


Abstract
Mutational rates are known to be variable along the mammalian genome but the extent of this non-random fluctuation and their causes are less well understood. Using 5509 human and mouse orthologous genes with known chromosome positions, it is shown here that there are extreme differences in synonymous evolutionary rates between different human chromosomes when distances are measured using maximum-likelihood techniques. In particular, the average synonymous rate of genes located in human chromosome 19 is extremely high (Ks = 1.243 substitutions/site) compared with the average of all genes (Ks = 0.729), and significantly different from all other human chromosomes. When genes are sorted according to mouse chromosomes no such large differences are found. Strikingly, almost all genes of human chromosome 19 have very high GC content in humans but not in the mouse orthologs. More generally, correlation analysis shows that genes with very high GC content in humans have experienced the highest synonymous divergencies from the mouse. It is likely that, in such genes, the known relaxation of the isochore structure in rodents has caused an increased accumulation of synonymous substitutions in the mouse lineage, whereas the regions with the highest GC content in the human genome are accordingly maintained by a strong selective pressure.
INTRODUCTION
It has long been known that the mammalian genome is not homogeneous in its GC composition, and that this base heterogeneity or isochore structure is correlated with other important genomic features such as the insertion of repetitive elements and gene density (1–3). More recently, it has been shown that mutational rates are also variable along the genome (4–7), pointing to a mosaic model of genomic evolution (8). The measurement of sequence divergence between different species makes the estimation of mutational rates possible. In this context, large-scale comparison of mouse and human genes can be very useful for learning about the genomic evolution of mammalian genes (9,10) while knowledge of their chromosome positions (11,12) will allow visualization of the extent of the mutational variation in the genome.
In order to compare orthologous genes of such divergent species, two issues must be addressed. First, around 200 rearrangements exist between human and mouse chromosomes (13,14), and thus whole chromosomes are not directly comparable; however, there are still large tracts of orthology with tens to hundreds of genes that have been linked since the rodent–primate split, which can be very useful to understand the variation of evolutionary rates along large segments of the genome. Secondly, the large sequence divergence between these two species makes the estimation of genetic distances by maximum-likelihood techniques necessary, which can take care of multiple substitutions and codon usage bias (15–17). Application of these techniques allows the estimation of accurate evolutionary rates of the fastest genes, which might be very important in understanding the mutational dynamics. It is shown here, using a large collection of human and mouse orthologous genes with known chromosome positions that, despite the shuffling which occurred between these two genomes, some human chromosomes are not a random collection of genes with respect to the divergence from the mouse orthologs and that, in particular, genes in chromosome 19 are extremely divergent from the mouse. This effect is not observed when genes are sorted by mouse chromosomes. The possibility is speculated that the different isochore structure that exists in rodents and primates might explain the particularly high evolutionary rates in some tracts of the genome.
MATERIALS AND METHODS
Mouse and human orthologous genes and their respective chromosome locations were taken from the Human-Mouse Homology Map database, Build 25 (http://www.ncbi.nlm.nih.gov/Homology/), which contained 7195 homology pairs. Gapped alignments of the genes were made by BLASTN 2.1.2 (18,19). In order to obtain reliably aligned positions, only the best-hit segment of the BLAST alignment (that generally contained most of the alignment), was extracted, and it was only considered if it was larger than 150 positions. In total, 5509 alignments in 23 human chromosomes (and 20 mouse chromosomes) were used. Only a few genes were available in the Y chromosome and therefore not analyzed. The total number of positions in all genes was 7 614 750 (1382 positions per gene).
