

Abstract
We show that in budding yeast large rDNA deletions arise frequently and cause an increase in telomeric and mating-type gene silencing proportional to repeat loss. Paradoxically, this increase in silencing is correlated with a highly specific down-regulation of SIR2, which encodes a deacetylase enzyme required for silencing. These apparently conflicting observations suggest that a large nucleolar pool of Sir2 is released upon rDNA loss and made available for telomeric and HM silencing, as well as down-regulation of SIR2 itself. Indeed, we present evidence for a reduction in the fraction of Sir2 colocalizing with the nucleolar marker Nop1, and for SIR2 autoregulation. Despite a decrease in the fraction of nucleolar Sir2, and in overall Sir2 protein levels, short rDNA strains display normal rDNA silencing and a lifespan indistinguishable from wild type. These observations reveal an unexpectedly large clonal variation in rDNA cluster size and point to the existence of a novel regulatory circuit, sensitive to rDNA copy number, that balances nucleolar and nonnucleolar pools of Sir2 protein.
Keywords: rDNA repeats, Sir2, telomere position effect, mating-type gene silencing, autoregulation
In all rapidly growing eukaryotic cells, the transcription of ribosomal DNA (rDNA) accounts for roughly half of all nuclear RNA synthesis (for review, see Reeder 1999; Warner 1999). To keep up with this demand, cells maintain multiple copies of rRNA genes that are transcribed by an RNA polymerase (RNA polymerase I; pol I) dedicated solely to this task. In the budding yeast Saccharomyces cerevisiae, the ∼150–200 rDNA copies are present in a single head-to-tail tandem array on chromosome XII. Transcription of rDNA is associated with a distinct crescent-shaped subnuclear structure, the nucleolus, which also appears to be the major site of ribosome assembly. Despite the high demand for rRNA, only about one-half of the rDNA copies are active, even in rapidly growing cells (Dammann et al. 1993, 1995; French et al. 2003). Many studies in both plant and animal species have revealed a similar picture, which raises the question of why cells maintain an rDNA copy number in apparent approximately twofold excess over that required to meet maximal transcriptional demands. One possible explanation is that the “extra” rDNA copies protect the locus from homologous recombination events that might reduce copy number below some threshold required for viability.
The maintenance of rDNA copy number must be related, at some level, to the control of recombination within the arrays, a problem that is best understood in S. cerevisiae. Both meiotic and mitotic recombination within the rDNA cluster in yeast is repressed by Sir2 (Gottlieb and Esposito 1989; Davis et al. 2000; Kobayashi et al. 2004), a deacetylase enzyme initially identified due to its requirement for gene silencing at both telomeres and silent mating-type (HM) loci (Klar et al. 1979; Shore et al. 1984; Rine and Herskowitz 1987; Gottschling et al. 1990). Interestingly, sir2 mutants have a shortened replicative life span, apparently because mother cells accumulate toxic levels of extra-chromosomal rDNA circles (ERCs) (Sinclair and Guarente 1997). Conversely, cells containing a single extra copy of SIR2 display a significantly increased lifespan (Kaeberlein et al. 1999).
Sir2, in addition to its effect on rDNA recombination, is also required for a form of transcriptional silencing that acts on RNA polymerase II-transcribed genes that have been either inserted or transposed into the rDNA repeat cluster (Bryk et al. 1997; Fritze et al. 1997; Smith and Boeke 1997). As is the case for rDNA recombination, the nucleolar silencing role of Sir2 is also independent of the other Sir proteins, and, in fact, is antagonized by Sir4 and Sir3 (Gotta et al. 1998; Smith et al. 1998; Cockell et al. 2000). This and other observations indicate that Sir2 is limiting for both rDNA and telomeric silencing, and that the two loci compete with each other for this limiting pool of Sir2. Sir2's localization and action within the rDNA depends on its binding to the nucleolar protein Net1 (Shou et al. 1999; Straight et al. 1999; Cuperus et al. 2000; Kasulke et al. 2002), which is itself essential for normal growth (unlike Sir2), probably because of a stimulatory effect on PolI (Shou et al. 2001).
In the present study, we uncovered clonal variants, in which a large number of rDNA repeats (typically ∼50% of the total) had been spontaneously lost from the rDNA locus. These “short rDNA” strains are relatively stable both mitotically and meiotically, and grow normally. They were identified because they display increased telomeric and mating-type gene silencing compared with isogenic siblings with normal rDNA size. By comparing the transcript profiles of short and normal rDNA strains, we found a consistent and significant difference in the expression of only two genes, one of which encodes the silencing factor Sir2, which is down-regulated in strains with short rDNA. Thus, although short rDNA strains contain less total Sir2 protein than wild type, they exhibit stronger silencing at both telomeres and HM mating-type loci. We present evidence that Sir2 protein down-regulates its own expression, and propose that the increase in telomeric and HMR silencing, as well as the decrease in SIR2 transcription in short rDNA strains, are due to an increased concentration of free nuclear Sir2 after release from the nucleolus. Interestingly, despite changes in Sir2 levels and distribution, short rDNA strains display no change in either rDNA silencing or lifespan. The reduction in total Sir2 levels in short rDNA strains is likely to be an adaptive response, because these strains are hypersensitive to Sir2 overexpression compared with strains with normal rDNA copy number.
Results
Mutations mapping to the rDNA locus suppress a telomeric silencing defect
In a study of mutations affecting mating-type gene (HM locus) and telomeric silencing (telomere position effect, TPE) (Laman et al. 1995; Sussel et al. 1995), we identified a hypomorphic allele of the ribonucleotide reductase 1 (RNR1) gene (referred to here as rnr1-12) that severely reduces TPE. In order to gain some insight into the relationship between RNR1 function and silencing, we conducted a screen for extragenic suppressors of the TPE defect of an rnr1-12 mutant. To this end, we constructed an rnr1-12 strain containing an ADE2 reporter gene at a novel telomere generated by truncation of chromosome VII–L telomere at the ADH4 locus (adh4::ADE2-Tel, hereafter referred to as ADE2-Tel). Whereas wild-type colonies display a sectored phenotype due to the variegated silencing of the ADE2 reporter (Gottschling et al. 1990), the rnr1-12 strain gives rise, for the most part, to uniformly white colonies, due to the reduction of telomeric silencing. Surprisingly, however, we observed that ∼1% of the colonies derived from an rnr1-12 ADE2-tel strain display a distinctly red/white sectored phenotype, indicative of restored telomeric silencing, which is typically stable upon restreaking (Fig. 1A). Remarkably, considering the high frequency at which they arise in unmutagenized cells, the TPE suppressors behave like recessive Mendelian alleles of the same gene, which are unlinked to either the telomeric reporter gene or the RNR1 locus (data not shown). We obtained only false positives in attempts to clone the suppressor gene by complementation of the mutant phenotype (e.g., a dominant-negative C-terminal fragment of SIR4 that causes derepression at ADE2-Tel), using either centromeric or 2-μm vector libraries of yeast genomic sequences (data not shown).
Figure 1.
