p140 interaction. In addition, RFC4, coding for the 37 kDa core subunit of RFC, was recently described as an E2F1-regulated gene in Zinc induced rat-1a fibroblasts (48). Moreover, it has been suggested that E2F1 is involved in various DNA repair activities by, for example, RAD51 (5). Here, we identified RAD52 as a direct target for the E2F transcription factor 1. The DNA damage repair protein RAD52 binds specifically to single-stranded tails present at sites of resected double-strand breaks (DSBs) and is known to play an important role in the early stages of genetic recombination and DSB repair (49). Given the number of known replication-related E2F1 targets, our data support the idea that virtually the entire process involved in the regulation of the G1/S transition and the activation of DNA replication is under the control of the RB/E2F pathway.
Furthermore, the E2F1 protein can act as an oncogene and participates in transformation (50). Viewed in the context of abnormal proliferation and transformation, centrosome duplication is a key requirement for correct segregation of chromosomes during cell division. A previous study indicated that centrosome duplication in mammalian somatic cells requires E2F and Cdk2-cyclin A (51). Interestingly, in our cDNA screen the expression of STK15 (also known as BTAK and aurora2), encoding a critical centrosome associated serine/threonine kinase, was found to be strongly induced by E2F1. STK15 amplification has been previously detected in multiple human cancer cell lines and primary tumors, and overexpression was shown to be associated with the induction of centrosome duplication–distribution abnormalities, aneuploidy and transformation (52). However, we did not observe an increase in expression intensity in the presence of CHX, suggesting that STK15 is an indirect target of E2F1. Since the RB pathway is disrupted in virtually all human cancers (3,5), deregulated E2F1 activity and subsequent STK15-induced centrosome amplification may contribute to the loss of cell growth control and development of cancer.
In addition, under deregulated conditions the activity of E2F1 is linked to events that determine cell fate through induction of apoptosis, thus protecting the organism against oncogenic transformation. Many studies have shown that E2F1 promotes apoptosis in several systems, either in association with p53 or alone (16,18,28,29). Three mechanisms of E2F1-induced apoptosis have been described so far: (i) activation of the CDKN2A transcript p14ARF, resulting in p53-dependent apoptosis (27); (ii) inhibition of survival signals, making tumor cells more sensitive to apoptotic signals such as TNFα (53); and (iii) induction of p73 expression, leading to the activation of p53-responsive target genes and programmed cell death (37,41). Although p73 has been discovered as a first target, it does not fully account for E2F1-induced death in a p53-negative system. Using RNA from inducible Saos-2 cells which are negative for p53, we detected an increase of six apoptosis related genes, such as the pro-apoptotic Bcl-2 family members BAD, BAK1 and BID, the caspase-8 and FADD-like apoptosis regulator CFLAR (FLIP), the PRKC apoptosis WT1 regulator (PAWR), and MAP3K14, also known as the NF-κB-inducing kinase (NIK) which had not been described as E2F1 targets before (Fig. 2). Together with the previous finding, indicating that the cytochrome c (cyt c)-binding protein Apaf1 is a target for the E2F1 factor (40), which can activate caspase-9 in a cyt c-dependent manner (54), our data clearly suggest a central role for E2F1 in apoptosis by both the mitochondrial pathway and the death receptor pathway through (i) release of cyt c from the mitochondria by regulating pro-apoptotic members of the Bcl-2 family; (ii) induction of the caspase-8 substrate BID, which amplifies death-receptor signaling via the mitochondria, resulting in enhanced cyt c release; and (iii) upon inhibition of death-receptor-mediated apoptosis by inducing the cellular FLICE-inhibitory protein (FLIP), which protects cells from procaspase-8-induced death (55). As mentioned above, deregulated E2F1 activity has previously been linked to TRAF2 degradation, leading to the inhibition of anti-apoptotic signaling via NF-κB (53). Interestingly, we identified the TRAF2-interacting protein NIK as a direct target of E2F1, thereby linking increased NIK activity to the inhibition of anti-apoptotic TNF-receptor signaling. Thus, it will be interesting to analyze whether increased NIK activity is responsible for TRAF2 degradation.
With regard to recent reports studying the role of E2F1 in tumor progression, it has been shown that overexpression of E2F1 is associated with increased tumor cell invasiveness and metastatic progression (56,57). Moreover, expression of the RB tumor suppressor gene inhibits tumor cell invasion in vitro (58). Here, we provide evidence that E2F1 indirectly up-regulates the expression of the membrane-type matrix metalloproteinase 16 (MMP16 or MT3-MMP) (Figs 2 and 3) which belongs to the group of MT-MMPs that activate MMP-2, a key regulator of invasion and metastasis (59).