The number of substitutions per synonymous (Ks) and non-synonymous (Ka) site (easily convertible to evolutionary rates dividing by the human/mouse divergence time) from the alignments were estimated by maximum-likelihood using a codon-based model of evolution (15) with the Codeml program of the PAML 3.0 package [Z. Yang (2000) Phylogenetic Analysis by Maximum Likelihood (PAML), Version 3.0. University College London, London, UK]. Equilibrium codon frequencies of the model were used as free parameters (CodonFreq = 3). To further avoid false orthologous gene pairs, only alignments where Ks < 5 and Ka < 0.5 were used. In addition, Ka and Ks were also estimated by the approximate Nei–Gojobori method (20) implemented in the same PAML package. The GC level was estimated as the G+C proportion at 4-fold degenerate sites (GC4, mostly refer to as GC throughout the paper) of each human or mouse gene. ANOVA, Tukey–Kramer tests and correlation coefficients were calculated using the JMP package (SAS Institute, Cary, NC). The spatial autocorrelation of Ks, Ka and GC4 was calculated with the Moran’s I statistic (21) as in Matassi et al. (5). To calculate the significance of I, the values of Ks, Ka and GC4 were randomly permuted 10 000 times (or 1000 times in the pool of all chromosomes) and I recalculated. The significance level is the proportion of times that the value of I after permutation was larger than I for the original data set. Programs for parsing the BLASTN alignments and for calculating GC4, window averages and the Moran’s I statistics and its significance were written in Perl.
RESULTS AND DISCUSSION
Visualization of the fluctuation of base composition and evolutionary rates in human chromosomes
Maximum-likelihood estimates of synonymous (Ks) and non-synonymous (Ka) rates, and the GC content of the genes in each species (measured at 4-fold degenerate sites), were calculated for 5509 human and mouse orthologous genes. In order to visualize and to be able to easily compare the extent of the evolutionary rate variation along the genome and in different chromosomes, sliding and overlapping window averages of these measurements were used. Figure 1 shows the corresponding graphs for five representative chromosomes. Due to random fluctuations, the plot of the rates for single genes is very noisy (data not shown) and thus only windows of 5 Mb that contained three or more genes were plotted. The overlap of 1 Mb of such windows allows visualization of the average rates in all areas of the genome, but it should be taken into account that, in this type of plot, only variations that extend over >5 Mb should be considered. With this in mind, it becomes apparent that both Ks and Ka as well as the GC content in mouse and human genes are very variable along the genome. For example, it is possible to detect some genomic areas with very well conserved genes at the protein level in chromosome 4 (between 33 and 48 Mb) or very large areas with extremely high silent rates (chromosome 19 and particularly its short arm; see below). Thus, the amount of data already available allows visualization of the extent of the variation along the chromosomes and in the whole genome, and confirms that evolutionary rate bands (as well as isochores) constitute an important component of the genome.
Figure 1.

Sliding and overlapping window averages of synonymous rates, Ks, non-synonymous rates, Ka (in substitutions/site), and GC4 (in %) for human (purple squares) and mouse (green circles) genes in five representative human chromosomes. Windows were positioned every 1 Mb and span 5 Mb. Only windows that contained three or more genes were plotted.
Analysis of the variation of base composition and evolutionary rates in human chromosomes
An objective quantification of the mutational variability within each chromosome (intra-chromosomal variability) has been deduced from pools of chromosomes (7), but it is important to analyze individual chromosomes to better understand the differences among them. The extent to which genes with similar rates are clustered within each human chromosome can be efficiently analyzed by means of the Moran spatial autocorrelation statistics (I), which measures if values of a variable are randomly distributed in space or whether similar values tend to occur in proximity (5). For the present analysis, two genes were defined to be adjacent when they were located within 1 Mb. The I statistics shows that Ks and Ka are non-randomly distributed in most human chromosomes, but in several chromosomes there is no spatial autocorrelation for one or another rate (Table 1). In addition, the GC content of the human genes shows spatial autocorrelation in almost all chromosomes, even after Bonferroni correction, as expected for the well known heterogeneity of GC content in the genome. Clearly, different chromosomes show very different properties with respect to the spatial autocorrelation of these variables.
Table 1. Spatial autocorrelation (I) for Ks, Ka and GC4 in every individual chromosome and in the genome, and the probability value (P) for every estimate.