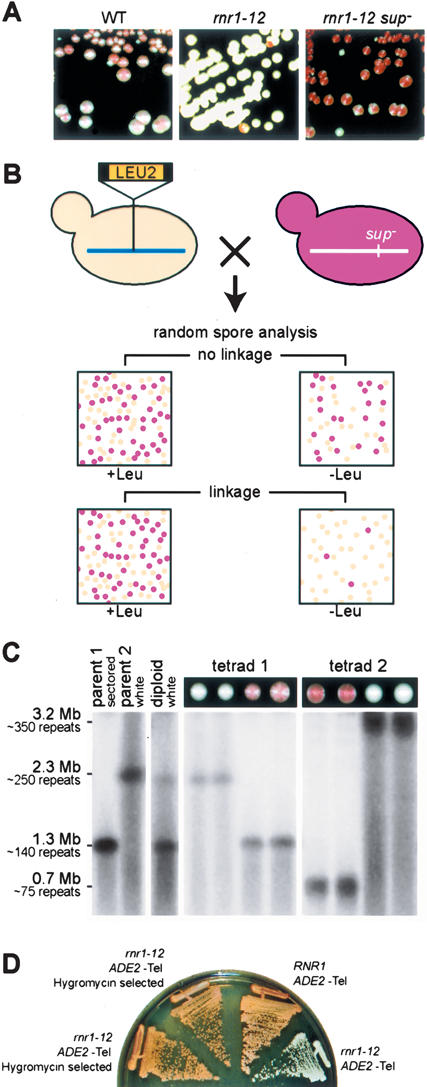
Spontaneous suppressors of telomeric position effect (TPE) loss in rnr1-12 mutants are due to large rDNA repeat deletions. (A) Wild-type (WT) cells with ADE2-Tel (left panel), rnr1-12 ADE2-Tel cells (middle panel), and TPE suppressors (rnr1-12 ADE2-Tel sup–), which are stable upon restreaking (right panel). (B) Strategy for genetic mapping of TPE suppressor mutations. The linkage between the LEU2 marker and the SUP gene is assessed by random spore analysis. Fifty percent of the Leu+ spores are sectored in the absence of linkage. This ratio decreases in the event of linkage. (C) The TPE suppressor phenotype segregates 2:2 in backcrosses to a parental strain and is associated with a short rDNA cluster. Analysis of two complete tetrads from a cross between YAM70 (parent 1, rnr1-12 sup– ADE2-Tel) and YAM97 (parent 2, rnr1-12 ADE2-Tel). DNA from the indicated strains was analyzed by PFGE of BamHI-digested plugs. The rDNA cluster was detected by Southern blotting using a radiolabeled rDNA probe. (D) rnr1-12 ADE2-Tel cells (YAM97) were subjected to Hygromycin-resistance selection for short rDNA. The TPE phenotypes of two such HygR clones (left) were then compared with otherwise isogenic RNR1 and rnr1-12 strains (right).
To identify the suppressor gene, we decided to determine its location on the yeast genetic map. For this purpose, we designed a genetic screen (Fig. 1B; Supplemental Material) that allowed us to rapidly score for linkage between the suppressor mutation and a LEU2 marker gene inserted at random genomic sites (Ross-Macdonald et al. 1999) by random spore analysis. We estimated that ∼300 genetic crosses might be required to map the suppressor locus to a resolution of ∼50 cM, and were thus surprised when we found that eight of the first 35 crosses analyzed showed an extremely tight linkage between the suppressor mutation and the LEU2 tag (>95% nonrecombinants). DNA sequencing of the region of LEU2 integration from several of these strains revealed that the tag was within the rDNA (RDN1) locus. Because the rDNA locus comprises ∼10%–13% of the yeast genome, it will be over-represented in the original insertion library and probably also amongst the homologous recombination events that generated our tester strains. The low frequency of meiotic recombination within the rDNA (Petes et al. 1978) might also explain the extremely tight linkage observed between the suppressor mutation and eight LEU2 inserts. In conclusion, the genetic analysis described above clearly indicates that the TPE suppressor phenotype is due to a genetic alteration of some sort that maps to the rDNA (RDN1) locus. Finally, although the situation described here is a special case, we note that a screen of this sort might be generally useful for identifying single-copy genes that prove difficult to clone by standard complementation procedures.
rDNA repeat deletion causes increased telomeric and mating-type gene silencing and occurs frequently in wild-type cells
We considered two possible explanations for an rDNA locus mutation that might explain restored TPE in the rnr1-12 ADE2-Tel strain. The first proposes that a sequence change has occurred in the rDNA. The recessive nature of the suppressors implies that such a change would have to have propagated to all of the repeats (Muscarella and Vogt 1993). An alternative explanation is that the rDNA cluster had either contracted or expanded, in the absence of any nucleotide sequence change, and that this copy-number change somehow influenced telomeric heterochromatin. We tested this latter hypothesis by examining rDNA repeat size in the suppressor mutants by Pulsed Field Gel Electrophoresis (PFGE). Analysis of tetrads from a backcross between a suppressor strain and a congenic (rnr1-12 ADE2-Tel) parent of the opposite mating type by PFGE clearly showed that the suppressor phenotype (red sectored colonies indicative of restored TPE) cosegregates (2:2) with reduced rDNA copy number, which is typically on the order of 30%–50% compared with “wild type” (Fig. 1C, tetrad 1; data not shown). Tetrad 1 in Figure 1C is typical of the majority of tetrads examined from this backcross (19/20), which displayed two haploid segregants with a red pigment intensity indistinguishable from the suppressor parental strain. However, one meiosis of 20 yielded two red spore colonies of increased pigment intensity (Fig. 1C, tetrad 2). PFGE analysis suggests that the diploid cell that gave rise to these four haploid spore colonies had undergone an unequal exchange event prior to meiosis, yielding one chromosome with a further shortened rDNA (0.7 Mbp) and another with a longer than normal locus (∼3.2 Mbp, though difficult to determine precisely with available markers). The fact that segregants with the shorter rDNA cluster display a stronger TPE phenotype (more intense red pigmentation) is indicative of a dose-dependent relationship between rDNA copy number and TPE (see below).
To test further the hypothesis that the shortened rDNA cluster in the suppressor mutants is the cause of increased TPE, we asked whether the forced reduction of rDNA copy number in rnr1-12 strains is sufficient to restore TPE. To do this, we made use of pRDN-Hyg1 (Chernoff et al. 1994), a high copy-number plasmid bearing a single copy of an rDNA repeat carrying a recessive mutation in the 18S subunit that confers hygromycin resistance. Selection by hygromycin in pRDN-Hyg1 transformants favors the growth of cells that have lost a significant fraction of their endogenous rDNA repeats. As shown in Figure 1D, rnr1-12 ADE2-Tel strains submitted to hygromycin selection, followed by plasmid loss (in order to avoid possible effects of plasmid-borne rDNA copies), display a TPE suppressor phenotype, showing that rDNA cluster size reduction alone is responsible for the TPE suppression phenomenon.
The results described above show that the rnr1-12 mutation causes a reduction in TPE that is frequently suppressed by large deletions within the rDNA repeat cluster. We wondered whether the rnr1-12 mutation itself was responsible for the high frequency of “suppressor” clones with rDNA deletions. Because wild-type cells already display considerable repression of the ADE2-Tel reporter, this assay is not useful for detecting increased silencing variants, and may explain why the rDNA deletion phenomenon described here has not been noted before. However, we knew from previous experiments that treatment with low doses (20 mM) of the RNR inhibitor hydroxyurea (HU) would phenocopy the rnr1-12 effect on HMR silencing (Laman et al. 1995), and we found that the same is true for TPE. This latter observation provides a convenient method to identify wild-type strains that still display strong ADE2-Tel silencing when RNR is inhibited; they should yield sectored colonies when plated on medium containing HU. To ask whether or not such variants appear spontaneously in the absence of HU, we used a replica-plating assay. Wild-type ADE2-Tel strains were first plated on YPAD medium, then, following a short period of growth, they were replica-plated on HU-containing YPAD plates. As expected, colonies on the YPAD master plate were mainly red/white sectored, whereas most of the colonies that arose on the plates containing HU were uniformly white. However, a small fraction (∼3%) of replicated colonies on the HU plates were red/white sectored (Fig. 2A). Clones that gave rise to sectored colonies on HU plates were identified on the YPAD master plate and the size of their rDNA was assessed by PFGE. As shown in Figure 2B, these clones had a shorter rDNA cluster than their nonsectoring siblings, and the intensity of the red sectoring phenotype on HU plates was again proportional to the loss of rDNA repeats. Because cells on the master plate had never been exposed to HU, this experiment shows that clones with reduced rDNA size arise frequently and spontaneously in wild-type cells and can be identified by their resistance to TPE reduction in the presence of HU.
Figure 2.