Furthermore, angiogenesis is an essential step in tumor progression. Thus, it has been speculated that the angiogenic phenotype may involve the inactivation of the RB-pathway related tumor suppressor genes such as p16INK4a and p53. Viewed in the above context, induction of the RB-related protein p130 was previously observed to inhibit angiogenesis in vivo, correlating with the down-regulation of vascular endothelial growth factor (VEGF) expression (60). In agreement, we found a significant induction in the expression of the vascular endothelial growth factor-B (VEGF-B), an effector of blood vessel growth during development and disease (61), and the fibroblast growth factor-2 (FGF-2), strongly suggesting an additional role of E2F1 in angiogenesis. It is interesting to note that the adenovirus E1A protein, which promotes cellular proliferation by deregulation of the RB pathway, stimulates FGF-2 production and promotes differentiation of primary endothelial cells (62).
Given the critical role of E2F for normal cell proliferation and tumorigenesis, a number of recent studies were aimed at identifying E2F target genes on a genome-wide scale. Whereas Kel et al. (63) used a computer-assisted approach to identify E2F-regulated promoters based on screening genome databases for sequences with high homology to an E2F consensus site, Weinmann et al. (64) cloned novel E2F target promoters by use of ChIP as an unbiased, in vivo approach. In contrast to these studies, which identified E2F-binding genomic sequences (in silico or in vivo), several other groups used DNA microarray analysis to identify E2F-regulated genes (40,48,65). The strength of this latter approach lies in the ability to assay a very large number of potential targets in an unbiased manner (65). However, all groups have used different strategies to induce E2F activity. Whereas Kalma et al. (48) used Zinc-inducible E2F-expressing Rat-1 fibroblasts, Ishida and colleagues (65) used murine cells in which high-level E2F expression was obtained by infection with adenoviral vectors. In contrast, we regulated E2F1 activity in human cells on a post-translational level to allow classification of identified targets as direct or indirect. However, despite considerable differences in the experimental approaches and cellular systems used, there appears to be substantial overlap in the identified target genes. Although every single study has its methodical limitations and needs to be considered with appropriate caution, the sum of all data underlines the strength of microarray-based analyses to define the gene regulatory function of a specific transcription factor such as E2F1.
As a key regulator of cell cycle progression, E2F drives cells into the cell cycle, thereby activating a number of cell cycle regulated genes. These can be genes that are directly induced by E2F1, such as cyclin E (CCNE1), where E2F1 binds directly to the Cyclin E promoter (66). Furthermore, E2F1 up-regulation results in the activation of other transcription factors such as c-myc, which in turn lead to the induction of a variety of ‘secondary’ genes, so-called indirect targets of E2F1. As an advantage of our approach we are able to discriminate direct and indirect targets by analyzing induction in the presence of CHX which inhibits the activation of indirect target genes. In our experimental setting, we found direct (e.g. RAD52, CCNE1) as well as indirect E2F1 targets (e.g. MMP16).
In summary, our present analysis provides a spectrum of novel E2F1 target genes involved in multiple cellular functions which are known to be E2F1 regulated such as cell cycle control, DNA replication and apoptosis. In addition, the identification of novel E2F1 target genes participating in the processes of angiogenesis, invasion and metastasis supports the view that E2F1 plays a central role in many aspects of cancer development. Given the fact that virtually all human tumors exhibit alterations in the RB pathway, understanding the role of E2F1 in the development of human malignancies will have a profound impact on the understanding of normal cell growth and possibly on the development of novel anti-cancer therapeutics.
Acknowledgments
ACKNOWLEDGEMENT
This work was supported by grant 10-1411-PüI of the Deutsche Krebshilfe (B.M.P.).