When all human chromosomes are pooled for the calculation, the spatial autocorrelation is particularly very high for Ks and GC and much smaller, but still highly significant, for Ka (Table 1), in basic agreement with earlier works that studied pools of human and mouse chromosomes (5,7). To show that the Moran’s I statistic can be a valid and very helpful measurement to analyze the aggregation of genes in the genome with respect to certain variables, the autocorrelation of Ks, Ka and GC was calculated with different definitions of linkage limits in the pool of all chromosomes (Fig. 2). When only genes within 250 kb are defined to be adjacent, the I statistics becomes much higher in all three variables, indicating a proper behavior of this statistics. From this point, the spatial autocorrelation decreases greatly for Ka, whereas Ks and GC retain some spatial structure even with genes separated >20 Mb.
Figure 2.

Spatial autocorrelation (Moran’s I) of synonymous rates, Ks, non-synonymous rates, Ka, and GC4 for the pool of all human chromosomes as a function of the linkage limit or maximum value to define two genes as being adjacent.
Extreme differences in mutational rates between human chromosomes: synonymous rates of chromosome 19 are significantly different from all other chromosomes
To analyze the inter-chromosomal variability, the means of the maximum-likelihood estimates of Ks, Ka, as well as the GC content for every human chromosome were calculated (Fig. 3). While average non-synonymous substitution values are only moderately different among chromosomes (ANOVA: F = 2.73, P < 0.0001), synonymous substitutions show large inter-chromosomal variation (ANOVA: F = 18.91, P < 0.0001). A similarly large inter-chromosomal variation occurs in the GC content of human chromosomes (ANOVA: F = 21.17, P < 0.0001). The plot of the means of these parameters for each chromosome makes more clear the large inter-chromosomal variations in Ks and GC, and the lack of it in Ka (Fig. 3). Most strikingly, the average of the synonymous rates for 344 genes in chromosome 19 is extremely high (1.243 substitutions/site) compared with the average of all genes (Ks = 0.729), and statistically different (P < 0.05) from all other individual chromosomes. This dramatic difference can be visualized in the complete separation of chromosome 19 from all others in the circles representation of the Tukey–Kramer test (Fig. 3E), and in the constantly high window values of Ks for this chromosome (Fig. 1). Human chromosome 19 is unusual in many other respects such as the high gene density (22), high expression levels (23), the large amount of duplicate regions in other chromosomes (11), and the high density of minisatellites (24). However, non-synonymous mutational rates are completely normal in this chromosome (Fig. 3D), indicating that false orthologs or erroneous gene identification are not the cause of the high synonymous rates. Moreover, when genes are sorted according to mouse chromosomes, no such big differences in Ks are found (Fig. 4). Additionally, two other short chromosomes, 16 and 22, also have high synonymous rates (Ks = 0.873 and 0.930, respectively), but not significantly different from several other chromosomes (seven and three chromosomes, respectively). Therefore, the extreme and significant difference from all other chromosomes is unique for silent rates of genes situated in human chromosome 19.
Figure 3.

GC4 for human genes (A), GC4 for mouse genes (B), the difference between GC4 in both species (C), non-synonymous rates, Ka (D), and synonymous rates, Ks (E), in every individual human chromosome. Average ± SE and the SD (when not negative) are shown. The purple line connects the mean values of each chromosome. The horizontal green line represents the mean of all chromosomes. The Tukey–Kramer test is a conservative test to evaluate if two means are significantly different. The multiple comparison circles allow visualization of the results of this test. Every circle is centered at the mean value of every chromosome and when two means are statistically different (P < 0.05) the circles do not intersect or intersect by an angle <90°.
Figure 4.

Synonymous rates, Ks, for the same genes as in Figure 3, ordered according to the mouse chromosomes. See legend to Figure 3 for the explanation of the symbols and tests used.
This analysis shows that inter-chromosomal differences of synonymous rates are much higher than previously thought. In a recent work (7), significant differences between chromosomes were found, but the large divergences for chromosome 19, statistically different from all other human chromosomes, were not detected. The large variations of Ks are detected here only with maximum-likelihood but not when distances are measured with approximate methods like the Nei–Gojobori method (data not shown). In the work by Lercher et al. (7), a similar approximate method was used to analyze inter-chromosomal differences in rates, thus explaining that the largest divergences were not detected. Since approximate methods cannot take multiple substitutions and biased codon usage into account, the measurements in the most divergent genes become saturated and the largest distances remain underestimated (15–17).