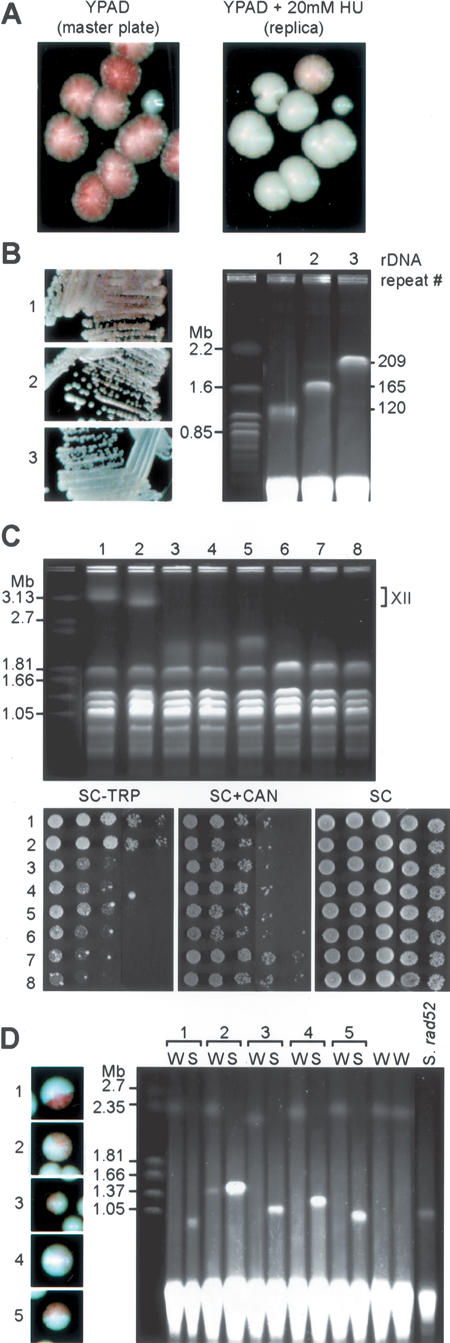
rDNA shortening occurs spontaneously in wild-type (WT) cells, leads to a dose-dependent increase in telomeric and HMR silencing, and arises through a single-step process. (A) ADE2-Tel (YAM235) cells were plated on YPAD medium and microcolonies (8 h growth at 30°C) were replica-plated onto YPAD + 20 mM hydroxyurea (HU). Most colonies growing on the HU-containing plate are white (mimicking the rnr1-12 phenotype), while a few (∼3%) remain sectored. Both the YPAD master plate (left) and the HU plate (right) were photographed after 3–4 d growth at 30°C. Colonies were replicated early to allow sufficient growth on HU plates for red sectors to appear, and as a result, a fraction of colonies failed to transfer. (B, right, lanes 1,2) Colonies showing a clear sectored phenotype on HU were identified on a YPAD master plate and the size of their rDNA locus was assessed by PFGE. (Lane 3) A colony that displayed the expected white color on HU plates was taken as a control. (Left) Cultures used for the chromosome preparation were restreaked on YPAD + 20 mM HU plates to confirm their TPE phenotype. (C) rDNA cluster shortening correlates with increased HMR, but not rDNA silencing. YAM295 (hmrΔE::TRP1 RDN1::ADE2-CAN1) was transformed with pRDN-Hyg1 and submitted to hygromycin selection as above. Individual colonies (lane 1, CCFY100 [hmrΔE::TRP1 RDN1::ADE2-CAN1 URA3-ChrVR-Tel]; lane 2, YAM295 were not selected on hygromycin; lanes 3–8, six independent hygromycin-selected clones of YAM295) were grown in liquid culture and analyzed by PFGE (top panel) to estimate the size of their rDNA locus on chromosome XII (“]”, position of “normal” chromosome XII in parental strains), and by fivefold serial dilution and spotting on medium lacking tryptophan (SC-TRP) or containing canavanine (SC + CAN) to measure HMR and rDNA silencing, respectively (bottom panel). An SC plate is shown as a growth control. (D) rDNA shortening arises as a single-step event. An individual white clone of YAM97 was spread onto YPAD plates. Half-sectored colonies were identified (labeled 1–5, left panel) and cells from the white (W) or red-sectored (S) halves of each colony were cultured independently. (Right panel) BamHI-digested plugs prepared from these cells were analyzed by PFGE. Two independent uniformly white colonies were analyzed as controls (W without numbers). DNA from a red/white sectored rad52Δ rnr1-12 ADE2-Tel isolate was analyzed in the right-most lane of this gel (S. rad52).
We next examined the effect of rDNA deletion on mating-type and rDNA silencing (again in an RNR1 wild-type background), by using the hygromycin selection procedure in a strain containing reporter genes at both HMR (hmrΔE::TRP1) and the rDNA (RDN1::ADE2-CAN1; silencing of the CAN1 gene relieves sensitivity to the toxic arginine analog canavanine). As before, we eliminated the pRDN-Hyg1 plasmid before performing silencing assays. As shown in Figure 2C, six independent hygromycin-resistant clones displayed a significant increase in HMR silencing (as indicated by a ∼50- to 200-fold decrease in colony formation in medium lacking tryptophan), whose magnitude correlated with the extent of rDNA loss. Thus, clones 6–8 have the shortest rDNA, resulting in a shortened chromosome XII that comigrates with chromosome IV (confirmed by Southern blot analysis, data not shown), and show the strongest HMR silencing. Significantly, these short rDNA strains show either no increase in rDNA silencing (clones 3–6), or for two of the largest deletions (clones 7 and 8) a modest increase (Fig. 2C, SC + CAN).
rDNA deletions arise in a single-step, RAD52-independent process
Two extreme hypotheses can be proposed to explain the generation of short rDNA strains, i.e., gradual repeat loss, where rDNA copy number eventually drops below a threshold required to detect a TPE change or a rapid, single-step deletion event. We attempted to address this issue by searching for colonies from a rnr1-12 ADE2-Tel strain, in which one half of the colony is uniformly white and the other half red/white sectored. Such colonies would be predicted to arise if a deletion event sufficient to lead to TPE restoration occurs following the first cell division after plating. Such colonies were readily identified, and appeared at frequencies of 0.065% (n = 16,700) and 0.047% (n = 23,520) in two separate experiments. We then analyzed the rDNA length in cells derived from both the white and red half-sectors of five independent colonies by PFGE (Fig. 2D). The results show very clearly that the red half-sectors, as expected, have short rDNA lengths, varying from ∼40%–60% that of white sectors. Strikingly, all of the white sectors from these colonies display rDNA lengths similar or identical to those of two independent uniformly white colonies (Fig. 2D). This suggests that in all of the cases examined, the short rDNA clones arose from a single-step deletion event in a chromosome with normal rDNA length, and argues against the idea that deletions arise through unequal sister-chromatid exchange, which had been proposed as a model to explain marker loss events in the rDNA (Petes 1980; Szostak and Wu 1980), since such an event would be expected to generate an even longer rDNA cluster in the white sector. Thus, although we have detected unequal exchange events in a diploid strain (see Fig. 1C), it seems that the rDNA deletion events that we observe in haploids arise through a different mechanism.
Restored TPE variants appear readily in rad52Δ rnr1-12 ADE2-Tel strains (data not shown), and they also have short rDNA arrays (Fig. 2D). Thus, the large deletion events that we detect in our TPE assay do not require the major RAD52-dependent recombination pathway, and are thus apparently unrelated either to a gene conversion pathway that controls rDNA repeat stability (Gangloff et al. 1996) or to a RAD52-dependent pathway that generates extra-chromosomal rDNA circles (Park et al. 1999).
Sir2 is down-regulated in short rDNA strains
We were initially surprised by the apparently normal growth of strains containing only half the normal rDNA content, and wondered whether these cells might have compensated for rDNA loss by a specific alteration of their transcriptional program. To test this idea, we generated otherwise isogenic short and normal rDNA strains by sporulation of a diploid heterozygous for rDNA size and measured total steady-state mRNA levels in four independent clones of each type using Affymetrix arrays. Notably, only two genes showed consistent changes in mRNA levels that correlated with rDNA size in all 16 pairwise comparisons, SIR2 and WSC4 (data not shown). Both of these genes are down-regulated approximately twofold in strains with an approximately twofold shorter rDNA locus. As pointed out above, Sir2 is known to regulate both recombination and gene silencing in the rDNA, silencing at telomeres and HM mating-type loci, and aging. Wsc4, one of four related proteins in yeast, is likely to be a plasma membrane-associated protein with a role in stress response(s), though its function is still poorly defined (Zu et al. 2001). The change in WSC4 message levels is unlikely to be a consequence of altered Sir2 levels, since WSC4 expression is unaffected in cells deleted for SIR2 (Wyrick et al. 1999).