REFERENCES
- 1.Hartwell L.H. and Kastan,M.B. (1994) Cell cycle control and cancer. Science, 266, 1821–1828. [DOI] [PubMed] [Google Scholar]
- 2.Pardee A.B. (1989) G1 events and regulation of cell proliferation. Science, 246, 603–608. [DOI] [PubMed] [Google Scholar]
- 3.Sherr C.J. (1996) Cancer cell cycles. Science, 274, 1672–1677. [DOI] [PubMed] [Google Scholar]
- 4.Sladek T.L. (1997) E2F transcription factor action, regulation and possible role in human cancer. Cell Prolif., 30, 97–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nevins J.R. (2001) The Rb/E2F pathway and cancer. Hum. Mol. Genet., 10, 699–703. [DOI] [PubMed] [Google Scholar]
- 6.Dyson N. (1998) The regulation of E2F by pRB-family proteins. Genes Dev., 12, 2245–2262. [DOI] [PubMed] [Google Scholar]
- 7.Nevins J.R. (1998) Toward an understanding of the functional complexity of the E2F and retinoblastoma families. Cell Growth Differ., 9, 585–593. [PubMed] [Google Scholar]
- 8.Helin K. (1998) Regulation of cell proliferation by the E2F transcription factors. Curr. Opin. Genet. Dev., 8, 28–35. [DOI] [PubMed] [Google Scholar]
- 9.Hiebert S.W., Chellappan,S.P., Horowitz,J.M. and Nevins,J.R. (1992) The interaction of RB with E2F coincides with an inhibition of the transcriptional activity of E2F. Genes Dev., 6, 177–185. [DOI] [PubMed] [Google Scholar]
- 10.Johnson D.G., Schwarz,J.K., Cress,W.D. and Nevins,J.R. (1993) Expression of transcription factor E2F1 induces quiescent cells to enter S phase. Nature, 365, 349–352. [DOI] [PubMed] [Google Scholar]
- 11.Slansky J.E. and Farnham,P.J. (1996) Introduction to the E2F family: protein structure and gene regulation. Curr. Top. Microbiol. Immunol., 208, 1–30. [DOI] [PubMed] [Google Scholar]
- 12.Müller H. and Helin,K. (2000) The E2F transcription factors: key regulators of cell proliferation. Biochim. Biophys. Acta, 1470, M1–M12. [DOI] [PubMed] [Google Scholar]
- 13.Lam E.W.-F. and LaThangue,N.B. (1994) DP and E2F proteins: Coordinating transcription with cell cycle progression. Curr. Opin. Cell Biol., 6, 859–866. [DOI] [PubMed] [Google Scholar]
- 14.Emili A. and Ingles,C.J. (1995) Promoter-dependent photocross-linking of the acidic transcriptional activator E2F-1 to the TATA-binding protein. J. Biol. Chem., 270, 13674–13680. [DOI] [PubMed] [Google Scholar]
- 15.Trouche D. and Kouzarides,T. (1996) E2F1 and E1A(12S) have a homologous activation domain regulated by RB and CBP. Proc. Natl Acad. Sci. USA, 93, 1439–1442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Qin X.Q., Livingston,D.M., Kaelin,W.G.,Jr and Adams,P.D. (1994) Deregulated transcription factor E2F-1 expression leads to S-phase entry and p53-mediated apoptosis. Proc. Natl Acad. Sci. USA, 9, 10918–10922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shan B. and Lee,W.H. (1994) Deregulated expression of E2F-1 induces S-phase entry and leads to apoptosis. Mol. Cell. Biol., 14, 8166–8173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wu X. and Levine,A.J. (1994) p53 and E2F-1 cooperate to mediate apoptosis. Proc. Natl Acad. Sci. USA, 91, 3602–3606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.DeGregori J., Leone,G., Miron,A., Jakoi,L. and Nevins,J.R. (1997) Distinct roles for E2F proteins in cell growth control and apoptosis. Proc. Natl Acad. Sci. USA, 94, 7245–7250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kowalik T.F., DeGregori,J., Schwarz,J.K. and Nevins,J.R. (1995) E2F1 overexpression in quiescent fibroblasts leads to induction of cellular DNA synthesis and apoptosis. J. Virol., 69, 2491–2500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Almasan A., Yin,Y., Kelly,R., Lee,Y.-H., Bradley,A., Li,W., Bertino,J.R. and Wahl,G.M. (1995) Deficiency of retinoblastoma protein leads to inappropriate S-phase entry, activation of E2F-responsive genes and apoptosis. Proc. Natl Acad. Sci. USA, 92, 5436–5440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Macleod K.F., Hu,Y. and Jacks,T. (1996) Loss of Rb activates both p53-dependent and independent cell death pathways in the developing mouse nervous system. EMBO J., 15, 6178–6188. [PMC free article] [PubMed] [Google Scholar]