High differences in GC content between human chromosomes and relationship between GC content and synonymous rates
The data in Figure 3A, largely coincident with previous in situ hybridization analysis (2,25), show that chromosomes 16, 19 and 22 maintain the highest density of GC-rich genes in humans, whereas the GC level is much lower in the corresponding mouse orthologous segments (Fig. 3B). Thus, these human chromosomes contain genes with the highest difference in GC content with respect to rodents, as it can be appreciated in the plot of the difference between them (Fig. 3C) and in the consistently smaller GC window values for mouse compared with those for human in these chromosomes (see chromosomes 19 and 22 in Fig. 1; chromosome 16, not shown, has a similar trend). Interestingly, these are also the human chromosomes with the highest synonymous mutations (Fig. 3E), as explained above. It should be noted that the difference in GC content does not account for the high Ks in these chromosomes; for example, in chromosome 19, the difference in GC content between mouse and human is ∼9.5%, corresponding to a difference of 0.095 substitutions/site (Fig. 3C), whereas the distance between orthologous genes in this chromosome is 1.243 substitutions/site (Fig. 3E) and the average of the genome is 0.729 substitutions/site. Thus, the increase in synonymous rates in chromosome 19 is five times bigger than the GC difference at 4-fold degenerate sites.
The tendencies in the GC content and synonymous rates of these three human chromosomes (16, 19 and 22) indicate that there is some kind of correlation between these two variables. Apart from these human chromosomes, there are no other large regions in the present data set with constantly high CG content in the sliding windows to test if this tendency is more general, because many of the other GC-rich isochores are present at telomeric locations (2), for which not many mouse and human orthologs are currently available. However, it seems that this correlation is not a unique feature of these chromosome tracts but it extends to the whole genome. When synonymous rates measured by maximum likelihood from all 5509 genes are compared with the GC content in the human genes (Fig. 5), it becomes apparent that synonymous rates have a much higher tendency to increase in genes with the highest GC4 content (>70%) than in genes with more moderate GC4 content (<70%). Again, this relationship is not detected when approximate methods are used to calculate evolutionary rates (data not shown). The large amount of data used here clearly indicates that the relationship between GC content and Ks is not lineal, which probably explains that correlation analyses carried out over all genes have been contradictory (4,5,26–29). Thus, the correlation coefficient for the set of genes with >70% GC4 is 0.37 (P < 0.0001) whereas the strength of this correlation is much weaker for the set of genes with <70% GC4, where the correlation coefficient is 0.14 (P < 0.0001).
Figure 5.
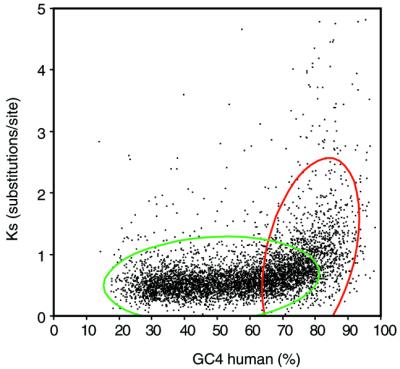
Correlation of GC4 in human genes and synonymous substitutions, Ks. Since the relationship of these variables is clearly non-lineal, the 95% bivariate normal ellipse is shown for genes above and below 70% GC content. For genes with <70% GC, the correlation coefficient is 0.14 and for genes >70% this coefficient is 0.37. The correlation is highly significant (P < 0.0001) in both groups of genes.
What are the causes of the high evolutionary rates in human chromosome 19? Possible implication of differences in mammalian isochores
The high evolutionary rates detected in the genes of human chromosome 19 may be an extreme manifestation of the variation of mutational rates (or repairing mechanisms) along both the mouse and human genomes (4–7). We still do not know whether mutational rates have been equal in rodents and primates and whether mutations are equally distributed along the human and mouse lineages (30). But if mutations occurred homogeneously along both lineages, a large number of mutations in certain chromosomal regions would correspond to the recent human evolutionary history, with all the important implications that this might have for the evolutionary analysis and population history of these genomic regions. This simple explanation, which involves homogeneous mutations along both lineages, cannot be discarded, although the best way to discover whether such extreme differences in mutations have occurred recently in the evolutionary history of these human chromosomal regions will be the comparative analysis of genes with more closely related species, like other primates.