We confirmed the decrease in SIR2 and WSC4 mRNA in short rDNA strains by a quantitative real-time PCR assay using actin (ACT1) mRNAs as a control (Fig. 3B). We next compared normal and short rDNA strains for total Sir2 protein level, using an antibody against native Sir2 to probe Western blots of whole-cell extracts. As shown in Figure 3C, short rDNA strains also have reduced Sir2 protein levels (approximately twofold) compared with otherwise identical strains with normal rDNA length, consistent with their reduction in steady-state SIR2 mRNA levels.
Figure 3.

rDNA shortening causes down-regulation of SIR2 transcript and protein. (A) Colonies derived from strain YAM274 (hmrΔE::TRP1 RDN1::mURA3-HIS3) were replica-plated from YPAD onto 20-mM HU plates. Two white clones (lanes 1,2) and two sectored clones (lanes 3,4) identified on the HU plate were isolated from the YPAD master plate, and their rDNA size was measured by PFGE following BamHI digestion. As the mURA3-HIS3 reporter carries a BamHI site, the rDNA appears as two bands, except in lane 4, where the clone has lost the reporter (confirmed by its Ura-, His-phenotype). The estimated rDNA repeat number is given above each lane. (B) SIR2 and WSC4 transcripts levels correlate with rDNA copy number. mRNA levels where measured by real-time PCR on cDNA prepared from the same cultures used in the PFGE analysis (shown above in A), using actin (ACT1) as an internal control and setting the SIR2:ACT1 and WSC4:ACT1 ratios to 1 in strain 1. (Error bars correspond to the standard deviation of measurements from five independent cDNA preparations made from the same RNA sample). (C, top panel) Sir2 protein level correlates with rDNA cluster size. Western blots of cultures prepared from the strains shown in A and B were probed with affinity-purified rabbit anti-Sir2 antiserum. (Bottom panel) For comparison, protein extracts from a SIR2 normal rDNA strain were mixed at different ratios, as indicated, with protein extract of a sir2Δ strain, maintaining the total extract amount constant. (Arrow) Position of Sir2 protein; (*) position of a cross-reacting protein that serves as loading control. (D) SIR2 down-regulation in short rDNA strains does not depend on the Sir2/3/4-silencing complex. Sir2 protein levels were measured by Western blot as before (top panel; except that RnaseH was used as a loading control) in a sir2Δ strain and in pairs of wild-type, sir3Δ and sir4Δ mutants, with (alternating) normal and short rDNA clusters, as shown by PFGE (bottom panel).
We then asked whether the increase in silencing itself that we observed in short rDNA strains was responsible for the concommitant Sir2 down-regulation. If so, we would expect to observe high and uniform levels of Sir2 in silencing-defective mutants regardless of their rDNA size. This is clearly not the case, since sir3Δ and sir4Δ short rDNA strains contain less Sir2 than their long rDNA siblings (Fig. 3D). From this experiment, we conclude that the increase of silencing in short rDNA strains is not responsible for Sir2 down-regulation.
Evidence for SIR2 autoregulation
The above data show that rDNA cluster deletion has at least two consequences, i.e., an increase in both telomeric and HM locus silencing and a decrease in total cellular levels of Sir2 mRNA and protein. Because previous studies have shown clearly that Sir2 is both necessary and limiting for silencing, these results pose a paradox. Since Sir2 appears to be highly concentrated within the nucleolus where it binds to multiple sites within the rDNA (Gotta et al. 1997; Cuperus et al. 2000; Buck et al. 2002; Huang and Moazed 2003), we hypothesized that rDNA shortening could be accompanied by the release of a portion of nucleolar-bound Sir2 into the nucleoplasm. This released pool of Sir2 could be responsible for the increase in telomeric and HM silencing, as well as the down-regulation of SIR2 in short rDNA strains.
To test this idea, we first examined Sir2 protein by indirect immunofluoresence on fixed whole cells, with costaining for the nucleolar marker Nop1, and compared normal and short rDNA strains (Fig. 4A). We estimated the ratio of nucleolar to nonnucleolar Sir2 by quantifying the proportion of total Sir2 colocalized with Nop1. Together with an overall decrease in Sir2 signal and Nop1 signal, we observe a diminution of the ratio of nucleolar to nonnucleolar Sir2 in short rDNA strains. This suggests that the decrease of Sir2 level in short rDNA strains is mostly due to a decrease of the nucleolar fraction, consistent with our idea that rDNA size reduction releases Sir2 into the nucleoplasm.
Figure 4.

Alteration in the nucleolar/nucleoplasmic Sir2 distribution in short rDNA strains and evidence for Sir2 transcriptional autoregulation. (A) Sir2 nucleolar staining is reduced in short rDNA strains. Nop1 (red) and Sir2 (green) were localized by indirect immunofluorescence in long (∼200 repeats, 34 cells analyzed) and short (∼135 repeats, 88 cells analyzed) rDNA strains. Merged signals are shown in the top panel. The median of the percentage of Sir2 colocalized with Nop1 is shown. (B) Sir2 protein levels are autoregulated. Normal (YAM274) and short (YAM278) rDNA strains were transformed either with a plasmid containing a copy of SIR2 driven by its own promoter, but tagged by fusion at its C terminus to GFP (Sir2-GFP), or with pRS415 (empty vector, Ø). Sir2 and Sir2-GFP proteins were detected by Western blotting with anti-Sir2 antiserum. (*) Nonspecific cross-reacting proteins that serve as loading controls. (C) Sir2 regulates its own promoter. (Top panels) A centromeric plasmid containing the GFP ORF expressed from the SIR2 promoter (pAM60) was introduced into normal (YAM274) and short (YAM279) rDNA strains, and a sir2Δ strain (YAM302). (Left) Activity of the SIR2 promoter was determined by a quantitative FACS measure of GFP fluorescence, reported here as arbitrary units (with standard deviation). (Right) The rDNA cluster size of two independent isolates of the indicated genotypes used in the GPF analysis was measured by PFGE. (Bottom panel) A SIR2 promoter-HIS3 fusion displays reduced activity in short rDNA strains, but increased expression in sir2Δ, compared with wild type. The ability of two independent isolates of the indicated genotypes to grow in the presence of 3-AT, an inhibitor of the HIS3 gene product, is shown.
Our model implies that Sir2 can negatively regulate its own expression, either directly or indirectly. To test this idea, we introduced a second, plasmid-borne copy of SIR2 into both short and normal rDNA cells. This additional copy of SIR2 is driven by its own promoter, but tagged by fusion at its C terminus to the Aequorea victoria green fluorescent protein (GFP). The exogenous Sir2-GFP protein, which is functional in all three forms of silencing (Cuperus et al. 2000) can be readily distinguished from the endogenous protein on Western blots because of its increased size. Strikingly, expression of the Sir2-GFP protein resulted in a reduction in the amount of endogenous Sir2, both in normal and short rDNA strains (Fig. 4B). Significantly, Sir2-GFP was expressed at lower levels in the short rDNA strain than in the normal rDNA strain, showing that it too was down-regulated as a consequence of rDNA deletion.
To ask whether the apparent auto-regulation of Sir2 protein levels described above operates at the level of the SIR2 promoter, and to examine this phenomenon in the context of a SIR2 loss-of-function mutation, we constructed a plasmid in which the SIR2 promoter drives the expression of GFP. This plasmid was transformed into normal and short rDNA strains, and into a sir2Δ strain. Levels of GFP in these three strains were measured by a quantitative FACS assay (Niedenthal et al. 1996), and their rDNA size by PFGE (Fig. 4C, top panels). This system clearly recapitulates the rDNA size-driven regulation of SIR2, since the short rDNA strains have reduced expression of the pSIR2-GFP construct compared with normal rDNA strains. Significantly, a sir2Δ mutant expresses higher levels of GFP than either short or normal rDNA strains, suggesting that Sir2 negatively regulates its own expression (either directly or indirectly) and supporting our model that down-regulation of total Sir2 levels in short rDNA strains is due to an increased level of “free” nuclear protein. The increased expression from the SIR2 promoter in a sir2Δ strain cannot be explained by rDNA copy number increase. In fact, rDNA copy number is reduced in sir2Δ mutants (and more heterodisperse) compared with wild-type cells (Fig. 4C, top, right; Kobayashi et al. 2004). A similar result was obtained using a chromosomally integrated HIS3 reporter gene placed under the control of the SIR2 promoter, where expression levels were quantified by the ability of cells to grow in the presence of 3-aminotriazole, a His3 inhibitor (Fig. 4C, bottom). Altogether, these data show that Sir2 can negatively regulate its own expression and we propose that this mechanism is responsible for the rDNA size-driven regulation of SIR2 promoter.