- 23.Hiebert S.W., Packham,G., Strom,D.K., Haffner,R., Oren,M., Zambetti,G. and Cleveland,J.L. (1995) E2F-1:DP-1 induces p53 and overrides survival factors to trigger apoptosis. Mol. Cell. Biol., 15, 6864–6874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kowalik T.F., DeGregori,J., Leone,G., Jakoi,L. and Nevins,J.R. (1998) E2F1-specific induction of apoptosis and p53 accumulation, which is blocked by Mdm2. Cell Growth Differ., 9, 113–118. [PubMed] [Google Scholar]
- 25.de Stanchina E., McCurrach,M.E., Zindy,F. Shieh,S.Y., Ferbeyre,G., Samuelson,A.V., Prives,C., Roussel,M.F., Sherr,C.J. and Lowe,S.W. (1998) E1A signaling to p53 involves the p19(ARF) tumor suppressor. Genes Dev., 12, 2434–2442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zindy F., Eischen,C.M., Randle,D.H., Kamijo,T., Cleveland,J.L., Sherr,C.J. and Roussel,M.F. (1998) Myc signaling via the ARF tumor suppressor regulates p53-dependent apoptosis and immortalization. Genes Dev., 12, 2424–2433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bates S., Phillips,A.C., Clark,P.A., Stott,F., Peters,G., Ludwig,R.L. and Vousden,K.H. (1998) p14ARF links the tumour suppressors RB and p53. Nature, 395, 124–125. [DOI] [PubMed] [Google Scholar]
- 28.Phillips A.C., Stewart,S., Ryan,K.M., Helin,K. and Vousden,K.H. (1997) Induction of DNA synthesis and apoptosis are separable functions of E2F-1. Genes Dev., 11, 1853–1863. [DOI] [PubMed] [Google Scholar]
- 29.Hsieh J.-K., Fredersdorf,S., Kouzarides,T., Martin,K. and Lu,X. (1997) E2F-1 induced apoptosis requires DNA binding but not transactivation and is inhibited by the retinoblastoma protein through direct interaction. Genes Dev., 11, 1840–1852. [DOI] [PubMed] [Google Scholar]
- 30.Holmberg C., Helin,K., Sehested,M. and Karlstrom,O. (1998) E2F-1 induced p53-independent apoptosis in transgenic mice. Oncogene, 17, 143–155. [DOI] [PubMed] [Google Scholar]
- 31.Haas-Kogan D.A., Kogan,S.C., Levi,D., Dazin,P., Ang,A., Fung,Y.K. and Israel,M.A. (1995) Inhibition of apoptosis by the retinoblastoma gene product. EMBO J., 14, 461–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Krek W., Xu,G. and Livingston,D.M. (1995) Cyclin A-kinase regulation of E2F-1 DNA binding function underlies suppression of an S phase checkpoint. Cell, 83, 1149–1158. [DOI] [PubMed] [Google Scholar]
- 33.Zacksenhaus E., Jiang,Z., Phillips,R.A. and Gallie,B. (1996) Dual mechanisms of repression of E2F1 activity by the retinoblastoma gene product. EMBO J., 15, 5917–5927. [PMC free article] [PubMed] [Google Scholar]
- 34.Field S.J., Tsai,F.Y., Kuo,F., Zubiaga,A.M., Kaelin,W.G.,Jr, Livingston,D.M., Orkin,S.H. and Greenberg,M.E. (1996) E2F-1 functions in mice to promote apoptosis and suppress proliferation. Cell, 85, 549–561. [DOI] [PubMed] [Google Scholar]
- 35.Yamasaki L., Jacks,T., Bronson,R., Goillot,E., Harlow,E. and Dyson,N.J. (1996) Tumor induction and tissue atrophy in mice lacking E2F-1. Cell, 85, 537–548. [DOI] [PubMed] [Google Scholar]
- 36.Asano M., Nevins,J.R. and Wharton,R. (1996) Ectopic expression induces S phase and apoptosis in Drosophila imaginal discs. Genes Dev., 10, 1422–1432. [DOI] [PubMed] [Google Scholar]
- 37.Stiewe T. and Pützer,B.M. (2000) Role of the p53 homolog p73 for E2F1-induced apoptosis. Nature Genet., 26, 464–469. [DOI] [PubMed] [Google Scholar]