In addition, there is a second possibility to explain the results found for human chromosome 19 and similar regions. It is related to the differences in isochore structures between mouse and human (31), and involves that mutations have not been equal in the mouse and human lineages. This possibility would also help to better understand the evolution of isochores. The existence of isochores or large fluctuations of base composition in the mammalian genome has long been known, and it has tried to be explained by either neutral mutations (there are regional mutational biases and isochores do not respond to any selective advantage) or natural selection (the isochore structure is beneficial, for example to increase thermal stability of DNA and RNA in GC-rich isochores of warm-blooded vertebrates) (2,32). Previous analysis of isochores led to some contradictory results and interpretations (32). However, the different isochore structure in mouse and human (31) allows an important prediction to be made with respect to both evolutionary rates and isochores. The isochore structure in rodents is much less heterogeneous than in all other mammals, i.e. high GC regions and low GC regions have less extreme values in rodents. In addition, maximum-likelihood estimation of ancestral GC content showed that the ancestral mammalian state was human-like, i.e. with large fluctuations in the isochore structure, and therefore that the rodent lineage experienced a homogenization of its isochores (33). If the isochore structure is maintained in mammals by negative selection, relaxation of this force in rodents would cause the largest homogenization in the ancestral regions with highest GC content. And, more importantly, the extremely biased base composition, high frequency of the hypermutable CpG dinucleotides and, in summary, a changed mode of evolution in these regions, would cause an accumulation of mutations in the mouse lineage that are no longer eliminated by the selective force which preserved the compositional bias. Other shifts in evolutionary modes have also been shown to lead to an increase in the mutational rates (34).
In complete agreement with the predictions that can be made by the selection hypothesis of isochore structure, the chromosomes with highest GC content in humans, and therefore those that experienced the highest homogenization in rodents (chromosomes 16, 19 and 22), are also the chromosomes with the highest accumulation of synonymous rates (Fig. 3). Considering the tentative estimation made about the nature of the ancestral mammalian genome (33), it seems that the main genomic changes happened in the mouse lineage, and therefore the increase of mutations in these divergent genes would have occurred mainly in this lineage (where probably some other mechanism accounts for the lack of isochores). On the contrary, the same orthologous genes in humans have very high GC content and are not homogenized by mutations: therefore, the large regions containing these genes must be preserved for some important selective advantage. In other words, a selective pressure would explain the high GC content in some large tracts of the human genome like chromosome 19, whereas the loss of such pressure in rodents, together with the change in mode of evolution, would explain a high number of substitutions in the same orthologous genes (located in diverse chromosomes and regions) of the rodent lineage. It has previously been argued, also under the selection hypothesis of isochore structure, that overall high mutation rates in rodents provide the explanation for the isochore homogenization that occurred in them (3,33). However, the fact that high rates are not homogeneously distributed but spatially aggregated in the genome (and correlated with high GC content in humans) indicates that these high rates might rather be the consequence of the isochore homogenization in rodents.
Without the action of selection, it would be difficult to explain the extremely high distance to mouse that occurs particularly in the regions of the human genome that have the strongest compositional bias. This selective pressure most surely affects at least the isochores with the highest GC content, like those in chromosome 19 or those containing genes with >70% GC, but it cannot be excluded that fluctuations of GC content at smaller scales may have other neutralist explanations. In addition, the selective advantage or function of the isochores is still unknown. Large-scale comparisons of more closely related species like human with chimpanzee and mouse with rat will tell us if the mutational rate in particular chromosome regions has been unequal or not in the rodent and primate lineages, and will give us further clues about the function of isochores. Thus, further work in other mammals should be carried out in order to better understand these important aspects of mammalian genomic evolution.