Short rDNA strains maintain a wild-type lifespan, but are hypersensitive to Sir2 overexpression
Several previous studies have demonstrated a notable effect of SIR2 gene dosage on replicative lifespan. For example, a single extra copy of SIR2 increases lifespan by ∼30% (Kaeberlein et al. 1999). Conversely, the deletion of one copy of SIR2 in diploid cells results in a significant (>20%) decrease in lifespan (Kaeberlein et al. 1999), as well as an approximately twofold decrease in total Sir2 protein levels and a roughly proportional decrease in rDNA silencing (Smith et al. 1998). Since the decrease in Sir2 levels in short rDNA strains (Fig. 3) and hemizygous diploids relative to their wild-type counterparts (Smith et al. 1998) is very similar, we decided to measure lifespan in our short rDNA strains. Somewhat unexpectedly, we find that a short rDNA strain has a lifespan indistinguishable from that of an otherwise isogenic normal rDNA strain (Fig. 5), in marked contrast to the previously reported comparison between wild-type and hemizygous SIR2+/– diploids (Kaeberlein et al. 1999). This constancy of lifespan in the short rDNA strains matches their rDNA-silencing phenotype (Fig. 2), and again distinguishes them from hemizygous SIR2+/– diploids (Smith et al. 1998). It is worth noting, however, that short rDNA strains are still able to respond to extra SIR2 gene dosage by an increase in their lifespan (Fig. 5). Taken together, these observations raise the intriguing possibility that Sir2 regulation in response to rDNA size variation has evolved to maintain a relatively constant level of rDNA silencing and fixed lifespan (see Discussion).
Figure 5.

Short rDNA maintain a fixed life span. YAM337 (∼150 rDNA repeats) was transformed with GLC343 to integrate an extra copy of the SIR2 gene at the LEU2 locus or with the corresponding empty vector (pRS305). Life spans were determined for YAM337, YAM337 + SIR2, and YAM338 (∼250 rDNA repeats; transformed with pRS305). Median life spans and the number of mothers examined was as follows: YAM338 19.5 (n = 58), YAM337 19.5 (n = 54), YAM337 + SIR2 (n = 50) 26.64. The rDNA size as well as Sir2 levels of all three strains are shown.
However, short rDNA strains do display a striking growth difference, compared with their normal counterparts, when Sir2 is overexpressed from a strong promoter. In the experiment shown in Figure 6, SIR2 transcription was driven by a galactose-inducible promoter from a high-copy plasmid (similar results were obtained using a centromeric plasmid and the constitutive ADH1 promoter; data not shown). Under these conditions, the short rDNA strain is unable to form colonies, whereas growth of the normal rDNA sibling is considerably less affected. This result suggests that short rDNA strains are less capable than normal strains of “buffering” excess Sir2, and supports the idea that the rDNA cluster is a sink for Sir2, from which the protein is released upon rDNA repeat deletion.
Figure 6.

Short rDNA strains are hypersensitive to the toxic effect of SIR2 overexpression. YAM307 (normal rDNA) and YAM306 (short rDNA) were transformed with pJG45-ySir2, which expresses SIR2 under control of the GAL10 promoter, and growth of three independent transformants of each strain was examined on noninducing (glucose) and inducing (galactose) medium, as indicated.
We note that deleting either SIR3 or SIR4 does not relieve Sir2 toxicity (data not shown), consistent with previous work (Holmes et al. 1997), and arguing against the idea that it is due to increased action of the Sir2/3/4-silencing complex either at subtelomeric regions (Ai et al. 2002) or elsewhere in the genome (Marcand et al. 1996; Kristjuhan et al. 2003). It remains possible that Sir2, in the absence of Sir3 and Sir4, can cause excessive deacetylation of histones or other substrates when present at elevated levels, thus leading to growth inhibition. We also note that Sir2 toxicity is not relieved by overexpression of TAR1 (data not shown), a protein-coding gene embedded within the rDNA repeat unit whose expression is subject to Sir2-mediated silencing (Coelho et al. 2002).
Discussion
Here, we describe several novel observations that reveal an unexpected relationship between rDNA cluster size regulation, gene silencing, and cellular levels of the Sir2 deacetylase. First, we show that large deletions of the rDNA cluster in yeast, that remove up to ∼50% of the locus, occur at a remarkably high frequency in a single-step, RAD52-independent process. Second, we show that short rDNA strains have increased telomeric and HM locus gene silencing relative to isogenic siblings. Finally, we show that rDNA repeat loss is associated with down-regulation of Sir2 and provide evidence that this occurs through an autoregulatory effect on SIR2 transcription.
Superficially, our finding that reduced rDNA cluster size leads to an increase in TPE fits well with previous studies showing that Sir2 is limiting for both rDNA and telomeric silencing (Smith et al. 1998; Cockell et al. 2000; Cuperus et al. 2000). Thus, one hypothesis to explain these observations is that rDNA deletion releases Sir2 normally sequestered in the nucleolus, and thus, increases its effective concentration in the nonnucleolar space within the nucleus, a claim supported by our immunofluorescence analysis (Fig. 4A). However, the surprising finding of reduced Sir2 expression under these conditions challenges this simple explanation. Taken together, our data point to the existence of an unanticipated rDNA “repeat number-sensing” mechanism that not only has an effect on telomeric and HM silencing, but also acts on SIR2 expression. The fact that TPE and HMR silencing increase, while total Sir2 levels decrease, suggests that the rise in nonnucleolar Sir2 following rDNA repeat loss is only partially compensated by Sir2 down-regulation and/or that other factors may be released from the nucleolus following rDNA loss to increase silencing. We thus propose a model in which Sir2 released from the nucleolus upon rDNA shortening is made available for HM locus and telomeric silencing, and for the direct or indirect repression of its own transcript (Fig. 7). This model accounts for the paradoxical observation of increased HM and telomeric silencing in conditions of overall reduction in Sir2 levels, and states that Sir2 negatively feeds back on its own promoter.
Figure 7.

Model for the regulation of Sir2 localization and SIR2 expression through changes in rDNA copy number.
We show directly that expression of an exogenous copy of SIR2, fused to GFP, results in down-regulation of the endogenous protein, in both normal and short rDNA strains. Furthermore, using both SIR2 promoter-driven GFP and HIS3 constructs, we provide evidence that the endogenous Sir2 protein negatively regulates expression of its own gene in a manner that is modulated by rDNA copy number. Our data are consistent with the idea that Sir2 represses its own transcription, directly or indirectly, but we cannot at present rule out the possibility that Sir2 mRNA might be regulated post-transcriptionally, through interactions with 5′ and/or 3′-untranslated sequences still present in the fusion constructs that we have tested. A global chromatin-binding analysis (Lieb et al. 2001) failed to identify the SIR2 promoter as an in vivo Sir2-binding site, and we also failed to detect Sir2 at its own promoter by direct ChIP analysis (A. Bhat and D. Shore, unpubl.), although we do detect strong enrichment of Sir2 at both telomeres and rDNA, arguing for an indirect role of Sir2 in SIR2 regulation. It is worth noting that Sir2 autoregulation is unable to maintain a constant level of protein in the face of (artificially) increased SIR2 gene dosage, which does, in fact, lead to increased levels of total cellular Sir2 protein (Fig. 5; Smith et al. 1998), increased rDNA and telomeric silencing (Smith et al. 1998; Cockell et al. 2000), and longer life span (Fig. 5; Kaeberlein et al. 1999).