- 38.Pützer B.M., Stiewe,T., Crespo,F. and Esche,H. (2000) Improved safety through tamoxifen-regulated induction of cytotoxic genes delivered by Ad vectors for cancer gene therapy. Gene Ther., 7, 1317–1325. [DOI] [PubMed] [Google Scholar]
- 39.Vigo E., Müller,H., Prosperini,E., Hateboer,G., Cartwright,P., Moroni,M.C. and Helin,K. (1999) CDC25A phosphatase is a target of E2F and is required for efficient E2F-induced S phase. Mol. Cell. Biol., 19, 6379–6395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Müller H., Bracken,A.P., Vernell,R., Moroni,M.C., Christians,F., Grassilli,E., Prosperini,E., Vigo,E., Oliner,J.D. and Helin,K. (2001) E2Fs regulate the expression of genes involved in differentiation, development, proliferation and apoptosis. Genes Dev., 15, 267–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Irwin M., Martin,M.C., Phillips,A.C., Seelan,R.S., Smith,D.I., Liu,W., Flores,E.R., Tsai,K.Y., Jacks,T., Vousden,K.H. and Kaelin,W.G.,Jr (2000) Role for the p53 homologue p73 in E2F-1-induced apoptosis. Nature, 407, 645–648. [DOI] [PubMed] [Google Scholar]
- 42.Lissy N.A., Davis,P.K., Irwin,M., Kaelin,W.G. and Dowdy,S.F. (2000) A common E2F-1 and p73 pathway mediates cell death induced by TCR activation. Nature, 407, 642–644. [DOI] [PubMed] [Google Scholar]
- 43.Ohtani K., Degregori,J. and Nevins,J.R. (1995) Regulation of the cyclin E gene by transcription factor E2F1. Proc. Natl Acad. Sci. USA, 92, 12146–12150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Inoshita S., Terada,Y., Nakashima,O., Kuwahara,M., Sasaki,S. and Marumo,F. (1999) Roles of E2F1 in mesangial cell proliferation in vitro. Kidney Int., 56, 2085–2095. [DOI] [PubMed] [Google Scholar]
- 45.Halaban R., Cheng,E., Smicun,Y. and Germino,J. (2000) Deregulated E2F transcriptional activity in autonomously growing melanoma cells. J. Exp. Med., 191, 1005–1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Uhlmann F., Cai,J., Flores-Rozas,H., Dean,F.B., Finkelstein,J., ÓDonnell,M. and Hurwitz,J. (1996) In vitro reconstitution of human replication factor C from its five subunits. Proc. Natl Acad. Sci. USA, 93, 6521–6526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gray F.C. and MacNeill,S.A. (2000) The Schizosaccharomyces pombe rfc3+ gene encodes a homologue of the human hRFC36 and Saccharomyces cerevisiae Rfc3 subunits of replication factor C. Curr. Genet., 37, 159–167. [DOI] [PubMed] [Google Scholar]
- 48.Kalma Y., Marash,L., Lamed,Y. and Ginsberg,D. (2001) Expression analysis using DNA microarrays demonstrates that E2F-1 up-regulates expression of DNA replication genes including replication protein A2. Oncogene, 20, 1379–1387. [DOI] [PubMed] [Google Scholar]
- 49.Van Dyck E., Stasiak,A.Z., Stasiak,A. and West,S.C. (1999) Binding of double-strand breaks in DNA by human Rad52 protein. Nature, 398, 728–731. [DOI] [PubMed] [Google Scholar]
- 50.Singh P., Wong,S.H. and Hong,W. (1994) Overexpression of E2F-1 in rat embryo fibroblasts leads to neoplastic transformation. EMBO J., 13, 3329–3338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Meraldi P., Lukas,J., Fry,A.M., Bartek,J. and Nigg,E.A. (1999) Centrosome duplication in mammalian somatic cells requires E2F and Cdk2-cyclin A. Nature Cell Biol., 1, 88–93. [DOI] [PubMed] [Google Scholar]
- 52.Zhou H., Kuang,J., Zhong,L., Kuo,W.-L., Gray,J.W., Sahin,A., Brinkley,B.R. and Sen,S. (1998) Tumour amplified kinase STK15/BTAK induces centrosome amplification, aneuploidy and transformation. Nature Genet., 20, 189–193. [DOI] [PubMed] [Google Scholar]
- 53.Phillips A.C., Ernst,M.K., Bates,S., Rice,N.R. and Vousden,K.H. (1999) E2F-1 potentiates cell death by blocking antiapoptotic signaling pathways. Mol. Cell, 4, 771–781. [DOI] [PubMed] [Google Scholar]