REFERENCES
- 1.Filipski J., Thiery,J.P. and Bernardi,G. (1973) An analysis of the bovine genome by Cs2SO4-Ag+ density gradient centrifugation. J. Mol. Biol., 80, 177–197. [DOI] [PubMed] [Google Scholar]
- 2.Bernardi G. (2000) Isochores and the evolutionary genomics of vertebrates. Gene, 241, 3–17. [DOI] [PubMed] [Google Scholar]
- 3.Bernardi G. (2000) The compositional evolution of vertebrate genomes. Gene, 259, 31–43. [DOI] [PubMed] [Google Scholar]
- 4.Wolfe K.H., Sharp,P.M. and Li,W.H. (1989) Mutation rates differ among regions of the mammalian genome. Nature, 337, 283–285. [DOI] [PubMed] [Google Scholar]
- 5.Matassi G., Sharp,P.M. and Gautier,C. (1999) Chromosomal location effects on gene sequence evolution in mammals. Curr. Biol., 9, 786–791. [DOI] [PubMed] [Google Scholar]
- 6.Williams E.J. and Hurst,L.D. (2000) The proteins of linked genes evolve at similar rates. Nature, 407, 900–903. [DOI] [PubMed] [Google Scholar]
- 7.Lercher M.J., Williams,E.J. and Hurst,L.D. (2001) Local similarity in evolutionary rates extends over whole chromosomes in human–rodent and mouse–rat comparisons: implications for understanding the mechanistic basis of the male mutation bias. Mol. Biol. Evol., 18, 2032–2039. [DOI] [PubMed] [Google Scholar]
- 8.Koop B.F. (1995) Human and rodent DNA sequence comparisons: a mosaic model of genomic evolution. Trends Genet., 11, 367–371. [DOI] [PubMed] [Google Scholar]
- 9.Makalowski W., Zhang,J. and Boguski,M.S. (1996) Comparative analysis of 1196 orthologous mouse and human full-length mRNA and protein sequences. Genome Res., 6, 846–857. [DOI] [PubMed] [Google Scholar]
- 10.Makalowski W., Boguski,M.S., Baldwin,B.G., Sanderson,M.J., Jacobs,D.K. and Lindberg,D.R. (1998) Evolutionary parameters of the transcribed mammalian genome: an analysis of 2820 orthologous rodent and human sequences. Proc. Natl Acad. Sci. USA, 95, 9407–9412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. IHGSC (2001) Initial sequencing and analysis of the human genome. Nature, 409, 860–921. [DOI] [PubMed] [Google Scholar]
- 12.Venter J.C., Adams,M.D., Myers,E.W., Li,P.W., Mural,R.J., Sutton,G.G., Smith,H.O., Yandell,M., Evans,C.A., Holt,R.A. et al. (2001) The sequence of the human genome. Science, 291, 1304–1351. [DOI] [PubMed] [Google Scholar]
- 13.Burt D.W., Bruley,C., Dunn,I.C., Jones,C.T., Ramage,A., Law,A.S., Morrice,D.R., Paton,I.R., Smith,J., Windsor,D., Sazanov,A., Fries,R. and Waddington,D. (1999) The dynamics of chromosome evolution in birds and mammals. Nature, 402, 411–413. [DOI] [PubMed] [Google Scholar]
- 14.O’Brien S.J., Menotti-Raymond,M., Murphy,W.J., Nash,W.G., Wienberg,J., Stanyon,R., Copeland,N.G., Jenkins,N.A., Womack,J.E. and Marshall Graves,J.A. (1999) The promise of comparative genomics in mammals. Science, 286, 458–481. [DOI] [PubMed] [Google Scholar]
- 15.Goldman N. and Yang,Z. (1994) A codon-based model of nucleotide substitution for protein-coding DNA sequences. Mol. Biol. Evol., 11, 725–736. [DOI] [PubMed] [Google Scholar]
- 16.Dunn K.A., Bielawski,J.P. and Yang,Z. (2001) Substitution rates in Drosophila nuclear genes: implications for translational selection. Genetics, 157, 295–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Smith N.G. and Eyre-Walker,A. (2001) Nucleotide substitution rate estimation in enterobacteria: approximate and maximum-likelihood methods lead to similar conclusions. Mol. Biol. Evol., 18, 2124–2126. [DOI] [PubMed] [Google Scholar]