We do not yet know whether the increase in TPE and mating-type gene silencing observed in short rDNA strains confers any selective advantage to these cells. In fact, the observed down-regulation of Sir2 in these cells may have evolved to minimize this effect. Furthermore, we are unaware of conditions in which increased TPE enhances growth. Instead, one recent study indicates that stress-induced derepression of some subtelomeric genes, through a mechanism involving Sir3 phosphorylation, confers increased resistance to stress (Ai et al. 2002). Finally, we note that our microarray analysis failed to uncover any clear evidence for increased repression of native telomere-linked genes in short rDNA strains. Although this may be due to limitations of the analysis, an alternative explanation is that our TPE reporters are extremely sensitive to small changes in telomeric heterochromatin that may not have important physiological consequences.
Why, then, are Sir2 levels regulated as a function of rDNA size? Although we do not yet have a clear answer to this question, the extreme specificity of this effect suggests to us that it might serve some physiological function. Our model, supported by previous work showing that increased SIR2 gene dosage results in increased rDNA silencing (Smith et al. 1998), proposes that the rDNA “buffers” the amount of Sir2 in the cell. Therefore, one explanation is that the rDNA size-driven Sir2 regulation is used by the cell to minimize the variations of “free” Sir2 in the nucleus, and thus, to avoid the adverse effects of having too much or too little Sir2. For example, a short rDNA strain that would fail to down-regulate Sir2 expression, and thus, would not be able to buffer appropriately the excess of Sir2, might encounter adverse effects analogous to those that impair growth in the case of Sir2 overexpression. Conversely, in a long rDNA strain that fails to up-regulate Sir2, the rDNA might titrate Sir2 away from genomic loci where Sir2 is required, for example, at the HM loci, where Sir2 is crucial for proper mating-type determination. Another possibility is that reduction of Sir2 levels favors a more rapid return to the apparently “optimal” rDNA cluster size by relieving normal constraints on rDNA recombination. Several studies have shown that short rDNA strains ultimately expand their rDNA to the “long” or “normal” size, in a process mediated by the Replication Fork Block (RFB) element within NTS1 and Fob1 protein (Kobayashi et al. 1998, 2001; Wai et al. 2001; Kobayashi 2003). Sir2 also binds to the RFB region (Huang and Moazed 2003) and has been shown recently to repress unequal sister-chromatid exchange within the rDNA repeats (Kobayashi et al. 2004). Lowering Sir2 levels might therefore favor such events, ultimately leading to rDNA repeat expansion. However, our rDNA silencing data suggest that the per-rDNA repeat amount of Sir2 remains largely unchanged with rDNA size variations (Fig. 2C).
Finally, it is possible that the Sir2 regulation we observe here has evolved to control lifespan. There is now emerging evidence for complex post-translational regulation of Sir2 activity, either through PNC1 control of the inhibitor nicotinamide (Bitterman et al. 2002; Anderson et al. 2003; Gallo et al. 2004), or by modulation of NADH levels (Lin et al. 2004). The end result is a dramatic effect on lifespan, through a still poorly understood pathway that is regulated by calorie restriction. Here we found, somewhat surprisingly, that lifespan remains constant in short rDNA strains compared with their normal rDNA siblings, despite their reduced Sir2 content. Since short rDNA strains still respond to increased SIR2 gene dosage by an increase in lifespan, one can speculate that SIR2 down-regulation in these strains is a response designed to maintain lifespan at some optimal level. A more critical examination of this issue will obviously require a better understanding of the selective force (presumably) underlying lifespan regulation in yeast.
Although our experiments have focused on the altered expression and subnuclear localization of Sir2 caused by rDNA shortening, we think that it is likely that other factors are affected in a similar manner. We note, for example, that short rDNA strains display stronger HMR silencing than strains with increased SIR2 gene dosage (Fig. 2C; data not shown), suggesting that additional silencing factors might be released from the nucleolus upon rDNA shortening. One possible candidate is the Sir2-interacting nucleolar factor Net1, since the net1-1 mutant exhibits both increased HM silencing and distribution of Sir2 throughout the nucleus, but no change in Sir2 level (Kasulke et al. 2002). In summary, our data point to a regulatory mechanism driven by rDNA copy number per se that appears to alter the balance of Sir2 (and very likely other factors) within different compartments of the nucleus. We propose that this mechanism is superimposed on an additional mechanism that controls the overall concentration of Sir2 by autoregulation.
The finding reported here that the copy number of a repeated sequence can generate a signal that regulates gene expression in trans might have important implications for other paradigms involving a DNA counting mechanism. Sex determination in worms and flies, as well as X-inactivation in mammals, are two such examples. In mammalian embryos, chromosome counting leads to the inactivation of all but one X-chromosome. In Drosophila melanogaster and Caenorhabditis elegans, the ratio of X-chromosomes over autosomes is a determinant for adopting a male or a female/hermaphrodite developmental fate. This ratio is thought to be established by the balance between the products of X-linked genes (numerator genes) and autosomal genes (denominator genes). In light of our results, one could imagine that numerators and denominators are not necessarily transcribed genes. Instead, we speculate that denominator regions (nongenic) could titrate the products of numerator genes, and/or vice-versa. For example, in C. elegans denominator autosomal region(s) could sequester the SEX-1 numerator protein away from the xol-1 promoter, where SEX-1 acts as a repressor of the male developmental fate. In support of this idea, an exogenous array of xol-1 promoters is sufficient to titrate some of the SEX-1 protein and to rescue the male-specific lethality caused by the presence of two extra copies of sex-1 (Carmi et al. 1998).
Similarly, phenotypes associated with trisomy of chromosome 21 in humans (Down syndrome) are thought to be due to pleiotropic effects of the 1.5-fold overexpression of all or a subset of chromosome 21-linked genes (Antonarakis et al. 2001). As above, one could speculate that the extra chromosome might sequester some crucial factor(s), leading to gene misregulation elsewhere in the genome, and thus, participating in Down syndrome etiology. Indeed, many conserved nongenic sequences recently identified on chromosome 21 (Dermitzakis et al. 2002) could serve as binding sites for protein complexes. Taken together, our results suggest a novel way of thinking about DNA counting mechanisms.
Materials and methods
Strains, plasmids, and general methods
Yeast strains used (all derivatives of W303) are described in Supplementary Table 1. Plasmids and several general methods used are also described in the Supplemental Material.
Analysis of ribosomal DNA cluster size
Pulsed Field Gel Electrophoresis (PFGE) on yeast chromosomes was performed on a Bio-Rad DR-II Contour-clamped Homogenous Electric Field (CHEF) apparatus. The protocol for chromosomal preparations was modified after (Schedl et al. 1993) and information at http://www.cnb.uam.es/~montoliu/prot.html. Details are given in the Supplemental Material.
Microarray
Biotinylated cRNAs were prepared and hybridized to YG-S98 arrays according to the manufacturer's standard procedures (Affymetrix). Data analysis was performed using the Microarray Analysis Suite version 5.0 (MAS, Affymetrix) and GeneSpring Software (Silicon Genetics).
FACS analysis and GFP expression
FACS analysis of strains carrying a plasmid expressing GFP from the SIR2 promoter (pAM60) was carried out according to Niedenthal et al. (1996), except that the cells were grown in SC-Leu containing at least 20 mg/L of adenine. The mean fluorescence value of a clone carrying an empty vector was subtracted from mean fluorescence values of clones carrying pAM60. Measurements were done on three independent clones. The average of the background-corrected mean values are shown together with standard deviations. Each measurement was carried out on 10,000 cells, and the figure represents data from one typical experiment.
Immunofluorescence
Indirect immunofluorescence microscopy was performed as described (Gotta et al. 1997) using an LSM510 (Zeiss) confocal microscope and affinity purified antibodies (gift of F. Cubizolles and S. Perrod, University of Geneva, Geneva, Switzerland). Data were analyzed using Imaris4.0.5 software. Further details are available in the Supplemental Material.
Life-span analysis
Micromanipulation and life-span analysis were performed as described (Kaeberlein et al. 1999; Park et al. 2002) with slight modifications as described in the Supplemental Material.