- 54.Hu Y., Benedict,M.A., Wu,D., Inohara,N. and Nunez,G. (1998) Bcl-XL interacts with Apaf-1 and inhibits Apaf-1-dependent caspase-9 activation. Proc. Natl Acad. Sci. USA, 95, 4386–4391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Krueger A., Schmitz,I., Baumann,S., Krammer,P.H. and Kirchhoff,S. (2001) Cellular FLICE-inhibitory protein splice variants inhibit different steps of caspase-8 activation at the CD95 death-inducing signaling complex. J. Biol. Chem., 276, 20633–20640. [DOI] [PubMed] [Google Scholar]
- 56.Zhang S.Y., Liu,S.C., Johnson,D.G. and Klein-Szanto,A.J.P. (2000) E2F-1 gene transfer enhances invasiveness of human head and neck carcinoma cell lines. Cancer Res., 60, 5972–5976. [PubMed] [Google Scholar]
- 57.Banerjee D., Richchard,G., Liefshitz,A., Danenberg,K., Danenberg,P.C., Danenberg,P.V., Klimstra,D., Jhanwar,S., Cordon-Cardo,C., Fong,Y., Kemeny,N. and Bertino,J.R. (2000) Levels of E2F-1 expression are higher in lung metastasis of colon cancer as compared with hepatic metastasis and correlate with levels of thymidylate synthase. Cancer Res., 60, 2365–2367. [PubMed] [Google Scholar]
- 58.Li J., Hu,S.X., Perng,G.S., Zhou,Y., Xu,K., Zhang,C., Seigne,J., Benedict,W.F. and Wu,H.J. (1996) Expression of the retinoblastoma (RB) tumor suppressor gene inhibits tumor cell invasion in vitro. Oncogene, 13, 2379–2386. [PubMed] [Google Scholar]
- 59.Ueno H., Nakamura,H., Inoue,M., Imai,K., Noguchi,M., Sato,H., Seiki,M. and Okada,Y. (1997) Expression and tissue localization of membrane-types 1, 2 and 3 matrix metalloproteinases in human invasive breast carcinoma. Cancer Res., 57, 2055–2060. [PubMed] [Google Scholar]
- 60.Claudio P.P., Stiegler,P., Howard,C.M., Bellan,C., Minimo,C., Tosi,G.M., Rak,J., Kovatich,A., De Fazio,P., Micheli,P., Caputi,M., Leoncini,L., Kerbel,R., Giordano,G.G. and Giordano,A. (2001) RB2/p130 gene-enhanced expression down-regulates vascular endothelial growth factor expression and inhibits angiogenesis in vivo. Cancer Res., 61, 462–468. [PubMed] [Google Scholar]
- 61.Bellomo D., Headrick,J.P., Silins,G.U., Paterson,C.A., Thomas,P.S., Gartside,M., Mould,A., Cahill,M.M., Tonks,I.D., Grimmond,S.M., Townson,S., Wells,C., Little,M., Cummings,M.C., Hayward,N.K. and Kay,G.F. (2000) Mice lacking the vascular endothelial growth factor-B gene (Vegfb) have smaller hearts, dysfunctional coronary vasculature and impaired recovery from cardiac ischemia. Circ. Res., 86, E29–E35. [DOI] [PubMed] [Google Scholar]
- 62.Giampietri C., Levrero,M., Felici,A., Dálessio,A., Capogrossi,M.C. and Gaetano,C. (2000) E1A stimulates FGF-2 release promoting differentiation of primary endothelia cells. Cell Death Differ., 7, 292–301. [DOI] [PubMed] [Google Scholar]
- 63.Kel A.E., Kel-Margoulis,O.V., Farnham,P.J., Bartley,S.M., Wingender,E. and Zhang,M.Q. (2001) Computer-assisted identification of cell cycle-regulated genes: new targets for E2F transcription factors. J. Mol. Biol., 309, 99–120. [DOI] [PubMed] [Google Scholar]
- 64.Weinmann A.S., Bartley,S.M., Zhang,T., Zhang,M.O. and Farnham,P.J. (2001). Use of chromatin immunoprecipitation to clone novel E2F target promoters. Mol. Cell. Biol., 21, 6820–6832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ishida S., Huang,E., Zuzan,H., Spang,R., Leone,G., West,M. and Nevins,J.R. (2001). Role for E2F in control of both DNA replication and mitotic functions as revealed from DNA microarray analysis. Mol. Cell. Biol., 21, 4684–4699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Geng Y., Eaton,E.N., Picon,M., Roberts,J.M., Lundberg,A.S., Sardet,C. and Weinberg,R.A. (1996). Regulation of cyclin E transcription by E2Fs and retinoblastoma protein. Oncogene, 12, 1173–1180. [PubMed] [Google Scholar]