- 18.Altschul S.F., Gish,W., Miller,W., Myers,E.W. and Lipman,D.J. (1990) Basic local alignment search tool. J. Mol. Biol., 215, 403–410. [DOI] [PubMed] [Google Scholar]
- 19.Tatusova T.A. and Madden,T.L. (1999) BLAST 2 sequences, a new tool for comparing protein and nucleotide sequences. FEMS Microbiol. Lett., 174, 247–250. [DOI] [PubMed] [Google Scholar]
- 20.Nei M. and Gojobori,T. (1986) Simple methods for estimating the numbers of synonymous and nonsynonymous nucleotide substitutions. Mol. Biol. Evol., 3, 418–426. [DOI] [PubMed] [Google Scholar]
- 21.Moran P. (1950) Notes on continuous stochastic phenomena. Biometrika, 37, 17–23. [PubMed] [Google Scholar]
- 22.Dehal P., Predki,P., Olsen,A.S., Kobayashi,A., Folta,P., Lucas,S., Land,M., Terry,A., Ecale Zhou,C.L., Rash,S. et al. (2001) Human chromosome 19 and related regions in mouse: conservative and lineage-specific evolution. Science, 293, 104–111. [DOI] [PubMed] [Google Scholar]
- 23.Caron H., van Schaik,B., van der Mee,M., Baas,F., Riggins,G., van Sluis,P., Hermus,M.C., van Asperen,R., Boon,K., Voute,P.A., Heisterkamp,S., van Kampen,A. and Versteeg,R. (2001) The human transcriptome map: clustering of highly expressed genes in chromosomal domains. Science, 291, 1289–1292. [DOI] [PubMed] [Google Scholar]
- 24.Wright F.A., Lemon,W.J., Zhao,W.D., Sears,R., Zhuo,D., Wang,J.P., Yang,H.Y., Baer,T., Stredney,D., Spitzner,J., Stutz,A., Kruhe,R. and Yuan,B. (2001) A draft annotation and overview of the human genome. Genome Biol., 2, RESEARCH0025.1–RESEARCH0025.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Saccone S., Federico,C., Solovei,I., Croquette,M.F., Della Valle,G. and Bernardi,G. (1999) Identification of the gene-richest bands in human prometaphase chromosomes. Chromosome Res., 7, 379–386. [DOI] [PubMed] [Google Scholar]
- 26.Bernardi G., Mouchiroud,D. and Gautier,C. (1993) Silent substitutions in mammalian genomes and their evolutionary implications. J. Mol. Evol., 37, 583–589. [DOI] [PubMed] [Google Scholar]
- 27.Wolfe K.H. and Sharp,P.M. (1993) Mammalian gene evolution: nucleotide sequence divergence between mouse and rat. J. Mol. Evol., 37, 441–456. [DOI] [PubMed] [Google Scholar]
- 28.Smith N.G. and Hurst,L.D. (1999) The effect of tandem substitutions on the correlation between synonymous and nonsynonymous rates in rodents. Genetics, 153, 1395–1402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bielawski J.P., Dunn,K.A. and Yang,Z. (2000) Rates of nucleotide substitution and mammalian nuclear gene evolution: approximate and maximum-likelihood methods lead to different conclusions. Genetics, 156, 1299–1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li W.H. (1997) Molecular Evolution. Sinauer Associates, Sunderland, MA, pp. 220–224.
- 31.Mouchiroud D., Gautier,C. and Bernardi,G. (1988) The compositional distribution of coding sequences and DNA molecules in humans and murids. J. Mol. Evol., 27, 311–320. [DOI] [PubMed] [Google Scholar]
- 32.Eyre-Walker A. and Hurst,L.D. (2001) The evolution of isochores. Nature Rev. Genet., 2, 549–555. [DOI] [PubMed] [Google Scholar]
- 33.Galtier N. and Mouchiroud,D. (1998) Isochore evolution in mammals: a human-like ancestral structure. Genetics, 150, 1577–1584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zischler H., Geisert,H. and Castresana,J. (1998) A hominoid-specific nuclear insertion of the mitochondrial D-loop: implications for reconstructing ancestral mitochondrial sequences. Mol. Biol. Evol., 15, 463–469. [DOI] [PubMed] [Google Scholar]