Acknowledgments
We thank O. Schaad, M. Docquier, and P. Descombes (of the NCCR “Frontiers in Genetics” genomics platform) for theircontributions to DNA array experiments and analysis; A. Taddei, T. Laroche, and S. Gasser for expert help with microscopy; Ashwin Bhat for sharing unpublished results; K. Shimada, E. Nagoshi, and L. Bjergbaek for help with FACS analysis; P. Damay for anti-Sir2 antibody; T. Kobayashi, T. Horiuchi, M. Snyder, S. Gasser, and S. Schawalder for generous gifts of plasmids, strains, or other reagents; R. Rothstein, A. Taddei, S. Gasser, U. Schibler, B. Meyer, D. Pauli, and M. Snyder for helpful discussion; S. Grossi, A. Bianchi, F. Gachon, A. Taddei, and all members of the Shore lab for comments on the manuscript; and Nicolas Roggli for expert assistance with the artwork. This work was supported by the Swiss National Science Foundation, the NCCR program “Frontiers in Genetics” (Swiss National Science Foundation), the Swiss Cancer League, and the Canton of Geneva.
Supplemental material is available at http://www.genesdev.org.
Article and publication are at http://www.genesdev.org/cgi/doi/10.1101/gad.340205.
References
- Ai W., Bertram, P.G., Tsang, C.K., Chan, T.F., and Zheng, X.F. 2002. Regulation of subtelomeric silencing during stress response. Mol. Cell 10: 1295–1305. [DOI] [PubMed] [Google Scholar]
- Anderson R.M., Bitterman, K.J., Wood, J.G., Medvedik, O., and Sinclair, D.A. 2003. Nicotinamide and PNC1 govern lifespan extension by calorie restriction in Saccharomyces cerevisiae. Nature 423: 181–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antonarakis S.E., Lyle, R., Chrast, R., and Scott, H.S. 2001. Differential gene expression studies to explore the molecular pathophysiology of Down syndrome. Brain Res. Brain Res. Rev. 36: 265–274. [DOI] [PubMed] [Google Scholar]
- Bitterman K.J., Anderson, R.M., Cohen, H.Y., Latorre-Esteves, M., and Sinclair, D.A. 2002. Inhibition of silencing and accelerated aging by nicotinamide, a putative negative regulator of yeast sir2 and human SIRT1. J. Biol. Chem. 277: 45099–45107. [DOI] [PubMed] [Google Scholar]
- Bryk M., Banerjee, M., Murphy, M., Knudsen, K.E., Garfinkel, D.J., and Curcio, M.J. 1997. Transcriptional silencing of Ty1 elements in the RDN1 locus of yeast. Genes & Dev. 11: 255–269. [DOI] [PubMed] [Google Scholar]
- Buck S.W., Sandmeier, J.J., and Smith, J.S. 2002. RNA polymerase I propagates unidirectional spreading of rDNA silent chromatin. Cell 111: 1003–1014. [DOI] [PubMed] [Google Scholar]
- Carmi I., Kopczynski, J.B., and Meyer, B.J. 1998. The nuclear hormone receptor SEX-1 is an X-chromosome signal that determines nematode sex. Nature 396: 168–173. [DOI] [PubMed] [Google Scholar]
- Chernoff Y.O., Vincent, A., and Liebman, S.W. 1994. Mutations in eukaryotic 18S ribosomal RNA affect translational fidelity and resistance to aminoglycoside antibiotics. EMBO J. 13: 906–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cockell M.M., Perrod, S., and Gasser, S.M. 2000. Analysis of Sir2p domains required for rDNA and telomeric silencing in Saccharomyces cerevisiae. Genetics 154: 1069–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coelho P.S., Bryan, A.C., Kumar, A., Shadel, G.S., and Snyder, M. 2002. A novel mitochondrial protein, Tar1p, is encoded on the antisense strand of the nuclear 25S rDNA. Genes & Dev. 16: 2755–2760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuperus G., Shafaatian, R., and Shore, D. 2000. Locus specificity determinants in the multifunctional yeast silencing protein Sir2. EMBO J. 19: 2641–2651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dammann R., Lucchini, R., Koller, T., and Sogo, J.M. 1993. Chromatin structures and transcription of rDNA in yeast Saccharomyces cerevisiae. Nucleic Acids Res. 21: 2331–2338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ____. 1995. Transcription in the yeast rRNA gene locus: Distribution of the active gene copies and chromatin structure of their flanking regulatory sequences. Mol. Cell. Biol. 15: 5294–5303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis E.S., Shafer, B.K., and Strathern, J.N. 2000. The Saccharomyces cerevisiae RDN1 locus is sequestered from interchromosomal meiotic ectopic recombination in a SIR2-dependent manner. Genetics 155: 1019–1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dermitzakis E.T., Reymond, A., Lyle, R., Scamuffa, N., Ucla, C., Deutsch, S., Stevenson, B.J., Flegel, V., Bucher, P., Jongeneel, C.V., et al. 2002. Numerous potentially functional but non-genic conserved sequences on human chromosome 21. Nature 420: 578–582. [DOI] [PubMed] [Google Scholar]
- French S.L., Osheim, Y.N., Cioci, F., Nomura, M., and Beyer, A.L. 2003. In exponentially growing Saccharomyces cerevisiae cells, rRNA aynthesis is setermined by the aummed RNA polymerase I loading rate rather than by the number of active genes. Mol. Cell. Biol. 23: 1558–1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fritze C.E., Verschueren, K., Strich, R., and Easton Esposito, R. 1997. Direct evidence for SIR2 modulation of chromatin structure in yeast rDNA. EMBO J. 16: 6495–6509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallo C.M., Smith Jr., D.L., and Smith, J.S. 2004. Nicotinamide clearance by Pnc1 directly regulates Sir2-mediated silencing and longevity. Mol. Cell. Biol. 24: 1301–1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gangloff S., Zou, H., and Rothstein, R. 1996. Gene conversion plays the major role in controlling the stability of large tandem repeats in yeast. EMBO J. 15: 1715–1725. [PMC free article] [PubMed] [Google Scholar]
- Gotta M., Strahl-Bolsinger, S., Renauld, H., Laroche, T., Kennedy, B.K., Grunstein, M., and Gasser, S.M. 1997. Localization of Sir2p: The nucleolus as a compartment for silent information regulators. EMBO J. 16: 3243–3255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gotta M., Palladino, F., and Gasser, S.M. 1998. Functional characterization of the N terminus of Sir3p. Mol. Cell. Biol. 18: 6110–6120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottlieb S. and Esposito, R.E. 1989. A new role for a yeast transcriptional silencer gene, SIR2, in regulation of recombination in ribosomal DNA. Cell 56: 771–776. [DOI] [PubMed] [Google Scholar]
- Gottschling D.E., Aparicio, O.M., Billington, B.L., and Zakian, V.A. 1990. Position effect at S. cerevisiae telomeres: Reversible repression of Pol II transcription. Cell 63: 751–762. [DOI] [PubMed] [Google Scholar]
- Holmes S.G., Rose, A.B., Steuerle, K., Saez, E., Sayegh, S., Lee, Y.M., and Broach, J.R. 1997. Hyperactivation of the silencing proteins, Sir2p and Sir3p, causes chromosome loss. Genetics 145: 605–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J. and Moazed, D. 2003. Association of the RENT complex with nontranscribed and coding regions of rDNA and a regional requirement for the replication fork block protein Fob1 in rDNA silencing. Genes & Dev. 17: 2162–2176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaeberlein M., McVey, M., and Guarente, L. 1999. The SIR2/3/4 complex and SIR2 alone promote longevity in Saccharomyces cerevisiae by two different mechanisms. Genes & Dev. 13: 2570–2580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasulke D., Seitz, S., and Ehrenhofer-Murray, A.E. 2002. A role for the Saccharomyces cerevisiae RENT complex protein Net1 in HMR silencing. Genetics 161: 1411–1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klar A., Fogel, S., and MacLeod, K. 1979. MAR1-A regulation of HMα and HMa loci in Saccharomyces cerevisiae. Genetics 93: 37–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi T. 2003. The replication fork barrier site forms a unique structure with Fob1p and inhibits the replication fork. Mol. Cell. Biol. 23: 9178–9188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi T., Heck, D.J., Nomura, M., and Horiuchi, T. 1998. Expansion and contraction of ribosomal DNA repeats in Saccharomyces cerevisiae: Requirement of replication fork blocking (Fob1) protein and the role of RNA polymerase I. Genes & Dev. 12: 3821–3830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi T., Nomura, M., and Horiuchi, T. 2001. Identification of DNA cis elements essential for expansion of ribosomal DNA repeats in Saccharomyces cerevisiae. Mol. Cell. Biol. 21: 136–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi T., Horiuchi, T., Tongaonkar, P., Vu, L., and Nomura, M. 2004. SIR2 regulates recombination between different rDNA repeats, but not recombination within individual rRNA genes in yeast. Cell 117: 441–453. [DOI] [PubMed] [Google Scholar]
- Kristjuhan A., Wittschieben, B.O., Walker, J., Roberts, D., Cairns, B.R., and Svejstrup, J.Q. 2003. Spreading of Sir3 protein in cells with severe histone H3 hypoacetylation. Proc. Natl. Acad. Sci. 100: 7551–7556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laman H., Balderes, D., and Shore, D. 1995. Disturbance of normal cell cycle progression enhances the establishment of transcriptional silencing in Saccharomyces cerevisiae. Mol. Cell. Biol. 15: 3608–3617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieb J.D., Liu, X., Botstein, D., and Brown, P.O. 2001. Promoter-specific binding of Rap1 revealed by genome-wide maps of protein–DNA association. Nat. Genet. 28: 327–334. [DOI] [PubMed] [Google Scholar]
- Lin S.J., Ford, E., Haigis, M., Liszt, G., and Guarente, L. 2004. Calorie restriction extends yeast life span by lowering the level of NADH. Genes & Dev. 18: 12–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcand S., Buck, S.W., Moretti, P., Gilson, E., and Shore, D. 1996. Silencing of genes at nontelomeric sites in yeast is controlled by sequestration of silencing factors at telomeres by Rap 1 protein. Genes & Dev. 10: 1297–1309. [DOI] [PubMed] [Google Scholar]
- Muscarella D.E. and Vogt, V.M. 1993. A mobile group I intron from Physarum polycephalum can insert itself and induce point mutations in the nuclear ribosomal DNA of Saccharomyces cerevisiae. Mol. Cell. Biol. 13: 1023–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niedenthal R.K., Riles, L., Johnston, M., and Hegemann, J.H. 1996. Green fluorescent protein as a marker for gene expression and subcellular localization in budding yeast. Yeast 12: 773–786. [DOI] [PubMed] [Google Scholar]
- Park P.U., Defossez, P.A., and Guarente, L. 1999. Effects of mutations in DNA repair genes on formation of ribosomal DNA circles and life span in Saccharomyces cerevisiae. Mol. Cell. Biol. 19: 3848–3856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park P.U., McVey, M., and Guarente, L. 2002. Separation of mother and daughter cells. Methods Enzymol. 351: 468–477. [DOI] [PubMed] [Google Scholar]
- Petes T.D. 1980. Unequal meiotic recombination within tandem arrays of yeast ribosomal DNA genes. Cell 19: 765–774. [DOI] [PubMed] [Google Scholar]
- Petes T.D., Hereford, L.M., and Botstein, D. 1978. Simple Mendelian inheritance of the repeating yeast ribosomal DNA genes. Cold Spring Harb. Symp. Quant. Biol. 42: 1201–1207. [DOI] [PubMed] [Google Scholar]
- Reeder R.H. 1999. Regulation of RNA polymerase I transcription in yeast and vertebrates. Prog. Nucleic Acid Res. Mol. Biol. 62: 293–327. [DOI] [PubMed] [Google Scholar]
- Rine J. and Herskowitz, I. 1987. Four genes responsible for a position effect on expression from HML and HMR in Saccharomyces cerevisiae. Genetics 116: 9–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross-Macdonald P., Coelho, P.S., Roemer, T., Agarwal, S., Kumar, A., Jansen, R., Cheung, K.H., Sheehan, A., Symoniatis, D., Umansky, L., et al. 1999. Large-scale analysis of the yeast genome by transposon tagging and gene disruption. Nature 402: 413–418. [DOI] [PubMed] [Google Scholar]
- Schedl A., Larin, Z., Montoliu, L., Thies, E., Kelsey, G., Lehrach, H., and Schutz, G. 1993. A method for the generation of YAC transgenic mice by pronuclear microinjection. Nucleic Acids Res. 21: 4783–4787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shore D., Squire, M., and Nasmyth, K.A. 1984. Characterization of two genes required for position effect control of mating type. EMBO J. 3: 2817–2823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shou W., Seol, J.H., Shevchenko, A., Baskerville, C., Moazed, D., Chen, Z.W., Jang, J., Charbonneau, H., and Deshaies, R.J. 1999. Exit from mitosis is triggered by Tem1-dependent release of the protein phosphatase Cdc14 from nucleolar RENT complex. Cell 97: 233–244. [DOI] [PubMed] [Google Scholar]
- Shou W., Sakamoto, K.M., Keener, J., Morimoto, K.W., Traverso, E.E., Azzam, R., Hoppe, G.J., Feldman, R.M., De-Modena, J., Moazed, D., et al. 2001. Net1 stimulates RNA polymerase I transcription and regulates nucleolar structure independently of controlling mitotic exit. Mol. Cell 8: 45–55. [DOI] [PubMed] [Google Scholar]
- Sinclair D.A. and Guarente, L. 1997. Extrachromosomal rDNA circles—a cause of aging in yeast. Cell 91: 1033–1042. [DOI] [PubMed] [Google Scholar]
- Smith J.S. and Boeke, J.D. 1997. An unusual form of transcriptional silencing in yeast ribosomal DNA. Genes & Dev. 11: 241–254. [DOI] [PubMed] [Google Scholar]
- Smith J.S., Brachmann, C.B., Pillus, L., and Boeke, J.D. 1998. Distribution of a limited Sir2 protein pool regulates the strength of yeast rDNA silencing and is modulated by Sir4p. Genetics 149: 1205–1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straight A.F., Shou, W., Dowd, G.J., Turck, C.W., Deshaies, R.J., Johnson, A.D., and Moazed, D. 1999. Net1, a Sir2-associated nucleolar protein required for rDNA silencing and nucleolar integrity. Cell 97: 245–256. [DOI] [PubMed] [Google Scholar]
- Sussel L., Vannier, D., and Shore, D. 1995. Suppressors of defective silencing in yeast: Effects on transcriptional repression at the HMR locus, cell growth and telomere structure. Genetics 141: 873–888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szostak J.W. and Wu, R. 1980. Unequal crossing over in the ribosomal DNA of Saccharomyces cerevisiae. Nature 284: 426–430. [DOI] [PubMed] [Google Scholar]
- Wai H., Johzuka, K., Vu, L., Eliason, K., Kobayashi, T., Horiuchi, T., and Nomura, M. 2001. Yeast RNA polymerase I enhancer is dispensable for transcription of the chromosomal rRNA gene and cell growth, and its apparent transcription enhancement from ectopic promoters requires Fob1 protein. Mol. Cell. Biol. 21: 5541–5553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warner J.R. 1999. The economics of ribosome biosynthesis in yeast. Trends Biochem. Sci. 24: 437–440. [DOI] [PubMed] [Google Scholar]
- Wyrick J.J., Holstege, F.C., Jennings, E.G., Causton, H.C., Shore, D., Grunstein, M., Lander, E.S., and Young, R.A. 1999. Chromosomal landscape of nucleosome-dependent gene expression and silencing in yeast. Nature 402: 418–421. [DOI] [PubMed] [Google Scholar]
- Zu T., Verna, J., and Ballester, R. 2001. Mutations in WSC genes for putative stress receptors result in sensitivity to multiple stress conditions and impairment of Rlm1-dependent gene expression in Saccharomyces cerevisiae. Mol. Genet. Genomics 266: 142–155. [DOI] [PubMed] [Google Scholar]